

Abstract
Bovine viral diarrhea virus (BVDV) is a widespread virus in beef and dairy herds. BVDV has been grouped into two genotypes, genotype 1 and genotype 2. In this study, the relative synonymous codon usage (RSCU) values, effective number of codon (ENC) values and nucleotide content were investigated, and a comparative analysis of codon usage patterns for open reading frames (ORFs) of 22 BVDV genomes, including 14 of genotype 1 and 8 of genotype 2, was carried out. A high A+U content and low codon bias were found in BVDV genomes. Depending on the RSCU data, it was found that there was a significant variation in bias of codon usage between the two genotypes, and a geographic factor exists only in genotype-1 of BVDV. The RSCU data have a negative correlation with general average hydrophobicity (GRAVY), aromaticity and nucleotide content. Furthermore, the overall abundance of C and U has no effect on the synonymous codon usage patterns. In contrast, the A and G content showed a significant correlation with the nucleotide content at the third position. In addition, the codon usage patterns of BVDV are similar to those of 22 conserved genes of Bos taurus. Taken together, the genetic characteristics of BVDV possibly result from interactions between natural seclection and mutation pressure.
Electronic supplementary material
The online version of this article (doi:10.1007/s00705-010-0848-0) contains supplementary material, which is available to authorized users.
Keywords: Codon Usage, Bovine Viral Diarrhea Virus, Classical Swine Fever Virus, Codon Usage Bias, Synonymous Codon Usage
Introduction
In 18 out of 20 amino acids (excluding Met and Trp), the degeneracy of the genetic code allows multiple codons to encode the same amino acid, resulting in codon usage bias in genes [7, 24]. Codon usage analysis has been applied to prokaryotes and eukaryotes, such as Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Caenorhabditis elegans and human beings [4, 16, 25, 27]. Some reports have shown that codon usage bias had a high correlation to tRNA abundance, GC content, mRNA secondary structure, exon splicing constraints, translation rate and gene expression level [12, 18, 26] . The study of codon usage can provide some evidence about the molecular evolution of the viruses. It can also enrich our understanding about the relationship between viruses and their hosts by analyzing their codon usage patterns.
BVDV is a member of the genus Pestivirus within the family Flaviviridae. The genus also includes classical swine fever virus (CSFV) and Border disease virus (BDV) of sheep [3, 20]. Based on a comparison of the 5’ untranslated region (UTR) and the Npro- and E2-encoding sequences [23, 30], BVDV can be divided into two different genotypes: BVDV-1 and BVDV-2 [21, 22]. The genome of each genotypes contains a single positive-stranded RNA with a size of approximately 12.3 kb, consisting of a single large open reading frame (ORF) flanked by 5’ and 3’ untranslated regions [6, 8]. The BVDV strains can grow in epithelial cell cultures with cytopathic (CP) or noncytopathic (NCP) effect [17].
Since BVDV is highly genetically variable, little information about synonymous codon usage patterns of BVDV genomes has been acquired to date [13, 29]. To our knowledge, this is the first report of codon usage analysis of BVDV. In this study, we analyzed the codon usage data and base composition of 22 available complete ORFs of BVDV to obtain some clues to the features of genetic evolution of this virus.
Materials and methods
Sequence data
A total of 22 BVDV genomes, consisting of 14 strains of genotype 1 and 8 strains of genotype 2, were used to analyze the relevant factors of synonymous codon usage patterns and nucleotide contents in this study. The genotype, phenotype, country of isolation and GenBank accession numbers of these strains are listed in Table 1. In addition, 22 different well-conserved genes of Bos taurus were selected to examine the relationship between codon preferences in the host and the viruses (Table 2). All of the abovementioned coding sequences were downloaded from NCBI (http://www.ncbi.nlm.nih.gov/Genbank/).
Table 1.
Information about the 22 BVDV genomes used in this study
| No. | Strain | Genotype | Country of isolation | Phenotype | Accession no. |
|---|---|---|---|---|---|
| 1 | 1-NADL | 1 | USA | CP | M311821 |
| 2 | ILLNC | 1 | USA | NCP | U86600 |
| 3 | ILLC | 1 | USA | CP | U86599 |
| 4 | C24 V | 1 | England | CP | AF091605 |
| 5 | NS3 | 1 | USA | CP | AF268278 |
| 6 | C413 | 2 | USA | NCP | AF002227 |
| 7 | KS86-1cp | 1 | Japan | CP | AB078952 |
| 8 | KS86-1ncp | 1 | Japan | NCP | AB078950 |
| 9 | Osloss | 1 | France | CP | M96687 |
| 10 | p4515 | 2 | Canada | NCP | AY149216 |
| 11 | VEDEVAC | 1 | Hungary | CP | AJ585412 |
| 12 | SD-1 | 1 | USA | NCP | M96751 |
| 13 | New York’93 | 2 | USA | CP | AF502399 |
| 14 * | ZM-95 | 1 | China | CP | AF526381 |
| 15 | 1373 | 2 | USA | CP | AY145967 |
| 16 | Singer_Arg | 1 | Argentina | CP | DQ088995 |
| 17 | KE9 | 1 | Germany | NCP | EF101530 |
| 18 | P11Q | 2 | Canada | NCP | AY149215 |
| 19 | XJ-04 | 2 | China | CP | FJ527854 |
| 20 | JZ05-1 | 2 | China | CP | GQ888686 |
| 21 | Hokudai-Lab/09 | 2 | Japan | Unknown | AB567658 |
| 22 | CP7 | 1 | Greece | NCP | U63479 |
* Strain 14 was isolated from swine
Table 2.
