

Abstract
Using viral metagenomics, the complete genome sequence of an infectious bronchitis virus (IBV) strain (named ahysx-1) from a fecal sample from a healthy chicken in Anhui province, China, was determined. The genome sequence of ahysx-1 was found to be very similar to that of IBV strain ck/CH/LLN/131040 (KX252787), except for the spike gene region, which is similar to that of a turkey coronavirus strain (EU022526), suggesting that ahysx-1 is a recombinant. Recombination analysis and phylogenetic analysis based on the genomic sequences of ahysx-1 and other related strains confirmed that ahysx-1 appears to be a recombinant resulting from a recombination event that occurred between a chicken coronavirus and a turkey coronavirus. Further studies need to be performed to determine whether this recombinant IBV strain is pathogenic and whether it is transmitted between chickens and turkeys.
Electronic supplementary material
The online version of this article (10.1007/s00705-019-04488-3) contains supplementary material, which is available to authorized users.
Keywords: Viral metagenomics, Coronavirus, Infectious bronchitis virus, Genome recombination
Coronaviruses (CoVs) are enveloped positive-sense RNA viruses that belong to the family Coronaviridae. They have been found in a wide variety of animals and can cause respiratory, enteric, hepatic and neurological diseases [1, 2]. Based on their genetic and serological properties, CoVs are classified into four genera, Alphacoronavirus, Betacoronavirus, Gammacoronavirus and Deltacoronavirus [3]. Avian infectious bronchitis virus (IBV), a member of the genus Gammacoronavirus, can affect the respiratory tract, gut, kidney and reproductive systems of chickens and cause a decrease in both meat and egg production and is therefore responsible for substantial economic losses to the poultry industry.
CoVs have an unsegmented, single-stranded, positive-sense RNA genome of 28-32 kb, including a 5’ cap and a 3’ poly(A) tail. Like other CoVs, the 5’ two thirds of the IBV genome encodes the 1a and 1ab polyproteins, which are proteolytically cleaved by virus-encoded papain-like and 3C-like proteinases into at least 15 nonstructural proteins (nsp2–nsp16) [4]. Like most CoVs, the remaining one third of the genome of IBV encodes four major structural proteins: the spike (S) glycoprotein, the small envelope (E) protein, the membrane (M) glycoprotein, and the nucleocapsid (N) protein. Generally, the S1 subunit of the S glycoprotein is responsible for attachment of the virion to the host cell membrane by interacting with sialic acid, initiating the infection, while the S2 subunit mediates fusion of the virion and cellular membranes by acting as a class I viral fusion protein [5, 6]. Two or three small accessory proteins (genes 3, 4 and 5) that vary in number and sequences among IBV strains are interspersed among the genes coding for the structural proteins. The 5’ and 3’ untranslated regions (UTRs) usually contain important structural elements and are involved in replication and/or translation [7].
In November of 2016, 10 fecal samples were collected 10 apparently healthy chickens on a commercial chicken farm in Anhui province, China. These samples were subjected to viral nucleic acid detection using the viral metagenomic method. All fecal samples were collected using disposable materials and shipped on dry ice. The samples were diluted in phosphate-buffered saline (PBS) to make a 10% suspension and mixed thoroughly by vortexing. Fecal suspensions were then centrifuged for 10 min at 15,000 × g. One hundred μL of each supernatant was then collected, and the samples were pooled. The pooled supernatant was filtered through a 0.45-mm filter (Millipore) to remove eukaryotic and prokaryotic particles and then treated with a mixture of nuclease enzymes to reduce the concentration of nonviral nucleic acids [8]. Total nucleic acid was extracted using a QIAamp MinElute Virus Spin Kit (QIAGEN) according to the manufacturer’s instructions. A reverse transcription reaction using reverse transcriptase (SuperScript IV, Invitrogen) was performed using the enriched viral nucleic acid preparation and 100 pmol of random hexamerprimer, followed by a round of DNA synthesis using Klenow fragment polymerase (New England Biolabs). A library was then constructed using a Nextera XT DNA Sample Preparation Kit (Illumina) and sequenced on an Illumina MiSeq platform with 250-bp paired-end reads with dual barcoding. Bioinformatics analysis was performed as described previously [9, 10]. Briefly, paired-end reads of 250 bp generated by Illumina MiSeq were debarcoded, and the sequencing data were processed using in-house analysis pipeline running on a 32-node Linux cluster. Reads that were identical from bases 5 to 55 were considered duplicates, and only one copy of these duplicates was randomly kept. Low-quality tails of each read were trimmed with Phred with a quality score threshold of 10. Adaptors were removed using VecScreen with default parameters. The cleaned reads were assembled de novo within each barcode using the ENSEMBLE assembler [9]. Contigs and unassembled reads were then matched against a customized viral protein database using BLASTx with an E-value cutoff of < 10−5. Putative viral hits are then matched against an in-house non-virus non-redundant (NVNR) protein database to remove false positive viral hits. Contigs with no significant similarity to viral proteins in the BLASTx search were used to search the vFam database [11] using HMMER3 [12–14] to detect remote similarities to viral proteins.
