

Abstract
Human metapneumovirus (HMPV) is an important respiratory virus implicated in respiratory infections. The purpose of this study was to develop a one-step real-time RT-PCR assay that can detect all four lineages of HMPV and to identify the HMPV lineages circulating in Pune, India. Conserved regions of the nucleoprotein gene were used to design real-time primers and a probe. A total of 224 clinical samples that were positive for different respiratory viruses (including 51 samples that were positive for HMPV) were tested using the real time RT-PCR assay, and the specificity of the assay was observed to be 100 %. Using in vitro-synthesized RNA, the sensitivity of the assay was ascertained to be 100 copies of the target gene per reaction. Phylogenetic analysis of the nucleoprotein (N) and attachment glycoprotein (G) genes confirmed that this assay detected all lineages of HMPV. A2, B1 and B2 strains were observed during the study period. Our assay is highly sensitive and specific for all known lineages of HMPV, making it a valuable tool for rapid detection of the virus. A2 and B2 were the predominant subtypes circulating in Pune, Western India.
Keywords: Reverse Transcription Polymerase Chain Reaction, Amino Acid Level, Respiratory Virus, Human Metapneumovirus, Reverse Transcription Polymerase Chain Reaction Assay
Introduction
Human metapneumovirus (HMPV), first isolated from children with acute lower-respiratory-tract infections (ALRTI) in the Netherlands in 2001, is an enveloped, non-segmented RNA virus that belongs to the family Paramyxoviridae and the genus Metapneumovirus [1]. Subsequently, it has been reported globally [2–8]. It frequently causes both upper and lower acute respiratory tract infections (ARTIs) in people of all age groups [9–19] and in immunocompromised patients [20, 21]. Several groups have described reverse transcription polymerase chain reaction (RT-PCR)-based assays to detect HMPV targeting the nucleoprotein, fusion or polymerase genes [22–29].
MPV has been classified into two broad groups: group A, with subgroups A1 and A2, and group B, with subgroups B1 and B2 [30–32]. More recently, the A2 group has been split into two clades (A2a and A2b) [33]. However, there is limited information on the prevalence and genetic diversity of human metapneumovirus (HMPV) strains circulating in India [34–36].
It is important to establish diagnostic HMPV assays which detect all subtypes of the virus. This study describes the development of real time RT-PCR assay for the detection of all four HMPV lineages (A1, A2, B1, B2) in respiratory specimens and genotyping of circulating strains from July 2009 to August 2011 in Pune, western India.
Materials and methods
Clinical samples
Clinical samples (nasal/throat swab) referred to the National Institute of Virology (NIV), Pune, for diagnosis of H1N1pdm09 from July 2009 to August 2011 were retrospectively used in this study with the approval of NIV’s Human Ethics Committee [110(01)/EC-I/1056] as per Indian Council of Medical Research (ICMR) guidelines. This included outpatient and admitted cases in Pune, Maharashtra, India. A total of 224 clinical specimens were tested to measure the sensitivity and specificity of the assay.
RNA extraction
RNA was extracted using a MagMax-96 viral RNA isolation kit according to the manufacturer’s instructions. Fifty microlitres of clinical specimen was used as starting material, and the RNA was finally eluted in 50 μl of elution buffer.
Primer and probe design
Primers and probe were designed using the HMPV sequences available in the NCBI database. Representative sequences of nucleoprotein genes of prototype sequences of A1, A2, B1 and B2 subtypes [31, 36] were aligned, and primers were designed from conserved regions of the consensus sequence to ensure that all lineages would be amplified and detected using this assay. Primers were checked for cross-reactivity, self-dimerization, hairpin formation and secondary structure formation using NCBI BLAST, OligoCalc software (http://www.basic.northwestern.edu/biotools/oligocalc.html) and OligoAnalyzer. Primer and probe sequences are given in Table 1. Sequencing primers were designed for the nucleoprotein (N) and attachment glycoprotein (G) genes as shown in Table 1.
Table 1.