Information about 22 conserved genes of Bos taurus
| No. | Gene | Product | Accession no. |
|---|---|---|---|
| 1 | Pepck-C | Phosphoenolpyruvate carboxykinase | AY145503 |
| 2 | CAT | Catalase | NM_001035386 |
| 3 | USP14 | Ubiquitin carboxyl-terminal hydrolase 14 | NM_001075189 |
| 4 | ADAM22 | Disinterin and metalloproteinase | NM_001191131 |
| 5 | USP16 | Ubiquitin carboxyl-terminal hydrolase 16 | NM_001076867 |
| 6 | LTF | Lactotransferrin precursor | NM_180998 |
| 7 | GSK3B | Glycogen synthase kinase-3 beta | NM_001101310 |
| 8 | GHR | Growth hormone receptor precursor | NM_176608 |
| 9 | CRLF3 | Cytokine receptor-like factor 3 | NM_001192650 |
| 10 | PPP3CA | Serine/threonine-protein phosphatase 2B catalytic subunit alpha isoform | NM_174787 |
| 11 | GPX6 | Glutathione peroxidase 6 | NM_001163142 |
| 12 | TLR1 | Toll-like receptor 1 | NM_001046504 |
| 13 | TLR2 | Toll-like receptor 2 | AY634629 |
| 14 | TLR3 | Toll-like receptor 3 precursor | NM_001008664 |
| 15 | TLR4 | Toll-like receptor 4 | AY634630 |
| 16 | TLR5 | Toll-like receptor 5 | GQ248711 |
| 17 | TLR6 | Toll-like receptor 6 | NM_001001159 |
| 18 | TLR7 | Toll-like receptor 7 | NM_001033761 |
| 19 | TLR8 | Toll-like receptor 8 | NM_001033937 |
| 20 | TLR9 | Toll-like receptor 9 precursor | NM_183081 |
| 21 | XCR 1 | Hemokine (C motif) receptor 1 | GU936968 |
| 22 | BOLA-DQA1 | BOLA-DQA 1 protein | BC112472 |
Calculation of relative synonymous codon usage
To investigate the patterns of synonymous codon usage (RSCU) without the confounding influence of amino acid composition among all BVDV samples, the RSCU values of codons in the ORFs of BVDV were calculated according to a formula described in previous reports [25, 32]:
 |
where g ij is the observed number of the ith codon for the jth amino acid, which has ni types of synonymous codons. A codon with an RSCU value of more than 1.0 has a positive codon usage bias, while a value of less than 1.0 has a negative codon usage bias. When the RSCU value is equal to 1.0, it means that this codon is chosen equally and randomly.
The effective number of codons
The effective number of codons (ENC) is used to measure deviation from expected random codon usage of BVDV and is independent of hypotheses involving natural selection [5]. The ENC values range from 20 to 61. The larger the codon preference in a gene is, the smaller the ENC value is. In an extremely biased gene where only one codon is used for each amino acid, this value would be 20, and if all codons were used equally, it would be 61 [28, 31]. The formulas for ENC are as follows:
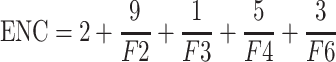 |
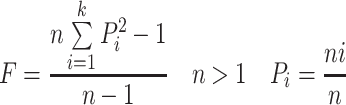 |
The n is the observed number of codons used, k is the number of synonymous codons, and P i is the usage frequency of the ith codon (n i /n). ENC is influenced by the amino acid content of the gene and its length.
The fraction of each codon within its synonymous family
Codon frequency normalizes the codon observations to a fraction for each codon within its synonymous family [1]. To examine the degree of similarity in codon usage between BVDV and that of its host animal (Bos taurus), the fraction of each codon (a total of 59 standard codons, excluding the synonymous single codon for AUG [Met], UGG [Trp] and the three termination codons) within its synonymous family of 22 ORFs of BVDV and 22 genes of Bos taurus was compared.
Statistical analysis
Principal component analysis (PCA) was conducted to analyze the major trend in codon usage pattern among BVDV samples. This is a statistical method that performs linear mapping to extract optimal features from an input distribution in the mean squared error and can be used by self-organizing neural networks to form unsupervised neural preprocessing modules for classification problems [15]. In order to minimize the effect of amino acid composition on codon usage, each ORF is represented as a 59-dimensional vector, and each dimension corresponds to the RSCU value of one sense codon excluding AUG (Met), UGG (Trp) and the three stop codons.
A Spearman’s rank correlation analysis was used to identify relationships among nucleotide content, RSCU and principal component factors of BVDV. A linear least-square regression was conducted to evaluate the correlation between the fraction of synonymous codons in BVDV and that in the genes of Bos taurus. General average hydrophobicity (GRAVY) and aromaticity scores were used to investigate hydrophobic properties of the targeted proteins. Both scores of each protein were obtained using the software Codon W 1.2.4.
