

Abstract
A novel recombinase polymerase amplification (RPA)-based method for detection of canine parvovirus type 2 (CPV-2) was developed. Sensitivity analysis showed that the detection limit of RPA was 10 copies of CPV-2 genomic DNA. RPA amplified both CPV-2a and -2b DNA but did not amplify the template of other important dog viruses (CCoV, PRV or CDV), demonstrating high specificity. The method was further validated with 57 canine fecal samples. An outstanding advantage of RPA is that it is an isothermal reaction and can be performed in a water bath, making RPA a potential alternative method for CPV-2 detection in resource-limited settings.
Keywords: Canine parvovirus type 2, Molecular diagnosis, Recombinase polymerase amplification, Polymerase chain reaction
Canine parvovirus type 2 (CPV-2), a linear singe-stranded DNA virus, is a member of the genus Parvovirus within the family Parvoviridae. It was first recognized as a canine pathogen in the late 1970s [1]. So far, three genetic variants, i.e., CPV-2a, -2b and -2c, have been identified [2]. CPV-2 is a highly contagious pathogen of domestic dogs and other members of the family Canidae [3]. Animals infected with CPV-2 develop acute gastroenteritis characterized by loss of appetite, vomiting, fever, diarrhea (from mucoid to haemorrhagic) and leucopenia with high morbidity and mortality [2, 4]. CPV-2 is shed in the feces of infected animals and spreads through the fecal-oral route [5].
Early and rapid diagnosis is crucial for the prevention of CPV-2 spread and disease management. A number of gene-amplification-based assays have been developed for CPV-2 diagnosis, such as polymerase chain reaction (PCR), nested PCR, real-time PCR, loop-mediated isothermal amplification, and insulated isothermal PCR (iiPCR) [6–10]. Recombinase polymerase amplification (RPA), a novel isothermal gene amplification technique, employs three core enzymes: a recombinase, a single-stranded DNA-binding protein (SSB), and a strand-displacing polymerase [11–13]. The reaction initiates with the recombinase binding to the primer to form filaments (protein-DNA complex) that are capable of pairing the primer with a homologous sequence in the target DNA. After filaments recombine with homologous DNA, the other strand of target DNA is displaced, where SSB binds to prevent the dissociation of the primer. Finally, the strand-displacing polymerase adds bases to the 3’ end of the primer and primer extension occurs. When opposing primers are used, exponential amplification typically runs to completion in about 30 minutes. RPA has been explored for the molecular detection of diverse pathogens, e.g., bacteria, fungi, parasites and viruses [12–17], as it possesses superiority in speed, portability and accessibility [17]. In this study, a novel RPA-based method was developed for the detection of CPV-2.
CPV-2a, CPV-2b, canine distemper virus (CDV), and canine coronavirus (CCoV) were propagated in Madin-Darby canine kidney (MDCK) cells (Sigma, Shanghai, China), and pseudorabies virus (PRV) was propagated in baby Syrian hamster kidney (BHK-21) cells (ATCC, Manassas, USA). Viral DNA and RNA were extracted using a TIANamp Virus DNA Kit (Tiangen Biotech Co., Ltd., Beijing, China) and TRIzol Reagent (Invitrogen, Beijing, China), respectively, according to the manufacturers’ instructions and quantified using an ND-1000 spectrophotometer (NanoDrop, Wilmington, USA). One hundred ng of viral RNA was reverse transcribed using a PrimeScript II 1st Strand cDNA Synthesis Kit (Takara, Dalian, China) following the manufacturer’s instructions. The synthesized cDNA was purified using a cDNA purification kit (Takara, Dalian, China) and quantified using an ND-1000 spectrophotometer. Fifty-seven clinical fecal swab samples were collected from veterinary clinics and dog husbandry farms from 2012 to 2014 in Hebei Province, China. Ten mg of faeces was emulsified in 1 ml of sterile phosphate-buffered saline and centrifuged at 10,000 × g for 10 min at 4 °C. The supernatant was collected and used for viral DNA extraction using a TIANamp Virus DNA Kit. The viral DNA extracted from 10 mg of each fecal sample was eluted in 20 µl of nuclease-free water. All DNA and cDNA templates were stored at -20 °C until the assay was done.
