

Abstract
Despite advances in genetic and proteomic techniques, a complete portrait of the proteome and its complement of dynamic interactions and modifications remains a lofty, and as of yet unrealized objective. Specifically, traditional biological and analytical approaches have not been able to address key questions relating to the interactions of proteins with small molecules, including drugs, drug candidates, metabolites, or protein post-translational modifications. Fortunately, chemists have bridged this experimental gap through the creation of bioorthogonal reactions. These reactions allow for the incorporation of chemical groups with highly selective reactivity into small molecules or protein modifications without perturbing their biological function, enabling the selective installation of an analysis tag for downstream investigations. The introduction of chemical strategies to parse and enrich subsets of the “functional” proteome has empowered mass spectrometry (MS)-based methods to delve more deeply and precisely into the biochemical state of cells and its perturbations by small molecules. In this Primer, we discuss how one of the most versatile bioorthogonal reactions, “click chemistry,” has been exploited to overcome limitations of biological approaches to enable the selective marking and functional investigation of critical protein/small-molecule interactions and post-translational modifications in native biological environments.
eTOC blurb
eToC blurb – Parker and Pratt
In this Primer, Pratt & Parker overview the biological applications of “click chemistry,” a class of small molecule reactions that can efficiently conjugate substrates to specific biomolecules. Focusing on proteomic investigations, they provide insight into the biological questions where it may be most useful, including studies of post-translational modifications and protein-protein, protein-drug, and protein-metabolite interactions.
Introduction
Explosive advances in genome sequencing technologies have greatly enhanced our understanding of the full protein complement of the genome (the proteome) and have provided valuable insight into numerous physiological processes and disease mechanisms. However, such methods alone do not capture the myriad post-translational events and molecular interactions that can affect protein function. This dramatic contextual and temporal variability of the proteome is part of what sets it apart from the relatively stable genome, limiting the reach of genome-based strategies to study proteins and their interactions in native environments.
Proteomic methods, on the other hand, enable the measurement of multiple properties for thousands of proteins, including their abundance, tissue distribution, subcellular localization, various post-translational modifications (PTMs), and other protein-protein interactions (Figure 1). Significant advances in enrichment techniques, ionization methods and instrument resolution have greatly facilitated the identification, characterization and quantitation of PTMs, while herculean proteomic studies have mapped networks rich with thousands of new candidate protein-protein interactions (PPIs) (Hein et al., 2015; Huttlin et al., 2015). However, despite these advances, traditional proteomic methods alone do not illuminate the complete network of interactions and modifications that proteins may undergo, nor do they evaluate protein activity or functional state.
Figure 1. Biological studies enabled by proteomics.
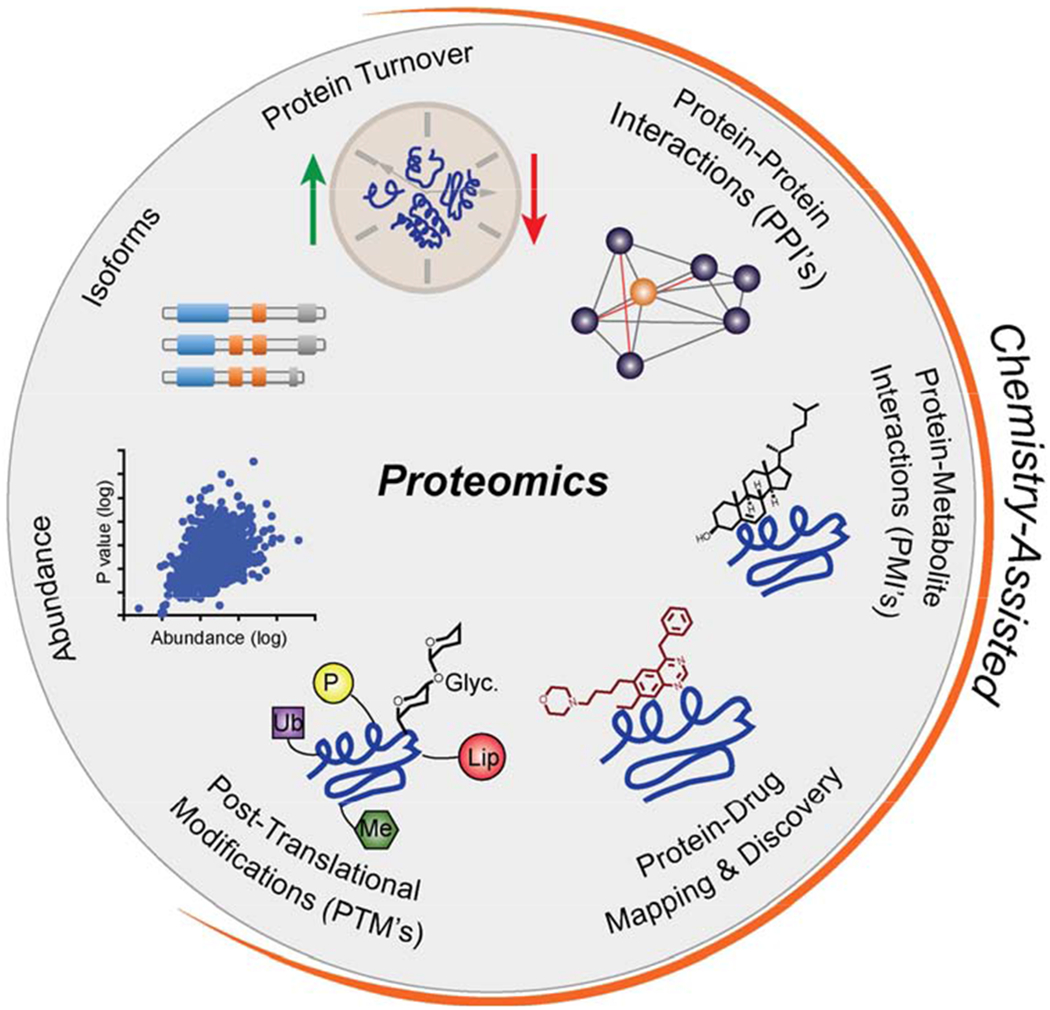
Highlighted in orange are applications that can be integrated with chemical methods to aide in proteomic investigations
Thus, a set of challenges emerged which have sparked shared interests between chemists and biologists: 1) the selective capture of dynamic interactions between proteins and small molecules, 2) direct assessment of the functional state of proteins in native environments, and 3) the selective enrichment of proteins with specific modifications. Here, we discuss the integration of chemistry with proteomic methods to address these challenges. In particular, we highlight how biorthogonal chemistry, specifically azide-alkyne click chemistry, has been exploited to selectively label, measure and identify proteins, their interactions and modifications in native environments.
Bioorthogonal Chemistry
The ability to selectively label, enrich and detect proteins, their interactions and their modifications in their native systems is key to fundamental biological research as well as drug discovery. Traditional biological approaches, including the genetic incorporation of affinity tags and the creation of selective antibodies, have been transformative for the detection of proteins. However, for various reasons, both biophysical and technical, they struggle to address the interactions of proteins, particularly with small molecules, as well as many protein post-translational modifications. These challenges have prompted the development of biorthogonal reactions that enable the selective chemical modification of proteins in complex, native environments utilizing complementary functional groups that fulfil the following criteria: (1) functional groups are not present in living systems, (2) are inert towards native biological components, and (3) react with one another under biocompatible conditions (e.g., in aqueous environment, physiological pH and temperature, favorable kinetics).