Results
The characteristics of synonymous codon usage in BVDV
In order to investigate the extent of codon usage bias in BVDV, all RSCU values of different codons in 22 BVDV strains were calculated. There is only one preferred codon, AGU, with U at the third position; all of the remaining preferred codons end with A, C or G (Table 3). Moreover, the BVDV genome is A+U-rich, with the A+U content ranging from 53.63 to 55.11, with a mean value of 54.46 and S.D. of 0.35, but most of preferentially used codons are G/C-ended codons (G/C-ended: A/T-ended = 10:8), suggesting that the percentage of G+C at the third position may influence the pattern of synonymous codon usage (Table 4). The values of ENC among these BVDV ORFs are similar, which vary from 50.69 to 52.6, with a mean value of 51.43 and an S.D. of 0.46. The data showed that the extent of codon preference in BVDV genes remained basically stable.
Table 3.
Synonymous codon usage in BVDV ORFs
| AA a | Codon | RSCU b | AA | Codon | RSCU b |
|---|---|---|---|---|---|
| Phe | UUU | 0.988 | Gln | CAA | 1.068 |
| UUC | 1.012 | CAG | 0.932 | ||
| Leu | UUA | 0.951 | His | CAU | 0.819 |
| UUG | 1.187 | CAC | 1.181 | ||
| CUU | 0.566 | Asn | AAU | 0.853 | |
| CUC | 0.564 | AAC | 1.147 | ||
| CUA | 1.348 | Lys | AAA | 1.050 | |
| CUG | 1.388 | AAG | 0.95 | ||
| Val | GUU | 0.636 | Asp | GAU | 0.968 |
| GUC | 0.813 | GAC | 1.032 | ||
| GUA | 1.108 | Glu | GAA | 1.074 | |
| GUG | 1.396 | GAG | 0.926 | ||
| Ser c | UCU | 0.747 | Arg | AGA | 2.586 |
| UCC | 0.787 | AGG | 2.718 | ||
| UCA | 1.39 | CGU | 0.114 | ||
| UCG | 0.246 | CGC | 0.169 | ||
| AGU | 1.622 | CGA | 0.15 | ||
| AGC | 1.208 | CGG | 0.264 | ||
| Pro | CCU | 0.99 | Cys | UGU | 0.977 |
| CCC | 0.862 | UGC | 1.023 | ||
| CCA | 1.747 | Tyr | UAU | 0.908 | |
| CCG | 0.401 | UAC | 1.092 | ||
| Thr | ACU | 0.911 | Ala | GCU | 1.032 |
| ACC | 1.066 | GCC | 1.143 | ||
| ACA | 1.609 | GCA | 1.539 | ||
| ACG | 0.414 | GCG | 0.285 | ||
| Gly | GGU | 0.634 | Ile | AUU | 0.550 |
| GGC | 0.745 | AUC | 0.875 | ||
| GGA | 1.026 | AUA | 1.576 | ||
| GGG | 1.376 |
The preferentially used codons for each amino acid are indicated in bold
a Amino acid
b The RSCU value is a mean value of each codon for a particular amino acid
c The preferentially used codon ends in U
Table 4.
Nucleotide content of 22 BVDV genomes
| No. | A% | G% | U% | C% | A3% | C3% | G3% | U3% | A+U% | G+C% | C3/G3% | GRAVY | Aromaticity | ENC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 32.22 | 25.79 | 21.98 | 20.01 | 40.5 | 17.2 | 22.4 | 19.9 | 54.2 | 45.8 | 47.6 | -0.2505 | 0.08751 | 51.947 |
| 2 | 32.26 | 25.68 | 22.04 | 20.02 | 32.1 | 23.3 | 23 | 21.6 | 54.3 | 45.7 | 46.3 | -0.2757 | 0.08876 | 52.114 |
| 3 | 32.28 | 25.84 | 21.75 | 20.13 | 42 | 16.1 | 22.3 | 19.6 | 54 | 46 | 46.9 | -0.2864 | 0.08489 | 52.124 |
| 4 | 32.24 | 25.86 | 21.81 | 20.09 | 34.4 | 21.6 | 22.8 | 21.2 | 54 | 46 | 46.9 | -0.2439 | 0.08646 | 51.435 |
| 5 | 31.65 | 25.95 | 21.98 | 20.41 | 35.1 | 21.1 | 22.8 | 21 | 49.5 | 50.5 | 48.5 | -0.2219 | 0.08492 | 52.593 |