The library from 10 chicken fecal samples that was sequenced on the MiSeq platform generated a total of 3,391,272 raw sequence reads, which were deposited in the Short Read Archive of the GenBank database under accession no. SRX6816578 and were assembled de novo. The contigs and unassembled reads were then compared to the GenBank viral database using BLASTx, and it was found that this library contained abundant sequences reads showing similarities to putative mammalian or avian viruses belonging to the families Coronaviridae (17,209 reads), Picornaviridae (7185 reads), Circoviridae (642 reads), Picobirnaviridae (362 reads), Caliciviridae (81 reads), Adenoviridae (79 reads), and Astroviridae (26 reads). By assembling the 17,209 reads showing similarity to coronaviruses, a nearly complete genome sequence was obtained, which was 27,718 bp in length, excluding the 24-bp poly(A) tail. The nearly complete genome sequence was deposited in the GenBank database under accession no. MK142676 and the strain name ahysx-1. This sequence contains 10 open reading frames (ORFs) (Fig. 1A). A BLASTn search indicated that although 87% of the genome of ahysx-1 shows 98% sequence identity to infectious bronchitis virus strain ck/CH/LLN/131040 (KX252787), the spike (S) genes of these two strains were only 58% identical. A BLASTn search based on the S gene sequence showed that ahysx-1 shared 79% sequence identity with turkey coronavirus isolate TCoV-ATCC (EU022526), which was isolated from the small intestine of a turkey [15], suggesting that ahysx-1 is a recombinant.
Fig. 1.

Recombination and phylogenetic analysis of the infectious bronchitis virus from chicken. a Genome organization of the infectious bronchitis virus strain ahysx-1. b BOOTSCAN evidence for the recombination origin on the basis of pairwise distance, modeled with a window size 200, step size 20, and 100 Bootstrap replicates. c Neighbor joining tree constructed using the nucleotide sequence of gene 1a and 1b. d Neighbor joining tree constructed using the S gene. e Neighbor joining tree constructed using the nucleotide sequence of gene 3, 4, 5, and 6. f The overlapping reads coverage across the boundaries of S gene. The S gene region was highlighted in black, while the sequences flanking the boundaries of S gene were marked with dark gray. The coverage depth is shown in blue
To test whether strain ahysx-1 is a recombinant, the complete genome sequences of the five infectious bronchitis virus strains with complete genome sequences to which the non-spike regions of ahysx-1 showed the closest relationship based on a BLASTn search and four strains with complete genome sequences, including three turkey coronaviruses and one guinea fowl coronavirus, to which the S region of ahysx-1 shared the highest sequence similarity based on a BLASTn search, were retrieved from GenBank. These complete genome sequences, together with that of ahysx-1, were aligned using the Clustal W program [16]. Possible recombination sites and potential parental sequences were identified using Recombination Detection Program 4.0 (RDP 4.0), which includes seven different test methods: RDP, GENECONV, BootScan, MaxChi, Chimaera, SiSCan, and 3Seq [17] (Supplemental Fig. 1). The results obtained using all seven recombination detection methods indicated with a high degree of confidence (p-value < 6.2 × 10−15) that ahysx-1 is a potential recombinant resulting from a recombination event occurring between a member of the lineage represented by the five infectious bronchitis virus strains as the major parent and a member of the lineage including the three turkey coronaviruses and the guinea fowl coronavirus as the minor parent. A manual bootscan analysis comparing the complete genome sequence of ahysx-1 to the complete genome sequences of a representative major parent (KX252781) and a representative minor parent (EU022526) using the program RDP4.0 was then performed, and this confirmed the potential recombination event. In the non-spike regions, ahysx-1 showed higher sequence similarity to the major parental strain, while in the spike region, ahysx-1 displayed higher sequence similarity to the minor parental strains (Fig.1B). To confirm this result, phylogenetic analysis was performed separately, using only the non-spike region preceding the S gene, the S gene, and the non-spike region following the S gene to compare ahysx-1 and the other nine reference strains used in the recombination analysis. All phylogenetic trees were constructed based on an alignment of nucleotide sequences, using the maximum-likelihood (ML) method with 1,000 bootstrap resamplings of the alignment data and visualized using the program MEGA7.0 [18]. The resulting trees are shown in Fig. 1C, D and E. The trees based on the non-spike regions (Fig. 1C and D) show that ahysx-1 clustered closely with the five chicken infectious bronchitis virus strains, while the tree based on the S gene showed that ahysx-1 clustered with the three turkey coronaviruses and the guinea fowl coronavirus, sharing 74.8%-75.9% sequence identity in pairwise sequence alignments (Fig. 1E). The above data indicate that ahysx-1 might be a recombinant that was produced by recombination between coronaviruses from a chicken and a turkey.