These reactions involve a two-step labeling strategy. First, the selective tagging of any protein requires the introduction of a bioorthogonal functional group to directly onto to the target(s) of interest (Figure 2, step 1). The relatively small size of the chemical groups required for a bioorthogonal reaction allows them to be incorporated into small molecules, like drugs or metabolites, without perturbing their ability to interact or modify proteins. Following this installation of a “probe”, the second step involves the use of a bioorthogonal reaction to selectively install a visualization or enrichment tag (Figure 2, step 2), enabling subsequent analysis (Figure 2, step 3) via gel-based assays, various imaging techniques, or liquid chromatography mass spectrometry (LC-MS) methods. There are well over 20 unique bioorthogonal transformations available (Figure 3), which offer a range of reactivities, orthogonality, and utility in various applications. These reactions and their various applications have been the topic of numerous comprehensive and recent reviews, and thus will not be detailed here (Lang and Chin, 2014b; McKay and Finn, 2014; Patterson et al., 2014b; Prescher and Bertozzi, 2005; Sletten and Bertozzi, 2009). Our objective is to discuss one of the most widely-used bioorthogonal transformations, the cycloaddition reaction between an alkyne and azide functional group (Figure 3E, and 3F), often referred to just as a click reaction, and its utility in proteomic investigations of small molecule-protein interactions and protein post-translational modifications.
Figure 2. General strategies to introduce bioorthogonal functionality to study proteins.
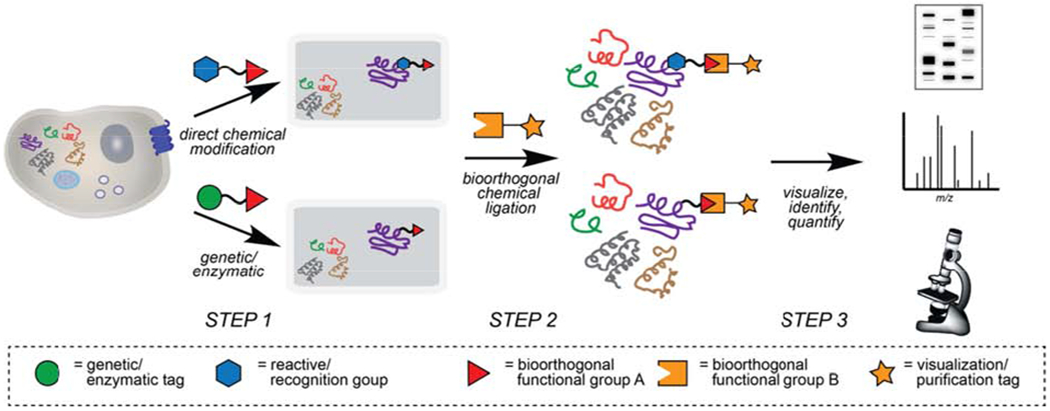
Step 1) Bioorthogonal reactive groups are first introduced onto protein targets through one of two general strategies, either i) direct chemical modification of endogenous proteins with chemical probes (top path), or ii) through a genetic tag, such as unnatural amino acid mutagenesis with functionalized amino acids and orthogonal tRNA/AARS pairs or enzymatic ligation of acceptor peptides with bioorthogonal motifs on engineered enzymes (bottom path). Step 2) Chemical tags possessing complementary bioorthogonal groups are selectively ligated to modified proteins. Step 3) Marked proteins are subsequently visualized, quantified and/or identified using a variety of techniques.
Figure 3.
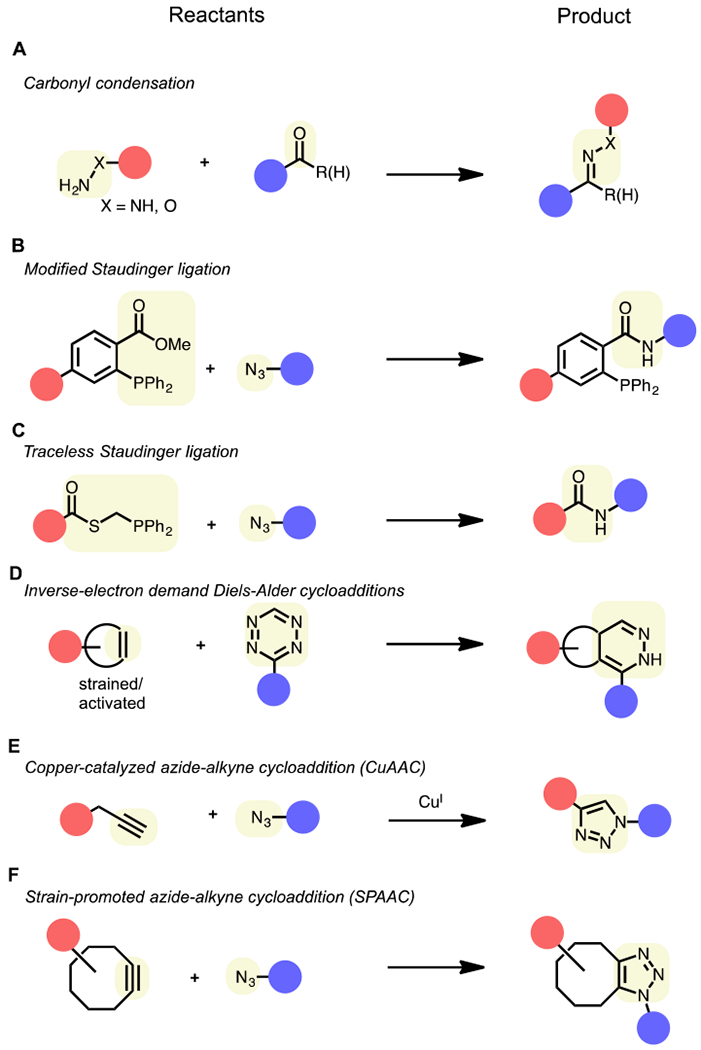
Common bioorthogonal chemical reactions. Red and blue circles correspond to the two “reactants” of a bioorthogonal reaction: 1) proteins labeled with a bioorthogonal reactive group and 2) complementary reactive tag for protein identification and/or visualization. Highlighted in yellow are the bioorthogonal reactive groups (left) and corresponding ligation product (right).
An Introduction to Click Chemistry – The Azide-Alkyne Cycloaddition
The term click chemistry, coined by Sharpless and coworkers, is used to describe chemical reactions that are selective, modular, wide in scope and high yielding (Kolb et al., 2001). The Cu(I)-catalyzed “click” reaction between an azide and terminal alkyne to form a 1,2,3-triazole (Figure 3E) ranks as one of the most widely utilized bioorthogonal reactions to date. Although the reaction between these functionalities was first reported in the 1960s (Huisgen, 1963), its uncatalyzed form requires heat for it to proceed at observable rates, making it unsuitable for applications outside of the round-bottom flask. The copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) was first reported independently by Sharpless (Rostovtsev et al., 2002) and Meldal in 2002 (Tornoe et al., 2002), where it was discovered that Cu(I) dramatically enhances that reaction rate, enabling its use at room temperature. In 2003, Finn and coworkers reported the first implementation of CuAAC in biological systems, where it was used to modify purified Cowpea Mosaic Virus (CPMV) particles with fluorescent dyes (Wang et al., 2003). Soon after, Benjamin Cravatt and coworkers optimized CuAAC conditions for reactions in cell lysates, laying the groundwork for applications in more complex biological systems and for its use as a biorthogonal reaction in proteomics (Speers et al., 2003).