| 6 | 32.95 | 25.15 | 22.16 | 19.75 | 43.5 | 15 | 22.2 | 19.3 | 55.1 | 44.9 | 46 | -0.2148 | 0.08853 | 50.923 |
| 7 | 32.45 | 25.77 | 22.02 | 19.76 | 33.6 | 22.2 | 22.9 | 21.3 | 54.5 | 45.5 | 45.7 | -0.2701 | 0.08625 | 51.521 |
| 8 | 32.55 | 25.63 | 22.17 | 19.65 | 32.9 | 22.8 | 22.9 | 21.5 | 54.7 | 45.3 | 45.7 | -0.2325 | 0.08671 | 51.019 |
| 9 | 31.82 | 25.85 | 22.03 | 20.31 | 39 | 18.3 | 22.5 | 20.2 | 53.8 | 46.2 | 46.8 | -0.2336 | 0.08579 | 51.956 |
| 10 | 32.85 | 25.24 | 22.19 | 19.72 | 46.6 | 12.7 | 22 | 18.6 | 54.8 | 45.2 | 46 | -0.2176 | 0.08766 | 51.072 |
| 11 | 32.16 | 25.69 | 22.15 | 20 | 36.7 | 20 | 22.7 | 20.7 | 54.3 | 45.7 | 46.3 | -0.2489 | 0.08789 | 51.950 |
| 12 | 32.23 | 25.76 | 21.98 | 20.03 | 39.7 | 17.7 | 22.5 | 20.1 | 54.2 | 45.8 | 47.2 | -0.2407 | 0.0862 | 51.455 |
| 13 | 32.87 | 25.19 | 22.12 | 19.82 | 44.3 | 14.4 | 22.2 | 19.1 | 55 | 45 | 46.1 | -0.217 | 0.08740 | 51.026 |
| 14 | 32.05 | 26.29 | 21.92 | 19.74 | 35.9 | 20.5 | 22.7 | 20.8 | 50.6 | 49.4 | 47.4 | -0.2428 | 0.08818 | 51.146 |
| 15 | 32.98 | 25.13 | 22.1 | 19.78 | 45.1 | 13.9 | 22.1 | 18.9 | 55.1 | 44.9 | 45.8 | -0.219 | 0.08817 | 50.761 |
| 16 | 32.46 | 25.58 | 21.89 | 20.07 | 37.4 | 19.4 | 22.6 | 20.5 | 54.3 | 45.7 | 47.3 | -0.2376 | 0.08569 | 51.549 |
| 17 | 32.09 | 25.8 | 22.16 | 19.95 | 38.2 | 18.9 | 22.6 | 20.4 | 54.2 | 45.8 | 46.2 | -0.2414 | 0.08697 | 51.651 |
| 18 | 32.78 | 25.47 | 21.99 | 19.76 | 45.8 | 13.3 | 22.1 | 18.8 | 54.8 | 45.2 | 46.2 | -0.2137 | 0.08676 | 51.072 |
| 19 | 32.94 | 25.31 | 22.15 | 19.6 | 47.4 | 12.2 | 22 | 18.5 | 55.1 | 44.9 | 45.5 | -0.2107 | 0.08750 | 50.85 |
| 20 | 32.59 | 25.56 | 22.06 | 19.79 | 48.1 | 11.6 | 21.9 | 18.3 | 54.6 | 45.4 | 46.7 | -0.2244 | 0.08750 | 50.86 |
| 21 | 32.39 | 25.93 | 22.09 | 19.59 | 42.8 | 15.5 | 22.3 | 19.4 | 54.5 | 45.5 | 46.5 | -0.2311 | 0.08673 | 50.69 |
| 22 | 32.11 | 25.62 | 22.38 | 19.89 | 41.3 | 16.6 | 22.4 | 19.7 | 54.5 | 45.5 | 45.9 | -0.2474 | 0.08728 | 51.97 |
Genetic relationship based on synonymous codon usage
Principal component analysis was carried out to identify the codon usage bias among ORFs. From this, we could detect one major trend in the first axis (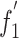 ), which accounted for 26.51% of the total variation, and another major trend in the second axis (
), which accounted for 26.51% of the total variation, and another major trend in the second axis (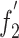 ), which accounted for 13.02% of the total variation. A plot of the
), which accounted for 13.02% of the total variation. A plot of the 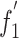 and the
and the  of each gene is shown in Supplementary Fig.1. Compared with the scattered groups of BVDV genotype 1, all BVDV genotype 2 strains aggregated more tightly to some degree. Interestingly, it seems that there is a clear geographical demarcation in the BVDV-1 groups.
of each gene is shown in Supplementary Fig.1. Compared with the scattered groups of BVDV genotype 1, all BVDV genotype 2 strains aggregated more tightly to some degree. Interestingly, it seems that there is a clear geographical demarcation in the BVDV-1 groups.