To computationally identify the boundaries of the distinct portion of the S gene in the recombinant, the clean raw reads were remapped to the complete genome sequence of ahysx-1, which revealed evident overlapping read coverage across the boundaries (Fig. 1F). We also remapped the clean raw reads to the complete genome sequence of infectious bronchitis virus strain (KX252781) which had 98% sequence identity to ahysx-1 over the non-spike gene region, and the results indicated that there was high depth in the non-spike regions, but no sequence reads mapped to the S gene region (Supplemental Fig. 3). The clean raw reads were also mapped to the complete genome sequence of the representative minor parent (EU022526), which showed that few sequence reads mapped to the whole genome sequence, which may due to the low sequence similarity between ahysx-1 and EU022526. The remapping results demonstrated that the recombinant was present in the fecal sample.
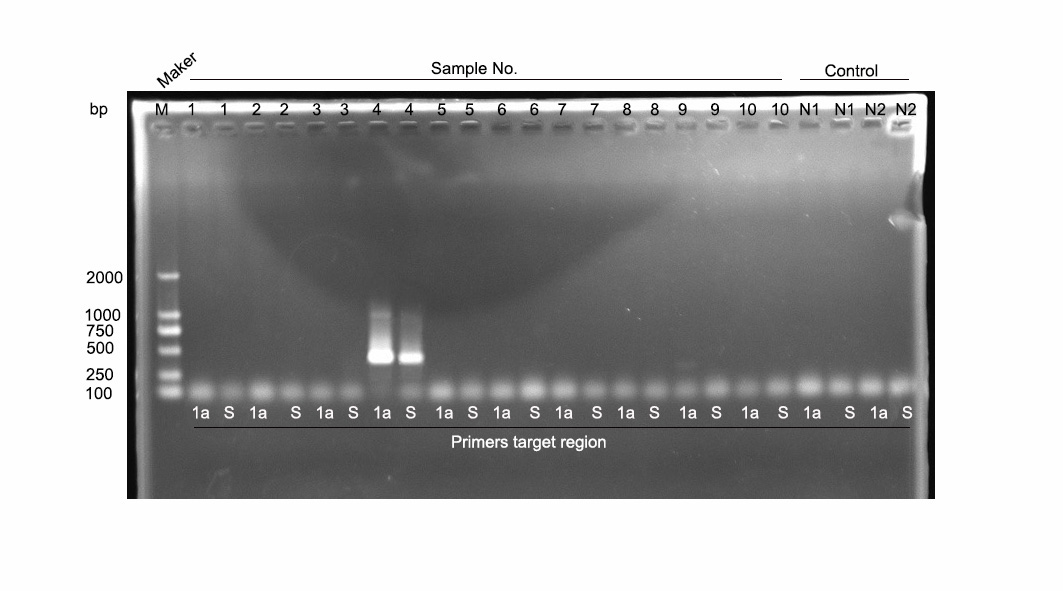
To confirm the presence of the recombinant in the fecal samples, PCR screening was conducted on the 10 fecal samples that were included in the library, using two sets of nested primers designed based on the 1a and S regions of ahysx-1, respectively (primer sequences and PCR conditions are shown in Supplemental Table 1). The results indicated that only one sample was positive with both sets of primers (Supplemental Fig. 2), and Sanger sequencing of the resulting PCR products showed that they were identical to the corresponding fragments of ahysx-1.
Homologous recombination within the genome, especially within the S gene, is an important feature of the evolution of coronaviruses that can affect pathogenicity and even host specificity and tissue tropism. In the case of IBV, this has led to the emergence of several new serotypes [19, 20] and contributed to disease outbreaks in flocks. Most of the IBV recombinants reported previously resulted from recombination events occurring between different IBVs from chickens. In this study, although the non-spike regions showed high sequence similarity (98% identity) to IBV isolates from chickens, the S gene of the recombinant showed rather low similarity (58% identity) to IBV from chickens but higher similarity (> 75% identity) to turkey coronaviruses, suggesting that this recombinant might have the potential for cross-species transmission between chickens and turkeys.