CuAAC Considerations
A potential liability of CuAAC is the cytotoxicity of the Cu(I) catalyst, which initially limited the reaction to in vitro or ex vivo applications (e.g., cell lysates) and prompted the development various solutions to mitigate Cu-mediated toxicity. The development of several biocompatible ligands (Chan et al., 2004) has not only accelerated CuAAC reactions but also reduced the amount of Cu required for the click reaction to occur, minimizing unwanted side effects induced by Cu(I) and enabling the use of CuAAC in living cells and organisms (del Amo et al., 2010). Picolyl azide reagents have also been developed, which internally chelate Cu, thus accelerating CuACC in the absence of exogenous ligands (Uttamapinant et al., 2012). To obviate the need of Cu(I) altogether, Bertozzi and coworkers turned to ring strain to accelerate the reaction between alkynes and azides (Agard et al., 2004; Baskin et al., 2007). These click reactions, known as strain-promoted alkyne-azide cycloaddition (SPAAC, Figure 3F), capitalize on the inherent strain of cyclooctyne rings to promote the reaction of various cyclooctyne derivatives with azides under ambient conditions without any additional catalysts. Such improvements have enabled the labeling of proteins and other biological molecules with azido tags in live cells and organisms, including C. elegans, zebrafish and mice (Chang et al., 2009; Laughlin et al., 2008; Laughlin and Bertozzi, 2009), although cyclooctyne motifs are larger and synthetically more challenging to incorporate compared to terminal alkynes. We do not discuss them here, but other biorthogonal reactions (Figure 3) may have unique advantages over CuAAC for specific applications, such as those that would benefit from alternative or improved reaction speeds and those that may require in situ reactivity under physiological conditions such as live-cell imaging (Lang and Chin, 2014a; Murrey et al., 2015; Rutkowska et al., 2016). For example, inverse electron Diels-Alder reactions (Figure 3D) have gained popularity for a variety of applications due to their favorable kinetics, high selectivity, and catalyst-free nature (Oliveira et al., 2017). However, obstacles associated with their relatively large, hydrophobic reactive groups, which could influence biological interactions, and challenges associated with the synthesis of tetrazine and dienophile reactive groups limits their broad implementation. For simplicity in this article, click chemistry will be used to reference reactions between azide and alkyne functionalities to ligate two components together.
There are a few important technical considerations when performing the CuAAC reaction (Pickens et al., 2018). First, the choice of buffer system used to generate cell lysates can affect the efficiency of the reaction (Darabedian and Pratt, 2019). In general, buffers with primary amines, like Tris (tris(hydroxymethyl)aminomethane), should be avoided as they inhibit CuAAC. Accordingly, two of the most widely used buffers for CuAAC reactions are PBS and triethanolamine (TEA), while one of the best buffers is HEPES (4-(2-hydroxyethyl-1-piperazineethanesulfonic acid). Second, the orientation of the CuAAC reaction is key, as this reaction can be performed in two orientations: an azide-probe and alkyne-tag or an alkyne-probe and azide-tag. In order to increase the rate of the CuAAC reaction, the tags are used in large excess compared to the probe-labeled proteins. The mechanism of CuAAC involves first the activation of the alkyne, and when the alkyne-tag is present in excess, this can result in subsequent reaction with protein nucleophiles, most notably cysteine residues. Therefore, the ideal orientation contains an alkyne-probe and corresponding azide-tag to minimize non-specific proteomic labeling (Speers and Cravatt, 2004). Unfortunately, due to synthetic limitations, this may not always be achieved. If an azide-probe is to be used, either PBS or TEA should be used as a buffer, as HEPES results in significantly higher cysteine reactivity of alkyne-tags. Similarly, it has been shown that certain cyclooctyne SPAAC reagents can also react with cysteine residues, which can be avoided with thorough blocking of free thiols via alkylation (Kim et al., 2013; van Geel et al., 2012). Lastly, there are commercially available click-reaction kits; however, caution should be used as the components are often incompletely described and may contain reagents incompatible with desired workflows, and furthermore, premixed reagents can deteriorate overtime, making these kits unreliable. Given the simplicity and robustness of this bioorthogonal reaction, we recommend acquiring the individual reagents and their fresh preparation prior to click-based applications.
Proteomics
A significant portion of mass spectrometry (MS)-based proteomics utilize a bottom-up workflow, in which proteins are characterized and measured by the analysis of peptides generated from proteins after proteolysis (Zhang et al., 2013). When performed on a mixture of proteins (e.g. cell or tissue lysates), it is most commonly referred to as “shotgun” proteomics (Wolters et al., 2001). A typical shotgun proteomics experiment (Figure 4) begins with the preparation, extraction or isolation of the protein mixture derived from cells, tissue or biofluids. Considering proteins are part of a complex network of interacting biomolecules and sometimes are of low abundance, they often require isolation, separation or enrichment from their native environment. This is particularly true in instances where specific protein modifications or interactions are being investigated. Once isolated, proteins are subjected to selective digestion using a variety of proteases, often trypsin, resulting in the formation of a heterogenous mixture of proteolytic peptides. This digest is subsequently subjected to liquid chromatography to physically separate peptides, most often exploiting differences in peptide charge and hydrophobicity. This separation step enhances proteome coverage and increase the resolution of each peptide by reducing the likelihood of coeluting and co-ionizing species. The resulting separated peptides are then analyzed by MS and assigned by database searching against an appropriate database (e.g. SwissProt) with a search engine (e.g. Sequest or Mascot) in order to identify enriched proteins and permit a more in-depth analysis. Numerous methods, including isotopic labels (e.g. SILAC (Ong et al., 2002), ICAT (Gygi et al., 1999), iTRAQ (Ross et al., 2004), TMT (Dayon et al., 2008)) and various label free approaches (Cox et al., 2014; Gillet et al., 2012; Venable et al., 2004), have been developed for the relative quantitation of proteins across entire proteomes.
Figure 4.
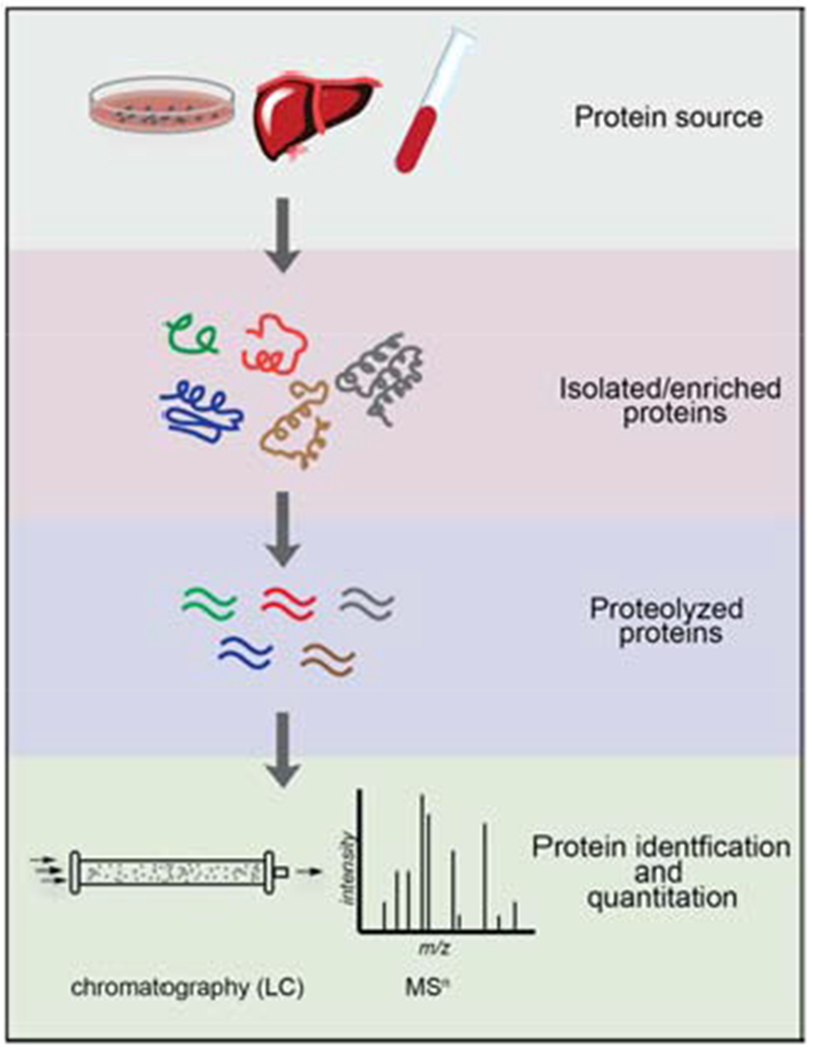
General “bottom-up” proteomics experimental workflow.