Compositional properties of all BVDV genomes
Natural selection and mutation pressure are thought to be the main factors that account for codon usage variation in different organisms. The A%, U%, C%, G% and (C+G)% were compared with A3%, C3%, G3%, U3%, (G+C) 3%, respectively. An interesting and complex correlation was observed. In detail, the (C+G)3% values have highly significant correlations with the A%, U%, C%, G% and (C+G)% values, indicating that (C+G)3% may reflect an interaction between mutation pressure and natural selection. In contrast, the U% and C% values did not correlate with the A3%, U3%, G3% and C3% values (Table 5). Both cases suggest that nucleotide constraints possibly influence synonymous codon usage in BVDV. Correlation analysis was used to analyze the relationships among ENC values, (G+C)3% values and (C+G)% values. A highly significant correlation was observed between ENC and (C+G)% (Spearman r = 0.765, p < 0.01), while significant correlation was also observed between ENC and (G+C)3% (Spearman r = 0.534, 0.01 < p<0.05), indicating that codon usage bias is influenced by nucleotide constraints. In addition, the correlation between the 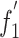 value and A%, C%, G%, U%, A3%, C3%, G3%, U3%, (G+C)%, (G+C) 3% values of each strain was also analyzed. A significant correlation was found between nucleotide composition and synonymous codon usage to some extent (Table 6). The analysis revealed that most of the codon usage bias among ORFs of BVDV strains was directly related to base composition. We found that
value and A%, C%, G%, U%, A3%, C3%, G3%, U3%, (G+C)%, (G+C) 3% values of each strain was also analyzed. A significant correlation was found between nucleotide composition and synonymous codon usage to some extent (Table 6). The analysis revealed that most of the codon usage bias among ORFs of BVDV strains was directly related to base composition. We found that 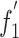 also had a significant negative correlation with the general average hydrophobicity (GRAVY) of each protein (Spearman r = -0.737, p < 0.01), and negative correlation with the aromaticity of each protein (Spearman r = –0.455, p = 0.033 < 0.05), indicating that the expressed sequences are hydrophilic, since they accomplish their functions in the aqueous media of the cell.
also had a significant negative correlation with the general average hydrophobicity (GRAVY) of each protein (Spearman r = -0.737, p < 0.01), and negative correlation with the aromaticity of each protein (Spearman r = –0.455, p = 0.033 < 0.05), indicating that the expressed sequences are hydrophilic, since they accomplish their functions in the aqueous media of the cell.
Table 5.
Correlation analysis between the A, U, C, G content and the A 3, U 3, C 3, G 3 content in all ORFs
| A3% | U3% | G3% | C3% | (G+C) 3% | |
|---|---|---|---|---|---|
| A | r = 0.627** | r = -0.626** | r = -0.631** | r = -0.689** | r = -0.666** |
| U | r = 0.263 N | r = -0.265 N | r = -0.232 N | r = -0.259 N | r = -0.608** |
| G | r = -0.610** | r = 0.608** | r = 0.607** | r = 0.607** | r = 0.614** |
| C | r = -0.416 N | r = 0.417 N | r = 0.407 N | r = 0.414 N | r = 0.695** |
| G+C | r = -0.429* | r = 0.422 N | r = 0.424* | r = 0.426* | r = -0.757** |
** Means p < 0.01
* Means 0.01 < p < 0.05
Table 6.
Correlation analysis between 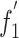 and nucleotide content
and nucleotide content
| A% | C% | G% | U% | G + C% | GRAVY | ||
|---|---|---|---|---|---|---|---|

|
r | -0.736** | 0.555** | 0.74** | -0.463* | 0.494* | -0.737** |
| p | <0.01 | <0.01 | <0.01 | <0.05 | 0.019 | <0.001 | |
| A 3 % | C 3 % | G 3 % | U 3 % | C 3 /G 3 % | Aromaticity | ||
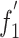
|
r | -0.829 | 0.808 | 0.817 | 0.83 | 0.513* | -0.455* |
| p | <0.01 | <0.01 | <0.01 | <0.01 | 0.015 | 0.033 | |
** Means p < 0.01
* Means 0.01 < p < 0.05
Effect of other factors on codon usage
As shown in Figure 1, a plot of actual ENC values against both the (G+C)3% and the expected ENC value provides a useful display of the main features of codon usage patterns. The curve indicates the expected codon usage if it is influenced only by the (G+C)3% value of the genome:
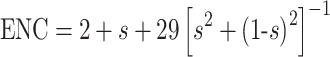 |
where s represents the given (G+C)3% value [31]. However, all of the points with low ENC values lying below the expected curve suggest that although codon usage bias is influenced by mutational pressure, certain other factors must have an influence on the variation of codon usage in these genes. Therefore, we performed another correlation analysis on  in principal component analysis between GRAVY and the aromaticity score of each protein (Table 6).
in principal component analysis between GRAVY and the aromaticity score of each protein (Table 6).
Fig. 1.
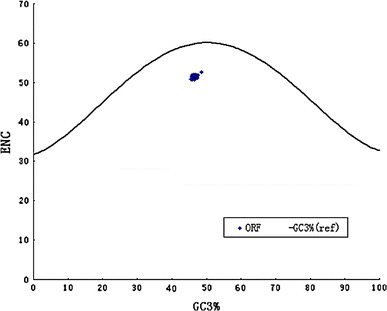
The relationship between the effective number of codons (ENC) and the GC content of the third codon position (GC3)
Comparison of codon usage in BVDV and its host
A plot of average proportions of codons within its synonymous family in BVDV (excluding strain no. 14, which was isolated from swine) and Bos taurus was conducted to explore the relationship between BVDV and its host in codon usage. When two factors are both less than or equal to 0.15, it is defined as a low frequency of usage; and when one factor is greater than or equal to twice of the other factor, it is considered a great difference in frequency. The plot gave a clear linear relationship between BVDV and Bos taurus, showing that the virus and host had very similar patterns of codon usage (r2 = 0.697). The patterns indicate that the least frequently used codons in the host were also the non-preferred codons of the viruses, such as UCG (Ser), CCG (Pro), ACG (Thr), CGU, CGC, CGA, CGG (Arg) and GCG (Ala), and some highly scattered codons including CUA (Leu), AGG (Arg), AUA and AUU (Ile). Linear regression analysis was also performed to investigate the relationship of codon usage patterns between strain 14 and the other BVDV strains. There was no significant difference between the two patterns (P<0.05).
