

Abstract
The ubiquitin proteasome system plays important role in virus infection. A previous study showed that the proteasome inhibitor MG132 could potentially affect hepatitis E virus (HEV) replication. In this study, we found that MG132 could inhibit HEV and hepatitis C virus (HCV) replication-related luciferase activity in subgenomic models. Furthermore, treatment with MG132 in a HEV infectious model resulted in a dramatic reduction in the intracellular level of HEV RNA. Surprisingly, MG132 concurrently inhibited the expression of a luciferase gene used as a control as well as a wide range of host genes. Consistently, the total cellular RNA and protein content was concurrently reduced by MG132 treatment, suggesting a nonspecific antiviral effect.
Keywords: Ubiquitin proteasome system, MG132, Hepatitis E virus
Introduction
The ubiquitin proteasome system (UPS), which serves as a major pathway for protein degradation and modification in eukaryotic cells, can be utilized by many types of viruses [1–3]. Previous studies have demonstrated that UPS can regulate viral RNA-dependent RNA polymerase (RdRp), which mediates viral RNA synthesis [1, 4–6]. In addition, UPS can also regulate ubiquitylation and degradation of some viral structural proteins [7–9] and thus represents a potential antiviral target.
Hepatitis E virus (HEV) is a single-strand positive-sense RNA virus that belongs to the family Hepeviridae. It is a small non-enveloped virus with a 7.2-kb RNA genome, which is capped at the 5′ termini and polyadenylated at the 3′ termini [10]. Outbreaks of hepatitis E occur periodically throughout the developing world. It typically causes an acute and self-limiting infection, but fulminant hepatitis and high mortality (reaching 25 %) have been described in cases of pregnant women. In the western world, HEV mainly affects immunocompromised patients with a high risk of developing chronic hepatitis [11]. However, no proven medication is available to treat hepatitis E. A recent study reported potent antiviral effects of a well-known proteasome inhibitor, MG132, against HEV [12]. In a Renilla-luciferase-coupled HEV replication model, the authors showed that treatment with MG132 resulted in dramatic reduction of HEV-related luciferase activity [12]. These important findings have inspired us to further evaluate the effects of MG132 in two HEV cell culture models.
Materials and methods
In this study, two human hepatoma cell line (Huh7)-based HEV cell culture models were employed: a subgenomic HEV replicon containing Gaussia luciferase reporter (p6-Luc) in which the accumulation of secreted luciferase serves as a reporter for HEV replication, and a full-length infectious model (p6) in which Huh7 cells were electroporated with full-length HEV genomic RNA (Kernow-C1 p6 clone, GenBank accession number JQ679013) [13]. Two firefly luciferase cell models were also used: a cell line for normalization in which stable expression of luciferase is driven by a phosphoglycerate kinase (PKG) promoter (Huh7-PGK) and a hepatitis C virus (HCV, also a single-strand positive-sense RNA virus) subgenomic cell culture model (Huh7-ET) [14]. The Gaussia luciferase and firefly luciferase activity were measured as described previously [13] using a Lumi Star Optima luminescence counter (BMG Lab Tech, Offenburg, Germany). MTT assays were performed as described previously [15]. The absorbance of each well was read using a microplate absorbance reader (Bio-Rad) at a wavelength of 490 nm.
RNA was isolated using a Macherey-Nagel NucleoSpin RNA II Kit (Bioke, Leiden, The Netherlands) and quantified using a NanoDrop ND-1000 (Thermo, DE, USA). cDNA was prepared from total RNA using a cDNA Synthesis Kit (Takara Bio Inc.). HEV, GAPDH, RP2 (human retinitis pigmentosa 2), CyA (cyclophilin A), CyB (cyclophilin B), CD81 (cluster of differentiation 81) and IMPDH2 (inosine-5′-monophosphate dehydrogenase 2) were quantified by SYBR-Green-based real-time PCR. The HEV primer sequences were 5′-ATTGGCCAGAAGTTGGTTTTCAC-3′ (sense) and 5′-CCGTGGCTATAATTGTGGTCT-3′ (antisense), the primer sequences for the housekeeping gene GAPDH were 5′-TGTCCCCACCCCCAATGTATC-3′ (sense) and 5′-CTCCGATGCCTGCTTCACTACCTT-3′ (antisense), and the primers for the housekeeping gene RP2 were 5′-CCCATTAAACTCCAAGGCAA-3′ (sense) and 5′-AAGCTGAGGATGCTCAAAGG-3′ (antisense). The primer sequences for CyA were 5′-GGCAAATGCTGGACCCAACACA-3′ (antisense) and 5′-TGCTGGTCTTGCCATTCCTGGA-3′ (sense), and the primers for CyB were 5′-AACGCAGGCAAAGACACCAACG-3′ (antisense) and 5′-TCTGTCTTGGTGCTCTCCACCT-3′ (sense). The primers for CD81 were 5′-CTGCTTTGACCACCTCAGTGCT-3′ (antisense) and 5′-TGGCAGCAATGCCGATGAGGTA-3′ (sense), and the primers for IMPDH2 were 5′-AGTGGCTCCATCTGCATTACGC-3′ (antisense) and 5′-GGATTCCTCCATCAGCAATGACC-3′ (sense).