Proteomics and Click Chemistry
When integrated with proteomic methods, chemical tools, such as click chemistry, can be used to selectively label and enrich proteins for their subsequent identification and quantitation. Common applications of click chemistry include the mapping of small molecule-protein interactions as well as various post-translational modifications (PTMs). Although each of these applications of click chemistry are unique, the overall general workflow is similar to that of standard shotgun proteomics. Tissue, cells or lysates are first treated with a tag containing a click handle (either azide or alkyne) for the labeling of proteins of interest. This is most often done through direct chemical modification of endogenous proteins with chemical probes. Proteins may also be labeled through unnatural amino acid mutagenesis with functionalized amino acids or ligation of acceptor peptides with bioorthogonal motifs (Figure 2). Following protein labeling with a click moiety, cells or lysates are then subjected to click reaction conditions with a complementary click functional group linked to a tag for protein enrichment, such as biotin. Labeled proteins are subsequently enriched with an affinity matrix and washed extensively, typically with detergents, salts or denaturing agents. Enriched proteins are then subjected to the proteolysis, separation, and mass spectrometry workflow described above. In the sections below, we will discuss strategies in which bioorthogonal labeling and click chemistry tools can be used to study protein-small molecule interactions and PTMs.
Click Chemistry to Investigate Small Molecule-Protein Interactions
Targeted gene disruption and editing methods have been employed with great success to investigate protein function. However, such approaches are often weighted towards loss-of-function (LoF) perturbations and chronic removal of proteins can lead to compensatory mechanisms, which can potentially confound functional investigations. Further, a delay is required prior to examining the effects of genetic manipulation, making such approaches unsuitable to investigate temporally sensitive processes. Selective chemical probes can serve as powerful tools to directly investigate and modulate protein interactions and functions (Arrowsmith et al., 2015). Chemical probes can produce LoF, gain-of-function (GoF) and even neo-functional outcomes on protein targets (Belshaw et al., 1996; Lai and Crews, 2017; Lu et al., 2014; Schreiber, 2018), and often they provide exquisite dose-dependent control over the magnitude and kinetics of perturbation. Finally, quality chemical probes can create paths for further clinical development (Blagg and Workman, 2017; Garbaccio and Parmee, 2016). Despite the utility of small molecules for the study of protein function in living systems, the vast majority of proteins still lack useful chemical probes, and indeed, only ~650 human proteins are currently targeted by small molecules therapeutically (Santos et al., 2017). Key to the development of chemical probes is the complete understanding of their protein targets in native biological systems and the identification of proteins that are tractable to small molecule-based approaches.
Investigations of small molecule-protein interactions using chemical proteomics can be divided into two general categories: (i) protein-centric strategies, which aim to broadly profile features of proteins, such as enzymatic activity or protein ligandability (i.e. the ability to interact with small molecules), through the use broad-spectrum chemical probes to map interactions across the proteome, or (ii) chemical-centric strategies, which focus on characterizing protein targets of individual bioactive small molecules, such as drugs or metabolites (Figure 5A). Both categories utilize chemical probes that are designed from the outset to contain: 1) a recognition group that promotes interaction with specific subsets of proteins in the proteome, 2) a reactive group (either electrophilic or photoreactive) for covalent bonding and capture of interacting proteins, and 3) a reporter tag (fluorophores, biotin, or click-chemistry enabled handles like alkynes and azides) for the detection, enrichment, and identification or visualization of interacting proteins. Major strengths of such chemical proteomic-enabled probes are: 1) efficient enrichment and identification of low-abundance and low-affinity probe-interacting proteins enabled by the covalent trapping of these interactions; and 2) probe-protein interactions can be trapped and ultimately identified in native environments such as living cells, or in some cases, in vivo, thereby preserving labile interactions that might be disrupted by cell lysis and processing protocols, or even missed during investigations which are limited to in vitro studies with purified protein.
Figure 5.
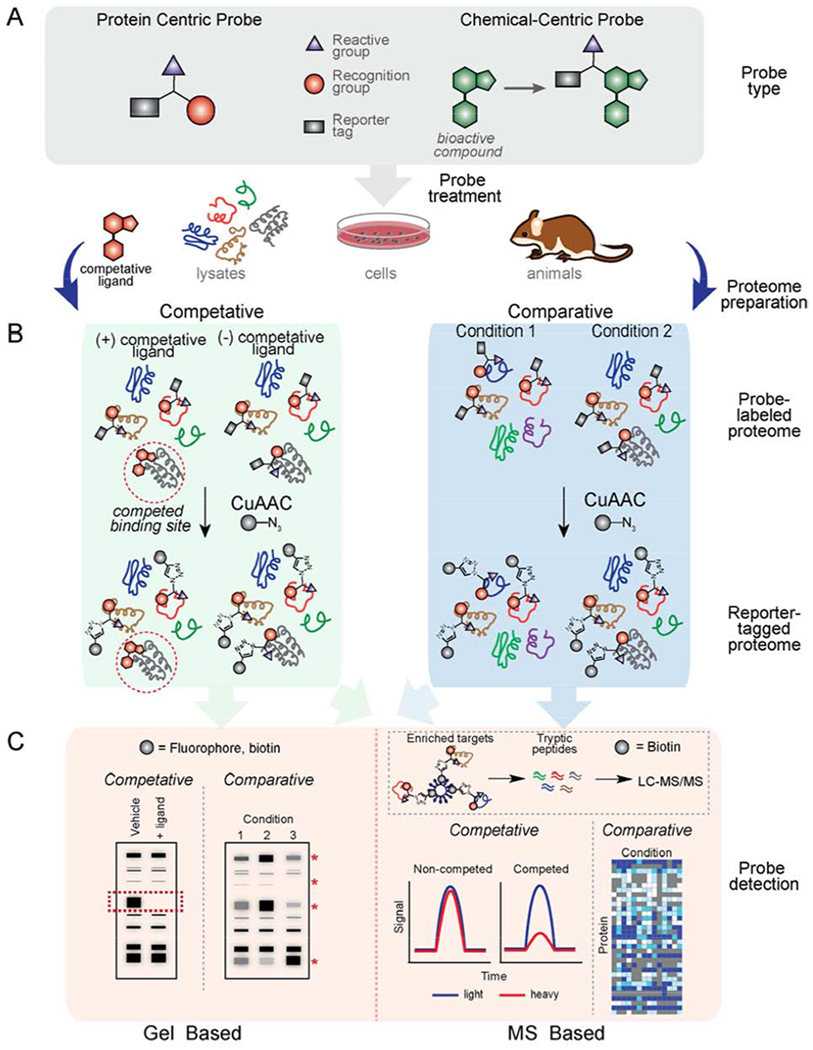
Chemoproteomic methods to map features of proteins or to characterize molecular interactions of bioactive compounds using chemical probes. (A) General design of click probes used in protein-centric or chemical-centric proteomic investigations. (B) Schematic of proteomic workflows. (C) Various readouts of chemoproteomic investigations.