Primer and probe sequences
| Serial no. | Gene | Primer/probe | Sequence 5’–3’ |
|---|---|---|---|
| 1 | NP | Forward (real- time) | 5’GCACTCAAAAGATACCCTAGRATRGAYATAC-3’ |
| 2 | NP | Reverse (real-time) | 5’-AATAAACTTTCTGCTTTGCTKCCTGTWGA-3’ |
| 3 | NP | Probe (Real time) | FAM-ATGAGCCTAAWGCTTTCCCATACTCAATGAA- BHQ1 |
| 4 | NP | T7HMPVNFP1 | TAATACGACTCACTATAGGGCATACAAACATGCTATATTAAAAGAGTCTC |
| 5 | NP | HMPVNFP2 | GAAGAAATAGACAAAGAAGCAAG |
| 6 | NP | HMPVNFP3 | AACAATATAATGCTAGGGCATGTATC |
| 7 | NP | HMPVNRP1 | ATTATTGGTGTGTCTGGTGCTGA |
| 8 | NP | HMPVNRP2 | CCAGATTCAGGACCCATTTCTC |
| 9 | NP | HMPVNRP3 | TTTTGCTGCTTCATTACCCATG |
| 10 | G | HMPVGAFP1 | GGGACAAGTGGCCATGGA |
| 11 | G | HMPVGAFP2 | AACAGCACTAAGTATGGCACT |
| 12 | G | HMPVGAFP3 | GCACAGCTGCAACCCAAAC |
| 13 | G | HMPVGARP1 | GGGTCTTCTTGCTTGGTTC |
| 14 | G | HMPVGARP2 | GATCGCTGCTTTGAGTTGTG |
| 15 | G | HMPVGBFP1 | GGGACAAGTAGTTATGGAGG |
| 16 | G | HMPVGBFP2 | CCAACTCAACAGTCCACAGAA |
| 17 | G | HMPVGBFP3 | AGCAGCACARGAAAAAGACC |
| 18 | G | HMPVGBRP1 | GGGTGRTGAGCTGGTGTG |
| 19 | G | HMPVGBRP2 | GTCTTTGTTCTGCTTKCACTTG |
| 20 | L | HMPVLRP | GGRAGATAGACATTRACAGTGG |
In vitro RNA synthesis
The entire nucleoprotein gene was amplified using T7HMPVNFP1 and HMPVNRP3 primers (Table 1). The forward primer was tagged with a universal T7 promoter sequence at its 5’ end. PCR amplification was performed on a GeneAmp PCR System 9700 using an Invitrogen Superscript III one step RT-PCR kit. The master mix for RT-PCR consisted of 25 μl 2x buffer, 2 μl enzyme mix, 20 μM forward primer, 20 μM reverse primer, and 5 μl of RNA template to make a 50-μl reaction. Thermal cycling conditions were 50 °C for 30 min for reverse transcription, initial denaturation at 94 °C for 10 min, 45 cycles of three steps – 15 s at 94 °C, 15 s at 52 °C, 60 s at 68 °C – and final extension at 68 °C for 5 min. In vitro-synthesized RNA was made using a T7 riboprobe kit (Promega, Leiden, The Netherlands) as per manufacturer’s instructions. In vitro-synthesized RNA was quantified using a Nanophotometer (IMPLEN, Germany), and the RNA copy number was calculated. The in vitro-transcribed RNA was then serially diluted tenfold and used to determine the analytical sensitivity of the real-time RT-PCR.
Standardization of real time RT-PCR
Real-time RT-PCR assay was performed on an Applied Biosystems ABI 7500 machine using an Invitrogen Superscript III one-step qRT-PCR kit (Invitrogen, California, USA). Different parameters of the assay, such as annealing temperature and time and primer and probe concentrations, were optimized. A standard curve was plotted using tenfold serially diluted in vitro-synthesized RNA, testing each concentration in duplicate. A typical 25-μl PCR reaction consisted of 10 μM forward and 10 μM reverse primer, 2.5 μM of TaqMan probe, 12.5 μl 2× buffer, 1 μl Superscript III enzyme, and 5 μl RNA template. Real-time RT-PCR thermal cycling conditions were as follows: 50 °C for 30 min for reverse transcription, initial denaturation at 94 °C for 5 min and 45 cycles of 15 s at 94 °C and 45 s at 55 °C.
Sequencing of nucleoprotein (N) and glycoprotein (G) genes
Sequencing primers were designed for the nucleoprotein (N) and attachment glycoprotein (G) genes. The glycoprotein sequence is highly variable, and hence, subgroup-specific primers were designed as shown in Table 1. The nucleoprotein gene was amplified as described earlier, and the PCR product was purified using a QIAquick PCR Purification Kit according to the manufacturer’s instructions. Sublineages were determined using BLAST analysis of N gene sequences. After determining the subgroup, the glycoprotein gene was amplified using a subgroup-specific forward primer (HMPVGAFP1 for A1 and A2) (HMPVGBFP1 for B1 and B2) and a reverse primer HMPVLRP (Table 1). PCR reaction setup and conditions for the G gene were the same as those used for the N genes. Internal primers (Table 1) were used to get overlapping sequences for N and G gene. Sequencing was performed for representative HMPV-positive samples on an ABI 3730XL 96 capillary sequencer using an Applied Biosystems RR100 Sequencing Kit (Applied Biosystems, Foster City, California, USA) according to manufacturer’s instructions. The GenBank accession numbers of the nucleoprotein gene and glycoprotein gene sequences obtained in this study are KC731484 to KC731527.