In summary, we have identified a recombinant IBV strain from a healthy chicken that contained a distinct S gene similar to those of turkey isolates. The putative recombination event was verified by phylogenetic analysis and analysis using the RDP4.0 program. Whether this recombinant IBV strain is pathogenic and whether it can cause cross-species infection between chickens and turkeys needs further investigation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
Funding
This work was partly supported by the National Key Research and Development Programs of China (no. 2017YFC1200201), Jiangsu Provincial Key Research and Development Projects (no. BE2017693), Jiangsu Social Development Project (no. BE2016663), National Natural Science Foundation of China (no. 81741062), Student’s Platform for Innovation and Entrepreneurship Training Program (no. 201810299587 W), and the Blood Systems Research Institute.
Compliance with ethical standards
Competing interests
All authors declare that they have no competing interests.
Ethical approval
This article does not contain any studies with animals performed by any of the authors.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yan Wang, Xuejiao Cui and Xu Chen contributed equally to this work.
Contributor Information
Yu Li, Email: liyu@ahau.edu.cn.
Wen Zhang, Email: z0216wen@yahoo.com.
References
- 1.Perlman S, Netland J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol. 2009;7:439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Menachery VD, Graham RL, Baric RS. Jumping species—a mechanism for coronavirus persistence and survival. Curr Opin Virol. 2017;23:1–7. doi: 10.1016/j.coviro.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woo PCY, Lau SKP, Lam CSF, et al. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavirus. J Virol. 2012;86:3995–4008. doi: 10.1128/JVI.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ziebuhr J, Thiel V, Gorbalenya AE. The autocatalytic release of a putative RNA virus transcription factor from its polyprotein precursor involves two paralogous papain-like proteases that cleave the same peptide bond. J Biol Chem. 2001;276:33220–33232. doi: 10.1074/jbc.M104097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bickerton E, Maier HJ, Stevenson-Leggett P, et al. The S2 Subunit of infectious bronchitis virus beaudette is a determinant of cellular tropism. J Virol. 2018;92:e01044–18. doi: 10.1128/JVI.01044-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shan D, Fang S, Han Z, et al. Effects of hypervariable regions in spike protein on pathogenicity, tropism, and serotypes of infectious bronchitis virus. Virus Res. 2018;250:104–113. doi: 10.1016/j.virusres.2018.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goebel SJ, Hsue B, Dombrowski TF, Masters PS. Characterization of the RNA components of a putative molecular switch in the 3’ untranslated region of the murine coronavirus genome. J Virol. 2004;78:669–682. doi: 10.1128/JVI.78.2.669-682.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang W, Li L, Deng X, et al. Viral nucleic acids in human plasma pools. Transfusion. 2016;56:2248–2255. doi: 10.1111/trf.13692. [DOI] [PubMed] [Google Scholar]
- 9.Deng X, Naccache SN, Ng T, et al. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res. 2015 doi: 10.1093/nar/gkv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang W, Yang S, Shan T, et al. Virome comparisons in wild-diseased and healthy captive giant pandas. Microbiome. 2017;5:90. doi: 10.1186/s40168-017-0308-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skewes-Cox P, Sharpton TJ, Pollard KS, DeRisi JL. Profile hidden Markov models for the detection of viruses within metagenomic sequence data. PLoS One. 2014;9:e105067. doi: 10.1371/journal.pone.0105067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson LS, Eddy SR, Portugaly E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics. 2010;11:431. doi: 10.1186/1471-2105-11-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009;23:205–211. [PubMed] [Google Scholar]
- 14.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao J, Wu C-C, Lin TL. Complete nucleotide sequence of polyprotein gene 1 and genome organization of turkey coronavirus. Virus Res. 2008;136:43–49. doi: 10.1016/j.virusres.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 17.Martin Martin, Martin B, Golden M. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1:vev003. doi: 10.1093/ve/vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamura K, Peterson D, Peterson N, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin S-Y, Chen H-W. Infectious bronchitis virus variants: molecular analysis and pathogenicity investigation. Int J Mol Sci. 2017;18:2030. doi: 10.3390/ijms18102030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackwood MW, Boynton TO, Hilt DA, et al. Emergence of a group 3 coronavirus through recombination. Virology. 2010;398:98–108. doi: 10.1016/j.virol.2009.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.