For Western blot, 100,000 cells were seeded in a 6-well plate and treated with MG132 for 48 h. Cell lysates (300 µl) were heated for 5 minutes at 95 °C followed by loading 30 µl of sample onto a 10 % sodium dodecyl sulfate polyacrylamide gel and separating by electrophoresis. Mouse β-actin antibody (1:1000) was used as primary antibody. For SDS-PAGE, after electrophoresis for 90 min at 120 V, the gel was stained in Coomassie brilliant blue solution and distained.
Results
Consistent with a previous study [12], treatment with 1 µM and 10 µM MG132 did not significantly impair cellular metabolic activity or viability as determined by MTT assay after 24 h (Fig. 1A) and 48 h (Fig. 1B), although a minor inhibitory effect was observed when cells were treated with 10 µM MG132 for 48 h. As expected, treatment with 1 µM MG132 potently inhibited HEV-replication-related Gaussia luciferase activity in the p6-Luc model (Fig. 1) after 24 h and 48 h. Furthermore, we tested this proteasome inhibitor in the Huh7-based hepatitis C virus subgenomic model (Huh7-ET). Consistently, MG132 inhibited HCV-coupled firefly luciferase activity (Fig. 1). Surprisingly, when Huh7-PGK cells were treated with MG132, the control firefly luciferase activity driven by the PGK promoter was also potently inhibited (Fig. 1). These results raised concerns regarding the specificity of the effect of MG132 on viral replication.
Fig. 1.

Nonspecific effects of the proteasome inhibitor MG132 on luciferase activity. Treatment with MG132 after 24 h (A) and 48 h (B) resulted in dramatic reduction of luciferase activity in subgenomic HEV replicon (p6-Luc) (mean ± SD, n = 12), HCV replicon (Huh7-ET) (mean ± SD, n = 4) and Huh7 cells constantly expressing a control luciferase gene under the control of the PGK promoter (Huh7-PGK) (mean ± SD, n = 4). MG132 treatment did not strongly affect cellular metabolic activity or viability, as determined by MTT assay (OD490 value) (mean ± SD, n = 4), although a minor inhibitory effect was observed after treatment at 10 µM for 48 h
To investigate further, the HEV infectious model (p6) was treated with MG132 for 48 h. The relative levels of HEV viral RNA and two host reference genes (GAPDH and RP2) were quantified by SYBR-based qRT-PCR. As shown in Fig. 2A, treatment with 1 or 10 µM MG132 resulted in a significant decrease in intracellular HEV RNA by 32 ± 19 % and 76 ± 24 % (mean ± SD, n = 6, p < 0.01), respectively. Strikingly, the expression levels of two references genes, GAPDH and RP2 were concurrently decreased. In addition, the expression of four other host genes that we tested, CyA, CyB, CD81 and IMPDH2, also decreased simultaneously (Fig. 2B). These results confirm that the effect of MG132 is nonspecific.
Fig. 2.
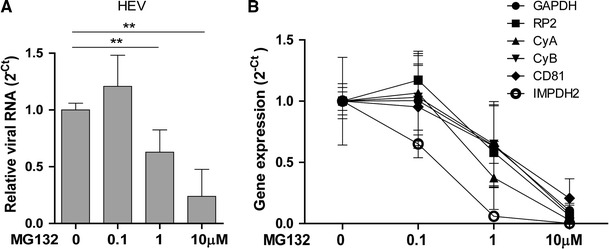
Significantly decrease in HEV viral RNA levels after MG132 treatment (A). (B) The expression of two reference genes (GAPDH, RP2) and four host genes (CyA, CyB, CD81 and IMPDH2) were concurrently inhibited by treatment with MG132. Relative gene expression was quantified by qRT-PCR. Data are presented as 2−Ct and normalized to the untreated control (mean ± SD, n = 6). **P < 0.01
Next, we measured the RNA concentration and total protein content of the cells after MG132 treatment and we found that MG132 treatment (1 µM and 10 µM) drastically reduced the total cellular RNA content (Fig. 3A). Furthermore, cells that were treated with MG132 and lysed showed reduced cellular protein expression. As shown in Fig. 3B, the protein level of internal reference β-actin was decreased after treatment with 1 µM and 10 µM MG132, and the total protein content was also reduced (Fig. 3C). However, the effects of MG132 at the protein level were less profound than that at the RNA level. These results suggest that MG132 inhibits expression and translation of a broad range of genes rather than having a specific effect on viral infection.