Sequence and phylogenetic analysis
Sequence analysis and tree building were performed using MEGA 5.2 software. Of the 51 samples that were positive for HMPV, 39 randomly selected samples were sequenced for the N gene, and 27 for the G gene. Phylogenetic trees were constructed using the maximum-likelihood method applying the Kimura 2-parameter model and checked using 1000 bootstrap replicates. Bootstrap percentages greater than or equal to 85 % are displayed. Sequences available in the NCBI database from India (IND, KOL), Canada (CAN), the Netherlands (NL), Argentina (Arg), Uruguay (UY) and the USA (HMPV, TN) were used as reference sequences. NetOGlyc software (http://www.cbs.dtu.dk/services/NetOGlyc/) was used to predict potential O-glycosylation sites for the attachment glycoprotein. The nucleotide and amino acid sequences of the N and G genes were aligned using ClustalW to check for percentage similarity between the circulating strains and the reference prototype strains.
Results
Sensitivity and specificity
Studies for assessing the detection limit of the assay were performed using in vitro-transcribed RNA from template of lineage A and B viruses. The standard curve was plotted using tenfold serial dilutions of the in vitro-synthesized RNA in duplicate from 5 × 106 copies per reaction to 0.5 copies per reaction to check the PCR efficiency of the assay, and it was found to be approximately 95 % (Fig. 1). The slope of the standard curve was found to be -3.434 (acceptable value = -3.3 to -3.8). The analytical sensitivity of the assay was 100 copies of in vitro-synthesized RNA per reaction for both lineages.
Fig.1.

Standard curve for real-time RT-PCR using in vitro-synthesized RNA
The sensitivity and specificity were determined by testing HMPV-positive samples (n=51), known positive samples of respiratory viruses other than HMPV (n=128), and samples that were negative for any of the respiratory viruses (n=45). Clinical specimens that were confirmed previously as positive by multiplex RT-PCR [37] were used in this study. A total of 224 samples were tested, as shown in Table 2. No cross-reactivity was observed with other respiratory viruses, and all samples that were positive for HMPV by multiplex RT-PCR were positive in the real-time RT-PCR assay. One known RSV-positive sample also tested positive for HMPV in the assay. Both RSV and HMPV were found to be present in the sample, as shown by sequencing, confirming a dual infection.
Table 2.
Different respiratory viruses tested by real-time RT-PCR
| Clinical samples positive for: | Total no. of samples tested | Real-time RT-PCR result |
|---|---|---|
| HMPV | 51 | Positive |
| Influenza virus type B | 12 | Negative |
| H1N109pdm | 14 | Negative |
| Seasonal H1N1 | 10 | Negative |
| Seasonal H3N2 | 15 | Negative |
| Respiratory syncytial virus (A/B) | 43 | Negative |
| Human rhino virus (A/B/C) | 11 | Negative |
| Parainfluenza virus (1/2/3/4) | 22 | Negative |
| Coronavirus 43 | 1 | Negative |
| Clinical samples negative for the above- mentioned respiratory viruses | 45 | Negative |
| Total number of samples | 224 |
Phylogenetic analysis
Phylogenetic analysis of the 39 nucleoprotein gene sequences showed homology with HMPV reference sequences, as shown in Fig. 2. Of the 39 samples randomly selected for sequencing, 19 were from 2009, 19 were from 2011, and only one was from 2010. Out of the 19 sequences from 2009, 10 (52.6 %) clustered in the A2 lineage and 9 (47.3 %) in the B2 lineage. During 2010, only one sample was positive for HMPV, and it clustered in A2. During 2011, one sequence clustered in the A1 lineage (5.2 %), 7 in A2 (36.8 %), 5 in B1 (26.3 %) and 6 in B2 (31.5 %). During 2009 and 2011, the A2 and B2 subgroups were both co-circulating, and B1 was detected only in 2011. There was no predominant subgroup that was responsible for infection in any particular year, as can be seen in the phylogenetic tree.
Fig. 2.