Biological investigations using chemoproteomic-enabled probes are most often conducted in two different formats: comparative or competitive profiling (Figure 5B). In comparative profiling, multiple probes or conditions can be directly compared to understand differences in protein enrichment, which might result from differences in probe recognition or reactivity. In addition, various experimental conditions (e.g. cell type, genetic variations, growth conditions, etc.) can be directly compared upon treatment with a common probe in order to assess the influences of external perturbations on corresponding probe-protein interactions. Competitive profiling, on the other hand, involves treating a biological system with several candidate small-molecule ligands (e.g. inhibitors, drugs, metabolites) and comparing probe-protein interactions relative to an untreated systems. With this method, such ligands can be screened against numerous proteins simultaneously in native proteomes, cells, or even animal models (Cisar et al., 2018; Speers and Cravatt, 2004) (Leung et al., 2003), thereby providing information about ligand targets, potency and selectivity. There are numerous excellent reviews covering chemical proteomic platforms, such as ABPP (Roberts et al., 2017b; Sanman and Bogyo, 2014; Yang and Liu, 2015), chemical proteomic inhibitor discovery (Niphakis and Cravatt, 2014) and various compound-centric profiling applications in detail (Drewes and Knapp, 2018; Rix and Superti-Furga, 2009; Wright and Sieber, 2016). Below, we discuss strategies in which click-chemistry enabled probes have been used to illuminate small molecule-protein interactions and other biology.
Protein-Centric Profiling
Activity-Based Protein Profiling
One of the earliest protein-centric profiling strategies, known as activity-based protein profiling (ABPP) pioneered by the Cravatt lab, aims to broadly profile the activities of enzymes in native biological systems. Applications of ABPP have historically involved the use of electrophilic chemical probes that mechanistically react with the active sites of members of related enzyme families, thereby serving as a reporter of their biochemical activity and as powerful tools for the discovery of new proteins with analogous enzymatic activity (Cravatt et al., 2008). Probe labeling is guided by active-site accessibility and the intrinsic reactivity of catalytic residues, properties that can be dynamically regulated within the cell. These probes enable global profiling of many enzymes in parallel, and in some cases, achieve family-wide coverage. Such broad coverage makes ABPP particularly useful for the discovery of dysregulated enzymes in disease settings via comparative profiling as well as for assessing the selectivity of inhibitors via competitive profiling. Numerous activity-based probes (ABPs) have been developed and deployed for many enzyme classes, including serine hydrolases (Kidd et al., 2001; Liu et al., 1999), cysteine proteases (Kato et al., 2005), metallohydrolases, phosphatases, deubiquinating enzymes (Hewings et al., 2018; Ward et al., 2016), kinases (Zhao et al., 2017), various oxidreductases and others. Further, ABPP has been used to map deregulated protein activities in cancer (Jessani et al., 2002; Jessani et al., 2005; Nomura et al., 2010) and other diseases (Barglow and Cravatt, 2004), to discover as well as optimize both covalent and non-covalent inhibitors for numerous enzymes (Niphakis and Cravatt, 2014), and also assist in the identification of targets and off-targets of bioactive compounds (van Esbroeck et al., 2017).
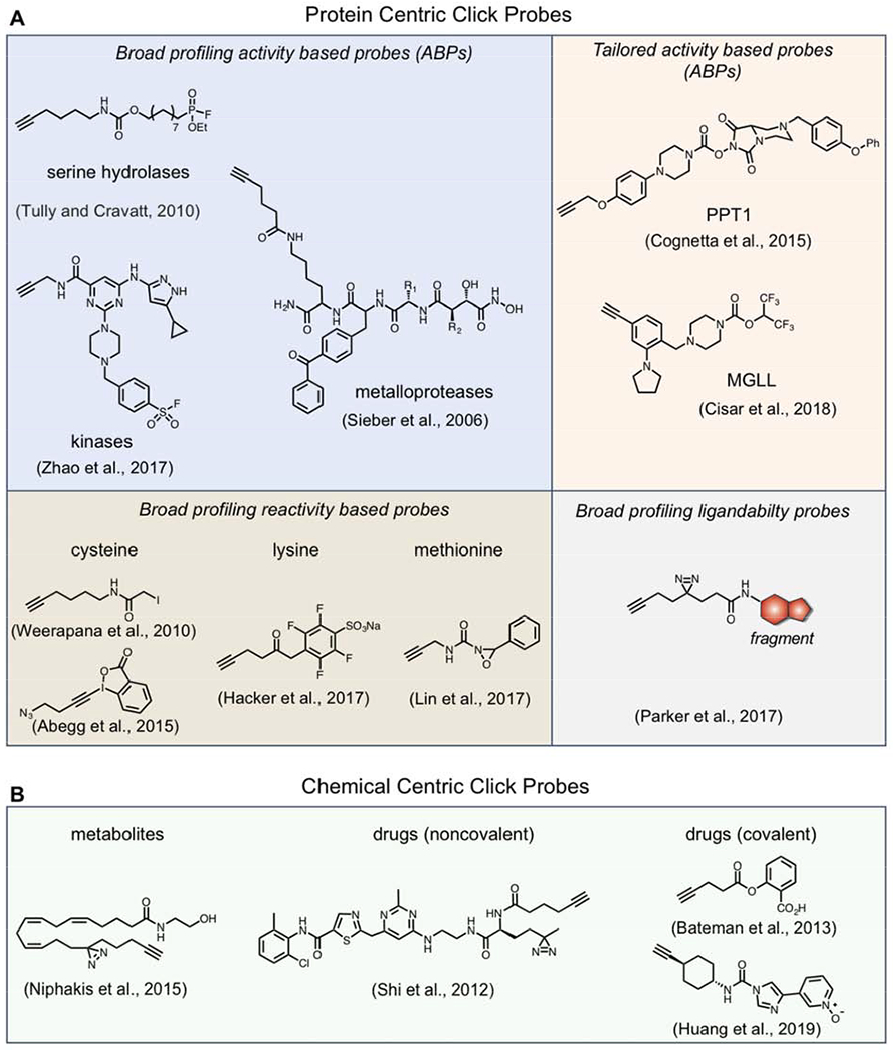
Although ABPs often utilize reporter groups such as biotin or fluorophores for the direct enrichment or visualization of labeled proteins, bypassing the need for additional conjugation steps, such bulky tags can hamper cellular uptake and tissue distribution, potentially prohibiting their use in living systems. The adoption of sterically minimized latent “clickable” tags can overcome these challenges and subsequently be reacted with probes, thereby providing a convenient modular system for detection, enrichment and identification of probe labeled protein targets directly from cells, tissues or even whole animals. The first class of enzymes targeted for ABPP studies were the serine hydrolases (SHs), a large and diverse family of enzymes that perform numerous roles in physiological and pathological processes. Although most ABPP studies for SHs are performed using biotin or rhodamine tagged fluorophosophonate (FP) (Simon and Cravatt, 2010), which mechanistically reacts with active-site serine residues, CuAAC has shown utility in the profiling of these enzymes with the generation of a “tag-free” SH probe (FP-alkyne, Figure 6) (Gillet et al., 2008; Tully and Cravatt, 2010). This probe was shown to label SHs with greater affinity than the reporter-tag functionalized FP probes, and importantly, the vastly improved cell permeability of FP-alkyne facilitates profiling of SH activity directly in living cells, enabling the development of a competitive ABPP platform to screen inhibitor selectivity directly within a cellular milieu instead of in lysates where information pertaining to subcellular localization is disrupted (Gillet et al., 2008).
