

Introduction:
The protective efficacy afforded by immunizing rhesus macaques (Macaca mulatta) with rhesus (Rh) cytomegalovirus (CMV) vaccine vectors expressing Simian Immunodeficiency Virus (SIV) antigens was both unpredicted and unprecedented. In multiple experiments encompassing a large number (>300) of animals, >50% of RhCMV-vector vaccinated monkeys were protected against SIV infection following repeated mucosal exposure to virulent SIV [1-6]. There are multiple elements of these studies that highlight the imperative for further study of RhCMV-vectored vaccines as a prelude to trials in humans. Protected monkeys manifest a rapid decline and ultimate clearance of SIV from the blood after acute viremia. Durably protected animals also clear reservoirs of SIV RNA and integrated proviral SIV DNA. Impressively, the phenotype of RhCMV vector-induced immune protection is unique and challenges current paradigms of viral vector-mediated immunity.
Protection is associated in all cases with antigen-specific, CD8 T effector cells recognizing epitopes presented by MHC class II and Mamu-E molecules. Comparable levels of protection are observed against other human pathogens such as Mycobacterium tuberculosis (MTb) [7], Plasmodium [8], and Ebola virus [9] when their respective antigens are expressed in RhCMV-vectored vaccines. Recognition of the unique and non-canonical phenotype of protected monkeys has prompted excitement that the results in monkeys can be translated to vaccine-mediated protection of humans against multiple pathogens. To replicate the successes observed in macaques, clinical trials are proposed using a human (H) CMV vector with a genetic backbone comparable to RhCMV.
The mechanisms by which RhCMV vectors can elicit protective immunity in macaques, however, remain to be fully elucidated. It is worth emphasizing that, to date, protection against SIV has only been generated by a particular RhCMV variant (RhCMV 68-1) used for the studies in macaques. RhCMV 68-1 has multiple genetic changes compared to the genomic sequence of wild-type (WT) RhCMV. The genetic structure of RhCMV 68-1 resulted from fortuitous and random genomic rearrangements that can occur within WT CMV genomes during serial passage of WT CMV in certain cultured cell types. The genetic alterations within RhCMV 68-1 took place well before RhCMV was envisioned as a vaccine vector. The constellation of genetic changes left behind within RhCMV 68-1 have bestowed it with novel and provocative capacities for immune protection. Fortunately, the hunt into how RhCMV 68-1 stimulates MHC class II- and Mamu-E-restricted CD8 T cell responses appears to be limited to the presence or absence of very few viral Open Reading Frames (ORFs). It is the specific absence of these specific ORF, or a subset thereof, that confers the remarkable phenotype of the RhCMV 68-1 vaccine vector. Accordingly, a reductionist approach should enable the identification of the individual or collective ORF contributing to this phenotype of immune protection.
The successes with RhCMV 68-1 in macaques are possible only because RhCMV profoundly modulates host immunity. Immune modulation is a defining hallmark for the natural histories of all species of CMV within their appropriate hosts. Alteration of host immunity enables the establishment and maintenance of lifelong viral persistence within healthy immune-competent hosts who devote an extraordinarily large component of their immune repertoire specifically to CMV. It also enables reinfection with the RhCMV 68-1 vaccine vectors in animals with naturally acquired RhCMV immunity from prior WT RhCMV infection. It appears likely that the random changes in the 68-1 RhCMV genome that occurred during serial passage peeled back one unbeknownst layer of RhCMV-directed immune modulation. This phenotype was revealed only after vaccination with RhCMV/SIV vectors and SIV challenge. A key element in defining the mechanistic basis for RhCMV-vectored protective immunity will be to address the roles of specific RhCMV ORF in the broader contexts of viral natural history and RhCMV-mediated immune modulation. Only when this knowledge base is gained can HCMV-vectored vaccines be optimally used in humans.
This review summarizes what is known about RhCMV 68-1 as a vaccine vector and salient issues related to how it might work as a vaccine vector. These are presented in the context of CMV natural history within immune competent hosts.
CMV Evolution:
CMV is a double-stranded DNA virus, whose genetic coding content and morphology taxonomically places it as a member of the order Herpesvirales (HV) of viruses [10]. CMV is considered to be a species-specific virus, such that HCMV only infects humans, and RhCMV infects only rhesus macaques (Macaca mulatta) or evolutionarily close Macaca species in vivo. It is estimated that the progenitor herpesvirus (HV) representative arose ~400 MYrs ago [10]. Examples of HV are found throughout vertebrate species, and there is one example in a non-vertebrate species in oysters (Ostreid herpesvirus 1) [11]. The hypothesis has been put forth that HV and T phage may have arisen from a common progenitor, based on common structural motifs in viral proteins [12]. Such a putative common origin for bacterial T phage and vertebrate HV would move the possible origin of HVs to >500 MYrs ago. This is contemporaneous with or prior to the evolution of progenitor vertebrates [10]. Viruses are defined as “submicroscopic, obligate intracellular pathogens”. As such, they must first infect a cell or host, and then coopt cellular machinery to produce sufficient progeny virions that enable transmission to the next susceptible cell or host. This requirement for coopting intracellular machinery to complete its life cycle sets a very high threshold for success. This is particularly relevant for overcoming physical, intrinsic, innate, and adaptive barriers to infection.
Charles Darwin wrote in On the Origin of Species that, “natural selection is daily and hourly scrutinising, throughout the world, every variation; rejecting that which is bad, preserving and adding up all that is good; silently and insensibly working [at] the improvement of each organic being” [13]. The fact that HVs in general, and CMV in particular, still exist to infect and occasionally cause disease in their appropriate host means that, through a >400 MYr process of Darwinian selection, CMV has to be considered as an exceedingly “fit” virus. To put this protracted evolution of HV into geologic perspective, the earliest known hominin to walk upright (Sahelanthropus) was 7 MYrs ago [14]. Since the origin of the progenitor HV, ancestral HVs co-evolved with their host species and descendants. During this protracted coexistence, the virus and host each affected the other’s genetic content, undoubtedly shaping the mechanisms of immune protection stimulated by current RhCMV/SIV vectors in macaques.
HCMV Genomic Coding Capacity:
Given the protracted co-evolution of CMV in hosts with increasing complex and sophisticated immune functionalities, CMV had to evolve counter strategies for almost any conceivable form of innate and adaptive immunity. This evolution was in addition to overcoming physical barriers associated with host-to-host transmission. For CMV, this change resulted in the evolution of a virally-encoded repertoire of ORFs that modulate a wide breadth of the innate and adaptive immunological processes. Alterations of host immune functions were essential to enable the successful propagation in the host species. The complexities of this immune modulation have been prominently brought forth with the mechanisms of protection elicited by RhCMV vectors expressing SIV antigens (see below).
The HCMV genome (~236 kbp) is the largest of any virus known to infect humans, encoding ≥165 ORFs and non-translated RNA transcripts [15]. As a member of HV, HCMV encodes 43 core ORFs expressed by all herpesviruses [16]. These core ORF express proteins involved in essential viral functions, including DNA replication, nucleic acid metabolism, capsid assembly and function, packaging of nascent viral genomes into nascent capsids, tegument proteins and envelope glycoproteins. Collectively, these ORFs enable HCMV to have a wide cell tropism (including fibroblasts, epithelial and endothelial cells, macrophages, and CD34+ myeloid progenitor cells). This broad host cell tropism magnifies the obstacles in the unfulfilled quest to develop HCMV vaccines despite over four decades of efforts [17].
Notably, HCMV devotes an impressive portion of its coding repertoire to functions that modulate cellular signaling, trafficking, activation, antigen presentation, and susceptibility to apoptosis. While the functional capabilities of many HCMV ORFs remain to be characterized, those that have been characterized include functions that modulate host signaling, antigen presentation, and apoptosis in cells that productively support CMV infection (e.g., endothelial and epithelial cells, macrophages, fibroblasts). In addition, HCMV encodes ORF that modulate the functionality of cell types that are not susceptible for productive CMV replication but are critical for innate and adaptive effector functions, including natural killer, dendritic, and T cells.
The identified HCMV ORFs that target host intrinsic, innate, and adaptive functions (Table 1), make up >33% of coding capacity. It is very possible that upwards of 50% of HCMV ORF have functions comparable to these, and as yet-to-be characterized, immune/cellular modulating functions. Collectively, the viral armamentarium of immune modulating proteins significantly skews host antiviral immune responses. Viral modulation of host immunity begins at the earliest virus-hosts interactions at the primary site of mucosal exposure and throughout lifelong virus-host homeostasis in persistently infected, healthy hosts. It is highly likely that the particulars of the earliest viral manipulation of host responses establish the ability to persist for the life of the infected host despite massive host antiviral immune responses (see below).
Table 1:
CMV Modulators of Intrinsic, Innate, Adaptive Host Responses
| ORF/Transcript | Function |
|---|---|
| β2.7 | Inhibition of apoptosis [83] |
| UL7 | Homolog of SLAM-Family Receptor CD229 [84] |
| UL8 | Inhibits production of proinflammatory factors by infected myeloid cells [85] |
| UL11 | Paralysis of T cells via inhibition of CD45 [86, 87] |
| UL16 | Evasion of natural killer cell (NK) cytotoxicity (NKG2D) [88] |
| UL18 | Evasion of NK, CTL cytotoxicity via binding to the ILT2 (CD85j) receptor [89-92] |
| UL21.5 | RANTES decoy receptor [93] |
| UL23 | Inhibition of interferonγ-induced STAT1-dependent transcription [94] |
| UL25 | UL25 miRNA-mediated down-regulation of cellular genes involved in cell cycle control [95] |
| UL26 | Modulation of NF-κB [96] |
| UL29/28 | Modification of NuRD complex, which stimulates the accumulation of IE RNAs [97] |
| UL33, 78 | G-protein-coupled receptor [98, 99] |
| UL36/37/38 | Inhibition of apoptosis; interaction with viperin [57, 100-103] |
| UL40 | Inducer of HLA-E surface expression [104]; inhibition of NK by HLA-E [92, 105-108] |
| UL42 | Negative regulator of cGAS/MITA-dependent antiviral response [109] |
| UL50 | Inhibition of ISGylation by causing proteasomal degradation of UBE1L [110] |
| UL76 | Induction of IL-8 [111] |
| UL82 | Suppression of DAXX-mediated inhibition of IE expression [112] |
| UL83 | Inhibition of NKp30[113] and IRF-3 [114] |
| UL97 | Phosphorylation of the retinoblastoma (Rb) p107 and p130 tumor suppressors [115] |
| UL111A | IL-10 [116-120]; LAcmvIL-10 [121] |
| UL112 | UL112 miRNA-mediated block of IKKα and IKKβ [122-124] |
| UL119/118 | Fc Receptor [125] |
| UL122 | IFNβ antagonist [126] |
| UL123 | Antagonist of IFN-responsive transcripts [127] |
| UL128 | β chemokine, chemoattractant; inhibits monocyte migration [73-77] |
| UL135 | Suppresses formation of the immunological synapse [92] |
| UL138 | Potentiator of TNFR Signaling[128, 129]; Latency [130] |
| UL141 | Evasion of NK cytotoxicity via inhibition of the NK-activating ligand CD 112 & 155 [92, 131, 132] |
| UL142 | Inhibition of NK cell lysis [92, 133] |
| UL144 | Tumor necrosis family receptor superfamily [134] |
| UL146/147 | α-chemokine [135] |
| UL148A | Downregulates the Activating NK Cell Ligand MICA to Avoid NK [92, 136, 137] |
| TRL11 | Fc Receptor [125, 138] |
| RL13 | Fc Receptor [139] |
| IRS1, TRS1 | Inhibition of Protein Kinase R [114, 140, 141] |
| US2, 11 | Dislocation of MHC I to cytosol [142-145] |
| US2 | Disruption of MHC II pathway [146] |
| US3 | Retention of class I in endoplasmic reticulum [147, 148] |
| US5 | US5 miRNA-mediated inhibition of IKKα and IKKβ [124] |
| US6 | Inhibition of TAP [149] |
| US10 | Degradation of HLA-G [150] |
| US12-21 | G-Protein-Coupled Receptors [151, 152] |
| US12, 14, 18, 20 | Inhibition of NK functions [153, 154] |
| US27/28 | β chemokine receptor [155-165] |
HCMV genes are temporally expressed in a sequential cascade with genes characterized as immediate-early (IE), early (E), and late (L), based on their kinetics of expression [18]. IE genes are expressed immediately after infection, in the absence of de novo viral gene expression. IE gene expression is essential to elicit subsequent expression of the E genes, the gene products of which are involved in replication of the viral genome. Expression of the L genes is intrinsically linked to replication of the viral genome. The relevance of the temporal pattern of gene expression is that the timing of ectopic gene expression in RhCMV-vectored vaccines may influence the type of immune responses elicited against the antigen encoded by the vector (described below).