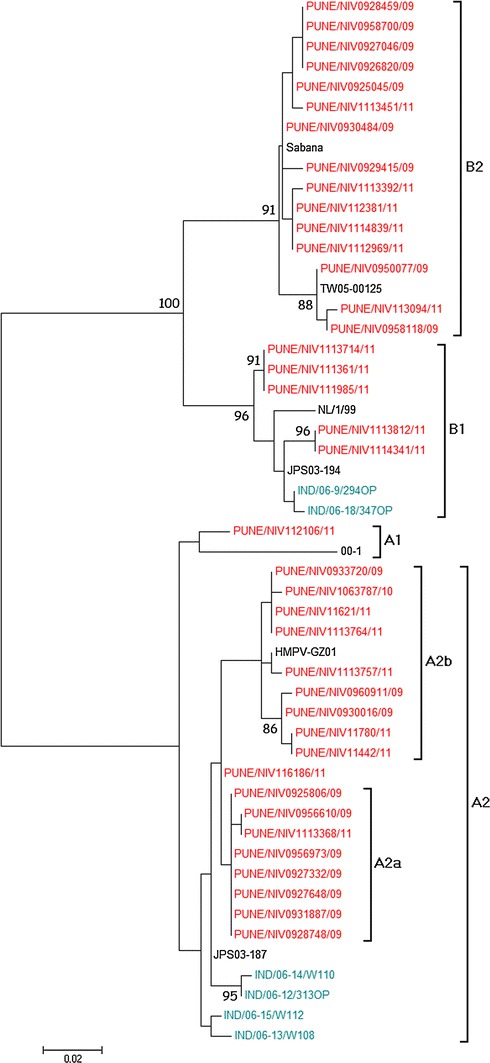
Phylogenetic analysis of the nucleoprotein (N) gene. A phylogenetic tree was constructed with MEGA 5.1 software using the maximum-likelihood method. Bootstrap probabilities greater than 85 % are shown at the branch nodes. Reference sequences for each genotype were obtained from GenBank
For the nucleoprotein gene, 96 % sequence homology was observed between the samples and reference sequence (CAN97-83) at the nucleotide level, and 97-99 % at the amino acid level, for the A2 genotype. Also, the sample sequences showed 100 % amino acid sequence identity to Indian strain IND/06-12/313OP. For the B1 lineage, sequence identities between the sample sequences and reference strains (NL/1/99) were between 92 and 98 % at the nucleotide level and 93-99 % at the amino acid level. The amino acid sequence identity between the sample sequences and Indian strain IND/06-9/294OP was between 91 and 100 %. For the B2 lineage, sequence identity between the sample sequences and reference strains (CAN98-75) was 98 % at the nucleotide level and between 96 and 100 % at the amino acid level. As expected, the nucleoprotein sequences were highly conserved, both at the nucleotide and amino acid level, showing 95-99 % identity between the sample sequences and reference sequences.
Twenty-seven randomly selected HMPV-positive samples were sequenced for the glycoprotein gene. Of the 27 sequences, 11 sequences aligned with the A2 subtype, 3 with the B1 subtype, and 13 with the B2 subtype (Fig. 3). Of the 27 sequences, 13 were from 2009, 13 were from 2011, and one was from 2010. From the two phylogenetic trees it was observed that the B1 subtype was observed only in 2011. However in 2011, the A2 and B2 subtypes were also found in co-circulation with the B1 subtype, as shown in Fig. 2.
Fig. 3.
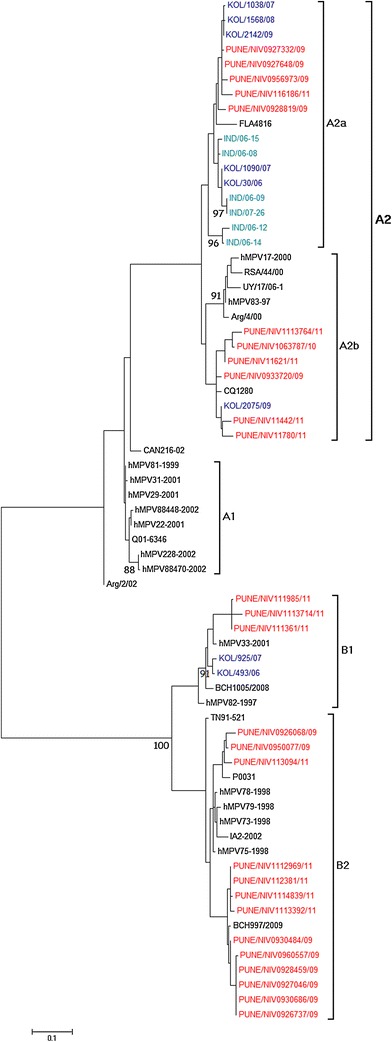
Phylogenetic analysis of the attachment glycoprotein (G) gene. A phylogenetic tree was constructed with MEGA 5.1 software using the maximum-likelihood method. Bootstrap probabilities greater than 85 % are shown at the branch nodes. Reference sequences for each genotype were obtained from GenBank
Glycoprotein gene sequence alignments of the A2 subtype showed 88 % sequence identity between sample and reference sequences (CAN97-83, NL/17/00, KOL/1038/07) at the nucleotide level and 82 % with CAN97-83 and KOL/1038/07, and 80 % with Ind/06-15 at the amino acid level. The B1 subtype sequences for the G gene showed 92 % sequence identity with NL1/99 at the nucleotide level and 89 % at the amino acid level, but only 87 % at the nucleotide level and 82.6 % at the amino acid level with a strain from eastern India (KOL/925/07). The sequence homologies are similar to those reported by Agrawal et al. [34]. The B2 subtype G gene sequences showed 93 % identity to the reference sequence HMPV73-1998 at the nucleotide level and 86 % to CAN98-75 at the amino acid level. Again, homology to an Indian strain (Ind/07-27) was lower, showing 77 % sequence identity at the amino acid level. The deduced G proteins of different HMPV strains had different lengths. Changes in the stop codon were observed among strains of different lineages. Those from subgroup A2a were 219 aa in length with the stop codon UAG (nt 658), A2b had 217 aa and utilized stop codon UAA at nt position 652, whereas those from subgroup B1 were 231 aa in length and terminated at nt position 694 using the stop codon UGA. Strains from the B2 subgroup had two different stop codons, resulting in a G protein with 208 aa using a UAG stop codon at nt position 625 and one with 238 aa using a UAA stop codon at nt position 715.