Figure 6.
Examples of click probes chemical proteomic investigations
Although SHs were subject of some of the earliest ABPP studies, CuAAC-enabled ABPs or “click-ABPs” are constantly being developed for diverse enzyme classes. Electrophilic click-ABPs have been developed for kinases (Zhao et al., 2017), protein arginine deiminases (PADs) (Nemmara et al., 2018; Slack et al., 2011), ubiquitin machinery (An and Statsyuk, 2013; Hewings et al., 2018), cytochrome P450 enzymes (Wright and Cravatt, 2007), and glycosidase enzymes (Tsai et al., 2013). For example, high-throughput methods for profiling kinase inhibitors have typically relied on enzymatic activity or competitive binding readouts on isolated kinases, removed from native environments. Recent chemical proteomic methods have been developed which quantify inhibitor binding to endogenous kinases in complex proteomes through the use of bead-immobilized kinase inhibitors (‘kinobeads’) (Bantscheff et al., 2007) or covalent ATP-biotin probes (Patricelli et al., 2007). These platforms enabled the profiling of 150-200 endogenous kinases in a single cell line, however, neither are cell permeable, requiring membrane dissolution and disruption of protein complexes during lysis, potentially altering kinase conformation and activity. To overcome this limitation, the Tauton lab, in a collaboration with colleagues at Pfizer, developed a cell-permeable sulfonyl fluoride-based click probe (Figure 6) that target a conserved lysine in the ATP binding site which captures 133 phylogenetically diverse kinases in cells, 50 of which were not captured by kinobeads, highlighting the utility and complementarity of click ABPs to other platforms (Zhao et al., 2017).
In addition to broad spectrum probes, which are suited for maximizing coverage across protein families, investigators can also tailor click ABPs to engage a limited number of proteins. By limiting probe reactivity with only a limited number of proteins the overall signal and resolution is increased, aiding in studies where broad reactivity can become confounding, such as in instances with proteins of low abundance which might be obscured by highly expressed proteins. Tailored probes are often derived from activity-based inhibitors discovered via competitive profiling with broad spectrum ABPs, which are subsequently derivatized with click groups (Figure 6). For example, a recent competitive ABPP screen against SHs led to the discovery of a selective and in vivo active covalent inhibitor of palmitoyl protein thioesterase (PPT1), mutations of which cause infantile neuronal ceroid lipofuscinosis. Subsequent modification of this inhibitor with an alkyne yielded a tailored ABP for the direct detection and enrichment PPT1 from cells and tissues, enabling direct functional and biochemical investigation (Cognetta et al., 2015). Click-ABPs have also been developed for in vivo applications. Recently, highly potent and orally available activity-based inhibitors were developed for monoacylglycerol lipase (MGLL), an SH that controls the content and signaling of the endogenous cannabinoid 2-archidonoylglycerol (2-AG) in the central nervous system. To assess in vivo selectivity, the authors modified their lead inhibitor ABX-1431 with an alkyne, and showed with oral treatment, exquisite proteome-wide selectivity, with their click-ABP and lead inhibitor engaging only one off-target (Cisar et al., 2018). ABX-1431 is currently in clinical trials for the treatment of Tourette syndrome and chronic motor tic disorder.
Activity-Based Protein Profiling of Amino Acid Reactivity
In addition to enzyme activity profiling, ABPP applications have been extended beyond enzyme active sites towards the global analysis of amino acid reactivity via general electrophilic chemical probes that react with nucleophilic amino acid side chains, such as cysteine and lysine. These reactivity-based probes bypass the requirement of enzyme activity-mediated probe capture, dramatically expanding the scope of ABPP beyond catalytic sites on enzymes but to all sites on proteins with reactive side chains. One of the first strategies to integrate reactivity-based probes with proteomics was through the utilization of electrophiles to label and enrich alkylation-sensitive proteinaceous cysteines. Iodoacetimide, acrylamide and maleimide reagents had long been used for the alkylation of cysteine residues on purified proteins. However, Gygi and coworkers developed electrophilic probes with isotopically encoded affinity tags (called ICATs), composed of a biotin enrichment handle, an isotopically labeled linker and a cysteine reactive group (iodoacetamide or maleimide), enabling the selective enrichment and quantitation of labeled proteins from lysates (Gygi et al., 1999). Though, the large size and presence of biotin impeded profiling of more diverse electrophiles and limited cell permeability. The advent of click chemistry and its integration with ABPP enabled the characterization of more diverse electrophilic compounds (Backus, 2019). For example, Weerapana and colleagues used tandem orthogonal proteolysis-ABPP (TOP-ABPP) (Weerapana et al., 2007) for the simultaneous identification of protein targets and exact sites of modification by a diverse set of electrophilic alkyne containing click-probes (Weerapana et al., 2008). In these experiments, lysates were treated with various electrophilic click probes and labeled lysates are then conjugated via CuAAC to a bioorthogonally cleavable Tobacco Etch Virus (TEV)-biotin purification tag, enabling the enrichment and release of captured peptides for the subsequent identification of probe-labeled reactive residues. These studies revealed high selectivity of α-halide electrophiles for cysteine thiols. In subsequent work, the use of isotopically encoded ‘heavy’ and ‘light’ TEV tags enabled quantitative profiling of reactive cysteines with an iodoacetamide alkyne (IAA, Figure 6) probe (Weerapana et al., 2010). This strategy, termed isotopic tandem orthogonal proteolysis – activity-based protein profiling (isoTOP-ABPP), has been used to identify thousands of reactive cysteines and to demonstrate that ‘hyper-reactive’ cysteines are highly enriched in protein functional sites. In a competitive format, isoTOP-ABPP has been used to identify the covalent modification of reactive cysteines with various electrophiles or modifications. For example, competitive isoTOP-ABPP has been used to identify sites on proteins modified by lipid-derived electrophiles, such as oxidative stress product 4-hydroxy-2-nonenal (HNE), resulting in modulation of endogenous proteins, such as ZAK kinase (Wang et al., 2014). Competitive isoTOP-ABPP has recently been used to screen electrophilic fragments in proteomes to broadly identify ligandable, and therefore potentially druggable, cysteine residues. These efforts lead to the discovery over 700 ligandable cysteines across 600+ proteins, most of which previously lacked chemical probes (Backus et al., 2016). Initial broad-reactive fragments identified in such studies have been used to develop lead covalent ligands of protein targets, including compounds that selectively target pro-caspase-8 (Backus et al., 2016) and the transcriptional regulator NR0B1 (Bar-Peled et al., 2017) to investigate their physiological and pathophysiological roles. Competitive isoTOP-ABPP has also been used to identify the targets of bioactive compounds, including clinically approved drugs (Blewett et al., 2016; Zaro et al., 2019), herbicides (Ford et al., 2017), phenotypic screening hits (Bateman et al., 2017; Roberts et al., 2017a) and natural products (Grossman et al., 2017), providing direct insights into their mechanism of action and biological consequences. Additional broadly-reactive cysteine click probes have been developed to improve upon the biological properties and residue selectivity of IAA. For instance, although IAA is broadly reactive, its cytotoxicity precludes live cell labeling. Weerapana and coworkers recently developed photo-caged bromomethylketone (Abo and Weerapana, 2015) and iodomethyl ketone (Abo et al., 2017) click probes that show minimal cytotoxicity and provide spatial and temporal control of electrophile activation directly in living cells. In addition, Abegg et al. demonstrated that hypervalent iodine ethynyl benziodoxolone-based click probes exhibit improved selectivity for cysteine residues over other nucleophilic residues, compared to IAA (Abegg et al., 2015).
