

Abstract
H9N2 avian influenza viruses sporadically infect humans worldwide. These viruses have also contributed internal genes to H5N1, H5N6, H7N9, and H10N8 viruses, which have been isolated from humans with infections and are a substantial public health threat. To investigate the potential pathogenic mechanism of the H9N2 virus, we performed serial lung-to-lung passage of an avirulent H9N2 avian influenza virus (A/Chicken/Shandong/416/2016 [SD/416]) in mice to increase the pathogenicity of this virus. We generated a mouse-adapted (MA) virus that exhibited increased viral titers in the lungs, caused severe lung damage in mice, and induced body weight loss in mice; however, the avirulent parental virus did not cause any clinical symptoms in infected mice. Global gene expression analysis was performed and indicated that the transcriptional responses of these viruses were distinct. The lungs of mice infected with the MA virus exhibited the downregulation of genes related to innate immunity and ubiquitin-mediated proteolysis, which was not seen in infections with the avirulent parental virus. These data indicated that the MA virus might evade immune surveillance and changed its replication capacity to increase the viral replication level and pathogenicity. Our study demonstrates that host factors play an important role in the adaptive evolution of influenza virus in new hosts.
Electronic supplementary material
The online version of this article (10.1007/s00430-020-00656-4) contains supplementary material, which is available to authorized users.
Keywords: Influenza virus, Adaptation, Pathogenicity, Gene expression analysis
Introduction
Influenza A virus is a segmented negative-sense RNA virus composed of 18 hemagglutinin (HA) subtypes (H1–18) and 11 neuraminidase (NA) subtypes (N1–11), based on serotype and evolutionary relationships. Except for the H17N10 and H18N11 viruses that were isolated from bats [1, 2], all influenza virus subtypes (H1–16, N1–9) have been detected in waterfowl, which are a mixing vessel for influenza viruses [3]. The H1, H2, and H3 subtypes can be stably transmitted to humans and have caused several pandemics in human history [4]. In addition, influenza viruses, such as H5N1, H5N6, H7N9, and H9N2, have sporadically infected humans directly from terrestrial animals, suggesting that other influenza virus subtypes also pose a potential threat as the cause of new pandemics through further evolution [5–7].
The H9N2 AIVs were first detected in poultry in the United States in 1966 [8]. In 1999, H9N2 viruses were isolated from two patients in Hong Kong, and since then, H9N2 viruses have been occasionally reported in humans [9]. Most significantly, H9N2 viruses function as a “vehicle” that deliver different subtypes of influenza viruses from avian species to humans [9]. H9N2 viruses contributed internal genes to H5N1, H5N6, H7N9, and H10N8 viruses isolated from humans with infections [10–14]. In our previous studies, the co-infection of a bird with H4N6 and H9N2 viruses led to a high frequency of reassortment and generated some reassortants that had higher virulence than that of the wild-type viruses in mammals [15].
In nature, the adaptive evolution of influenza viruses in new hosts occurs regularly, and the viruses acquire higher replication abilities, pathogenicity, transmissibility, antigenicity, and receptor-binding properties by undergoing adaptive evolution in the new host. In the laboratory, the serial passage of AIVs in mammalian hosts can result in adaptive viral changes. Currently, scientists have mainly studied the pathogenic mechanism of viral adaptability in regard to viral replication, transmission, and virulence. For example, Wang et al. reported that the combination of 147 L and 627 K in the PB2 protein was a major contributor to the adaptation and increased virulence of the H9N2 influenza virus in mice [16]. The PB1–K577E mutation is a marker of the mammalian adaptation of the H9N2 virus [17]. The NS1–T5S substitution has previously been observed in a mouse-adapted (MA) avian H9N2 subtype (A/Ck/Korea/163/04H9N2), implying that this mutation may be preferentially selected during the viral adaptation of avian influenza in mammals [18]. However, scientists rarely study the pathogenic mechanism of the interaction between an adapted virus and the new host.
Mice are the most common animal model used for pathogenicity studies of influenza virus. Many low-pathogenic avian influenza (LPAI) viruses have been shown to acquire high virulence after passage in mice [16, 17]. Here, we sought to explore the impact of mammalian adaptation on the pathogenesis of an H9N2 AIV by performing five blind serial lung-to-lung passages in mice. We obtained an MA virus with increased virulence. To study the pathogenic mechanisms of the adapted virus toward the host, we compared and analyzed the gene expression in the lung tissues of infected mice to obtain information concerning the host response to infection.
Materials and methods
Ethics statement
The mouse study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Ministry of Science and Technology of the People’s Republic of China. The protocols for animal studies were approved by the Committee on the Ethics of Animal Experiments of Liaocheng University (BRDW-XBS-17-04).
Virus and mouse experiments
H9N2 (A/Chicken/Shandong/416/2016 [SD/416]) was isolated from a chicken. Virus stocks were propagated in specific-pathogen-free (SPF) embryonated chicken eggs (Merial, Beijing, China) at 37 °C and stored at − 80 °C.
Adaptation of the SD/416 virus in mice
The adaptation of the SD/416 virus to the mouse lung was carried out by serial lung-to-lung passage. Three 6-week-old female BALB/c mice (Vital River, Beijing, China) were intranasally (i.n.) inoculated with 106 50% egg infective dose (EID50) of SD/416 and designated line A, line B, and line C. Mice were monitored for weight loss and survival and were sacrificed 3 days postinoculation (dpi). The lungs were harvested in 1 mL of PBS, and 0.05 mL of supernatant from the centrifuged homogenate was used to inoculate mice for the next passage. Passaging was performed five times.
According to the changes in mouse body weights, MA virus had acquired adaption in line C. Then the viruses in line C were plaque-purified and then cultured in eggs. After amplification, we performed next-generation sequencing. The results showed the monoclonal with identical sequences that were two amino acid alterations in PB2 (T129A/E627K), a single codon change in NP (T62S) and two changes in NS1 (T5S/S42A).