Following infection of cells, the pattern of HCMV gene expression can follow either of two profiles [18]. HCMV can go through the entire progression of gene expression resulting in the production of progeny virions capable of infecting other cells within the host (or transplacentally in pregnant women to fetuses), or excreted long-term in bodily fluids enabling horizontal transmission to close contacts of the infected individual. Alternatively, HCMV can enter into a state where gene expression is extremely limited, a hallmark of which is the absence of productive virion generation. Such a pattern is exhibited by CD34+ myeloid progenitor cells in which viral DNA can be detected, although viral transcription is restricted to a small but intriguing minority of viral ORFs. While circulating CD34+ cells do not exhibit a phenotype of productive gene expression, a shift to a productive pattern of gene expression can occur following allogeneic stimulation or differentiation. This biphasic pattern of cells either expressing or not expressing the full complement of HCMV-encoded ORF forms the foundation for why HCMV stimulates broad and durable host antiviral immune responses. This biphasic pattern also illustrates why HCMV is a clinically relevant cause of morbidity and mortality in certain settings, despite the general consideration that HCMV is a low pathogenic virus (described below).
HCMV Natural History:
Based on the extent of infection in humans, HCMV is highly efficient at person-to-person spread. HCMV is ubiquitous throughout the globe, with a global seroprevalence rate in the general population of 83% (range of regional rates: 66 - >90%) [19]. Primary infection can occur throughout life and in utero following exposure of mucosal surfaces to infectious bodily fluids (e.g., breast milk, saliva, urine, semen) or transplacentally from infected mother to fetus, respectively. HCMV is the most common congenital infection with an overall global frequency of ~0.6% of all pregnancies [20-23], with studies reporting a range of 0.2 – 6% [23-25]. Seroconversion to HCMV exhibits an age-related increase such that 50% seroprevalence rates are frequently observed by the time of puberty outside of the United States, and ~29 years within the United States, although seroprevalence is inversely related to socioeconomic status [26-33].
Host responses to an almost inevitable infection emphasize that the host is well adapted to HCMV infection. Primary infection with HCMV in those with a fully functional immune system is usually subclinical, although transient and self-limiting clinical sequelae (e.g., fever, mononucleosis) are observed in some individuals [34, 35]. Host immune responses are effective at restricting disease outcomes because of a massive and unprecedented devotion of the adaptive immune repertoire specifically to HCMV antigens. A large study involving 32 long-term healthy carriers of HCMV mapped T cell responses to the HCMV proteome. Using almost 14,000 overlapping peptides to HCMV ORF, Sylwester et al. observed that ~10% of memory CD4 and CD8 T cells are HCMV-specific, leaving 90% of the remaining immunological memory to everything else the host has encountered [36]. Moreover, HCMV stimulates a broad array of binding and neutralizing antibodies to multiple viral envelope glycoproteins, and other virion-associated and intracellular proteins. The clinical relevance of these responses is highlighted by the high incidence of HCMV disease in individuals lacking a fully functional immune system. Pathogenic outcomes are common in immune suppressed transplant recipients, immunologically immature and congenitally infected fetuses, and immune deficient and non-antiretroviral-treated HIV individuals. Apart from the clinical scenarios just mentioned, HCMV-specific immune responses can be viewed as protective. Accordingly, adaptive immune responses in long-term healthy carriers of HCMV have served as a template for vaccines designed to prevent primary HCMV infection [17, 37].
Nevertheless, HCMV-specific immune responses in HCMV-infected individuals must also be viewed as a signature of HCMV persistence in which reservoirs of HCMV are maintained for life despite the magnitude of host antiviral immunity. The vast majority of primary HCMV exposures comes from contact of mucosal surfaces with infectious HCMV particles contained within the bodily fluids from HCMV-infected individuals. As mentioned above, HCMV infects multiple cell types, and the virus disseminates systemically via cell-to-cell spread, blood, and probably the lymphatic system with the result that virus is asymptomatically shed in bodily fluids soon after primary infection [38]. Shedding generally declines in an age-related manner. Nevertheless, virus can be shed in high titers for years, particularly in healthy children infected in utero or early childhood, presenting an infectious threat to the fetuses borne by pregnant women. Moreover, HCMV can be episodically reactivated in breast milk during subsequent pregnancies of HCMV-infected women, enabling efficient transmission to neonates. The critical aspect of this response in relation to HCMV vaccine vectors is that HCMV is capable of reactivation with apparent impunity to potent antiviral effector functions (T, B, and natural killer cells). In summary, the long-term virus-host relationship is a détente in which host immunity protects against viral disease whilst at the same time, viral immune modulating functions ensure lifelong persistence in the face of these same protective immune responses.
The magnitude and duration of host immune responses support the argument that, even in the absence of infectious virus production, the infected host is persistently exposed to viral antigens. For CMVs, persistent antigen exposure to memory T cells leads to a phenomenon termed ‘memory inflation’ in which memory T cells expand in frequency, as opposed to undergoing an apoptosis-mediated contraction [39]. This activity leads to what can be viewed as mis-application of the terms ‘latency’ and ‘persistence’ to describe CMV. There is no doubt that, as mentioned above, CMV can latently infect certain cells (e.g., CD34+ myeloid progenitor cells). Such cells are phenotyped by the presence of cells harboring CMV genomes but with a greatly restricted gene expression profile and the concomitant absence of virion production. In this case, latency refers to a cellular description of the virus-host relationship. However, at the organismal level, the infected host is persistently exposed to viral antigens in that, somewhere within the infected host, a virally infected cell is expressing the full complement of viral gene products that may or may not result in the production of infectious virions. This latter state of the virus ensures both the persistence of the large host antiviral immune responses, and the continuous or episodic production of virus in bodily fluids.
It is worth noting that no one has ever described an immune-escape variant for HCMV in which there is genetic drift that yields a variant that is not susceptible to host-derived T/B-specific immune responses that otherwise would limit the replicative capacity of a putative parental version of the virus. This fact is a hallmark of RNA viruses, such as influenza or HIV, that lack proof-reading polymerases. Given the antigenic breadth of HCMV ORF that elicit cellular and antibody responses, and the lack of precise immune correlates of protection, this is not a straightforward experiment to perform for HCMV. Given that caveat, however, the absence of a described immune-escape variants for HCMV contrasts with the relatively rapid emergence of drug-resistant HCMV variants during in vivo or in vitro anti-HCMV prophylaxis [40]. One simplistic interpretation of this finding is that, given the absence of antiviral drug immunity as a selective pressure during HCMV evolution, the only ‘tool’ available to HCMV is occasional emergence of point mutations that confer resistance to the antiviral targets of the drug. A corollary to this is that HCMV doesn’t need emergence of immune-escape variants to persist because the virus has evolved a complex set of viral functions that modulate host adaptive and intrinsic immunity, yielding an extremely fit version of the virus in the context of lifelong immune surveillance.
A hint to the sophistication of viral immunomodulation is illustrated by a study evaluating the in vivo replicative potential of a RhCMV 68-1 variant in which a late gene, phosphoprotein 66 (pp65) has been deleted. The viral pp65 tegument protein is an immunodominant T cell antigen. Deletion of the pp65 gene results in a variant with profoundly increased replicative potential following inoculation of RhCMV-uninfected macaques, compared to the parental RhCMV 68-1 expressing pp65 [41]. The authors concluded that in essence “pp65 likely acts as an immunological brake during the initial stages of primary infection to limit viral replication and dissemination”.
The protracted evolution of HVs and CMV has bestowed another remarkable phenotype of HCMV, namely the ability to reinfect those with extant immunity to HCMV resulting from prior infection. HCMV reinfection has clinical ramifications by contributing to the “silent global burden” of congenital HCMV infection and sequelae in which the majority of congenital CMV infections result from reinfection in women with preconceptual HCMV immunity [22, 24]. The mechanisms of reinfection are not known, nor are strategies for how to prevent it. However, the pioneering studies using RhCMV vaccine vectors highlight that vectors expressing ectopic antigens provide immune protection against challenge following immunization in healthy rhesus macaques that have robust anti-RhCMV immune responses at the time of vaccination. Additional studies in the rhesus model clearly show that primary infection and reinfection is a function of viral-mediated modulation of immune surveillance mediated by NK and CD8 cells, respectively [3, 42].
Taken as a whole, the prolonged co-evolution of human and HCMV interactions have endowed a stable coexistence that ensures the survival of each species to live another day. It is against this backdrop that the unprecedented and fortuitous discovery of the immune modulating potential of RhCMV-vectored vaccines must be approached. A better mechanistic understanding can be gained to guide prevention and therapeutic strategies that can be translated into use for humans.