Reactivity-based click probes have also been developed for other nucleophilic amino acids. Lysine represents a particularly attractive target for click probe development due its abundance in proteins (~6% of all residues), particularly at enzyme active sites and PPIs as well as their frequent post translational modification. Over the past decades, numerous electrophiles have been shown to react with lysine residues, albeit to varying levels of reactivity and selectivity, however, it was only recently that lysine-reactive click probes were developed to assess lysine reactivity directly in human proteomes. Hacker et al. recently integrated isoTOP-ABPP with lysine profiling, finding that a sulfotetrafluorophenyl (STP, Figure 6) click probe displays exquisite selectivity for lysine over other side chains (Hacker et al., 2017). In this work, approximately 4,000 lysine residues were profiled and 100+ ligandable lysine residues were identified in competitive isoTOP-ABPP experiments with a small library of electrophiles. It was also shown that these interactions can be advanced to generate covalent inhibitors of enzymes and protein-protein interactions. Alternative electrophiles have been used for the broad profiling of lysine residues, including NHS ester-based click probes, however, these probes show substantial reactivity for other nucleophilic residues, such as serine, threonine, tyrosine, and cysteine (Ward et al., 2017). Various click probes have been used to profile other nucleophilic amino acid reactivity with aryl sulfonyl fluorides, which covalently react with various residues, including serine, lysine and tyrosine via sulfur (VI) fluoride exchange (SuFEx) reaction (Gu et al., 2013; Narayanan and Jones, 2015; Shannon et al., 2012). Recently, click probes with arylfluorosulfate electrophiles have been shown to have reduced proteomic reactivity relative to sulfonyl fluorides, and an apparent preference for proteinaceous tyrosine residues (Chen et al., 2016; Martin-Gago and Olsen, 2019). Highly selective click probes have recently been generated for intrinsically less nucleophilic methionine residues by cleverly utilizing its unique redox reactivity (Lin et al., 2017). Specifically, Lin and coworkers generated novel oxaziridine-based click probes (Figure 6) which were shown to achieve exceptional selective modification of methionine over all other nucleophilic residues and were used to investigate effects of methionine oxidation on protein function. Collectively, these studies highlight the impact of chemical intuition in the generation of selective reactivity-based probes for proteomic investigations.
Proteomic Profiling of Non-Covalent Binding Interactions
Not all ligandable sites on proteins possess nucleophilic residues that can be targeted by mechanistically-driven or electrophilic reactive groups and not all bioactive compounds possess protein-reactive moieties for covalent capture, thus necessitating the development of alternative modes of reactivity to covalently capture bound protein targets. Classical approaches to identify non-covalent small molecule-protein interactions, such as affinity chromatography, where compounds are immobilized to a solid support for enrichment from cell lysates, have been used identify target proteins of bioactive compounds for decades (Rix and Superti-Furga, 2009). While affinity purification is well suited for identifying targets that are abundant and have a high affinity for corresponding immobilized small molecules (Kd in the nM range), such methods are ineffective for low affinity interactions, low abundance protein targets, and for the broad discovery of ligandable proteins in native contexts. Photoaffinity labeling provides a practical solution as it allows transient reversible interactions between a probe and protein to be fixed in a residue-agnostic fashion upon UV-irradiation. Applications of photoaffinity probes date to the early 1960’s (Singh et al., 1962); however, recent pairing these reagents with modern chemical proteomics and their integration with ABPP principles has dramatically enhanced their impact on mapping small molecule-protein interactions and the discovery of ligandable proteins (Geurink et al., 2012; Lapinsky and Johnson, 2015; Smith and Collins, 2015).
There are many enzyme classes that do not possess protein-bound nucleophiles for catalysis. However, in certain instances, enzyme classes utilize conserved functional or structural features which may be exploited for ABP implementation. For example, metalloproteases (MPs), a large and diverse group of enzymes that play key roles in a variety of physiological and pathological processes, use a zinc-activated water molecule for catalysis. Considering this conserved mechanism, Saghatelian, Sieber and coworkers designed click ABPs (Figure 6) that specifically target MP active sites through the incorporation of a zinc-chelating hydroxamate group coupled to a photoactivatable benzophenone group for covalent capture and an alkyne handle for CuAAC mediated enrichment and detection of bound MPs (Saghatelian et al., 2004; Sieber et al., 2006). Similarly, histone deacetylases utilize zinc-activated water in their active sites, enabling the development of zinc chelating suberoylanilide hydroxamic acid (SAHA) based click ABPs to profile HDAC activity in cells (Salisbury and Cravatt, 2007, 2008). Additionally, cofactors as well as enzyme substrates or products can be used as recognition elements in photoaffinity based ABPs. For example, Cisar and coworkers described the development of photoreactive GTP click probes to identify known and previously unknown proteins that utilize GTP in cells (Cisar et al., 2013). Horning et al. described a set of S-adenosyl homocysteine (SAH) photoaffinity (diazirine and benzophenone) probes with a high selectivity for methyltransferases (MTs) in cells as well as MT inhibitor discovery via competitive ABPP (Horning et al., 2016). Interestingly, in this study, it was discovered that the use of an alkyne handle resulted in substantial UV-independent labeling under CuAAC conditions, necessitating the use of biotin, rather than click-based, enrichment handles, highlighting the importance of careful control experiments in ABP development and the potential, albeit rare, drawbacks of CuAAC in some proteomic investigations.
Despite the immense success of ABPP and similar chemical proteomic platforms, the vast majority of proteins still lack chemical tools to study their function. This is due, in part, to the fact that not all protein families have conserved functional or structural features that can be selectively targeted with broad profiling chemical probes. To begin to address this challenge, recent chemical proteomic studies have aimed not to directly profile protein activity or reactivity but to agnostically explore protein ligandability, independent of protein function, cellular location or structure. For example, Kambe et al. generated a structurally diverse library (~60 members) of fully functionalized chemical probes which possess diazirine and alkyne functionalities for target capture and enrichment, respectively (Kambe et al., 2014). Chemical proteomic profiling of these drug-like probes in live human cells revealed that such probes achieved proteome-wide structure-activity relationships (SAR) and that numerous proteins (~25) were selectively enriched, including well-established druggable proteins, such as enzymes, as well as much less pharmacologically accessed targets, such as scaffolding and adaptor proteins. To more broadly identify ligandable, and therefore potentially druggable proteins in cells, we recently integrated chemical proteomics with fragment-based ligand discovery (FBLD) to broadly interrogate proteome ligandability (Parker et al., 2017a; Wang et al., 2019). Small molecule fragments efficiently explore broad chemical space and can serve as practical leads for subsequent synthetic elaboration. The development of fully-functionalized fragment (FFF) probes (Figure 6), which are structurally diverse drug-like fragments appended to a retrieval tag composed of an alkyne and photoactivatable diazirine, enabled the identification of more than 2000 reversible fragment-protein interactions in cells. It was demonstrated that FFF probes have proteome-wide binding preferences and can be used to map sites of ligandability on endogenous proteins in living cells through the identification of probe-modified peptides. It was further established that fragment-protein interactions could be used to generate selective ligands that modulate functions of proteins in cells. For example, simple fragment leads identified in comparative profiling studies were subsequently utilized in competitive profiling experiments (Figure 5B) to generate first-in-class inhibitors for prostaglandin reductase 2 (PTGR2) and the multi-pass transmembrane protein SLC25A20. Additional studies lead to the identification of a gain-of-function ligand for the poorly characterized transmembrane protein progesterone receptor membrane component 2 (PGRMC2), which facilitated the subsequent characterization of PGRMC2 as a heme chaperone critical for adipocyte function (Galmozzi et al., 2019). Although this strategy yielded an abundance of potentially ligandable protein targets, due to the simple nature of fragments, discerning specific interactions (e.g. at a genuine binding pocket) from less/non-specific interactions required careful manual review. To overcome this challenge, a second generation library of FFF probes consisting of stereochemically matched fragments was constructed, which provide high confidence evidence of specific interactions between small molecule and proteins in cells (Wang et al., 2019).