Nucleotide sequences of CPV-2a (GenBank: M24003), -2b (GenBank: M38245) and -2c (GenBank: FJ005196) were aligned to identify conserved regions of the VP2 gene. RPA primers were designed as follows: forward, 5′-CAC TTA CTA AGA ACA GGT GAT GAA TTT GCT ACA G-3′; reverse, 5′-AGT TTG TAT TTC CCA TTT GAG TTA CAC CAC GTC T-3′. Both primer sequences were 100 % identical to their target sequences in the CPV-2a, -2b and -2c genomic DNA. The size of the RPA product was ~210 bp. Primers were synthesized by Sangon Biotech Co., Ltd (Shanghai, China).
RPA reactions were performed at 38 °C in a water bath in a total volume of 50 µl. The mixture contained 480 nM each RPA primer, 14 mM magnesium acetate and 1 μL of template, and the reaction was carried out using a TwistAmp Basic Kit (TwistDX, Cambridge, UK). The Kit provided reaction microtubes with a dried enzyme pellet. To optimize the reaction time, 103 copies of CPV-2a genomic DNA were amplified for different times, i.e., 10, 20, 30 and 40 min. The amplified products were purified using a Universal DNA Purification Kit (Tiangen Biotech Co., Ltd., Beijing, China) according to the manufacturer’s instructions. The purified products were electrophoresed in a 2.0 % (W/V) agarose gel, stained with Goldview (Dongsheng Biotech Co., Ltd., Guangzhou, China) and visualized under UV light. As shown in Figure 1, an RPA product with the expected size (~210 bp) was clearly visible after agarose gel electrophoresis and staining when the reaction done for as short a time as 10 minutes. Semi-quantification by measuring the DNA band density revealed that the DNA yield doubled when the reaction was done for 20 minutes (data not shown), but no significant difference was observed in the product yields of 20-min, 30-min and 40-min reactions (data not shown), which probably indicates that after 20 min of reaction, dNTPs or other components in the reaction system were completely consumed. All RPA reactions were done for 20 min for the rest of this study.
Fig. 1.
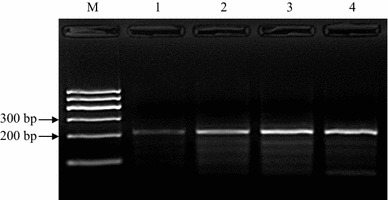
Optimization of RPA reaction time. CPV-2a genomic DNA was amplified with RPA for different lengths of time, and a clear DNA band with the expected size (~210 bp) could be visualized by agarose gel electrophoresis after a 10-min reaction. Semi-quantification by measuring the DNA band density revealed that the DNA yield doubled when the reaction was done for 20 min, but no significant difference was observed in the product yields of 20-min, 30-min and 40-min reactions. M, DNA marker; lanes 1–4, DNA products from reactions incubated for 10 min, 20 min, 30 min and 40 min, respectively
To determine RPA sensitivity, a tenfold serial dilution of CPV-2a genomic DNA was made to achieve concentrations ranging from 105 to 100 copies/μL. One μL of each DNA dilution was amplified with RPA as well as conventional PCR. The PCR primer sequences were as follows: forward, 5’-GAA TCT GCT ACT CAG CCA AC-3’; reverse, 5’-GTG CAC TAT AAC CAA CCT CAG C-3’. The size of PCR product was ~600 bp. One µl of template was mixed with the 2× Taq PCR MasterMix (Tiangen Biotech Co., Ltd, Beijing, China), and PCR was done as follows: stage 1, 95 °C for 10 min; stage 2, 40 cycles of 95 °C for 15 s, 60 °C for 30 s and 72 °C for 45 s; stage 3, 72 °C for 10 min. The PCR product was analyzed by agarose gel electrophoresis as well. As shown in Figure 2A, the detection limit of RPA was 10 copies/reaction, while the detection limit of PCR was 100 copies/reaction (panel B). Five independent reactions were performed, and similar results were observed.
Fig. 2.