Mouse experiments and lung tissue collection
Groups of 14 6-week-old female BALB/c mice were anesthetized with CO2 and i.n. inoculated with 106 EID50/mL test viruses in a volume of 0.05 mL. Three mice were euthanized at 3 dpi, and the nasal turbinates, lungs, kidneys, spleens, and brains were collected for virus titration in SPF-embryonated chicken eggs. Organs of each mouse were titrated separately in SPF-embryonated chicken eggs. For histological examinations, three other mouse lungs were collected from mice at 3 dpi after virus inoculation and were fixed in 10% phosphate-buffered formalin. The sections were stained with hematoxylin–eosin (H & E). For expression microarray analysis, groups of three mice were euthanized at 3 dpi, and their lung tissues were immediately harvested, frozen, and stored in liquid nitrogen. The remaining five mice in each group were monitored daily for 14 days for weight loss and survival. Mice that lost more than 25% of their initial body weight were recognized as being at the experimental endpoint and were humanely euthanized.
Expression microarray analysis and bioinformatics
Total RNA isolation and mRNA amplification were performed with equal amounts of total RNA isolated from the frozen lungs of influenza virus and mock-infected mice. Expression oligonucleotide arrays were performed using RNA isolated from the lung tissues of three individual animals per group at 3 dpi. Total genome gene expression was detected using an Illumina Mouse WG-6 Expression Bead chip (San Diego, CA, USA). To evaluate gene expression changes in response to infection, we used a random variance model (RVM) to filter differentially expressed genes (DEGs), because the RVM t test performs much better than the standard t test when identifying significantly different genes between groups. To select for genes that were the most relevant to infection, a P value of < 0.05 was considered significantly different [19, 20]. The detailed information of DEGs is described in Supplementary table 1.
Gene ontology (GO) analysis was performed using MAS 3.0 software, which is based on the Database for Annotation, Visualization, and Integrated Discovery [21], to analyze DEG functions. Fisher’s exact test was used, and the threshold for statistical significance was set at P < 0.01. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was used to identify significantly different regulatory pathways. Fisher’s exact test and the Chi-square test were used to identify the significant pathways. The threshold for statistical significance was set at P < 0.01. The detailed information of GO analysis and pathway analysis is described in Supplementary tables 2 and 3, respectively.
Confirmation of tag-mapped genes by qRT–PCR
Total RNA of qRT-PCR and total RNA of expression microarray analysis were a batch of samples. cDNA was synthesized by First-Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Microarray results were validated by qRT-PCR using SYBR green-based (TAKARA, Japan) detection with an ABI PRISM 7500 cycler (Applied Biosystems, Foster City, CA). To confirm DEGs, six genes (LY86, TLR13, CYP2A5, TCER2A, MAP3K6, and CLECL2A) were selected at random for quantitative RT-PCR assays. The GAPDH gene was used for expression normalization. RT-qPCR data were analyzed using the standard 2−ΔΔCT method and presented as the fold expression normalized to the reference GAPDH gene. The expression of six genes (LY86, TLR13, CYP2A5, TCER2A, MAP3K6, and CLECL2A) by qRT-PCR agreed with the results from Tag-seq analysis (Fig. 4).
Fig. 4.

qRT-PCR validation of DEGs. a–f These genes, which included LY86 (a), TLR13 (b), CYP2A5 (c), TCER2A (d), MAP3K6 (e), and CLECL2A (f), were previously identified or reported. Fold change refers to qRT-PCR and DEGs
Statistical analyses
The statistical significance of differences between experimental groups was evaluated using analysis of variance (one-way ANOVA and Newman–Keuls) in the GraphPad Prism five software package (GraphPad Software, La Jolla, CA, USA). P values less than 0.05 were considered statistically significant.
Accession numbers
All sequences were deposited in the GenBank database under the accession numbers (MN857550–MN857557).
Results
Adaptation of the wild-type SD/416 virus in mice
To acquire the MA virus, we performed serial passaging of an avirulent H9N2 virus [A/Chicken/Shandong/416/2016 (SD/416)] in mice, beginning with the intranasal inoculation of 106 EID50 of virus per mouse. The survival of infected animals was monitored, and weight changes in the mice were recorded every day. The mice infected with SD/416 did not show any clinical symptoms of disease. From three independent series of sequential lung-to-lung passages of virus in BALB/c mice, line A and line B of mice did not detect clinical symptoms and loss of body weight. In line C, the infected mice began to lose body weight at the third passage. The body weight loss at the fifth passage was up to 17.7% of the initial body weight, and the mice showed clinical symptoms of disease (Fig. S1). These results showed that the MA virus had acquired adaption at passage five in line C.
Enhanced virulence of the MA virus in mice
We compared the pathogenicity of the SD/416 and MA viruses in mice. Mice infected with the adapted virus lost 20.8% of their body weight, and one mouse died at 5 dpi (50% mean lethal dose (MLD50), > 6.38 log EID50) (Fig. 1a, b, and Fig. S2). In contrast, the body weight of the mice infected with the SD/416 virus continued to increase, and no morbidity or mortality was observed in mice infected with the SD/416 virus (Fig. 1b). The MA viral titers in infected mouse lungs and nasal turbinates reached 104.75 and 101.83 EID50/mL, respectively (Fig. 1c). In contrast, SD/416 lung titers reached 102.3 EID50/mL, and virus was not detected in the turbinates (Fig. 1c). Mouse kidneys, spleens, and brains were free of virus for both SD/416- and MA-infected mice. However, MA-infected mice incurred severe pathological damage, including focal pulmonary consolidation, the necrosis of alveolar epithelial cells, edema, and interstitial broadening as well as inflammatory cell infiltration (Fig. 2a). The lung damage of the mice infected by the MA virus was much more severe than that of the mice infected with SD/416 (Fig. 2a–c).