Origin of RhCMV 68-1 As the Prototypical Vector Backbone:
Several factors of CMV natural history guided the use of it as a viral vaccine vector to induce protective immune responses against heterologous pathogens. These included the facts that HCMV (1) is considered a low pathogenic virus, (2) can reinfect hosts with existing HCMV immunity, (3) establishes a lifelong persistent infection, (4) stimulates inordinately large T cell responses, and (4) has a large genome that can be readily engineered in vitro. The RhCMV model of HCMV persistence and pathogenesis [43] has been used as a proof-of-principal, nonhuman primate (NHP) host because (1) RhCMV infection in rhesus macaques strongly recapitulates HCMV infection in immune competent and immune impaired individuals, (2) the RhCMV genome had been molecularly engineered into a Bacterial Artificial Chromosome (BAC), and (3) there are tractable models for vaccination against clinically relevant human pathogens, particularly HIV (SIV) and MTb. BACs enable propagation of very large viral genomes, such as CMV, in a plasmid form in bacteria [44]. Importantly, for the development of CMV-vaccine vectors, BACs enable the power of bacterial recombination systems to engineer precise antigen cassettes into the viral genome and subsequent regeneration of engineered viruses in cultured mammalian cells bearing the site-specific changes. Notably, at the time the RhCMV genome was engineered into a BAC [45], the NHP model was in its relative infancy. There were few characterized viral isolates available for molecular cloning.
What was available at that time serendipitously gave rise to the paradigm-shifting mechanisms of immune protection observed with the current iteration of the RhCMV viral vaccine. The 68-1 variant of RhCMV was available through the American Type Culture Collection (ATCC). RhCMV 68-1 (ATCC VR-677) was isolated from the urine of a healthy rhesus macaque in 1968 after passage on MRC-5 human foreskin fibroblasts (ATCC CCL-171) [46]. No rhesus-derived fibroblast cells were in existence at that time. The use of human fibroblasts was notable for what is now belatedly recognized as frequent genomic lability when CMV is cultured on this cell type.
Although wild-type (WT) HCMV exhibits a broad cell tropism in vivo, HCMV was historically passaged in cultured fibroblasts. This was due, most likely, to the relative ease of establishing primary fibroblast cells in culture and the robustness of viral replication kinetics in these cultured cells. What was not appreciated at the time was that both human and NHP CMV isolates rapidly undergo genetic alterations (point mutations, deletions, and/or genetic rearrangements) during serial passage in fibroblasts. In retrospect, it became evident that RhCMV 68-1 underwent a comparable process of genetic changes after passage in fibroblasts. This action resulted in the loss of a subset of the full complement of viral ORF in the original WT RhCMV 68-1 [47] before it was engineered into a BAC (RhCMV 68-1/BAC). Notably, the particular loss of ORF radically altered the vaccine-mediated immune responses afforded via expression of ectopic antigens [45, 48, 49].
Coding Capacity of RhCMV68-1 vs RhCMV WT:
The prototypic RhCMV/SIV vectors are based on the fibroblast-adapted variant of RhCMV 68-1, which emerged in tissue culture from the original WT RhCMV. RhCMV 68-1/SIV mediates protection against SIV challenge in 50-60% of vaccinated macaques due to the unique stimulation of effector memory CD8 T cells [1-6]. Notably, RhCMV 68-1-derived vectors also elicit CD8 T cells recognizing antigens restricted by MHC class II and Mamu-E instead of MHC class I [4, 6]. The remarkable phenotype of RhCMV 68-1/SIV vectors (i.e., >50% protection against SIV challenge and unconventional CD8 T cell responses) appears to be the result of the particular alterations that spontaneously occurred to the 68-1 genome during passage in rhesus and human fibroblasts (Table 2). Partial restoration of the full WT coding capacity into the RhCMV 68-1/BAC variant (yielding RhCMV 68-1.2) completely abrogates the stimulation of MHC class II and Mamu-E-restricted CD8 T cell responses to SIV antigens expressed within the context of RhCMV 68-1.2/SIV vectors [4]. The stark difference in the induction of immunity to SIV between RhCMV 68-1 and RhCMV 68-1.2 reflects the collective or individual effects of only four differences in coding content between the two variants. It is important to emphasize that it will be critical to determine whether RhCMV 68-1.2 also does, or does not, confer protection against SIV challenge. RhCMV 68-1.2 variants expressing Mtb antigens significantly protects vaccinated macaques against Mtb challenge, comparable to the level of protection afforded by RhCMV 68-1/Mtb vectors [7]. RhCMV 68-1.2/Mtb vectors do not stimulate unconventional T cell responses to Mtb antigens.
Table 2:
% Infected
Quantification of the percentages of (A) blood vessels containing RhCMV-infected endothelial cells (Endo), (B) fibroblasts (Fibro) infected with RhCMV, and (C) macrophages (Mø) infected with RhCMV are presented. See Assaf et al. for details [71].
:significantly different
Figure 1 presents the partial coding capacity of RhCMV WT compared to that of RhCMV 68-1 and the repaired variant of RhCMV 68-1 (i.e., RhCMV 68-1.2). Adaptation of the original 68-1 isolate to passage in fibroblasts led to a complex genomic alteration that deleted segments A and E, and inverted segments B and C, relative to the original orientation in RhCMV WT [47-51]. Because of this event, RhCMV 68-1 lacks the RhUL128 and RhUL130 ORF but does retain the RhUL131 ORF. The RhUL128/UL130/UL131 ORFs are expressed from a polycistronic transcript originating from a promoter upstream of RhUL131 [52]. It is not known whether the residual RhUL131 is transcribed utilizing a cryptic polyadenylation site in RhCMV 68-1. RhUL128, RhUL130, and RhUL131 encode orthologs of HCMV UL128, UL130, and UL131, all of which are components of a pentameric protein complex (together with glycoproteins H and L) that is essential for tropism in epithelial and endothelial cells [53-55] (Table 2). Predictably, based on precedent with HCMV, RhCMV 68-1 is severely attenuated for growth in rhesus epithelial cells in vitro, compared to growth of RhCMV WT [52, 56]. Engineering both RhUL218 and RhUL130 back into RhCMV 68-1 (generating RhCMV 68-1.2) restores epithelial cell tropism [52], confirming the functional equivalence of RhUL128/130/131 with their HCMV orthologs.
Figure 1.
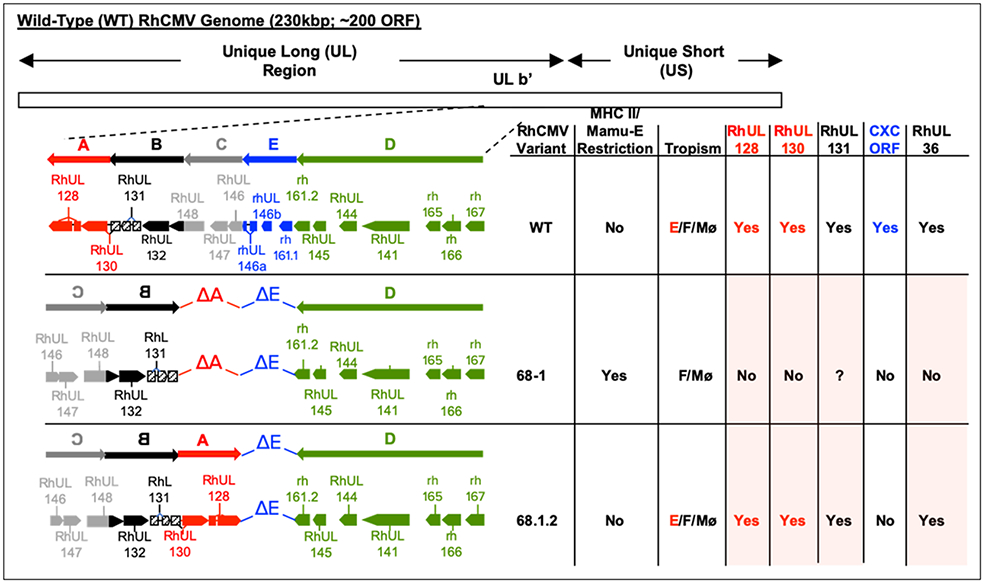
Genomic coding capacity of the ULB’ regions of RhCMV variants: Wild Type (WT), RhCMV 68-1 (68-1), and RhCMV 68-1.2 (68-1.2) [47-52]. The ULb’ region of WT RhCMV has been described as having five segments: A (red), B (black), C (grey), D (green), and E (blue). The direction of the arrows represents the orientation of transcription. Fibroblast-adapted 68-1 deleted segments A and E, and inverted segments B and C, relative to the orientation in WT RhCMV. Segment A was engineered back into 68-1 in addition to restoring an intact RhUL36 ORF, generating the 68-1.2 variant [52]. RhCMV ORF with HCMV orthologs are prefaced with ‘Rh’. RhCMV ORF without an HCMV ortholog are prefaced with ‘rh’. SIV Protection: see text for details; MHC II/Mamu-E Restriction:indicates whether the particular variant generates MHC class II or Mamu-E-restricted CD8 T cell responses (see text for detail); Tropism: E= endothelial and epithelial cells; F=fibroblasts; Mø=macrophages. RhUL128/RhUL130/RhUL131: indicates whether the variant encodes the ORF (Yes/No); ?: although 68-1 contains the RhUL131 ORF, it is unknown whether it is translated; CXC: indicates whether the variant encodes the CXC-like ORF encoded within segment E (RhUL146a, RhUL146b, and rh161.1). RhUL36: indicates whether a full-length RhUL36 ORF is present (Yes or No). Shaded boxes: indicates the four genetic differences between RhCMV 68-1 and RhCMV 68-1.2 (see text for detail).
Serial passage of RhCMV 68-1 in fibroblasts also resulted in the emergence of a variant with a frameshift mutation in the RhUL36 ORF [57]. It was this variant, containing the genomic rearrangement depicted in Fig. 1 and the UL36 mutation, that was iteratively engineered into the BAC [45, 58], from which the vaccine vectors were derived. RhUL36, like its UL36 ortholog in HCMV, encodes a viral inhibitor of caspase-8-induced apoptosis (vICA) and has a role in cell death suppression [57]. The frameshift mutation in RhCMV 68-1 prematurely truncates the protein and renders it non-functional for anti-apoptotic activity [57]. It is not clear whether the absence of a functional vICA impairs replication of RhCMV 68-1 in vitro. There is evidence that RhCMV 68-1 variants expressing a functional vICA may have marginally enhanced replication in rhesus fibroblasts and epithelial cells [52].
The rearrangement of ULb’ in RhCMV 68-1 deleted three additional ORFs beyond the deletion of RhUL128 and RhUL130 (i.e., RhUL146a, RhUL146b, rh161.1) and prematurely truncated one other ORF (rh161.2) (Fig.1). RhUL146a, RhUL146b, rh161.1, and rh161.2 potentially produce CXC-like alpha chemokines based on predicted protein sequences [50]. However, no functional activities have been reported for these ORF. They do not appear to directly contribute to RhCMV-vectored immune protection since these ORF were not repaired in RhCMV 68-1.2 (Fig. 1) [52], although they prominently affect the pattern of acute host responses to viral infection in vivo (discussed below). Their functional absence in RhCMV 68-1 may unmask a novel phenotype produced by the additional absence of RhUL128, RhUL130, RhUL131, and RhUL36, or a particular subset therein.