Fig. 3.
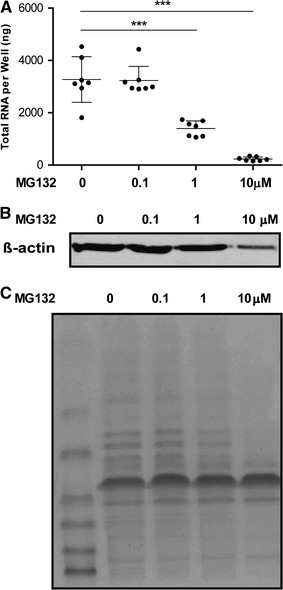
Decrease in cellular RNA and protein content after MG132 treatment. (A) Total intracellular RNA in cells was dramatically reduced by MG132 treatment after 48 h. RNA concentration was determined using a NanoDrop 2000 spectrophotometer (mean ± SD, n = 7). (B) Protein expression of internal reference β-actin was inhibited by MG132 (in particular with 10 µM) treatment after 48 h. The same volumes of cell lysates were loaded, and the protein level was determined by Western blot. (C) Total intracellular protein was reduced by MG132 treatment (in particular with 10 µM) after 48 h. The same volumes of cell lysate were loaded, and the gel was stained in Coomassie brilliant blue solution and distained. **P < 0.01 ***P < 0.001
Discussion
There is substantial evidence suggesting that the cellular UPS is associated with viral infection. RdRp, the essential enzyme for viral replication, can be regulated by UPS in turnip yellow mosaic virus (TYMV) [1], Sindbis virus [4], hepatitis A virus (HAV) [5] and HCV [6] infections. Virus-encoded proteases cleave viral polyprotein proteolytically but can also mediate the processing of many host proteins [16]. Mature 3C proteases of HAV and encephalomyocarditis virus (EMCV) have been shown to be subject to rapid, ubiquitin-mediated protein degradation [17, 18]. As a combat strategy, some viral proteases have been shown to contain de-ubiquitinating enzyme activity. Papain-like cysteine proteases of SARS coronavirus [19], HEV [20] and foot-and-mouth disease virus (FMDV) [21] have the ability to hydrolyze ubiquitinating substrates. Therefore, modulating the UPS represents as a potential antiviral strategy.
Treatment with the proteasome inhibitor MG132 has been shown to decrease the titer of porcine circovirus type 2 (PCV2) at an early stage of infection [22]. Treatment with MG132 has also been shown to decrease the activity of Renilla luciferase expressed from an HEV replicon [12]. However, our study raised concerns regarding the specificity of the effect of MG132 on HEV replication. Although we confirmed the inhibitory effects on luciferase activity in both the HEV and HCV replicon models, MG132 also inhibited constitutively expressed luciferase in control cells. Furthermore, in the full-length HEV model, although MG132 treatment reduced HEV RNA levels, it also simultaneously inhibited the expression of reference genes and other host genes. We further demonstrated that MG132 dramatically decreases the levels of total intracellular RNA and protein, which explains its nonspecific effect on viral infection.
It is not surprising that inhibition of this system could exert variety of effects on cell physiology, since the UPS plays an essential role in the processing of cellular proteins. Proteasomes promptly degrade ubiquitylated proteins [23], and some of these proteins are important mediators of cell-cycle progression and apoptosis [24]. MG132 has been shown to induce the expression of death receptor 5 (DR5), a receptor for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), resulting in enhanced sensitivity to TRAIL-induced apoptosis in cancer cells [25, 26]. Thus, inhibition of this major intracellular protein degradation pathway could nonspecifically affect viral infection, but we do not fully exclude that UPS may also specifically modulate certain viruses [2].
In summary, this study demonstrated that inhibition of HEV infection by the proteasome inhibitor MG132 is nonspecific. Thus, we should be careful in interpreting data regarding the effects and mechanisms of proteasome inhibitors on viral infection. Although proteasome inhibitors are in the preclinical phase of testing as anticancer agents [24, 27], we would call for caution in developing proteasome-targeted antiviral therapies.