Fig. 1.

Virulence of SD/416 and MA viruses in mice. Groups of five BALB/c mice were i.n. inoculated with 106 EID50 of the SD/416 or MA virus or with PBS (a, b). a Body weight changes in mice infected with 106 EID50 of the SD/416 or MA virus or with PBS. b Percent survival of mice infected with 106 EID50 of the SD/416 or MA virus or with PBS. c Viral titers in the lungs and nasal turbinates of infected mice at 3 dpi. Groups of three BALB/c mice were i.n. inoculated with 106 EID50 of the SD/416 or MA virus or with PBS. Lungs and nasal turbinates were collected at 3 days p.i. for virus titration in eggs (c). Each value represents the mean of triplicates. Asterisk denotes statistical significance (**P < 0.01; ***P < 0.001) for one-way ANOVA and Newman–Keuls to SD/416, respectively
Fig. 2.

Histopathologic analysis of lesions in the lungs of mice infected with SD/416 and MA viruses. Mice were i.n. inoculated with 106 EID50 of the test virus, and the lungs were collected for pathological study. The lungs of SD/416 virus-inoculated animals showed no obvious pathological symptoms (H&E staining) (a), the lungs of MA virus-inoculated mice showed severe pathological lesions (H&E staining) (b), and the lungs of control animals showed no pathological symptoms (H&E staining) (c). Images a–c were taken at ×200 magnification
We then sequenced by the next generation and compared the complete genomes of both viruses. We found two amino acid alterations in PB2 (T129A/E627K), a single codon change in NP (T62S) and two changes in NS1 (T5S/S42A). The E627K mutation was previously associated with an increase in virulence and adaptation to mammals [22, 23].
Host gene expression analysis
To investigate the potential pathogenic mechanism of the H9N2 MA virus towards the new host, we performed mRNA expression analysis of lung tissues from infected and control mice during acute infection (3 dpi). We identified 975 differentially expressed transcripts, which included 390 upregulated and 585 down-regulated genes, between the uninfected controls and the SD/416-infected mouse lungs (Table S1). We also found 2511 differentially expressed transcripts, which included 756 upregulated and 1755 down-regulated genes, between the control and the MA virus-infected mouse lungs (Table S1). Compared to the control mice, MA- and SD/416-infected mice shared 117 upregulated genes and 323 down-regulated genes (Fig. 3).
Fig. 3.
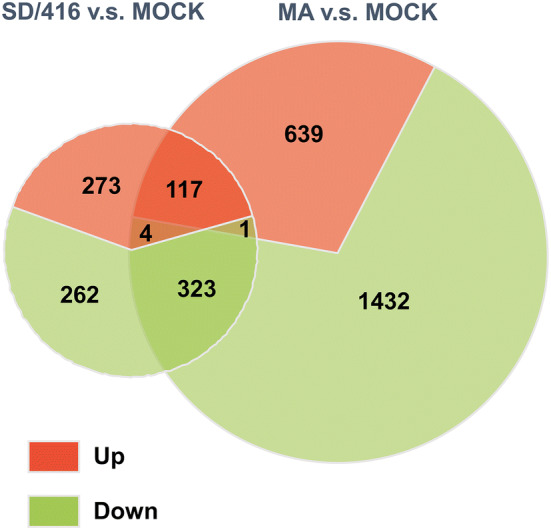
Venn diagram of the significant DEGs between the SD/416 and MA viral subtypes
To validate the results of RNA-Seq, six genes were chosen for quantitative RT-PCR (qRT-PCR) analysis. Figure 4 displays the gene expression patterns derived from the RNA-Seq and qRT -PCR experiments. For six genes, i.e., LY86, TLR13, CYP2A5, TCER2A, MAP3K6, and CLECL2A, the qRT-PCR expression profiles completely agreed with the RNA-Seq data (Fig. 4). These data suggested that the results of the RNA-Seq analysis were reliable indicators of the overall changes in the gene expression.
MA viral infections impact the innate immune response in the lung
Three mice infected with MA or SD/416 were euthanized at 3 dpi to harvest mouse lung tissues for expression microarray analysis. To examine the biological functions of the DEGs, we used GO enrichment analysis to examine the genes of the SD/416 and MA-infected mouse lungs (Table 1). This analysis method enriched the key genes and the surrounding genes of related biological functions, and in this study, we only focused on the key genes. We found that the transcriptional activity of the innate immune response and proteolysis were altered (Tables S2 and S3). The key genes of the innate immune response were down-regulated by infections with the MA virus compared with the control (Table S2). In addition to GO enrichment analysis and pathway analysis, key DEGs were assembled into a functional pathway, generating a global view of the host–pathogen immune response to influenza virus infection (Fig. 5). Genes were color coded according to the signaling pathways and gene expression differences (Fig. 5). Compared to the control group, the genes of MA-infected mouse lungs encoded three pattern-recognition receptors, including RIG-I-like receptors (RLRs), Toll-like receptors (TLRs), and NOD-like receptors (NLRs) (Table S3). We found changes in TLR3 and TLR7 that have been implicated in the defense against influenza virus infections [24, 25]. The expression levels of the TLR genes were reduced in response to the MA virus infection, suggesting that the MA virus inhibits TLR signaling through TLR3 and TLR7. The CYLD and TANK genes, which are RLR family members, were down-regulated in MA-infected mouse lungs but were not down-regulated in response to SD/416 infection (Fig. 5). CYLD is a negative regulator of RIG-I-mediated innate antiviral responses [26]. TANK interacts with MAVS, TRIF, and TBK1 to regulate RLR- and TLR-mediated interferon production [27]. We also found that the NLR signaling group members CASP1 and CASP8 decreased after MA viral infections (Fig. 5). To validate the results of the innate immune response in the lung, two genes (IRF5 and TLR2) that are down-regulated in both SD/416 and MA virus-infected mouse lungs were chosen for qRT-PCR analysis (Fig. S3a, b). Overall, the MA virus inhibited TLR, RLR, and NLR signaling more than the SD/416 virus, which may be related to the greater pathogenicity of the MA virus.