RhCMV 68-1 vs RhCMV WT Natural History:
Despite the genomic changes in RhCMV 68-1, this variant is capable of infecting and systemically disseminating in the host following intravenous, subcutaneous (SC), and oral routes of viral inoculation in both RhCMV-infected and RhCMV-uninfected (naïve) rhesus macaques [5, 59]. RhCMV 68-1 is pathogenic in SIV-coinfected animals [60] and in fetal macaques [61]. Notably, from a vaccine perspective, RhCMV 68-1 infection of immune competent macaques is subclinical [59], and RhCMV 68-1 establishes a lifelong persistence noted by the long-term presence of virally infected cells [62] and duration of immune responses to ectopic antigens [1, 2, 4-9].
RhCMV 68-1 is profoundly impacted in three critical aspects of viral natural history compared to RhCMV WT. All three are relevant for extension of HCMV-vectored vaccines in humans. The first relates to potential horizontal spread of the vaccine vector from the vaccine recipient to close, unvaccinated contacts. RhCMV WT is endemic in breeding populations of rhesus macaques, and there are multiple genetic variants in circulation [63, 64]. Seroprevalence of RhCMV WT infection approaches 100% by 1-year of age due to persistent, high-titer shedding of RhCMV WT in bodily fluids (e.g., saliva, urine) of infected animals [65, 66]. Shedding frequencies (i.e., detection of viral DNA) of 80% are observed in animals up to four years after primary infection, and 25% in animals infected more than 13 years [66]. WT-like variants of RhCMV, containing a full complement of ORF and passed on rhesus epithelial cells in culture, exhibit a characteristic shedding profile after inoculation into naïve monkeys; virus is persistently shed in urine and saliva beginning 8-10 weeks after oral or SC inoculation [67]. In contrast, mucosal shedding of RhCMV 68-1 is orders of magnitude lower and sporadic following inoculation of either naïve or RhCMV-infected macaques, compared to inoculation with RhCMV WT [5, 68-70]. The simplest interpretation of the attenuated phenotype of RhCMV 68-1 in vivo is that the full complement of ULb’-encoded ORF, particularly UL128/UL130/UL131, is essential for WT-like levels of shedding in bodily fluids [69]. A logical extrapolation of the attenuated growth in vivo of RhCMV 68-1 is that the protective immunity elicited with RhCMV 68-1/SIV vectors, compared to the absence of protection conferred with RhCMV 68-1.2/SIV[4], is related in some fashion to impaired epi/endothelial cell tropism with RhCMV 68-1.
The second distinctive phenotype of RhCMV 68-1 infection in vivo highlights the impact RhCMV infection in vivo has on the innate immune responses to acute infection. in vivo observations of the acute patterns of RhCMV 68-1 and RhCMV WT infections provide operational insight into how changes in tropism might alter how unconventional CD8 T cell responses are generated. One study analyzed skin biopsies of the inoculation sites from RhCMV-uninfected monkeys inoculated SC seven days previously with RhCMV 68-1, or RhCMV WT. RhCMV WT had been serially passed on primary monkey kidney epithelial cells and contained a full-length ULb’ region [71]. Biopsies of the inoculation site (at 7 days) were immunohistochemically co-stained for RhCMV antigen and cell type markers for endothelial cells (CD131), fibroblasts (vimentin), and macrophages (Mø; CD68), and quantified for the levels of infection in each cell type. The results show that RhCMV WT infect all three cell types (Table 2), whereas infection with RhCMV 68-1 is restricted to just fibroblasts and Mø (other cell types were not analyzed). A comparable analysis with RhCMV 68-1.2 has not been performed, but it is assumed that a similar pattern of cell tropism would be observed as for RhCMV WT based on the in vitro cell tropism for RhCMV68-1.2 [52]. Given that caveat, and given the observation that RhCMV 68-1 infects a subset of the cell types infected by RhCMV 68-1.2 (Table 2), the negative impact on unconventional CD8 T cell responses resulting from infection of endothelial cells with RhCMV68-1.2 is dominant over what would otherwise be positive effects resulting from infection of the same cell types infected with RhCMV 68-1 (i.e., fibroblasts and Mø). Put another way, infection of endothelial and epithelial cells by RhCMV vectors expressing RhUL128/130/131 acts as a dominant-negative influence on generation of protective immunity in the other cell types. Conversely, restriction of RhCMV 68-1 infection to just Mø and fibroblasts enables the decryption of protective immune responses. The mechanistic basis for how this is accomplished remains unresolved. It presumably involves distinct SIV antigen processing and presentation by the particular infected cell types to APC prior to APC trafficking to the draining lymph node to engage naïve T cells. If so, this finding further implies that generation of MHC class II-presented antigen in infected Mø takes precedence over the presentation of MHC class I-presented antigen in fibroblasts since fibroblasts do not express MHC class II.
The third vital distinction between infection with RhCMV WT and RhCMV 68-1 critically emphasize that it is the specific absence of the full coding potential in RhCMV 68-1 that enables the emergence of the unconventionally restricted CD8 T cells. RhCMV WT has not been evaluated as a vaccine vector. However, Hansen et al. evaluated CD8 T cells responses to the RhCMV immediate-early-1 (IE-1) protein in animals naturally exposed to RhCMV WT infection [4]. The CD8 T cell responses to the RhCMV IE-1 protein in these animals (naturally infected with RhCMV WT) were compared to a second set of animals that were also naturally infected with RhCMV WT but which were subsequently infected with RhCMV 68-1/SIV vectors. In animals only infected with RhCMV WT, IE-1-specific CD8 T cells were strictly MHC class I-restricted (7 – 8 independent epitopes/animal). No MHC class II-restricted epitopes were detected (Mamu-E was not evaluated in this study). In marked contrast in animals superinfected with RhCMV 68-1/SIV vectors, there was a profound change in the restriction of CD8 T cell epitopes. These animals exhibited on average ~10 IE-1 epitopes restricted by MHC class I and Mamu-E (7 – 12/animal) and 26 MHC class II-restricted epitopes (25 – 27/animal). At least, three major implications arise from this study. The results (1) prove that the absence of specific ORF in RhCMV 68-1 generate unconventional CD8 T cell responses, which are otherwise undetectable in the context of RhCMV WT infection; (2) highlight that RhCMV 68-1 can elicit de novo novel IE-1-specific CD8 T cells responses in animals with prior specificities arising from RhCMV WT infection. In other words, RhCMV 68-1 enables re-education of a host with existing immunological memory. And, (3) this study offers proof-of-principal that RhCMV 68-1 can theoretically generate post-infection immune treatments against a wide variety of persistent infections via the generation of otherwise cryptic epitope specificities. This particular study [4], however, did not evaluate whether there were virological or immunological changes in the parameters of RhCMV WT infection that would suggest that the MHC class II-restricted responses stimulated greater immunological control over RhCMV persistence.
There is an alternative, and non-mutually exclusive, hypothesis about how RhCMV 68-1 might stimulate protective immunity. This suggestion focuses on the effects that RhCMV induces on the surrounding microenvironment of the infected cell. RhCMV profoundly alters the immediate inflammatory environment around the infected cell, the extent of which is dependent on the coding content of the variant used for inoculation. RhCMV WT, expressing the full coding content, stimulates a mixed lymphocytic infiltrate that prominently includes polymorphonuclear leukocytes (PMN) and mononuclear cells seven days after subcutaneous inoculation (Fig. 2 left). The recruitment of PMN to the site of infection recapitulates what is seen in vivo with endemic RhCMV WT that has never been passed in culture [71], presumably due to the CXC-like ORF in segment E within ULb’ (Fig.1). In contrast, SC inoculation with RhCMV 68-1 stimulates a strictly mononuclear infiltrate (Fig. 2 right). Even in the absence of the CXC-like ORF, the microenvironment is still bathed in viral mediators acting in trans on host cells.
Figure 2.
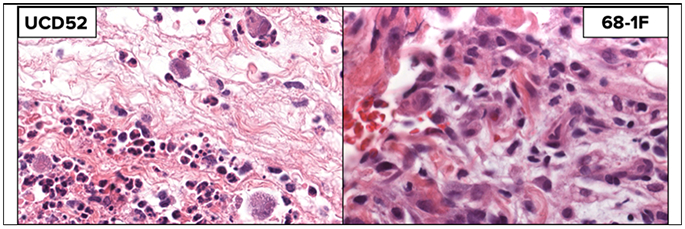
Skin biopsies (stained with H & E) seven days after SC inoculation with RhCMV WT (left) or RhCMV 68-1 (right).
A previous study compared inflammatory responses at seven days for RhCMV-uninfected animals inoculated subcutaneously (SC) with either RhCMV 68-1 or a variant of RhCMV 68-1 in which the viral IL-10 ORF (vIL-10) had been deleted (RhCMV 68-1ΔvIL-10) [72]. Compared to inoculation with RhCMV 68-1, the inflammatory response at the site of inoculation with RhCMV 68-1ΔvIL-10 was noted for increased mononuclear cellularity and a prominent relative increase in CD68+ Mø. This study concludes that vIL-10, “alters the earliest host responses to viral antigens by dampening the magnitude and specificity of innate effector cells to primary RhCMV infection. In addition, there is a commensurate reduction in the quality and quantity of early and long-term, RhCMV-specific adaptive immune responses” [72]. By extension, the same processes should be operative for RhCMV 68-1-based SIV vaccine vectors [1-5], through the functionalities of vIL-10 and particularly additional viral modulatory factors (Table 1) since both RhCMV 68-1 and RhCMV 68-1.2 express vIL-10. Because there are only ≤4 differences in coding content between these two RhCMV variants (Fig. 1), only a limited number of experiments are required to rule in/out their contribution(s) to the induction/inhibition of unconventional CD8 T cell epitopes. There are two ORF that warrant immediate attention.
HCMV UL128 has been reported to act as a CC chemokine with multiple functionalities, including acting as a chemoattractant for monocytes and PBMC, stimulating proliferation of PBMC through activation of the MAPK/ERK signaling pathway, and altering monocyte responsiveness to chemokines through the internalization of different chemokine receptors [73-77]. The UL128 ortholog of rat CMV also exhibits chemokine activity [78]. No functional activities have been reported for RhUL128, apart from those characterizing its role in formation of the RhCMV pentamer complex an alpha CC chemokine-like motif [50]. Moreover, RhCMV UL128 is readily secreted from cells expressing it in the absence of other members of the pentamer complex (Fig. 3), whereas neither RhUL130 or RhUL131 were secreted without co-expression of ≥1 other of the pentameric complex proteins. While such a result does not indicate functionality, its presence in the extracellular milieu would be consistent with a putative chemokine.