Discussion
Natural selection is a phenomenon that alters the behavior and fitness of living organisms within a given environment. It is the driving force of evolution. Mutation pressure is the change in some gene frequencies due to the repeated occurrence of the same mutations. There are not many biologically realistic situations where mutation pressure is the most important evolutionary process. However, for RNA viruses, the mutation rate is sometimes high enough that mutation pressure needs to be considered.
It is well established that synonymous codon usage reveals genetic information about some viral genomes [10, 14]. In this study, the evidence suggests that the synonymous codon usage bias in BVDV genes is low (mean ENC = 51.43, greater than 40). Therefore, together with published data on codon usage bias of some RNA viruses, such as influenza A H5N1 virus and SARS coronovirus, with mean values of 50.91 and 48.99, respectively [10, 33], the low frequency of codon usage bias for RNA viruses is similar to some degree. Bahir et al. also reported that there is a strong resemblance in codon usage between viruses and their host cells [2]. This suggests that the characteristics of low codon bias may assist BVDV to replicate efficiently in the host cells.
The general association between codon usage indices and composition constraints shows that mutation pressure plays an important role in determining codon usage variation in BVDV. This is supported by the highly significant correlation between codon usage indices (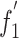 ) and A%, U%, G%, C%, A3%, U3%, G3% and C3% values (Table 6). The relationship between authentic ENC values and (G+C)3% is weaker than that of the expected values (Fig. 2). We suggest that mutation pressure is one of the main factors responsible for the variation of synonymous codon usage in genomes of BVDV. Further analysis showed that these C3% values of BVDV isolates were low, with an average C3 content of 17.47% and an S.D. of 3.05, but it is interesting that six preferential codons are all ended with C (Table 3). Meanwhile, the U3% value is higher than the C3% value (mean U3%: mean C3% = 19.97:17.47), but only one U-ended codon, AGU, is used as a preferentially used codon. This indicates that natural selection is possibly involved in the patterns of synonymous codon usage. No correlation was found between C%, or U% and A3%, U3%, G3%, or C3% (Table 5), suggesting that nucleotide constraints are involved in codon usage patterns due to low U% and C% values. Aromaticity is one of the factors in variations in amino acid usage [19]. The
) and A%, U%, G%, C%, A3%, U3%, G3% and C3% values (Table 6). The relationship between authentic ENC values and (G+C)3% is weaker than that of the expected values (Fig. 2). We suggest that mutation pressure is one of the main factors responsible for the variation of synonymous codon usage in genomes of BVDV. Further analysis showed that these C3% values of BVDV isolates were low, with an average C3 content of 17.47% and an S.D. of 3.05, but it is interesting that six preferential codons are all ended with C (Table 3). Meanwhile, the U3% value is higher than the C3% value (mean U3%: mean C3% = 19.97:17.47), but only one U-ended codon, AGU, is used as a preferentially used codon. This indicates that natural selection is possibly involved in the patterns of synonymous codon usage. No correlation was found between C%, or U% and A3%, U3%, G3%, or C3% (Table 5), suggesting that nucleotide constraints are involved in codon usage patterns due to low U% and C% values. Aromaticity is one of the factors in variations in amino acid usage [19]. The 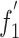 values had a negative correlation with the aromaticity of each protein (Table 5). In this study, the degree of aromaticity had a negative correlation with codon usage bias of BVDV, suggesting that natural selection may be involved in BVDV evolution.
values had a negative correlation with the aromaticity of each protein (Table 5). In this study, the degree of aromaticity had a negative correlation with codon usage bias of BVDV, suggesting that natural selection may be involved in BVDV evolution.
Fig. 2.
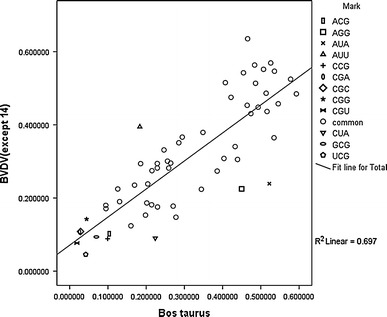
A plot of average proportions of codons of BVDV within its synonymous family (excluding strain no. 14, which was isolated from swine) and Bos taurus (excluding the codons for Trp, Met and the three termination codons)
BVDV was first reported in 1946 [11], and the scattered model of all 14 strains of BVDV-1 may imply that there is more diversity among BVDV-1 strains with the development of evolution (Supplementary Fig. 1). Three BVDV-1 strains isolated from Asia were different from other BVDV-1 strains, implying that the strains isolated from Asia were distantly related to American or European strains. However, the strains from American were more closely related to those from Europe than to those from Asia. The low diversity in BVDV-2 might result from the limited number of samples. It is most likely that the codon usage bias in BVDV is related to genotype and geographic factors.