Table 1.
Top 30 selected biological functions of the SD/416 and MA-infected mouse lungs
| Biological function | −log10 (p value) | Total no. of molecules in function | No. of molecules in function with increased expression | |
|---|---|---|---|---|
| SD/416 | MA | |||
| Cell cycle | 66.49 | 565 | 63 | 121 |
| Cell division | 45.73 | 333 | 49 | 78 |
| Protein transport | 45.22 | 538 | 22 | 96 |
| Transport | 43.91 | 1712 | 77 | 238 |
| Mitosis | 37.85 | 241 | 40 | 61 |
| Response to DNA damage stimulus | 32.59 | 361 | 15 | 67 |
| DNA repair | 30.88 | 291 | 14 | 59 |
| Phosphorylation | 26.67 | 684 | 38 | 83 |
| DNA replication | 25.94 | 123 | 10 | 37 |
| Transcription, DNA-dependent | 25.11 | 1756 | 18 | 181 |
| mRNA processing | 24.57 | 278 | 0 | 51 |
| Regulation of transcription DNA-dependent | 23.84 | 1852 | 22 | 193 |
| Protein phosphorylation | 23.03 | 611 | 37 | 73 |
| Metabolic process | 22.78 | 991 | 49 | 125 |
| Negative regulation of apoptotic process | 22.77 | 480 | 31 | 79 |
| Apoptotic process | 20.01 | 540 | 23 | 64 |
| Innate immune response | 19.62 | 180 | 17 | 37 |
| Vesicle-mediated transport | 18.77 | 190 | 0 | 37 |
| Ion transport | 18.75 | 570 | 30 | 73 |
| Positive regulation of cell proliferation | 18.19 | 382 | 22 | 71 |
| RNA splicing | 17.10 | 212 | 0 | 37 |
| Biological_process | 17.00 | 10237 | 0 | 222 |
| Cell differentiation | 12.55 | 636 | 33 | 88 |
| Signal transduction | 11.77 | 2212 | 0 | 71 |
| Multicellular organismal development | 11.01 | 954 | 35 | 101 |
| Proteolysis | 8.76 | 584 | 17 | 88 |
| Transmembrane transport | 8.48 | 385 | 19 | 23 |
| Drug metabolic process | 8.37 | 14 | 5 | 6 |
| G-protein-coupled receptor signaling pathway | 8.29 | 1850 | 0 | 24 |
| Oxidation–reduction process | 7.57 | 706 | 42 | 89 |
Fig. 5.

Influenza virus infection induces the production of innate immune response components that recognize viral nucleic acids. The gene expression of SD/416 (upper half) and MA (lower half) viruses. The same colors indicate functionally related gene classes. Genes depicted in white are those that either are not observed or are not differentially expressed during infection. All colored genes represent down-regulated genes
The MA virus alters ubiquitin-mediated proteolysis
Three mice infected with MA or SD/416 were euthanized at 3 dpi to harvest mouse lung tissues for expression microarray analysis. As mentioned above, MA viral infections alter the expression of proteolytic genes (Table 1). The pathway analysis of the genes involved with proteolysis revealed that these genes were involved in ubiquitin-mediated proteolysis in MA virus-infected mice compared with the control (Table S2). This pathway was generally absent in SD/416-infected mice compared with the control (Table S3). Eighteen genes were induced with the MA virus infection, and only two genes were induced with SD/416 infection. These 18 genes were members of E1 (SAE1 and UBA6), E2 (UBE2A, UBE2C, UBE2J1, and UBE2N), and E3 (WWP2, HERC4, RCHY1, FANCL, CUL2, TCEB2, BIRC3, RBX1, CDC16, CDC20, ANAPC4, and CDC27) (Fig. 6). Further research on the biological functions of these genes showed that approximately one-third of the genes (RBX1, UBE2N, WWP2, RCHY1, UBE2J1, and HERC4) [28–33] play roles in antiviral innate immunity, while others (CDC16, SAE1 and CUL2) [34, 35] are related to viral replication. To validate the results of ubiquitin-mediated proteolysis in the lung, two genes (UBE2A and CDC20) that are down-regulated in both SD/416 and MA virus-infected mouse lungs were chosen for qRT-PCR analysis (Figs. S3C, D). This suggests that the MA virus had a stronger replication ability and higher virulence, and these characteristics were related to the downregulation of the ubiquitin–proteasome system (UPS).
Fig. 6.

Significant differences in the expression of genes related to the UPS distinguish SD/416- and MA-infected mice at 3 dpi. The gene expression of SD/416 (upper half) and MA (lower half) viruses. The same colors indicate functionally related gene classes. Genes depicted in white are those that are either not observed or are not differentially expressed during infection. All colored genes represent down-regulated genes
Discussion
Previous studies have investigated the pathogenesis of adapted viruses from the perspective of the virus itself, but have rarely studied the interaction between the adapted virus and the new host. In the current study, we acquired an adapted virus by performing five passages of a virus in mice. The viral titers in the MA virus-infected lungs were almost 1000-fold greater than those in the SD/416-infected lungs, and the MA virus could kill one in five infected mice (MLD50 > 6.38 log EID50). The transcriptome data indicated that the MA virus activated the innate immune response and ubiquitin-mediated proteolysis. The differences in the host transcriptional responses to infection mirrored the clinical manifestations of the disease, with a clear response profile distinguishing the MA virus from SD/416. This study described the pathogenesis of influenza virus resulting from the interaction between the adapted virus and the new host.