Acknowledgments
The authors would like to thank Dr. Suzanne U. Emerson (National Institute of Allergy and Infectious Diseases, NIH, USA) for generously providing the plasmids to generate subgenomic and full-length HEV genomic RNA. The authors also thank the Netherlands Organization for Scientific Research (NWO/ZonMw) for a VENI grant (No. 916-13-032), the European Association for the Study of the Liver (EASL) for a Sheila Sherlock Fellowship, the Dutch Digestive Foundation (MLDS) for a career development grant (No. CDG 1304), the Daniel den Hoed Foundation for a Centennial Award grant (to Q. Pan), and the China Scholarship Council for funding PhD fellowships for L. Xu (201306300027) and X. Zhou (No. 201206150075).
Abbreviations
- UPS
Ubiquitin proteasome system
- HEV
Hepatitis E virus
- HCV
Hepatitis C virus
- RdRp
RNA-dependent RNA polymerase
- PGK
Phosphoglycerate kinase
- TYMV
Turnip yellow mosaic virus
- HAV
Hepatitis A virus
- RP2
Human retinitis pigmentosa 2
- CyA
Cyclophilin A
- CyB
Cyclophilin B
- CD81
Cluster of differentiation 81
- IMPDH2
Inosine-5′-monophosphate dehydrogenase 2
References
- 1.Camborde L, Planchais S, Tournier V, Jakubiec A, Drugeon G, Lacassagne E, Pflieger S, Chenon M, Jupin I. The ubiquitin-proteasome system regulates the accumulation of Turnip yellow mosaic virus RNA-dependent RNA polymerase during viral infection. Plant Cell. 2010;22(9):3142–3152. doi: 10.1105/tpc.109.072090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi AG, Wong J, Marchant D, Luo H. The ubiquitin-proteasome system in positive-strand RNA virus infection. Rev Med Virol. 2013;23(2):85–96. doi: 10.1002/rmv.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Isaacson MK, Ploegh HL. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe. 2009;5(6):559–570. doi: 10.1016/j.chom.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Groot RJ, Rumenapf T, Kuhn RJ, Strauss EG, Strauss JH. Sindbis virus RNA polymerase is degraded by the N-end rule pathway. Proc Natl Acad Sci USA. 1991;88(20):8967–8971. doi: 10.1073/pnas.88.20.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Losick VP, Schlax PE, Emmons RA, Lawson TG. Signals in hepatitis A virus P3 region proteins recognized by the ubiquitin-mediated proteolytic system. Virology. 2003;309(2):306–319. doi: 10.1016/S0042-6822(03)00071-0. [DOI] [PubMed] [Google Scholar]
- 6.Gao G, Luo H. The ubiquitin-proteasome pathway in viral infections. Can J Physiol Pharmacol. 2006;84(1):5–14. doi: 10.1139/y05-144. [DOI] [PubMed] [Google Scholar]
- 7.Yuksek K, Chen WL, Chien D, Ou JH. Ubiquitin-independent degradation of hepatitis C virus F protein. J Virol. 2009;83(2):612–621. doi: 10.1128/JVI.00832-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shirakura M, Murakami K, Ichimura T, Suzuki R, Shimoji T, Fukuda K, Abe K, Sato S, Fukasawa M, Yamakawa Y, Nishijima M, Moriishi K, Matsuura Y, Wakita T, Suzuki T, Howley PM, Miyamura T, Shoji I. E6AP ubiquitin ligase mediates ubiquitylation and degradation of hepatitis C virus core protein. J Virol. 2007;81(3):1174–1185. doi: 10.1128/JVI.01684-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fan Z, Zhuo Y, Tan X, Zhou Z, Yuan J, Qiang B, Yan J, Peng X, Gao GF. SARS-CoV nucleocapsid protein binds to hUbc9, a ubiquitin conjugating enzyme of the sumoylation system. J Med Virol. 2006;78(11):1365–1373. doi: 10.1002/jmv.20707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamar N, Bendall R, Legrand-Abravanel F, Xia NS, Ijaz S, Izopet J, Dalton HR. Hepatitis E. Lancet. 2012;379(9835):2477–2488. doi: 10.1016/S0140-6736(11)61849-7. [DOI] [PubMed] [Google Scholar]
- 11.Zhou X, de Man RA, de Knegt RJ, Metselaar HJ, Peppelenbosch MP, Pan Q. Epidemiology and management of chronic hepatitis E infection in solid organ transplantation: a comprehensive literature review. Rev Med Virol. 2013;23(5):295–304. doi: 10.1002/rmv.1751. [DOI] [PubMed] [Google Scholar]