The remarkable similarity in the codon usage patterns between the viruses and Bos taurus reveals that natural selective pressure gives BVDV higher adaptability to its host. This adaptability makes it possible for the virus to survive in the host cell and to use the components of the cell to produce more of itself. However, there is no evidence that the viruses are generally adapted to the codon usage patterns of their host (AUU, CUA, AGG, and AUA), and this is consistent with mutational bias theory [1]. Although it has been reported that isolate 14 was first found in swine, its nucleotide content is similar to that of strains originating from cattle, suggesting that strain 14 is also a possible cattle-origin virus.
In this study, our analysis reveals that codon usage bias in BVDV is low, and mutation pressure is the main factor that affects codon usage variation in BVDV. Other factors, including base composition, genotype, geography, GRAVY, and even aromaticity may also significantly influence codon usage bias.
Although our study provides a basic understanding of the codon usage patterns of BVDV and the roles played by mutation pressure and natural selection, a more comprehensive analysis is needed to reveal more information about codon usage bias variation within BVDV viruses and the other responsible factors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig.1 A plot of the values of the first axis (2 6.51%) and the second axis (13.02%) of each ORF in principal component analysis. The strains isolated form the same continent are shown in the same color.
Acknowledgements
This work was supported in parts by grants by National Natural Science foundation of China (No. 30700597 and No. 31072143). This study was also supported by the International Science & Technology Cooperation Program of China (No. 2010DFA32640) and the Science and Technology Key Project of Gansu Province (No. 0801NKDA034)
Footnotes
M. Wang and J. Zhang contributed equally to this work.
References
- 1.Adams MJ, Antoniw JF. Codon usage bias amongst plant viruses. Arch Virol. 2004;149:113–135. doi: 10.1007/s00705-003-0186-6. [DOI] [PubMed] [Google Scholar]
- 2.Bahir I, Fromer M, Prat Y, Linial M. Viral adaptation to host: a proteome-based analysis of codon usage and amino acid preferences. Mol Syst Biol. 2009;5:1–14. doi: 10.1038/msb.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becher P, Orlich M, Kosmidou A, Konig M, Baroth M, Thiel HJ. Genetic diversity of pestiviruses: identification of novel groups and implications for classification. Virology. 1999;262:64–71. doi: 10.1006/viro.1999.9872. [DOI] [PubMed] [Google Scholar]
- 4.Bulmer M. Codon usage and intragenic position. J Theor Biol. 1988;133:67–71. doi: 10.1016/S0022-5193(88)80024-9. [DOI] [PubMed] [Google Scholar]
- 5.Castillo-Davis CI, Hartl DL. Genome evolution and developmental constraint in Caenorhabditis elegans. Mol Biol Evol. 2002;19:728–735. doi: 10.1093/oxfordjournals.molbev.a004131. [DOI] [PubMed] [Google Scholar]
- 6.Colett MS, Larson R, Gold C, Strick D, Anderson DK, Purchio AF. Molecular cloning and nucleotide sequence of the pestivirus bovine viral diarrhea virus. Virology. 1988;165:191–199. doi: 10.1016/0042-6822(88)90672-1. [DOI] [PubMed] [Google Scholar]
- 7.Cutter AD, Charlesworth B. Selection intensity on preferred codons correlates with overall codon usage bias in Caenorhabditis remanei. Curr Biol. 2006;16:2053–2057. doi: 10.1016/j.cub.2006.08.067. [DOI] [PubMed] [Google Scholar]
- 8.Deng R, Brock KV. Molecular cloning and nucleotide sequence of a pestivirus genome, noncytopathic bovine viral diarrhea virus strain SD-1. Virology. 1992;191:867–869. doi: 10.1016/0042-6822(92)90262-N. [DOI] [PubMed] [Google Scholar]
- 9.Duret L. Evolution of synonymous codon usage in metazoans. Curr Opin Genet Dev. 2002;12:640–649. doi: 10.1016/S0959-437X(02)00353-2. [DOI] [PubMed] [Google Scholar]
- 10.Gu W, Zhou T, Ma J, Sun X, Lu Z. Analysis of synonymous codon usage in SARS Coronavirus and other viruses in the Nidovirales. Virus Res. 2004;101:155–161. doi: 10.1016/j.virusres.2004.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hillerton JE. Bovine spongiform encephalopathy: current status and possible impacts. J Dairy Sci. 1998;81:3042–3048. doi: 10.3168/jds.S0022-0302(98)75869-2. [DOI] [PubMed] [Google Scholar]
- 12.Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol. 1985;2:13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- 13.Jackova A, Novackova M, Pelletier C, Audeval C, Gueneau E, Haffar A, Petit E, Rehby L, Vilcek S. The extended genetic diversity of BVDV-1: typing of BVDV isolates from France. Vet Res Commun. 2008;32:7–11. doi: 10.1007/s11259-007-9012-z. [DOI] [PubMed] [Google Scholar]