The attachment glycoprotein of the B2 subtype has 41 potential O-glycosylation sites on average. The region spanning amino acids 77 to 208 (extracellular domain) was checked for potential glycosylation sites. G-values of ≥ 0.5 were considered to indicate potential O-glycosylation sites. The extracellular domain (amino acids 66-208) of the A2 subtype showed an average of 43 O-glycosylation sites. Also, A2 subtype sequences exhibited a higher ratio of serine to threonine than those belonging to the B2 subtypes.
Discussion
There are four distinct genetic lineages of HMPV, A1, A2, B1 and B2, which exhibit substantial diversity. Kuypers et al. [26] developed an assay in which detection of all known subtypes of HMPV was achieved by using two primer/probe sets for the fusion gene in a one-step real-time RT-PCR reaction. Each set was designed to amplify isolates belonging to subtype A or B but to be used together in one reaction mix. Pabbaraju et al. [27] developed a two-step real-time RT-PCR using a fusion gene with a lineage-specific primer-probe and a universal probe for detection of all HMPV lineages. Maertzdorf et al. [24] designed two sets of primers and probes within the nucleoprotein gene to detect all known genetic lineages of HMPV in a one-step real-time RT-PCR. The original NL-N assay developed by Maertzdorf et al. [24] performed poorly with subgroup B viruses, and it was further modified by Klemenc et al. [29] to improve its sensitivity for detecting all four lineages of HMPV. We have designed a primer-probe set using conserved regions of the nucleoprotein gene to detect all known genetic lineages of HMPV in a one-step real-time RT-PCR. Our assay detected 100 copies of in vitro-synthesized RNA per reaction for both lineages. A panel of 51 clinical samples that had previously been found to be positive for HMPV by multiplex PCR [37] was confirmed to be positive by our real-time RT-PCR assay. This assay was also concordant with an additional panel of samples that were negative for HMPV (Table 2). These results suggest that our one-step real-time RT-PCR assay is sensitive for detecting all genetic lineages and specific, as it did not exhibit cross-reactivity with other respiratory viruses.
Phylogenetic analysis of the nucleoprotein and glycoprotein genes in the present study showed that both group A and groups B HMPV were co-circulating in Pune, Western India, during the study period. Phylogenetic analysis reveals that in both 2009 and 2011, A2, B1 and B2 strains were seen in circulation, with predominance of the A2 and B2 subtypes. During the same period, the A2 lineage was predominantly detected in Southwest China [8], Okinawa, Japan [38] and New York, USA [39]. A2 and B2 strains were found to be co-circulating in our study population in 2009 and 2011, but the B1 subtype was seen only in 2011 in our study. Of all the samples sequenced, only one sequence belonging to A1 subtype was seen. Agrawal et al. [34] reported a higher prevalence of subgroup A2 (77 %) than subgroup B1 (23 %) during 2005-2007 in Kolkata, Eastern India, while Banerjee et al. [35] reported A2 (47.3 %) and B1 (47.3 %) in 2006 and B2 (5.2 %) in 2007 in Delhi, Northern India. In Korea, A2 was detected in 65.8 %, B1 in 8 % and B2 in 23.4 % of HMPV-positive cases during 2007-2010, B1was detected only in 2010 [40]. In Central and South America, subtypes A2 and B2 were the predominant strains circulating in 2008-2009 [41]. In Italy, B1 was the predominant subtype, followed by A2 and B2 in 2008-2009, and in 2009-2010, lineage A1 was the predominantly circulating lineage, followed by A2 [42]. The HMPV lineage A2 has been reported to cause HMPV epidemics globally [43].