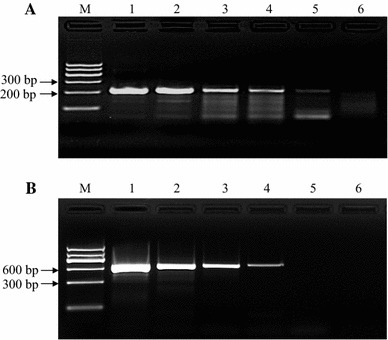
Sensitivity of RPA. Different amounts of CPV-2a genomic DNA were amplified by RPA (panel A) and conventional PCR (panel B). The detection limit for RPA was 10 copies/reaction. In contrast, the detection limit for conventional PCR was 100 copies/reaction. M, DNA marker; lane 1, 105 copies; lane 2, 104 copies; lane 3, 103 copies; lane 4, 102 copies; lane 5, 101 copies; lane 6, 100 copies. Five independent reactions were performed, and similar results were obtained
To determine RPA specificity, 10 ng of viral DNA or cDNA was amplified by RPA. As shown in Figure 3A, genomic DNA from CPV-2a and -2b was amplified, while templates from some important dog viruses, i.e., CDV, PRV and CCoV, were not (Fig. 3B). Viral DNA extracted from 57 clinical fecal samples was tested by RPA, and the results were compared with those obtained using conventional PCR. We observed that 50 out of 57 (83.3 %) samples were positive and seven were negative (16.7 %) when tested by PCR. Of the 50 samples that were positive by PCR, all were positive when assayed by RPA. Of the seven samples that were negative by PCR, three were negative and four were positive when tested by RPA. The overall diagnostic agreement between RPA and PCR was 92.9 % (53/57).
Fig. 3.
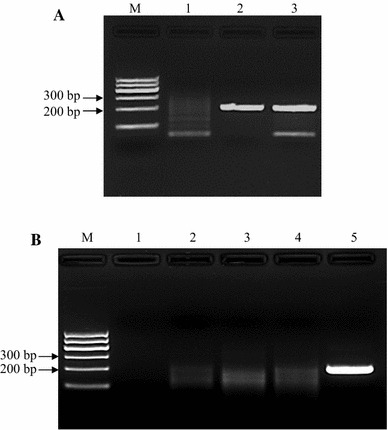
Specificity of RPA. Ten ng of viral DNA or cDNA was amplified with RPA. As shown in panel A, both CPV-2a and -2b genomic DNA were amplified. M, DNA marker; lane 1, no-template control; lane 2, CPV-2a; lane 3, CPV-2b. Panel B shows the test results of CCoV, PRV and CDV. None of these viruses were amplified by RPA. M, DNA marker; lane 1, no-template control; lane 2, CCoV; lane 3, PRV; lane 4, CDV; lane 5, CPV-2a as a positive control. Five independent reactions were performed, and similar results were observed
Kumar et al. reported that real-time PCR had a detection limit of 10 copies of CPV-2 genomic DNA/reaction [8], comparable to our method. We designed the RPA assay aiming to detect all CPV-2 variants. Indeed, both CPV-2a and -2b were successfully amplified with RPA (Fig. 3A). We were unable to obtain CPV-2c, which was thus not tested in this study. Nevertheless, the RPA primers were 100 % identical to the target sequences in CPV-2c genomic DNA, as shown by sequence alignment. RPA did not amplify the template of other important dog viruses, demonstrating high specificity. The validation experiment revealed that RPA had an excellent diagnostic agreement rate with PCR, which is indicative of the clinical applicability of RPA. It is known that low-temperature amplification usually creates a high background signal; hence, nonspecific bands generated from RPA are not uncommon [17]. However, we also observed that nonspecifically amplified bands can be easily differentiated by size difference and signal intensity (Figures 1, 2, 3).
Recently, an iiPCR method was reported for rapid and sensitive detection of CPV-2 [10]. Wilkes et al. found that iiPCR could detect as few as 13 copies of CPV-2b genomic DNA in about 1 hour. However, iiPCR requires a device, i.e., POCKIT™ Nucleic Acid Analyzer (POCKITTM; GeneReach USA, Lexington, MA, USA) that still costs about 10,000 US dollars and has a maximum capacity of running eight samples each time [10], a limit that RPA overcomes.
In conclusion, the RPA method developed in this study demonstrates high sensitivity and specificity for detection of CPV-2. In addition, a validation experiment showed its clinical applicability. An outstanding advantage for RPA is that it is performed in a water bath, making RPA a potential alternative for CPV-2 detection in resource-limited settings.