H9N2 influenza virus is an avian virus with low pathogenicity, so mandatory immunization is not required. Because of this, the H9N2 virus is easily amplified and disseminated in nature, increasing the probability of transmission to other birds and humans. In China, H9N2 infections in humans were documented in 1999, 2003, and 2013 [9]. India, Cambodia, Romania, the United States, Nigeria, and Vietnam have reported that poultry workers were serologically positive for H9N2 [36–43]. In addition, H9N2 virus can act as a “vehicle” to deliver different subtypes of AIVs to humans. The internal genes of H5N1, H5N6, H7N9, and H10N8 viruses, which pose a substantial threat to public health, came from H9N2 viruses [10–13]. The mechanisms through which the H9 avian influenza virus adapts to mammals and crosses the species barrier are a scientific problem that needs to be solved now. The “serial passage of virus” approach is an experimental method for adapting an AIV to mammals to make the virus pathogenic to mammals, which could simulate the phenomenon of viruses crossing the interspecies barrier in a natural environment [16]. Therefore, we chose a wild-type SD/416 virus that was avirulent in mice for our tests. Here, we gained a virulent H9N2 virus using the lung-to-lung passages in BALB/c mice, and the virulent virus acquired five amino acid alterations [PB2 (T129A/E627K), NP (T62S), and NS1 (T5S/S42A)]. The PB2/E627K substitution not only contributed to the increased virulence and transmission efficiency of viruses in mammals, but also affected virus escape [44, 45]. The NP protein plays an important role in both viral assembly and RNA synthesis, and the NP–T62S substitution located near the RNA-binding region which affected the formation of RNP [46]. The NS1 protein counteracts innate immune responses by several mechanisms. In this study, the amino acids residue S42A of the NS1 locates near the nuclear localization signals, and interacts with RIG-I, TRIM25 to affect the innate immune responses [47]. In addition, the NS1–T5S substitution was commonly appeared in the mouse-adapted virus [18]. This study indicated that H9N2 viruses can acquire pathogenicity under the appropriate circumstances and could pose a substantial public health threat.
Global transcriptome data have the potential to reveal the systemic changes in host gene expression that occur in the process of viral infection, which could be beneficial for understanding the interaction mechanisms and the pathogenesis of viruses and host [48, 49]. To study the interaction between the adapted virus and the new host, we used global transcriptome technology to analyze the gene expression in the lung of mice infected by SD/416 and MA viruses. In this study, the global transcriptome data showed that the gene expression patterns for protein-coding genes significantly changed after the lung tissue was infected by MA viruses. We used GO enrichment analysis to examine the biological functions of the DEGs and found that many biological functions had unclear effects on viral pathogenicity. However, the innate immune response and proteolysis were clearly related to the virulence of influenza virus.
Innate immunity is the first line of defense against invading pathogens and plays a pivotal role in viral infection. Microarray analysis revealed that the transcription of genes involved in the innate immune response was inhibited. This inhibition included the suppression of TLR, RLR, and NLR signaling in response to infection with the MA virus. This may be related to the adapted virus evading immune surveillance. Previous reports have demonstrated that the NS1 protein is a very potent inhibitor of immunity that allows influenza virus to efficiently escape immune surveillance and to establish an infection in the host [50]. Furthermore, the NS1 protein that contains S42A mutation can interact with RIG-I, TRIM25 to affect the innate immune responses [47]. The PB2 E627K mutation permits escape from a species-specific restriction factor that targets viral polymerases [44]. In addition, antigenic drift in NP enabled viral escape from recognition by cytotoxic T lymphocytes [51]. The results of our analysis suggest that adapted viruses evade immune surveillance by adaptive mutations.
Ubiquitin-mediated proteolysis plays key roles in regulating innate antiviral signaling pathways and the life cycle of influenza viruses [52]. In this study, some of the identified genes act as activators that regulate antiviral innate immunity. The downregulation of the expression of these genes can help viruses to escape immune surveillance. For instance, UBE2N as an activator that is essential for RIG-I-mediated MAVS aggregation in antiviral innate immunity [32]. In coronaviruses, RCHY1 interacts with the SARS unique domain to trigger the degradation of p53 and alters antiviral innate immunity [30]. The genes involved with ubiquitin-mediated proteolysis are involved in viral replication. CDC16 is an RAB1 GTPase-activating protein that mediates viral replication. SAE1 enzymes activate E1 by sumoylation and can restrict intracellular viral movement [35]. Perhaps, the ability of adapted viruses to replicate more is associated with the downregulation of these genes.
This study demonstrates that H9N2 viruses are a substantial threat to public health. Importantly, if a novel virus is generated via adaptation from birds to humans, this virus may be virulent in mammal. Further study is needed on the mechanism by which the adapted virus escapes immune surveillance and gains a higher ability to replicate. Therefore, these findings further underscore the pandemic potential of the H9N2 AIV and provide mechanistic insights into the replication and virulence of this influenza virus among mammalian hosts.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank the National Natural Science Foundation of China, the Natural Science Foundation of Shandong Province, and the State Key Laboratory of Veterinary Biotechnology. This work was supported by the National Natural Science Foundation of China (31702262 and 31702261), the Natural Science Foundation of Shandong Province (ZR2017BC057 and ZR2017BC048), and the State Key Laboratory of Veterinary Biotechnology (SKLVBF201906).
Compliance with ethical standards
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jing Guo, Email: guojing@lcu.edu.cn.
Xuyong Li, Email: lixuyong@lcu.edu.cn.