- 14.Jenkins GM, Holmes EC. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003;92:1–7. doi: 10.1016/S0168-1702(02)00309-X. [DOI] [PubMed] [Google Scholar]
- 15.Kanaya S, Kinouchi M, Abe T, Kudo Y, Yamada Y, Nishi T, Mori H, Ikemura T. Analysis of codon usage diversity of bacterial genes with a self-organizing map (SOM): characterization of horizontally transferred genes with emphasis on the E. coli O157 genome. Gene. 2001;276:89–99. doi: 10.1016/S0378-1119(01)00673-4. [DOI] [PubMed] [Google Scholar]
- 16.Karlin S, Mrazek J. What drives codon choices in human genes? J Mol Biol. 1996;262:459–472. doi: 10.1006/jmbi.1996.0528. [DOI] [PubMed] [Google Scholar]
- 17.Liebler-Tenorio EM, Ridpath JF, Neill JD. Distribution of viral antigen and development of lesions after experimental infection of calves with a BVDV 2 strain of low virulence. J Vet Diagn Invest. 2003;15:221–232. doi: 10.1177/104063870301500303. [DOI] [PubMed] [Google Scholar]
- 18.Liu YS, Zhou JH, Chen HT, Ma LN, Ding YZ, Wang M, Zhang J. Analysis of synonymous codon usage in porcine reproductive and respiratory syndrome virus. Infect Genet Evol. 2010;10:797–803. doi: 10.1016/j.meegid.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nayak KC. Mutational bias and Gene expression level shape codon usage in Thermobifida fusca YX. In Silico Biology. 2009;9:337–353. doi: 10.3233/ISB-2009-0421. [DOI] [PubMed] [Google Scholar]
- 20.Paton DJ, Sands JJ, Lowings JP, Smith JE, Ibata G, Edwards S. A proposed division of the pestivirus genus using monoclonal antibodies, supported by cross-neutralisation assays and genetic sequencing. Vet Res. 1995;26:92–109. [PubMed] [Google Scholar]
- 21.Qi F, Gustad T, Lewis TL, Berry ES. The nucleotide sequence of the 5’-untranslated region of bovine viral diarrhoea virus: its use as a probe in rapid detection of bovine viral diarrhoea viruses and border disease viruses. Mol Cell Probes. 1993;7:349–356. doi: 10.1006/mcpr.1993.1052. [DOI] [PubMed] [Google Scholar]
- 22.Ridpath JF, Bolin SR, Dubovi EJ. Segregation of bovine viral diarrhea virus into genotypes. Virology. 1994;205:66–74. doi: 10.1006/viro.1994.1620. [DOI] [PubMed] [Google Scholar]
- 23.Ridpath JF, Neill JD, Vilcek S, Dubovi EJ, Carman S. Multiple outbreaks of severe acute BVDV in North America occurring between 1993 and 1995 linked to the same BVDV2 strain. Vet Microbiol. 2006;114:196–204. doi: 10.1016/j.vetmic.2005.11.059. [DOI] [PubMed] [Google Scholar]
- 24.Sharp PM, Averof M, Lloyd AT, Matassi G, Peden JF. DNA sequence evolution: the sounds of silence. Philos Trans R Soc Lond B Biol Sci. 1995;349:241–247. doi: 10.1098/rstb.1995.0108. [DOI] [PubMed] [Google Scholar]
- 25.Sharp PM, Li WH. Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’ codons. Nucleic Acids Res. 1986;14:7737–7749. doi: 10.1093/nar/14.19.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sorensen MA, Kurland CG, Pedersen S. Codon usage determines translation rate in Escherichia coli. J Mol Biol. 1989;207:365–377. doi: 10.1016/0022-2836(89)90260-X. [DOI] [PubMed] [Google Scholar]
- 27.Stenico M, Lloyd AT, Sharp PM. Codon usage in Caenorhabditis elegans: delineation of translational selection and mutational biases. Nucleic Acids Res. 1994;22:2437–2446. doi: 10.1093/nar/22.13.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tao P, Dai L, Luo M, Tang F, Tien P, Pan Z. Analysis of synonymous codon usage in classical swine fever virus. Virus Genes. 2009;38:104–112. doi: 10.1007/s11262-008-0296-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vilcek S, Mojzisova J, Bajova V, Paulik S, Strojny L, Durkovic B, Hipikova V. A survey for BVDV antibodies in cattle farms in Slovakia and genetic typing of BVDV isolates from imported animals. Acta Vet Hung. 2003;51:229–236. doi: 10.1556/AVet.51.2003.2.11. [DOI] [PubMed] [Google Scholar]
- 30.Vilcek S, Strojny L, Durkovic B, Rossmanith W, Paton D. Storage of bovine viral diarrhoea virus samples on filter paper and detection of viral RNA by a RT-PCR method. J Virol Methods. 2001;92:19–22. doi: 10.1016/S0166-0934(00)00258-5. [DOI] [PubMed] [Google Scholar]
- 31.Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- 32.Zhou JH, Zhang J, Chen HT, Ma LN, Liu YS. Analysis of synonymous codon usage in foot-and-mouth disease virus. Vet Res Commun. 2010;34:393–404. doi: 10.1007/s11259-010-9359-4. [DOI] [PubMed] [Google Scholar]
- 33.Zhou T, Gu W, Ma J, Sun X, Lu Z. Analysis of synonymous codon usage in H5N1 virus and other influenza A viruses. Biosystems. 2005;81:77–86. doi: 10.1016/j.biosystems.2005.03.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.