Acknowledgments
This work was supported by Science and Technology Project Foundation of Animal Husbandry Bureau, Hebei Province, P.R. China (grant number 2014-3-03). The funding agency had no role in study design; in the collection, analysis and interpretation of data; in the writing of the report; or in the decision to submit the article for publication.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Appel MJ, Scott FW, Carmichael LE. Isolation and immunisation studies of a canine parco-like virus from dogs with haemorrhagic enteritis. Vet Rec. 1979;105:156–159. doi: 10.1136/vr.105.8.156. [DOI] [PubMed] [Google Scholar]
- 2.Hoelzer K, Parrish CR. The emergence of parvoviruses of carnivores. Vet Res. 2010;41:39. doi: 10.1051/vetres/2010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Desario C, Decaro N, Campolo M, Cavalli A, Cirone F, Elia G, Martella V, Lorusso E, Camero M, Buonavoglia C. Canine parvovirus infection: which diagnostic test for virus? J Virol Methods. 2005;126:179–185. doi: 10.1016/j.jviromet.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Macartney L, McCandlish IA, Thompson H, Cornwell HJ. Canine parvovirus enteritis 1: clinical, haematological and pathological features of experimental infection. Vet Rec. 1984;115:201–210. doi: 10.1136/vr.115.9.201. [DOI] [PubMed] [Google Scholar]
- 5.Decaro N, Desario C, Campolo M, Elia G, Martella V, Ricci D, Lorusso E, Buonavoglia C. Clinical and virological findings in pups naturally infected by canine parvovirus type 2 Glu-426 mutant. J Vet Diagn Invest. 2005;17:133–138. doi: 10.1177/104063870501700206. [DOI] [PubMed] [Google Scholar]
- 6.Uwatoko K, Sunairi M, Nakajima M, Yamaura K. Rapid method utilizing the polymerase chain reaction for detection of canine parvovirus in feces of diarrheic dogs. Vet Microbiol. 1995;43:315–323. doi: 10.1016/0378-1135(94)00102-3. [DOI] [PubMed] [Google Scholar]
- 7.Hirasawa T, Kaneshige T, Mikazuki K. Sensitive detection of canine parvovirus DNA by the nested polymerase chain reaction. Vet Microbiol. 1994;41:135–145. doi: 10.1016/0378-1135(94)90143-0. [DOI] [PubMed] [Google Scholar]
- 8.Kumar M, Nandi S. Development of a SYBR Green based real-time PCR assay for detection and quantitation of canine parvovirus in faecal samples. J Virol Methods. 2010;169:198–201. doi: 10.1016/j.jviromet.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun YL, Yen CH, Tu CF. Visual detection of canine parvovirus based on loop-mediated isothermal amplification combined with enzyme-linked immunosorbent assay and with lateral flow dipstick. J Vet Med Sci. 2014;76:509–516. doi: 10.1292/jvms.13-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilkes RP, Lee PY, Tsai YL, Tsai CF, Chang HH, Chang HF, Wang HT. An insulated isothermal PCR method on a field-deployable device for rapid and sensitive detection of canine parvovirus type 2 at points of need. J Virol Methods. 2015;220:35–38. doi: 10.1016/j.jviromet.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piepenburg O, Williams CH, Stemple DL, Armes NA. DNA detection using recombination proteins. PLoS Biol. 2006;4:e204. doi: 10.1371/journal.pbio.0040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyle DS, McNerney R, Teng Low H, Leader BT, Perez-Osorio AC, Meyer JC, O’Sullivan DM, Brooks DG, Piepenburg O, Forrest MS. Rapid detection of Mycobacterium tuberculosis by recombinase polymerase amplification. PLoS One. 2014;9:e103091. doi: 10.1371/journal.pone.0103091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakai K, Trabasso P, Moretti ML, Mikami Y, Kamei K, Gonoi T. Identification of fungal pathogens by visible microarray system in combination with isothermal gene amplification. Mycopathologia. 2014;178:11–26. doi: 10.1007/s11046-014-9756-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crannell ZA, Cabada MM, Castellanos-Gonzalez A, Irani A, White AC, Richards-Kortum R. Recombinase polymerase amplification-based assay to diagnose Giardia in stool samples. Am J Trop Med Hyg. 2014;92:583–587. doi: 10.4269/ajtmh.14-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyle DS, Lehman DA, Lillis L, Peterson D, Singhal M, Armes N, Parker M, Piepenburg O, Overbaugh J. Rapid detection of HIV-1 proviral DNA for early infant diagnosis using recombinase polymerase amplification. MBio. 2013;4:e00135-13. doi: 10.1128/mBio.00135-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abd EI, Wahed A, El-Deeb A, El-Tholoth M, Abd El Kader H, Ahmed A, Hassan S, Hoffmann B, Haas B, Shalaby MA, Hufert FT, Weidmann M. A portable reverse transcription recombinase polymerase amplification assay for rapid detection of foot-and-mouth disease virus. PLoS One. 2013;8:e71642. doi: 10.1371/journal.pone.0071642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaghloul H, El-shahat M. Recombinase polymerase amplification as a promising tool in hepatitis C virus diagnosis. World J Hepatol. 2014;6:916–922. doi: 10.4254/wjh.v6.i12.916. [DOI] [PMC free article] [PubMed] [Google Scholar]