References
- 1.Tong SX, Zhu XY, Li Y, Shi M, Zhang J, Bourgeois M, Yang H, Chen XF, Recuenco S, Gomez J, Chen LM, Johnson A, Tao Y, Dreyfus C, Yu WL, McBride R, Carney PJ, Gilbert AT, Chang J, Guo Z, Davis CT, Paulson JC, Stevens J, Rupprecht CE, Holmes EC, Wilson IA, Donis RO. New world bats harbor diverse influenza A viruses. Plos Pathog. 2013 doi: 10.1371/journal.ppat.1003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu Y, Wu Y, Tefsen B, Shi Y, Gao GF. Bat-derived influenza-like viruses H17N10 and H18N11. Trends Microbiol. 2014;22(4):183–191. doi: 10.1016/j.tim.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olsen B, Munster VJ, Wallensten A, Waldenstrom J, Osterhaus AD, Fouchier RA. Global patterns of influenza a virus in wild birds. Science. 2006;312(5772):384–388. doi: 10.1126/science.1122438. [DOI] [PubMed] [Google Scholar]
- 4.Taubenberger JK, Kash JC. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe. 2010;7(6):440–451. doi: 10.1016/j.chom.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, Chen J, Jie Z, Qiu H, Xu K, Xu X, Lu H, Zhu W, Gao Z, Xiang N, Shen Y, He Z, Gu Y, Zhang Z, Yang Y, Zhao X, Zhou L, Li X, Zou S, Zhang Y, Li X, Yang L, Guo J, Dong J, Li Q, Dong L, Zhu Y, Bai T, Wang S, Hao P, Yang W, Zhang Y, Han J, Yu H, Li D, Gao GF, Wu G, Wang Y, Yuan Z, Shu Y. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med. 2013;368(20):1888–1897. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- 6.Peiris M, Yuen KY, Leung CW, Chan KH, Ip PL, Lai RW, Orr WK, Shortridge KF. Human infection with influenza H9N2. Lancet. 1999;354(9182):916–917. doi: 10.1016/S0140-6736(99)03311-5. [DOI] [PubMed] [Google Scholar]
- 7.Claas EC, de Jong JC, van Beek R, Rimmelzwaan GF, Osterhaus AD. Human influenza virus A/HongKong/156/97 (H5N1) infection. Vaccine. 1998;16(9–10):977–978. doi: 10.1016/S0264-410X(98)00005-X. [DOI] [PubMed] [Google Scholar]
- 8.Homme PJ, Easterday BC. Avian influenza virus infections. I. characteristics of influenza A-Turkey-Wisconsin-1966 virus. Avian Dis. 1970;14(1):66–74. doi: 10.2307/1588557. [DOI] [PubMed] [Google Scholar]
- 9.Li X, Shi J, Guo J, Deng G, Zhang Q, Wang J, He X, Wang K, Chen J, Li Y, Fan J, Kong H, Gu C, Guan Y, Suzuki Y, Kawaoka Y, Liu L, Jiang Y, Tian G, Li Y, Bu Z, Chen H. Genetics, receptor binding property, and transmissibility in mammals of naturally isolated H9N2 Avian Influenza viruses. Plos Pathog. 2014;10(11):e1004508. doi: 10.1371/journal.ppat.1004508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He J, Liu BY, Gong L, Chen Z, Chen XL, Hou S, Yu JL, Wu JB, Xia ZC, Latif A, Gao R, Su B, Liu Y. Genetic characterization of the first detected human case of avian influenza A (H5N6) in Anhui Province, East China. Sci Rep. 2018;8(1):15282. doi: 10.1038/s41598-018-33356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guan Y, Shortridge KF, Krauss S, Chin PS, Dyrting KC, Ellis TM, Webster RG, Peiris M. H9N2 influenza viruses possessing H5N1-like internal genomes continue to circulate in poultry in southeastern China. J Virol. 2000;74(20):9372–9380. doi: 10.1128/JVI.74.20.9372-9380.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Q, Shi J, Deng G, Guo J, Zeng X, He X, Kong H, Gu C, Li X, Liu J, Wang G, Chen Y, Liu L, Liang L, Li Y, Fan J, Wang J, Li W, Guan L, Li Q, Yang H, Chen P, Jiang L, Guan Y, Xin X, Jiang Y, Tian G, Wang X, Qiao C, Li C, Bu Z, Chen H. H7N9 influenza viruses are transmissible in ferrets by respiratory droplet. Science. 2013;341(6144):410–414. doi: 10.1126/science.1240532. [DOI] [PubMed] [Google Scholar]
- 13.Chen H, Yuan H, Gao R, Zhang J, Wang D, Xiong Y, Fan G, Yang F, Li X, Zhou J, Zou S, Yang L, Chen T, Dong L, Bo H, Zhao X, Zhang Y, Lan Y, Bai T, Dong J, Li Q, Wang S, Zhang Y, Li H, Gong T, Shi Y, Ni X, Li J, Zhou J, Fan J, Wu J, Zhou X, Hu M, Wan J, Yang W, Li D, Wu G, Feng Z, Gao GF, Wang Y, Jin Q, Liu M, Shu Y. Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection: a descriptive study. Lancet. 2014;383(9918):714–721. doi: 10.1016/S0140-6736(14)60111-2. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Cui P, Zeng X, Jiang Y, Li Y, Yang J, Pan Y, Gao X, Zhao C, Wang J, Wang K, Deng G, Guo J. Characterization of avian influenza H5N3 reassortants isolated from migratory waterfowl and domestic ducks in China from 2015 to 2018. Transbound Emerg Dis. 2019;66(6):2605–2610. doi: 10.1111/tbed.13324. [DOI] [PubMed] [Google Scholar]
- 15.Li XY, Liu BT, Ma SJ, Cui P, Liu WQ, Li YB, Guo J, Chen HL. High frequency of reassortment after co-infection of chickens with the H4N6 and H9N2 influenza A viruses and the biological characteristics of the reassortants. Vet Microbiol. 2018;222:11–17. doi: 10.1016/j.vetmic.2018.06.011. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Sun Y, Xu Q, Tan Y, Pu J, Yang H, Brown EG, Liu J. Mouse-adapted H9N2 influenza A virus PB2 protein M147L and E627K mutations are critical for high virulence. PLoS ONE. 2012;7(7):e40752. doi: 10.1371/journal.pone.0040752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamiki H, Matsugo H, Kobayashi T, Ishida H, Takenaka-Uema A, Murakami S, Horimoto T. A PB1-K577E mutation in H9N2 influenza virus increases polymerase activity and pathogenicity in mice. Viruses. 2018 doi: 10.3390/v10110653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park KJ, Song MS, Kim EH, Kwon HI, Baek YH, Choi EH, Park SJ, Kim SM, Kim YI, Choi WS, Yoo DW, Kim CJ, Choi YK. Molecular characterization of mammalian-adapted Korean-type avian H9N2 virus and evaluation of its virulence in mice. J Microbiol. 2015;53(8):570–577. doi: 10.1007/s12275-015-5329-4. [DOI] [PubMed] [Google Scholar]