Figure 3.

The five RhCMV ORF comprising the pentameric complex essential for tropism in epithelial and endothelial cells were expressed individually, or in combinations from CEF cells infected with MVA vectors. The extracellular medium was analyzed for the presence of the RhCMV ORF. BR5 is a positive control MVA protein. (F Wussow, F Chiuppesi, D Diamond, personal communication).
The HCMV UL36-encoded vICA increases resistance to Fas-mediated apoptosis [79]. Mutations in UL36 occur in fibroblast-passaged strains of HCMV [80], similar to what occurs with RhCMV 68-1. vICA is distinct from the UL37-encoded anti-apoptotic protein, the mitochondria-localized inhibitor of apoptosis (vMIA). Discussing what selective pressures might have led to HCMV-encoding two anti-apoptotic functions, Slaletskaya et al. speculated that, “vICA and vMIA may have evolved to protect different infected cell populations of the host” [79]. Mechanistic linkage of deficient RhCMV vICA function and generation of unconventional T cell responses with RhCMV 68-1 is speculative. However, expression of both HCMV vICA and vMIA can attenuate cytotoxic T cell-mediated killing in an “HLA-Recognition independent manner” [81]. This finding demonstrates that both viral proteins can alter effector cell functionalities. It is not inconceivable, therefore, that the absence of RhCMV vICA in different cell types, could alter cell signaling pathways involved in antigen presentation [82].
A potential role of RhUL128 and RhUL36 in generating unconventional T cell responses is further piqued by a study investigating cross-species infection of cynomolgus macaques with RhCMV [62]. Whereas RhCMV 68-1 fails to infect cynomolgus macaques of Mauritian origin, RhCMV 68-1.2 variants, expressing either RhUL128 and RhUL130, or RhUL36, can successfully infect. The authors conclude that, “the results implicate cell tropism and evasion of apoptosis as critical determinants of CMV transmission” [62]. At the very least, this conclusion highlights that the presence/absence of these viral ORFs dramatically alters the phenotype of infection in vivo.
Finally, it is worth stressing that the mechanistic basis of protection with RhCMV 68-1/SIV vectors is afforded by the particulars of the ectopic antigen cassette used to drive expression of the SIV antigens [1-6]. The promoter for SIV antigen expression is the human EF1α promoter, which is constitutively expressed. In addition to the generation of unconventional T cell responses to SIV antigens, there is also the surprising result that no antibody responses to SIV antigens are detected in vaccinated, previously RhCMV WT-infected, animals. As mentioned above, HCMV and RhCMV genes are temporally expressed in a highly regulated cascade. Derivation of a RhCMV 68-1-based vaccine for Ebola virus (EBOV) used the endogenous RhCMV RhUL83 (pp65) promoter, which is expressed with late gene kinetics, to drive expression of the EBOV glycoprotein [9]. Protection against EBOV challenge was observed in 75% of vaccinated monkeys, but in this case, protection was associated with the induction of EBOV-specific antibodies. The mechanism by which EBOV-specific antibodies are stimulated may be a function of the timing of when the host is presented with antigen relative to the state of viral manipulation of the infected cell microenvironment. Importantly, the differential host responses highlight how CMV-vectored vaccines can be optimized to dial in the type of immune response best suited to the particular pathogen. Using a combination of constitutive and late gene promoters, it might be possible to incite the development of sustained antibody and effector memory T cell responses in vaccinated hosts.
Summary:
The “happy accident” that occurred during passage of RhCMV 68-1 in fibroblasts, resulting in loss of full coding capacity, opened up a novel window into how CMV species exploit host cell signaling, trafficking, activation, and restriction pathways. The studies on this virus highlight the power of nonhuman primate models to improve the human condition since this result was only possible in monkey models of human diseases. The startling discovery of how RhCMV can be harnessed to develop viral-vectored vaccines enables the development of new modalities, using HCMV vectors, to prevent and treat both long-time human scourges and newly emergent pathogens.
ACKNOWLEDGMENTS.
This work was supported by NIH Awards AI097629 (MRW and PAB), AI049342 (MRW and PAB), AI131568 (PAB and DJH-O), AI134618 (PAB and EES), AI063356 (PAB and DJD), and OD P51 OD011107 (CNPRC); the Margaret Deterding Infectious Disease Research Support Fund (PAB).
Literature Cited
- 1.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 2011; 473(7348):523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansen SG, Piatak M Jr., Ventura AB, Hughes CM, Gilbride RM, Ford JC, et al. Immune clearance of highly pathogenic SIV infection. Nature 2013; 502(7469):100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hansen SG, Powers CJ, Richards R, Ventura AB, Ford JC, Siess D, et al. Evasion of CD8+ T cells is critical for superinfection by cytomegalovirus. Science 2010; 328(5974):102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hansen SG, Sacha JB, Hughes CM, Ford JC, Burwitz BJ, Scholz I, et al. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 2013; 340(6135):1237874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hansen SG, Vieville C, Whizin N, Coyne-Johnson L, Siess DC, Drummond DD, et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med 2009; 15(3):293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansen SG, Wu HL, Burwitz BJ, Hughes CM, Hammond KB, Ventura AB, et al. Broadly targeted CD8(+) T cell responses restricted by major histocompatibility complex E. Science 2016; 351(6274):714–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansen SG, Zak DE, Xu G, Ford JC, Marshall EE, Malouli D, et al. Prevention of tuberculosis in rhesus macaques by a cytomegalovirus-based vaccine. Nat Med 2018. [DOI] [PMC free article] [PubMed]
- 8.Hansen SG, Womack J, Scholz I, Renner A, Edgel KA, Xu G, et al. Cytomegalovirus vectors expressing Plasmodium knowlesi antigens induce immune responses that delay parasitemia upon sporozoite challenge. PLoS ONE 2019; 14(1):e0210252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marzi A, Murphy AA, Feldmann F, Parkins CJ, Haddock E, Hanley PW, et al. Cytomegalovirus-based vaccine expressing Ebola virus glycoprotein protects nonhuman primates from Ebola virus infection. Scientific reports 2016; 6:21674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGeoch DJ, Rixon FJ, Davison AJ. Topics in herpesvirus genomics and evolution. Virus Res 2006; 117(1):90–104. [DOI] [PubMed] [Google Scholar]
- 11.Davison AJ, Trus BL, Cheng N, Steven AC, Watson MS, Cunningham C, et al. A novel class of herpesvirus with bivalve hosts. Journal of General Virology 2005; 86(Pt 1):41–53. [DOI] [PubMed] [Google Scholar]
- 12.Selvarajan Sigamani S, Zhao H, Kamau YN, Baines JD, Tang L. The structure of the herpes simplex virus DNA-packaging terminase pUL15 nuclease domain suggests an evolutionary lineage among eukaryotic and prokaryotic viruses. Journal of Virology 2013; 87(12):7140–7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darwin C On the Order of Species by Means of Natural Selection. London: J. Murray; 1859. [Google Scholar]
- 14.Le Fur S, Fara E, Mackaye HT, Vignaud P, Brunet M. Toros-Menalla (Chad, 7 Ma), the earliest hominin-bearing area: How many mammal paleocommunities? Journal of human evolution 2014; 69:79–90. [DOI] [PubMed] [Google Scholar]
- 15.Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C, et al. Genetic content of wild-type human cytomegalovirus. Journal of General Virology 2004; 85(Pt 5):1301–1312. [DOI] [PubMed] [Google Scholar]
- 16.Davison A Comparative analysis of the genomes In: Human Herpesviruses: Biology, Therapy and Immunoprophylaxis. Arvin A, Campadielli G, Moore P, et al. (editors). Cambridge: Cambridge University Press; 2007. pp. 16–26. [PubMed] [Google Scholar]
- 17.Nelson CS, Herold BC, Permar SR. A new era in cytomegalovirus vaccinology: considerations for rational design of next-generation vaccines to prevent congenital cytomegalovirus infection. NPJ vaccines 2018; 3:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reeves M, Sinclair J. Regulation of human cytomegalovirus transcription in latency: beyond the major immediate-early promoter. Viruses 2013; 5(6):1395–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zuhair M, Smit GSA, Wallis G, Jabbar F, Smith C, Devleesschauwer B, et al. Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis. Rev Med Virol 2019; 29(3):e2034. [DOI] [PubMed] [Google Scholar]
- 20.Barbosa NG, Yamamoto AY, Duarte G, Aragon DC, Fowler KB, Boppana S, et al. Cytomegalovirus (CMV) Shedding in Seropositive Pregnant Women from a High Seroprevalence Population: “The Brazilian Cytomegalovirus Hearing and Maternal Secondary Infection Study” (BraCHS). Clin Infect Dis 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boppana SB, Ross SA, Shimamura M, Palmer AL, Ahmed A, Michaels MG, et al. Saliva polymerase-chain-reaction assay for cytomegalovirus screening in newborns. N Engl J Med 2011; 364(22):2111–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Britt WJ. Maternal Immunity and the Natural History of Congenital Human Cytomegalovirus Infection. Viruses 2018; 10(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol 2007; 17(4):253–276. [DOI] [PubMed] [Google Scholar]
- 24.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. The “silent” global burden of congenital cytomegalovirus. ClinMicrobiolRev 2013; 26(1):86–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lanzieri TM, Dollard SC, Bialek SR, Grosse SD. Systematic review of the birth prevalence of congenital cytomegalovirus infection in developing countries. Int J Infect Dis 2014; 22:44–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cannon MJ, Schmid DS, Hyde TB. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev Med Virol 2010; 20(4):202–213. [DOI] [PubMed] [Google Scholar]
- 27.Lantos PM, Hoffman K, Permar SR, Jackson P, Hughes BL, Kind A, et al. Neighborhood Disadvantage is Associated with High Cytomegalovirus Seroprevalence in Pregnancy. Journal of racial and ethnic health disparities 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lantos PM, Hoffman K, Permar SR, Jackson P, Hughes BL, Swamy GK. Geographic Disparities in Cytomegalovirus Infection During Pregnancy. Journal of the Pediatric Infectious Diseases Society 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lantos PM, Maradiaga-Panayotti G, Barber X, Raynor E, Tucci D, Hoffman K, et al. Geographic and Racial Disparities in Infant Hearing Loss. Otolaryngology--head and neck surgery : official journal of American Academy of Otolaryngology-Head and Neck Surgery 2018:194599818803305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hotez PJ. Neglected infections of poverty in the United States of america. PLoS Negl Trop Dis 2008; 2(6):e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dowd JB, Zajacova A, Aiello A. Early origins of health disparities: burden of infection, health, and socioeconomic status in U.S. children. Soc Sci Med 2009; 68(4):699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fowler KB, Ross SA, Shimamura M, Ahmed A, Palmer AL, Michaels MG, et al. Racial and Ethnic Differences in the Prevalence of Congenital Cytomegalovirus Infection. J Pediatr 2018. [DOI] [PubMed] [Google Scholar]