This is the first study from India where the complete N gene from all circulating subtypes has been analysed. Sequence analysis of the G gene revealed homology to reference strains similar to that described previously [34]. The predicted G proteins of different HMPV strains had different lengths, which may be attributed to amino acid substitutions and insertions, deletions, and/or changes in the stop codon. Five different lengths of G proteins were observed in this study, with 217 or 219 amino acids in subgroup A2, 208 or 238 amino acids in the B2 subgroup, and 231 in B1. This was the first report showing an HMPV strain with 208 amino acids circulating in Pune, India. We need to further monitor HMPV cases from Pune and other parts of India to gain insights into the circulating patterns of this particular strain of HMPV. This will help us to better understand the mechanism of emergence of new strains of the virus.
Conclusion
In conclusion, we have developed a one-step real-time RT-PCR assay for the detection of HMPV from all four genetic lineages. Our assay is sensitive and highly specific and can be used for future implementation in a diagnostic setting. Phylogenetic analysis shows that the A2 and B2 lineages are predominantly circulating in Pune.
Acknowledgments
The authors would like to thank the Director for support, and the Indian Council of Medical Research (ICMR) for funding the study. We would like to acknowledge Mr. Santosh Kumar Jadhav from the bioinformatics group and the technical team of the influenza group for their help.
References
- 1.van den Hoogen BG, de Jong JC, Groen J, Kuiken T, de Groot R, Fouchier RA, Osterhaus AD. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med. 2001;7:719–724. doi: 10.1038/89098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peiris JS, Tang WH, Chan KH, et al. Children with respiratory disease associated with metapneumovirus in Hong Kong. Emerg Infect Dis. 2003;9:628–633. doi: 10.3201/eid0906.030009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ebihara T, Endo R, Kikuta H, Ishiguro N, Ishiko H, Hara M, et al. Human metapneumovirus infection in Japanese children. J Clin Microbiol. 2004;42:126–132. doi: 10.1128/JCM.42.1.126-132.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rao BL, Gandhe SS, Pawar SD, Arankalle VA, Shah SC, Kinikar AA. First detection of human metapneumovirus in children with acute respiratory infection in India: a preliminary report. J Clin Microbiol. 2004;42:5961–5962. doi: 10.1128/JCM.42.12.5961-5962.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ludewick HP, Abed Y, van Niekerk N, Boivin G, Klugman KP, Madhi SA. Human metapneumovirus genetic variability, South Africa. Emerg Infect Dis. 2005;11:1074–1078. doi: 10.3201/eid1107.050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mackay IM, Bialasiewicz S, Jacob KC, McQueen E, Arden KE, Nissen MD, et al. Genetic diversity of human metapneumovirus over 4 consecutive years in Australia. J Infect Dis. 2006;193:1630–1633. doi: 10.1086/504260. [DOI] [PubMed] [Google Scholar]
- 7.Lo Presti A, Cammarota R, Apostoli P, Cella E, Fiorentini S, Babakir-Mina M, Ciotti M, Ciccozzi M. Genetic variability and circulation pattern of human metapneumovirus isolated in Italy over five epidemic seasons. New Microbiol. 2011;34:337–344. [PubMed] [Google Scholar]
- 8.Zhang C, Du LN, Zhang ZY, Qin X, Yang X, Liu P, Chen X, Zhao Y, Liu EM, Zhao XD. Detection and genetic diversity of human metapneumovirus in hospitalized children with acute respiratory infections in Southwest China. J Clin Microbiol. 2012;50:2714–2719. doi: 10.1128/JCM.00809-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boivin G, Abed Y, Pelletier G, Ruel L, Moisan D, Cote S, et al. Virological features and clinical manifestations associated with human metapneumovirus: a new paramyxovirus responsible for acute respiratory-tract infections in all age groups. J Infect Dis. 2002;186:1330–1334. doi: 10.1086/344319. [DOI] [PubMed] [Google Scholar]
- 10.Jartti T, van den Hoogen B, Garofalo RP, Osterhaus AD, Ruuskanen O. Metapneumovirus and acute wheezing in children. Lancet. 2002;360:1393–1394. doi: 10.1016/S0140-6736(02)11391-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falsey AR, Erdman D, Anderson LJ, Walsh EE. Human metapneumovirus infections in young and elderly adults. J Infect Dis. 2003;187:785–790. doi: 10.1086/367901. [DOI] [PubMed] [Google Scholar]