- 19.Wright GW, Simon RM. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 2003;19(18):2448–2455. doi: 10.1093/bioinformatics/btg345. [DOI] [PubMed] [Google Scholar]
- 20.Clarke R, Ressom HW, Wang A, Xuan J, Liu MC, Gehan EA, Wang Y. The properties of high-dimensional data spaces: implications for exploring gene and protein expression data. Nat Rev Cancer. 2008;8(1):37–49. doi: 10.1038/nrc2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H, Li XY, Guo J, Li L, Chang C, Li YY, Bian C, Xu K, Chen HL, Sun B. The PB2 E627K mutation contributes to the high polymerase activity and enhanced replication of H7N9 influenza virus. J Gen Virol. 2014;95:779–786. doi: 10.1099/vir.0.061721-0. [DOI] [PubMed] [Google Scholar]
- 23.Cheng K, Yu Z, Chai H, Sun W, Xin Y, Zhang Q, Huang J, Zhang K, Li X, Yang S, Wang T, Zheng X, Wang H, Qin C, Qian J, Chen H, Hua Y, Gao Y, Xia X. PB2-E627K and PA-T97I substitutions enhance polymerase activity and confer a virulent phenotype to an H6N1 avian influenza virus in mice. Virology. 2014;468-470C:207–213. doi: 10.1016/j.virol.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 24.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303(5663):1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 25.Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, Chignard M, Si-Tahar M. Detrimental contribution of the toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PloS Pathog. 2006;2(6):e53. doi: 10.1371/journal.ppat.0020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedman CS, O'Donnell MA, Legarda-Addison D, Ng A, Cardenas WB, Yount JS, Moran TM, Basler CF, Komuro A, Horvath CM, Xavier R, Ting AT. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008;9(9):930–936. doi: 10.1038/embor.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian S, Fan W, Liu T, Wu M, Zhang H, Cui X, Zhou Y, Hu J, Wei S, Chen H, Li X, Qian P. Seneca valley virus suppresses host type I interferon production by targeting adaptor proteins MAVS, TRIF, and TANK for cleavage. J Virol. 2017 doi: 10.1128/JVI.00823-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding S, Mooney N, Li B, Kelly MR, Feng N, Loktev AV, Sen A, Patton JT, Jackson PK, Greenberg HB. Comparative proteomics reveals strain-specific beta-TrCP degradation via rotavirus NSP1 hijacking a host Cullin-3-Rbx1 complex. PloS Pathog. 2016;12(10):e1005929. doi: 10.1371/journal.ppat.1005929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paparisto E, Woods MW, Coleman MD, Moghadasi SA, Kochar DS, Tom SK, Kohio HP, Gibson RM, Rohringer TJ, Hunt NR, Di Gravio EJ, Zhang JY, Tian M, Gao Y, Arts EJ, Barr SD. Evolution-guided structural and functional analyses of the HERC family reveal an ancient marine origin and determinants of antiviral activity. J Virol. 2018 doi: 10.1128/JVI.00528-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma-Lauer Y, Carbajo-Lozoya J, Hein MY, Mueller MA, Deng W, Lei J, Meyer B, Kusov Y, von Brunn B, Bairad DR, Hunten S, Drosten C, Hermeking H, Leonhardt H, Mann M, Hilgenfeld R, von Brunn A. p53 down-regulates SARS coronavirus replication and is targeted by the SARS-unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc Natl Acad Sci USA. 2016;113(35):E5192–E5201. doi: 10.1073/pnas.1603435113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su WC, Chen YC, Tseng CH, Hsu PW, Tung KF, Jeng KS, Lai MM. Pooled RNAi screen identifies ubiquitin ligase itch as crucial for influenza A virus release from the endosome during virus entry. Proc Natl Acad Sci USA. 2013;110(43):17516–17521. doi: 10.1073/pnas.1312374110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi Y, Yuan B, Zhu W, Zhang R, Li L, Hao X, Chen S, Hou F. Ube2D3 and Ube2N are essential for RIG-I-mediated MAVS aggregation in antiviral innate immunity. Nat Commun. 2017;8:15138. doi: 10.1038/ncomms15138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng T, Deng L, Lu X, Pan W, Wu Q, Dai J. Ubiquitin-conjugating enzyme UBE2J1 negatively modulates interferon pathway and promotes RNA virus infection. Virol J. 2018;15(1):132. doi: 10.1186/s12985-018-1040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Westrich JA, Warren CJ, Klausner MJ, Guo K, Liu CW, Santiago ML, Pyeon D. Human papillomavirus 16 E7 stabilizes APOBEC3A protein by inhibiting cullin 2-dependent protein degradation. J Virol. 2018 doi: 10.1128/JVI.01318-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holscher C, Sonntag F, Henrich K, Chen Q, Beneke J, Matula P, Rohr K, Kaderali L, Beil N, Erfle H, Kleinschmidt JA, Muller M. The SUMOylation pathway restricts gene transduction by adeno-associated viruses. Plos Pathog. 2015;11(12):e1005281. doi: 10.1371/journal.ppat.1005281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang M, Fu CX, Zheng BJ. Antibodies against H5 and H9 Avian influenza among poultry workers in China. New Engl J Med. 2009;360(24):2583–2584. doi: 10.1056/NEJMc0900358. [DOI] [PubMed] [Google Scholar]