- 33.Inagaki K, Blackshear C, Palmer A, Hobbs CV. Risk Factors, Geographic Distribution, and Healthcare Burden of Symptomatic Congenital Cytomegalovirus Infection in the United States: Analysis of a Nationally Representative Database, 2000-2012. J Pediatr 2018; 199:118–123.e111. [DOI] [PubMed] [Google Scholar]
- 34.Britt W Human cytomegalovirus infections and mechanisms of disease In: Cytomegaloviruses: molecular biology and immunology. Reddehase MJ (editor). Wymondham, U.K.: Caister Academic Press; 2006. [Google Scholar]
- 35.Varani S, Landini MP. Cytomegalovirus-induced immunopathology and its clinical consequences. Herpesviridae 2011; 2(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med 2005; 202(5):673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diamond DJ, La Rosa C, Chiuppesi F, Contreras H, Dadwal S, Wussow F, et al. A fifty-year odyssey: prospects for a cytomegalovirus vaccine in transplant and congenital infection. Expert Rev Vaccines 2018; 17(10):889–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zanghellini F, Boppana SB, Emery VC, Griffiths PD, Pass RF. Asymptomatic primary cytomegalovirus infection: virologic and immunologic features. J Infect Dis 1999; 180:702–707. [DOI] [PubMed] [Google Scholar]
- 39.Baumann NS, Welten SPM, Torti N, Pallmer K, Borsa M, Barnstorf I, et al. Early primed KLRG1-CMV-specific T cells determine the size of the inflationary T cell pool. PLoS pathogens 2019; 15(5):e1007785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hage E, Wilkie GS, Linnenweber-Held S, Dhingra A, Suarez NM, Schmidt JJ, et al. Characterization of Human Cytomegalovirus Genome Diversity in Immunocompromised Hosts by Whole-Genome Sequencing Directly From Clinical Specimens. J Infect Dis 2017; 215(11):1673–1683. [DOI] [PubMed] [Google Scholar]
- 41.Malouli D, Hansen SG, Nakayasu ES, Marshall EE, Hughes CM, Ventura AB, et al. Cytomegalovirus pp65 limits dissemination but is dispensable for persistence. J Clin Invest 2014; 124(5):1928–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sturgill ER, Malouli D, Hansen SG, Burwitz BJ, Seo S, Schneider CL, et al. Natural Killer Cell Evasion Is Essential for Infection by Rhesus Cytomegalovirus. PLoS pathogens 2016; 12(8):e1005868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Früh K, Malouli D, Oxford K, Barry P. Non-Human-Primate Models of Cytomegalovirus Infection, Prevention, and Therapy In: CYTOMEGALOVIRUSES: From Molecular Pathogenesis to Therapy. Reddehase M (editor). Norfolk, UK: Caister Academic Press/Horizon; 2013. [Google Scholar]
- 44.Messerle M, Crnkovic I, Hammerschmidt W, Ziegler H, Koszinowski UH. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc Natl Acad Sci U S A 1997; 94(26):14759–14763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang WL, Barry PA. Cloning of the full-length rhesus cytomegalovirus genome as an infectious and self-excisable bacterial artificial chromosome for analysis of viral pathogenesis. Journal of Virology 2003; 77(9):5073–5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asher DM, Gibbs CJ Jr., Lang DJ, Gajdusek DC, Chanock RM. Persistent shedding of cytomegalovirus in the urine of healthy Rhesus monkeys. Proceedings of the Society for Experimental Biology and Medicine Society for Experimental Biology and Medicine 1974; 145(3):794–801. [DOI] [PubMed] [Google Scholar]
- 47.Gill RB, Jason Bowman J, Krogmann T, Wollenberg K, Asher DM, Cohen JI. Coding potential of UL/b’ from the initial source of rhesus cytomegalovirus Strain 68-1. Virology 2013; 447(1-2):208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hansen SG, Strelow LI, Franchi DC, Anders DG, Wong SW. Complete sequence and genomic analysis of rhesus cytomegalovirus. Jornal of Virology 2003; 77:6620–6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malouli D, Nakayasu ES, Viswanathan K, Camp DG 2nd, Chang WL, Barry PA, et al. Reevaluation of the coding potential and proteomic analysis of the BAC-derived rhesus cytomegalovirus strain 68-1. Journal of Virology 2012; 86(17):8959–8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oxford KL, Eberhardt MK, Yang KW, Strelow L, Kelly S, Zhou SS, et al. Protein coding content of the U(L)b’ region of wild-type rhesus cytomegalovirus. Virology 2008; 373(1):181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rivailler P, Jiang H, Cho YG, Quink C, Wang F. Complete nucleotide sequence of the rhesus lymphocryptovirus: genetic validation for an Epstein-Barr virus animal model. Journal of Virology 2002; 76(1):421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lilja AE, Shenk T. Efficient Replication of Rhesus Cytomegalovirus Variants in Multiple Rhesus and Human Cell Types. Proc Natl Acad Sci (USA) 2008; 105:19950–19955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, Sarasini A, et al. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. Journal of Virology 2004; 78(18):10023–10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. Journal of Virology 2006; 80(2):710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ryckman BJ, Rainish BL, Chase MC, Borton JA, Nelson JA, Jarvis MA, et al. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. Journal of Virology 2008; 82(1):60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yue Y, Kaur A, Lilja A, Diamond DJ, Walter MR, Barry PA. The susceptibility of primary cultured rhesus macaque kidney epithelial cells to rhesus cytomegalovirus strains. Journal of General Virology 2016; 97(6):1426–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goldmacher VS, Bartle LM, Skaletskaya A, Dionne CA, Kedersha NL, Vater CA, et al. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to bcl-2. Proceedings of the National Academy of Sciences 1999; 96:12536–12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang WL, Tarantal AF, Zhou SS, Borowsky AD, Barry PA. A recombinant rhesus cytomegalovirus expressing enhanced green fluorescent protein retains the wild-type phenotype and pathogenicity in fetal macaques. Journal of Virology 2002; 76(18):9493–9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lockridge KM, Sequar G, Zhou SS, Yue Y, Mandell CP, Barry PA. Pathogenesis of experimental rhesus cytomegalovirus infection. Journal of Virology 1999; 73(11):9576–9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sequar G, Britt WJ, Lakeman FD, Lockridge KM, Tarara RP, Canfield DR, et al. Experimental coinfection of rhesus macaques with rhesus cytomegalovirus and simian immunodeficiency virus: pathogenesis. Journal of Virology 2002; 76(15):7661–7671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tarantal AF, Salamat MS, Britt WJ, Luciw PA, Hendrickx AG, Barry PA. Neuropathogenesis induced by rhesus cytomegalovirus in fetal rhesus monkeys (Macaca mulatta). The Journal of infectious diseases 1998; 177(2):446–450. [DOI] [PubMed] [Google Scholar]
- 62.Burwitz BJ, Malouli D, Bimber BN, Reed JS, Ventura AB, Hancock MH, et al. Cross-Species Rhesus Cytomegalovirus Infection of Cynomolgus Macaques. PLoS pathogens 2016; 12(11):e1006014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barry PA, Strelow L. Development of breeding populations of rhesus macaques (Macaca mulatta) that are specific pathogen-free for rhesus cytomegalovirus. Comparative medicine 2008; 58(1):43–46. [PMC free article] [PubMed] [Google Scholar]
- 64.Vogel P, Weigler BJ, Kerr H, Hendrickx AG, Barry PA. Seroepidemiologic studies of cytomegalovirus infection in a breeding population of rhesus macaques. Lab Anim Sci 1994; 44(1):25–30. [PubMed] [Google Scholar]
- 65.Eberhardt MK, Deshpande A, Fike J, Short R, Schmidt KA, Blozis SA, et al. Exploitation of Interleukin-10 (IL-10) Signaling Pathways: Alternate Roles of Viral and Cellular IL-10 in Rhesus Cytomegalovirus Infection. Journal of Virology 2016; 90(21):9920–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oxford K, dela Pena-Ponce M, Jensen K, Eberhardt M, Spinner A, Van Rompay K, et al. The interplay between immune maturation, age, chronic viral infection and environment. Immunity Ageing 2015; 12:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deere JD, Chang WW-L, A. V, Schmidt KA, Deshpande A, Castillo LD, et al. Neutralization of rhesus cytomegalovirus IL-10 reduces horizontal transmission and alters long-term immunity. PNAS 2019; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abel K, Strelow L, Yue Y, Eberhardt MK, Schmidt KA, Barry PA. A heterologous DNA prime/protein boost immunization strategy for rhesus cytomegalovirus. Vaccine 2008; 26(47):6013–6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oxford KL, Strelow L, Yue Y, Chang WL, Schmidt KA, Diamond DJ, et al. Open Reading Frames Carried on UL/b’ Are Implicated in Shedding and Horizontal Transmission of Rhesus Cytomegalovirus in Rhesus Monkeys. Journal of Virology 2011; 85(10):5105–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yue Y, Kaur A, Eberhardt MK, Kassis N, Zhou SS, Tarantal AF, et al. Immunogenicity and protective efficacy of DNA vaccines expressing rhesus cytomegalovirus glycoprotein B, phosphoprotein 65-2, and viral interleukin-10 in rhesus macaques. Journal of Virology 2007; 81(3):1095–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Assaf BT, Mansfield KG, Westmoreland SV, Kaur A, Oxford KL, Diamond DJ, et al. Patterns of Acute Rhesus Cytomegalovirus (RhCMV) Infection Predict Long-Term RhCMV Infection. Journal of Virology 2012; 86(11):6354–6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chang WL, Barry PA. Attenuation of innate immunity by cytomegalovirus IL-10 establishes a long-term deficit of adaptive antiviral immunity. Proceedings of the National Academy of Sciences of the United States of America 2010; 107(52):22647–22652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Akter P, Cunningham C, McSharry BP, Dolan A, Addison C, Dargan DJ, et al. Two novel spliced genes in human cytomegalovirus. Journal of General Virology 2003; 84(Pt 5):1117–1122. [DOI] [PubMed] [Google Scholar]
- 74.Gao H, Tao R, Zheng Q, Xu J, Shang S. Recombinant HCMV UL128 expression and functional identification of PBMC-attracting activity in vitro. Arch Virol 2013; 158(1):173–177. [DOI] [PubMed] [Google Scholar]