- 12.Greensill J, McNamara PS, Dove W, Flanagan B, Smyth RL, Hart CA. Human metapneumovirus in severe respiratory syncytial virus bronchiolitis. Emerg Infect Dis. 2003;9:372–375. doi: 10.3201/eid0903.020289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mullins JA, Erdman DD, Weinberg GA, et al. Human metapneumovirus among children hospitalized for acute respiratory illness. Emerg Infect Dis. 2004;10:700–705. doi: 10.3201/eid1004.030555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson JL, Lee BE, Bastien N, Li Y. Seasonality and clinical features of human metapneumovirus infection in children in Northern Alberta. J Med Virol. 2005;76:98–105. doi: 10.1002/jmv.20329. [DOI] [PubMed] [Google Scholar]
- 15.Kahn JS. Epidemiology of human metapneumovirus. Clin Microbiol Rev. 2006;19:546–557. doi: 10.1128/CMR.00014-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams JV, Wang CK, Yang CF, Tollefson SJ, House FS, Heck JM, et al. The role of human metapneumovirus in upper respiratory tract infections in children: a 20-year experience. J Infect Dis. 2006;193:387–395. doi: 10.1086/499274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El Sayed Zaki M, Raafat D, El-Metaal AA, Ismail M. Study of human metapneumovirus-associated lower respiratory tract infections in Egyptian adults. Microbiol Immunol. 2009;53:603–608. doi: 10.1111/j.1348-0421.2009.00162.x. [DOI] [PubMed] [Google Scholar]
- 18.Liao RS, Appelgate DM, Pelz RK. An outbreak of severe respiratory tract infection due to human metapneumovirus in a long-term care facility for the elderly in Oregon. J Clin Virol. 2012;53:171–173. doi: 10.1016/j.jcv.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 19.Widmer K, Zhu Y, Williams JV, Griffin MR, Edwards KM, Talbot HK. Rates of hospitalizations for respiratory syncytial virus, human metapneumovirus, and influenza virus in older adults. J Infect Dis. 2012;206:56–62. doi: 10.1093/infdis/jis309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Souza JS, Watanabe A, Carraro E, Granato C, Bellei N. Severe metapneumovirus infections among immunocompetent and immunocompromised patients admitted to hospital with respiratory infection. J Med Virol. 2013;85:530–536. doi: 10.1002/jmv.23477. [DOI] [PubMed] [Google Scholar]
- 21.Syha R, Beck R, Hetzel J, Ketelsen D, Grosse U, Springer F, Horger M. Humane metapneumovirus (HMPV) associated pulmonary infections in immunocompromised adults-initial CT findings, disease course and comparison to respiratory-syncytial-virus (RSV) induced pulmonary infections. Eur J Radiol. 2012;81:4173–4178. doi: 10.1016/j.ejrad.2012.06.024. [DOI] [PubMed] [Google Scholar]
- 22.Cote S, Abed Y, Boivin G. Comparative evaluation of real-time PCR assays for detection of the human metapneumovirus. J Clin Microbiol. 2003;41:3631–3635. doi: 10.1128/JCM.41.8.3631-3635.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mackay IM, Jacob KC, Woolhouse D, Waller K, Syrmis MW, Whiley DM, et al. Molecular assays for detection of human metapneumovirus. J Clin Microbiol. 2003;41:100–105. doi: 10.1128/JCM.41.1.100-105.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maertzdorf J, Wang CK, Brown JB, Quinto JD, Chu M, de Graaf M, et al. Real-time reverse transcriptase PCR assay for detection of human metapneumoviruses from all known genetic lineages. J Clin Microbiol. 2004;42:981–986. doi: 10.1128/JCM.42.3.981-986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bouscambert-Duchamp M, Lina B, Trompette A, Moret H, Motte J, Andreoletti L. Detection of human metapneumovirus RNA sequences in nasopharyngeal aspirates of young French children with acute bronchiolitis by real-time reverse transcriptase PCR and phylogenetic analysis. J Clin Microbiol. 2005;43:1411–1414. doi: 10.1128/JCM.43.3.1411-1414.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuypers J, Wright N, Corey L, Morrow R. Detection and quantification of human metapneumovirus in pediatric specimens by real-time RT-PCR. J Clin Virol. 2005;33:299–305. doi: 10.1016/j.jcv.2004.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pabbaraju K, Wong S, McMillan T, Lee BE, Fox JD. Diagnosis and epidemiological studies of human metapneumovirus using real-time PCR. J Clin Virol. 2007;40:186–192. doi: 10.1016/j.jcv.