- 12.Karpe YA, Meng XJ. Hepatitis E virus replication requires an active ubiquitin-proteasome system. J Virol. 2012;86(10):5948–5952. doi: 10.1128/JVI.07039-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou X, Wang Y, Metselaar HJ, Janssen HL, Peppelenbosch MP, Pan Q. Rapamycin and everolimus facilitate hepatitis E virus replication: revealing a basal defense mechanism of PI3K-PKB-mTOR pathway. J Hepatol. 2014;61:746–754. doi: 10.1016/j.jhep.2014.05.026. [DOI] [PubMed] [Google Scholar]
- 14.Pan Q, de Ruiter PE, Metselaar HJ, Kwekkeboom J, de Jonge J, Tilanus HW, Janssen HL, van der Laan LJ. Mycophenolic acid augments interferon-stimulated gene expression and inhibits hepatitis C virus infection in vitro and in vivo. Hepatology. 2012;55(6):1673–1683. doi: 10.1002/hep.25562. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Zhou X, Debing Y, Chen K, Van Der Laan LJ, Neyts J, Janssen HL, Metselaar HJ, Peppelenbosch MP, Pan Q. Calcineurin inhibitors stimulate and mycophenolic acid inhibits replication of hepatitis E virus. Gastroenterology. 2014;146(7):1775–1783. doi: 10.1053/j.gastro.2014.02.036. [DOI] [PubMed] [Google Scholar]
- 16.Lloyd RE. Translational control by viral proteinases. Virus Res. 2006;119(1):76–88. doi: 10.1016/j.virusres.2005.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gladding RL, Haas AL, Gronros DL, Lawson TG. Evaluation of the susceptibility of the 3C proteases of hepatitis A virus and poliovirus to degradation by the ubiquitin-mediated proteolytic system. Biochem Biophys Res Commun. 1997;238(1):119–125. doi: 10.1006/bbrc.1997.7251. [DOI] [PubMed] [Google Scholar]
- 18.Lawson TG, Gronros DL, Evans PE, Bastien MC, Michalewich KM, Clark JK, Edmonds JH, Graber KH, Werner JA, Lurvey BA, Cate JM. Identification and characterization of a protein destruction signal in the encephalomyocarditis virus 3C protease. J Biol Chem. 1999;274(14):9904–9980. doi: 10.1074/jbc.274.14.9871. [DOI] [PubMed] [Google Scholar]
- 19.Barretto N, Jukneliene D, Ratia K, Chen Z, Mesecar AD, Baker SC. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol. 2005;79(24):15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karpe YA, Lole KS. Deubiquitination activity associated with hepatitis E virus putative papain-like cysteine protease. J Gen Virol. 2011;92(Pt 9):2088–2092. doi: 10.1099/vir.0.033738-0. [DOI] [PubMed] [Google Scholar]
- 21.Wang D, Fang L, Li P, Sun L, Fan J, Zhang Q, Luo R, Liu X, Li K, Chen H, Chen Z, Xiao S. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J Virol. 2011;85(8):3758–3766. doi: 10.1128/JVI.02589-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng S, Yan W, Gu W, He Q. The ubiquitin-proteasome system is required for the early stages of porcine circovirus type 2 replication. Virology. 2014;456–457:198–204. doi: 10.1016/j.virol.2014.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78(5):761–771. doi: 10.1016/S0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 24.Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4(5):349–360. doi: 10.1038/nrc1361. [DOI] [PubMed] [Google Scholar]
- 25.Yoshida T, Shiraishi T, Nakata S, Horinaka M, Wakada M, Mizutani Y, Miki T, Sakai T. Proteasome inhibitor MG132 induces death receptor 5 through CCAAT/enhancer-binding protein homologous protein. Cancer Res. 2005;65(13):5662–5667. doi: 10.1158/0008-5472.CAN-05-0693. [DOI] [PubMed] [Google Scholar]
- 26.He Q, Huang Y, Sheikh MS. Proteasome inhibitor MG132 upregulates death receptor 5 and cooperates with Apo2L/TRAIL to induce apoptosis in Bax-proficient and -deficient cells. Oncogene. 2004;23(14):2554–2558. doi: 10.1038/sj.onc.1207351. [DOI] [PubMed] [Google Scholar]
- 27.Elliott PJ, Zollner TM, Boehncke WH. Proteasome inhibition: a new anti-inflammatory strategy. J Mol Med (Berl) 2003;81(4):235–245. doi: 10.1007/s00109-003-0422-2. [DOI] [PubMed] [Google Scholar]