- 75.Straschewski S, Patrone M, Walther P, Gallina A, Mertens T, Frascaroli G. Protein pUL128 of human cytomegalovirus is necessary for monocyte infection and blocking of migration. Journal of Virology 2011; 85(10):5150–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tao R, Xu J, Gao H, Zhao W, Shang S. Characteristics and functions of human cytomegalovirus UL128 gene/protein. Acta virologica 2014; 58(2):103–107. [DOI] [PubMed] [Google Scholar]
- 77.Zheng Q, Tao R, Gao H, Xu J, Shang S, Zhao N. HCMV-encoded UL128 enhances TNF-alpha and IL-6 expression and promotes PBMC proliferation through the MAPK/ERK pathway in vitro. Viral Immunol 2012; 25(2):98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vomaske J, Denton M, Kreklywich C, Andoh T, Osborn JM, Chen D, et al. Cytomegalovirus CC chemokine promotes immune cell migration. Journal of Virology 2012; 86(21):11833–11844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Skaletskaya A, Bartle LM, Chittenden T, McCormick AL, Mocarski ES, Goldmacher VS. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc Natl Acad Sci U S A 2001; 98(14):7829–7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Patterson CE, Shenk T. Human cytomegalovirus UL36 protein is dispensable for viral replication in cultured cells. Journal of Virology 1999; 73(9):7126–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Proff J, Walterskirchen C, Brey C, Geyeregger R, Full F, Ensser A, et al. Cytomegalovirus-Infected Cells Resist T Cell Mediated Killing in an HLA-Recognition Independent Manner. Frontiers in microbiology 2016; 7:844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brutkiewicz RR. Cell Signaling Pathways That Regulate Antigen Presentation. J Immunol 2016; 197(8):2971–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McSharry BP, Tomasec P, Neale ML, Wilkinson GW. The most abundantly transcribed human cytomegalovirus gene (beta 2.7) is non-essential for growth in vitro. Journal of General Virology 2003; 84(Pt 9):2511–2516. [DOI] [PubMed] [Google Scholar]
- 84.Engel P, Perez-Carmona N, Alba MM, Robertson K, Ghazal P, Angulo A. Human cytomegalovirus UL7, a homologue of the SLAM-family receptor CD229, impairs cytokine production. Immunology and cell biology 2011; 89(7):753–766. [DOI] [PubMed] [Google Scholar]
- 85.Perez-Carmona N, Martinez-Vicente P, Farre D, Gabaev I, Messerle M, Engel P, et al. A Prominent Role of the Human Cytomegalovirus UL8 Glycoprotein in Restraining Proinflammatory Cytokine Production by Myeloid Cells at Late Times during Infection. Journal of Virology 2018; 92(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gabaev I, Steinbruck L, Pokoyski C, Pich A, Stanton RJ, Schwinzer R, et al. The human cytomegalovirus UL11 protein interacts with the receptor tyrosine phosphatase CD45, resulting in functional paralysis of T cells. PLoS pathogens 2011; 7(12):e1002432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zischke J, Mamareli P, Pokoyski C, Gabaev I, Buyny S, Jacobs R, et al. The human cytomegalovirus glycoprotein pUL11 acts via CD45 to induce T cell IL-10 secretion. PLoS pathogens 2017; 13(6):e1006454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rolle A, Mousavi-Jazi M, Eriksson M, Odeberg J, Soderberg-Naucler C, Cosman D, et al. Effects of human cytomegalovirus infection on ligands for the activating NKG2D receptor of NK cells: up-regulation of UL16-binding protein (ULBP)1 and ULBP2 is counteracted by the viral UL16 protein. J Immunol 2003; 171(2):902–908. [DOI] [PubMed] [Google Scholar]
- 89.Saverino D, Ghiotto F, Merlo A, Bruno S, Battini L, Occhino M, et al. Specific recognition of the viral protein UL18 by CD85j/LIR-1/ILT2 on CD8+ T cells mediates the non-MHC-restricted lysis of human cytomegalovirus-infected cells. J Immunol 2004; 172(9):5629–5637. [DOI] [PubMed] [Google Scholar]
- 90.Leong CC, Chapman TL, Bjorkman PJ, Formankova D, Mocarski ES, Phillips JH, et al. Modulation of natural killer cell cytotoxicity in human cytomegalovirus infection: The role of endogenous class I major histocompatibility complex and a viral class I homolog. J Exp Med 1998; 187(10):1681–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cosman D, Fanger N, Borges L. Human cytomegalovirus, MHC class I and inhibitory signalling receptors: more questions than answers. Immunol Rev 1999; 168:177–185. [DOI] [PubMed] [Google Scholar]
- 92.Patel M, Vlahava VM, Forbes SK, Fielding CA, Stanton RJ, Wang ECY. HCMV-Encoded NK Modulators: Lessons From in vitro and in vivo Genetic Variation. Frontiers in immunology 2018; 9:2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang D, Bresnahan W, Shenk T. Human cytomegalovirus encodes a highly specific RANTES decoy receptor. Proc Natl Acad Sci U S A 2004; 101(47):16642–16647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Feng L, Sheng J, Vu GP, Liu Y, Foo C, Wu S, et al. Human cytomegalovirus UL23 inhibits transcription of interferon-gamma stimulated genes and blocks antiviral interferon-gamma responses by interacting with human N-myc interactor protein. PLoS pathogens 2018; 14(1):e1006867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Grey F, Tirabassi R, Meyers H, Wu G, McWeeney S, Hook L, et al. A Viral microRNA Down-Regulates Multiple Cell Cycle Genes through mRNA 5’UTRs. PLoS Pathog 2010; 6(6):e1000967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Goodwin CM, Munger J. The IkappaB Kinases Restrict Human Cytomegalovirus Infection. Journal of Virology 2019; 93(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Terhune SS, Moorman NJ, Cristea IM, Savaryn JP, Cuevas-Bennett C, Rout MP, et al. Human Cytomegalovirus UL29/28 Protein Interacts with Components of the NuRD Complex Which Promote Accumulation of Immediate-Early RNA. PLoS pathogens 2010; 6(6):e1000965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chee MS, Bankier AT, Beck S, Bohni R, Brown CM, Cerny R, et al. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr Topics Microbiol Immunol 1990; 154:125–169. [DOI] [PubMed] [Google Scholar]
- 99.Margulies BJ, Browne H, Gibson W. Identification of the human cytomegalovirus G protein-coupled receptor homologue encoded by UL33 in infected cells and enveloped virus particles. Virology 1996; 225:111–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McCormick AL, Skaletskaya A, Barry PA, Mocarski ES, Goldmacher VS. Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virol 2003; 316:221–233. [DOI] [PubMed] [Google Scholar]
- 101.Terhune S, Torigoi E, Moorman N, Silva M, Qian Z, Shenk T, et al. Human Cytomegalovirus UL38 Protein Blocks Apoptosis. Journal of Virology 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Seo JY, Yaneva R, Hinson ER, Cresswell P. Human Cytomegalovirus Directly Induces the Antiviral Protein Viperin to Enhance Infectivity. Science 2011. [DOI] [PubMed]
- 103.Rodriguez-Sanchez I, Schafer XL, Monaghan M, Munger J. The Human Cytomegalovirus UL38 protein drives mTOR-independent metabolic flux reprogramming by inhibiting TSC2. PLoS pathogens 2019; 15(1):e1007569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tomasec P, Braud VM, Rickards C, Powell MB, McSharry BP, Gadola S, et al. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science 2000; 287(5455):1031. [DOI] [PubMed] [Google Scholar]
- 105.Sullivan LC, Westall GP, Widjaja JM, Mifsud NA, Nguyen TH, Meehan AC, et al. The Presence of HLA-E-Restricted, CMV-Specific CD8+ T Cells in the Blood of Lung Transplant Recipients Correlates with Chronic Allograft Rejection. PLoS ONE 2015; 10(8):e0135972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yawata N, Selva KJ, Liu YC, Tan KP, Lee AW, Siak J, et al. Dynamic change in natural killer cell type in the human ocular mucosa in situ as means of immune evasion by adenovirus infection. Mucosal immunology 2016; 9(1):159–170. [DOI] [PubMed] [Google Scholar]
- 107.Heatley SL, Pietra G, Lin J, Widjaja JM, Harpur CM, Lester S, et al. Polymorphism in human cytomegalovirus UL40 impacts on recognition of human leukocyte antigen-E (HLA-E) by natural killer cells. J Biol Chem 2013; 288(12):8679–8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Prod’homme V, Tomasec P, Cunningham C, Lemberg MK, Stanton RJ, McSharry BP, et al. Human cytomegalovirus UL40 signal peptide regulates cell surface expression of the NK cell ligands HLA-E and gpUL18. J Immunol 2012; 188(6):2794–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fu YZ, Guo Y, Zou HM, Su S, Wang SY, Yang Q, et al. Human cytomegalovirus protein UL42 antagonizes cGAS/MITA-mediated innate antiviral response. PLoS pathogens 2019; 15(5):e1007691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee MK, Kim YJ, Kim YE, Han TH, Milbradt J, Marschall M, et al. Transmembrane Protein pUL50 of Human Cytomegalovirus Inhibits ISGylation by Downregulating UBE1L. Journal of Virology 2018; 92(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Costa H, Nascimento R, Sinclair J, Parkhouse RM. Human cytomegalovirus gene UL76 induces IL-8 expression through activation of the DNA damage response. PLoS pathogens 2013; 9(9):e1003609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Saffert RT, Kalejta RF. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. Journal of Virology 2006; 80(8):3863–3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Arnon TI, Achdout H, Levi O, Markel G, Saleh N, Katz G, et al. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat Immunol 2005; 6(5):515–523. [DOI] [PubMed] [Google Scholar]
- 114.Abate DA, Watanabe S, Mocarski ES. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. Journal of Virology 2004; 78(20):10995–11006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Iwahori S, Umana AC, VanDeusen HR, Kalejta RF. Human cytomegalovirus-encoded viral cyclin-dependent kinase (v-CDK) UL97 phosphorylates and inactivates the retinoblastoma protein-related p107 and p130 proteins. J Biol Chem 2017; 292(16):6583–6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chang WL, Baumgarth N, Yu D, Barry PA. Human cytomegalovirus-encoded interleukin-10 homolog inhibits maturation of dendritic cells and alters their functionality. Journal of Virology 2004; 78(16):8720–8731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kotenko SV, Saccani S, Izotova LS, Mirochnitchenko OV, Pestka S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proceedings of the National Academy of Sciences 2000; 97(4):1695–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lockridge KM, Zhou SS, Kravitz RH, Johnson JL, Sawai ET, Blewett EL, et al. Primate cytomegaloviruses encode and express an IL-10-like protein. Virol 2000; 268(2):272–280. [DOI] [PubMed] [Google Scholar]