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hopkins MJ, Redmond C, Shaw JM, Hart IJ, Hart CA, Smyth RL, et al. Detection and characterisation of human metapneumovirus from children with acute respiratory symptoms in northwest England, UK. J Clin Virol. 2008;42:273–279. doi: 10.1016/j.jcv.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 29.Klemenc J, Asad Ali S, Johnson M, Tollefson SJ, Talbot HK, Hartert TV, Edwards KM, Williams JV. Real-time reverse transcriptase PCR assay for improved detection of human metapneumovirus. J Clin Virol. 2012;54:371–375. doi: 10.1016/j.jcv.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van den Hoogen BG, Bestebroer TM, Osterhaus AD, Fouchier RA. Analysis of the genomic sequence of a human metapneumovirus. Virology. 2002;295:119–132. doi: 10.1006/viro.2001.1355. [DOI] [PubMed] [Google Scholar]
- 31.van den Hoogen BG, Herfst S, Sprong L, Cane PA, Forleo-Neto E, de Swart RL, et al. Antigenic and genetic variability of human metapneumoviruses. Emerg Infect Dis. 2004;10:658–666. doi: 10.3201/eid1004.030393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boivin G, Mackay I, Sloots TP, Madhi S, Freymuth F, Wolf D, et al. Global genetic diversity of human metapneumovirus fusion gene. Emerg Infect Dis. 2004;10:1154–1157. doi: 10.3201/eid1006.031097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huck B, Scharf G, Neumann-Haefelin D, Puppe W, Weigl J, Falcone V. Novel human metapneumovirus sublineage. Emerg Infect Dis. 2006;12:147–150. doi: 10.3201/eid1201.050772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agrawal AS, Roy T, Ghosh S, Chawla-Sarkar M. Genetic variability of attachment (G) and Fusion (F) protein genes of human metapneumovirus strains circulating during 2006-2009 in Kolkata, Eastern India. Virol J. 2011;8:67. doi: 10.1186/1743-422X-8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Banerjee S, Sullender WM, Choudekar A, John C, Tyagi V, Fowler K, Lefkowitz EJ, Broor S. Detection and genetic diversity of human metapneumovirus in hospitalized children with acute respiratory infections in India. J Med Virol. 2011;83:1799–1810. doi: 10.1002/jmv.22176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piyaratna R, Tollefson SJ, Williams JV. Genomic analysis of four human metap-neumovirus prototypes. Virus Res. 2011;160:200–205. doi: 10.1016/j.virusres.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choudhary ML, Anand SP, Heydari M, Rane G, Potdar VA, Chadha MS, Mishra AC. Development of a multiplex one step RT-PCR that detects eighteen respiratory viruses in clinical specimens and comparison with real time RT-PCR. J Virol Methods. 2013;189:15–19. doi: 10.1016/j.jviromet.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nidaira M, Taira K, Hamabata H, Kawaki T, Gushi K, Mahoe Y, Maeshiro N, Azama Y, Okano S, Kyan H, Kudaka J, Tsukagoshi H, Noda M, Kimura H. Molecular epidemiology of human metapneumovirus from 2009 to 2011 in Okinawa, Japan. Jpn J Infect Dis. 2012;65:337–340. doi: 10.7883/yoken.65.337. [DOI] [PubMed] [Google Scholar]
- 39.Lamson DM, Griesemer S, Fuschino M, St. George K. Phylogenetic analysis of human metapneumovirus from New York State patients during February through April 2010. J Clin Virol. 2012;53:256–258. doi: 10.1016/j.jcv.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 40.Kim HR, Cho AR, Lee MK, Yun SW, Kim TH. Genotype variability and clinical features of human metapneumovirus isolated from Korean children, 2007 to 2010. J Mol Diagn. 2012;14:61–64. doi: 10.1016/j.jmoldx.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 41.Garcia J, Sovero M, Kochel T, Laguna-Torres VA, Gamero ME, Gomez J, Sanchez F, Arango AE, Jaramillo S, Halsey ES. Human metapneumovirus strains circulating in Latin America. Arch Virol. 2012;157:563–568. doi: 10.1007/s00705-011-1204-8. [DOI] [PubMed] [Google Scholar]
- 42.Apostoli P, Zicari S, Lo Presti A, Ciccozzi M, Ciotti M, Caruso A, Fiorentini S. Human metapneumovirus-associated hospital admissions over five consecutive epidemic seasons: evidence for alternating circulation of different genotypes. J Med Virol. 2012;84:511–516. doi: 10.1002/jmv.23213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, Ren L, Guo L, Xiang Z, Paranhos-Baccala G, et al. Evolutionary Dynamics Analysis of Human Metapneumovirus Subtype A2: Genetic Evidence for Its Dominant Epidemic. PLoS ONE. 2012;7:e34544. doi: 10.1371/journal.pone.0034544. [DOI] [PMC free article] [PubMed] [Google Scholar]