- 37.Pawar SD, Tandale BV, Raut CG, Parkhi SS, Barde TD, Gurav YK, Kode SS, Mishra AC. Avian influenza H9N2 seroprevalence among poultry workers in Pune, India, 2010. PLoS ONE. 2012 doi: 10.1371/journal.pone.0036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blair PJ, Putnam SD, Krueger WS, Chum C, Wierzba TF, Heil GL, Yasuda CY, Williams M, Kasper MR, Friary JA, Capuano AW, Saphonn V, Peiris M, Shao HX, Perez DR, Gray GC. Evidence for avian H9N2 influenza virus infections among rural villagers in Cambodia. J Infect Public Heal. 2013;6(2):69–79. doi: 10.1016/j.jiph.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okoye J, Eze D, Krueger WS, Heil GL, Friary JA, Gray GC. Serologic evidence of avian influenza virus infections among Nigerian agricultural workers. J Med Virol. 2013;85(4):670–676. doi: 10.1002/jmv.23520. [DOI] [PubMed] [Google Scholar]
- 40.Coman A, Maftei DN, Krueger WS, Heil GL, Friary JA, Chereches RM, Sirlincan E, Bria P, Dragnea C, Kasler I, Gray GC. Serological evidence for avian H9N2 influenza virus infections among Romanian agriculture workers. J Infect Public Heal. 2013;6(6):438–447. doi: 10.1016/j.jiph.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 41.Gray GC, Ferguson DD, Lowther PE, Heil GL, Friary JA. A national study of US bird banders for evidence of avian influenza virus infections. J Clin Virol. 2011;51(2):132–135. doi: 10.1016/j.jcv.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q, Ju L, Liu P, Zhou J, Lv X, Li L, Shen H, Su H, Jiang L, Jiang Q. Serological and virological surveillance of avian influenza A virus H9N2 subtype in humans and poultry in Shanghai, China, between 2008 and 2010. Zoonoses Public Hlth. 2015;62(2):131–140. doi: 10.1111/zph.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uyeki TM, Nguyen DC, Rowe T, Lu XH, Hu-Primmer J, Huynh LP, Hang NLK, Katz JM. Seroprevalence of antibodies to avian influenza A (H5) and A (H9) viruses among market poultry workers, Hanoi, Vietnam, 2001. PLoS ONE. 2012 doi: 10.1371/journal.pone.0043948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mehle A, Doudna JA. Adaptive strategies of the influenza virus polymerase for replication in humans. Proc Natl Acad Sci USA. 2009;106(50):21312–21316. doi: 10.1073/pnas.0911915106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Min JY, Santos C, Fitch A, Twaddle A, Toyoda Y, DePasse JV, Ghedin E, Subbarao K. Mammalian adaptation in the PB2 gene of avian H5N1 influenza virus. J Virol. 2013;87(19):10884–10888. doi: 10.1128/JVI.01016-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang T, Wang TC, Zhao PS, Liang M, Gao YW, Yang ST, Qin C, Wang CY, Xia XZ. Antisense oligonucleotides targeting the RNA binding region of the NP gene inhibit replication of highly pathogenic avian influenza virus H5N1. Int Immunopharmacol. 2011;11(12):2057–2061. doi: 10.1016/j.intimp.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 47.Jiao P, Tian G, Li Y, Deng G, Jiang Y, Liu C, Liu W, Bu Z, Kawaoka Y, Chen H. A single-amino-acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice. J Virol. 2008;82(3):1146–1154. doi: 10.1128/JVI.01698-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma W, Belisle SE, Mosier D, Li X, Stigger-Rosser E, Liu Q, Qiao C, Elder J, Webby R, Katze MG, Richt JA. 2009 pandemic H1N1 influenza virus causes disease and upregulation of genes related to inflammatory and immune responses, cell death, and lipid metabolism in pigs. J Virol. 2011;85(22):11626–11637. doi: 10.1128/JVI.05705-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zou W, Chen D, Xiong M, Zhu J, Lin X, Wang L, Zhang J, Chen L, Zhang H, Chen H, Chen M, Jin M. Insights into the increasing virulence of the swine-origin pandemic H1N1/2009 influenza virus. Sci Rep. 2013;3:1601. doi: 10.1038/srep01601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernandez-Sesma A. The influenza virus NS1 protein: inhibitor of innate and adaptive immunity. Infect Disord Drug Targets. 2007;7(4):336–343. doi: 10.2174/187152607783018754. [DOI] [PubMed] [Google Scholar]
- 51.Voeten JT, Bestebroer TM, Nieuwkoop NJ, Fouchier RA, Osterhaus AD, Rimmelzwaan GF. Antigenic drift in the influenza A virus (H3N2) nucleoprotein and escape from recognition by cytotoxic T lymphocytes. J Virol. 2000;74(15):6800–6807. doi: 10.1128/JVI.74.15.6800-6807.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luo H. Interplay between the virus and the ubiquitin-proteasome system: molecular mechanism of viral pathogenesis. Curr Opin Virol. 2016;17:1–10. doi: 10.1016/j.coviro.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.