- 119.Spencer JV, Lockridge KM, Barry PA, Lin G, Tsang M, Penfold ME, et al. Potent immunosuppressive activities of cytomegalovirus- encoded interleukin-10. Journal of Virology 2002; 76(3):1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jenkins C, Abendroth A, Slobedman B. A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. Journal of Virology 2004; 78(3):1440–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Young VP, Mariano MC, Tu CC, Allaire KM, Avdic S, Slobedman B, et al. Modulation of the Host Environment by Human Cytomegalovirus with Viral Interleukin 10 in Peripheral Blood. J Infect Dis 2017; 215(6):874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stern-Ginossar N, Elefant N, Zimmermann A, Wolf DG, Saleh N, Biton M, et al. Host immune system gene targeting by a viral miRNA. Science 2007; 317(5836):376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nachmani D, Lankry D, Wolf DG, Mandelboim O. The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat Immunol 2010; 11(9):806–813. [DOI] [PubMed] [Google Scholar]
- 124.Hancock MH, Hook LM, Mitchell J, Nelson JA. Human Cytomegalovirus MicroRNAs miR-US5-1 and miR-UL112-3p Block Proinflammatory Cytokine Production in Response to NF-kappaB-Activating Factors through Direct Downregulation of IKKalpha and IKKbeta. mBio 2017; 8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Atalay R, Zimmermann A, Wagner M, Borst E, Benz C, Messerle M, et al. Identification and expression of human cytomegalovirus transcription units coding for two distinct Fcgamma receptor homologs. J Virol 2002; 76(17):8596–8608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Taylor RT, Bresnahan WA. Human cytomegalovirus immediate-early 2 gene expression blocks virus-induced Beta interferon production. Journal of Virology 2005; 79(6):3873–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Paulus C, Krauss S, Nevels M. A human cytomegalovirus antagonist of type I IFN-dependent signal transducer and activator of transcription signaling. Proc Natl Acad Sci U S A 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Le VT, Trilling M, Hengel H. The Cytomegaloviral Protein pUL138 Acts as Potentiator of TNF Receptor 1 Surface Density to Enhance ULb’-encoded modulation of TNF-{alpha} Signaling. Journal of Virology 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Montag C, Wagner JA, Gruska I, Vetter B, Wiebusch L, Hagemeier C. The Latency-Associated UL138 Gene Product of Human Cytomegalovirus Sensitizes Cells to TNF{alpha} Signaling by Upregulating TNF{alpha} Receptor 1 Cell Surface Expression. Journal of Virology 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Goodrum F, Reeves M, Sinclair J, High K, Shenk T. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 2007; 110(3):937–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tomasec P, Wang EC, Davison AJ, Vojtesek B, Armstrong M, Griffin C, et al. Downregulation of natural killer cell-activating ligand CD155 by human cytomegalovirus UL141. Nat Immunol 2005; 6(2):181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Prod’homme V, Sugrue DM, Stanton RJ, Nomoto A, Davies J, Rickards CR, et al. Human Cytomegalovirus UL141 Promotes Efficient Downregulation of the Natural Killer cell Activating Ligand CD112. Journal of General Virology 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wills MR, Ashiru O, Reeves MB, Okecha G, Trowsdale J, Tomasec P, et al. Human cytomegalovirus encodes an MHC class I-like molecule (UL142) that functions to inhibit NK cell lysis. J Immunol 2005; 175(11):7457–7465. [DOI] [PubMed] [Google Scholar]
- 134.Benedict CA, Butrovich KD, Lurain NS, Corbeil J, Rooney I, Schneider P, et al. Cutting edge: a novel viral TNF receptor superfamily member in virulent strains of human cytomegalovirus. J Immunol 1999; 162(12):6967–6970. [PubMed] [Google Scholar]
- 135.Penfold MET, Dairaghi DJ, Duke GM, Saederup N, Mocarski ES, Kemble GW, et al. Cytomegalovirus encodes a potent a chemokine. Proc Natl Acad Sci (USA) 1999; 96:9839–9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dassa L, Seidel E, Oiknine-Djian E, Yamin R, Wolf DG, Le-Trilling VTK, et al. The HCMV protein UL148A Downregulates the Activating NK Cell Ligand MICA to Avoid NK Cell Attack. Journal of Virology 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nguyen CC, Siddiquey MNA, Zhang H, Li G, Kamil JP. Human Cytomegalovirus Tropism Modulator UL148 Interacts with SEL1L, a Cellular Factor That Governs Endoplasmic Reticulum-Associated Degradation of the Viral Envelope Glycoprotein gO. Journal of Virology 2018; 92(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lilley BN, Ploegh HL, Tirabassi RS. Human cytomegalovirus open reading frame TRL11/IRL11 encodes an immunoglobulin G Fc-binding protein. Journal of Virology 2001; 75(22):11218–11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cortese M, Calo S, D’Aurizio R, Lilja A, Pacchiani N, Merola M. Recombinant human cytomegalovirus (HCMV) RL13 binds human immunoglobulin G Fc. PLoS ONE 2012; 7(11):e50166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hakki M, Geballe AP. Double-stranded RNA binding by human cytomegalovirus pTRS1. J Virol 2005; 79(12):7311–7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hakki M, Marshall EE, De Niro KL, Geballe AP. Binding and nuclear relocalization of PKR by human cytomegalovirus TRS1. J Virol 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jones TR, Sun L. Human cytomegalovirus US2 destabilizes major histocompatibility complex class I heavy chains. Journal of Virology 1997; 71:2970–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Machold RP, Wiertz EJ, Jones TR, Ploegh HL. The HCMV gene products US11 and US2 differ in their ability to attack allelic forms of murine major histocompatibility complex (MHC) class I heavy chains. J Exp Med 1997; 185:363–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wiertz E, Hill A, Tortorella D, Ploegh H. Cytomegaloviruses use multiple mechanisms to elude the host immune response. Immunol Lett 1997; 57(1-3):213–216. [DOI] [PubMed] [Google Scholar]
- 145.Tirosh B, Iwakoshi NN, Lilley BN, Lee AH, Glimcher LH, Ploegh HL. Human Cytomegalovirus Protein US11 Provokes an Unfolded Protein Response That May Facilitate the Degradation of Class I Major Histocompatibility Complex Products. Journal of Virology 2005; 79(5):2768–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tomazin R, Boname J, Hegde NR, Lewinsohn DM, Altschuler Y, Jones TR, et al. Cytomegalovirus US2 destroys two components of the MHC class II pathway, preventing recognition by CD4+ T cells. Nat Med 1999; 5:1039–1043. [DOI] [PubMed] [Google Scholar]
- 147.Ahn K, Angulo A, Ghazal P, Peterson PA, Yang Y, Fruh K. Human cytomegalovirus inhibits antigen presentation by a sequential multistep process. Proc Natl Acad Sci U S A 1996; 93:10990–10995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jones TR, Wiertz EJ, Sun L, Fish KN, Nelson JA, Ploegh HL. Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. Proceedings of the National Academy of Sciences 1996; 93:11327–11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ahn K, Gruhler A, Galocha B, Jones TR, Wiertz EJ, Ploegh HL, et al. The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 1997; 6:613–621. [DOI] [PubMed] [Google Scholar]
- 150.Park B, Spooner E, Houser BL, Strominger JL, Ploegh HL. The HCMV membrane glycoprotein US10 selectively targets HLA-G for degradation. J Exp Med 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Das S, Skomorovska-Prokvolit Y, Wang FZ, Pellett PE. Infection-Dependent Nuclear Localization of US17, a Member of the US12 Family of Human Cytomegalovirus-Encoded Seven-Transmembrane Proteins. J Virol 2006; 80(3):1191–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Luganini A, Di Nardo G, Munaron L, Gilardi G, Fiorio Pla A, Gribaudo G. Human cytomegalovirus US21 protein is a viroporin that modulates calcium homeostasis and protects cells against apoptosis. Proc Natl Acad Sci U S A 2018; 115(52):E12370–e12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Fielding CA, Weekes MP, Nobre LV, Ruckova E, Wilkie GS, Paulo JA, et al. Control of immune ligands by members of a cytomegalovirus gene expansion suppresses natural killer cell activation. eLife 2017; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Charpak-Amikam Y, Kubsch T, Seidel E, Oiknine-Djian E, Cavaletto N, Yamin R, et al. Human cytomegalovirus escapes immune recognition by NK cells through the downregulation of B7-H6 by the viral genes US18 and US20. Scientific reports 2017; 7(1):8661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Billstrom MA, Johnson GL, Avdi NJ, Worthen GS. Intracellular signaling by the chemokine receptor US28 during human cytomegalovirus infection. J Virol 1998; 72:5535–5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Billstrom MA, Lehman LA, Worthen GS. Depletion of extracellular RANTES during human cytomegalovirus infection of endothelial cells. Am J Respir Cell Mol Biol 1999; 21:163–167. [DOI] [PubMed] [Google Scholar]
- 157.Gao J-L, Murphy PM. Human cytomegalovirus open reading frame US28 encodes a functional b chemokine receptor. J Biol Chem 1994; 269(46):28539–28542. [PubMed] [Google Scholar]
- 158.Kuhn DE, Beall CJ, Kolattukudy PE. The cytomegalovirus US28 protein binds multiple CC chemokines with high affinity. Biochem Biophys Res Commun 1995; 211:325–330. [DOI] [PubMed] [Google Scholar]
- 159.Neote K, DiGregorio D, Mak JY, Horuk R, Schall TJ. Molecular cloning, functional expression, and signaling characteristics of a CC chemokine receptor. Cell 1993; 72:415–425. [DOI] [PubMed] [Google Scholar]
- 160.Pleskoff O, Treboute C, Alizon M. The cytomegalovirus-encoded chemokine receptor US28 can enhance cell-cell fusion mediated by different viral proteins. Journal of Virology 1998; 72(8):6389–6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Streblow DN, Soderberg-Naucler C, Vieira J, Smith P, Wakabayashi E, Ruchti F, et al. The human cytomegalovirus chemokine receptor US28 mediates vascular smooth muscle cell migration. Cell 1999; 99:511–520. [DOI] [PubMed] [Google Scholar]
- 162.Penfold ME, Schmidt TL, Dairaghi DJ, Barry PA, Schall TJ. Characterization of the rhesus cytomegalovirus US28 locus. Journal of Virology 2003; 77(19):10404–10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Boeck JM, Stowell GA, O’Connor CM, Spencer JV. The Human Cytomegalovirus US27 Gene Product Constitutively Activates Antioxidant Response Element-Mediated Transcription through Gbetagamma, Phosphoinositide 3-Kinase, and Nuclear Respiratory Factor 1. Journal of Virology 2018; 92(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Amarandi RM, Luckmann M, Melynis M, Jakobsen MH, Fallah Z, Spiess K, et al. Ligand-selective small molecule modulators of the constitutively active vGPCR US28. European journal of medicinal chemistry 2018; 155:244–254. [DOI] [PubMed] [Google Scholar]
- 165.Krishna BA, Humby MS, Miller WE, O’Connor CM. Human cytomegalovirus G protein-coupled receptor US28 promotes latency by attenuating c-fos. Proc Natl Acad Sci U S A 2019; 116(5):1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]