Chemical Centric Profiling
Chemical centric approaches (Figure 5) have been used for decades to deconvolute the targets of bioactive compounds, such as drugs, phenotypic screening hits, natural products and metabolites. An important feature of chemical centric proteomic profiling methods is that they are inherently target agnostic and functionally unbiased, minimizing the risk of overlooking unexpected off-targets that might be missed using target- or function-centric characterization assays (Drewes and Knapp, 2018). Classically, chemical centric proteomic methods have involved the immobilization of bioactive compounds to a solid support followed by incubation with cell or tissue lysates to capture and subsequently identify bound protein targets. Competition experiments are often conducted with free active and structurally similar but inactive control compounds to identify and prioritize selective protein targets over non-selective binders. Pioneering work using affinity based methods include the identification of the targets of macrocyclic immunosuppressants and the discovery of HDACs (Harding et al., 1989; Liu et al., 1991; Taunton et al., 1996). However, as discussed above, these approaches are limited when binders have lower affinities and such methods do not preserve intact cellular environments. The use of minimally perturbing, cell permeable click handles and covalent capture groups can mitigate these challenges.
Photoaffinity-based click probes are likely the most common form of chemical proteomic tools to capture and identify noncovalent targets of bioactive small molecules (Lapinsky and Johnson, 2015). Generation of these probes involves the synthetic installation of click groups (azide or alkyne) and photoaffinity groups (e.g. diazirines, benzophenone, aryl azides) in such a way that is minimally perturbing to the bioactivity of the compound, a process that can sometimes require extensive SAR investigations and even entire new synthetic routes. The recent development of modular “all-in-one” tags that contain i) functional groups for compound attachment, ii) an azide or alkyne for CuAAC and iii) photo-crosslinking groups for covalent capture, has assisted simple installation of these groups often in one synthetic step (Li et al., 2013; Li et al., 2014). Cell permeable click photoaffinity probes have been developed to characterize targets of various clinically approved drugs and inhibitors (Figure 6), including kinase inhibitors (Li et al., 2013; Shi et al., 2012), γ-secretase inhibitors (Ballard et al., 2014), β-secretase inhibitors (Zuhl et al., 2016), antibiotics (Eirich et al., 2011), nonsteroidal anti-inflammatory drugs (NSAIDs) (Gao et al., 2018), and epigenetic modulating compounds (Li et al., 2014; Montgomery et al., 2014) as well as natural products (Wright and Sieber, 2016). In many cases, these studies uncover targets that were previously unassociated with the compound’s phenotypic effects but turn out to be functionally relevant molecular targets. For example, photoaffinity click probes of a BACE1 β-secretase inhibitor revealed that cathepsin D (CatD) is a common off-target of several clinical BACE inhibitors, which may contribute to observed ocular toxicity in vivo (Zuhl et al., 2016). Interestingly, the authors observed much greater CatD inhibition in cells compared to cell-free assays, highlighting the importance of in-cell target engagement assays. In recent studies, a photoaffinity click probe made based on the diterpenoid ester ingenol mebutate (IngMeb), the active ingredient in the topical drug Picato, revealed 28 previously unknown IngMeb targets in cells, including the mitochondrial transporter SLC25A20, which IngMeb was found to functionally inhibit (Parker et al., 2017b). Photoaffinity click probes can also be used to reveal new protein interactions of important metabolites such as lipids (Niphakis et al., 2015) and sterols (Hulce et al., 2013). Given that metabolites likely engage functionally relevant sites on proteins, such metabolite-derived click probes can serve as valuable profiling tools to map drug interactions across the metabolite-interaction proteome and even serve as tools to develop modulators of metabolite-binding protein function (Niphakis et al., 2015). Numerous bioactive compounds can also engage protein targets covalently. As discussed above, broad reactive ABPs can be used in a competitive format to identify targets of the covalent compound that also overlap with the ABP. Alternatively, target retrieval probes can be generated by appending a click handles directly to the bioactive compound, enabling CuAAC enrichment of captured targets. For example, click probes have been used to assess the target landscape of covalent kinase inhibitors (Gushwa et al., 2012; Lannine et al., 2014; Niessen et al., 2017) and various bioactive natural products (Wright and Sieber, 2016). Click probes have also recently been used to identify covalent targets of reactive drug metabolites, including that of the fatty acid amide hydrolase (FAAH) inhibitor BIA 10-2474 (Figure 6), a clinical candidate that failed Phase I trials due to serious neurotoxicity which led to the death of one human volunteer (Bateman et al., 2013; Huang et al., 2019; van Esbroeck et al., 2017). This approach has even been extended to identify in vivo targets of the reactive metabolites of hepatotoxic drugs including troglitazone, clozapine, and acetaminophen (Whitby et al., 2017).
Click Chemistry to Study Protein Post-Translational Modifications
The proteome is much more diverse than the corresponding genome as the result, in part, of PTMs. A wide range of both enzymatic and chemical modifications can be found on various amino acids and play key roles is nearly all biological processes. Therefore, identifying the protein substrates of PTMs, as well as the specific sites of modification when possible, is critical for a complete understanding of cellular biochemistry. Unfortunately, PTMs are typically very difficult to observe in shotgun proteomics experiments for several reasons. For example, PTMs are often substoichiometric and can be highly dynamic. Certain modifications are also chemically labile and can be lost during peptide ionization in the mass spectrometer, while others can inhibit peptide fragmentation and sequencing. Additionally, some PTMs are large and heterogeneous, making their recognition by proteomics-software challenging. Given these considerations, PTM selective enrichment is necessary prior to MS analysis. For certain PTMs, this can be accomplished using the biophysical properties of the modification or more traditional biological tools like antibodies. For example, phosphorylated peptides can be enriched using immobilized metal ion affinity chromatography (IMAC) or metal oxide matrices, and these technologies have allowed for the identification and quantitation of thousands of phosphorylation sites across the human proteome (Sharma et al., 2014). A similar number of lysine acetylation sites were identified after enrichment using an anti-acetyllysine antibody (Choudhary et al., 2009). Unfortunately, the properties of many PTMs are not amenable to direct enrichment and many antibodies are not truly pan-selective and will therefore fail to enrich certain modified peptide sequences.
Given these challenges, click chemistry has been particularly powerful for the analysis of PTMs (Chuh and Pratt, 2015) (Chuh et al., 2016) (Grammel and Hang, 2013). In general, these approaches can be divided into three basic categories (Figure 7). The first relies on the unique chemical reactivity of certain PTMs or protein residues for the direct installation of the click probe. Second, cellular metabolism can be exploited to “sneak” azides or alkynes into PTMs using endogenous biosynthetic pathways, an approach we have termed metabolic chemical reporters (MCRs). Finally, enzymes can be exploited to modify certain PTMs ex vivo with click groups. In the metabolic and enzymatic strategies, the use of click chemistry is particularly important and often necessary. More specifically, the biosynthetic machinery and enzymes responsible for PTM installation or elaboration are able to tolerate small structural changes, like the addition of an azide or alkyne, but not larger groups like biotin that would allow for direct detection.
Figure 7.
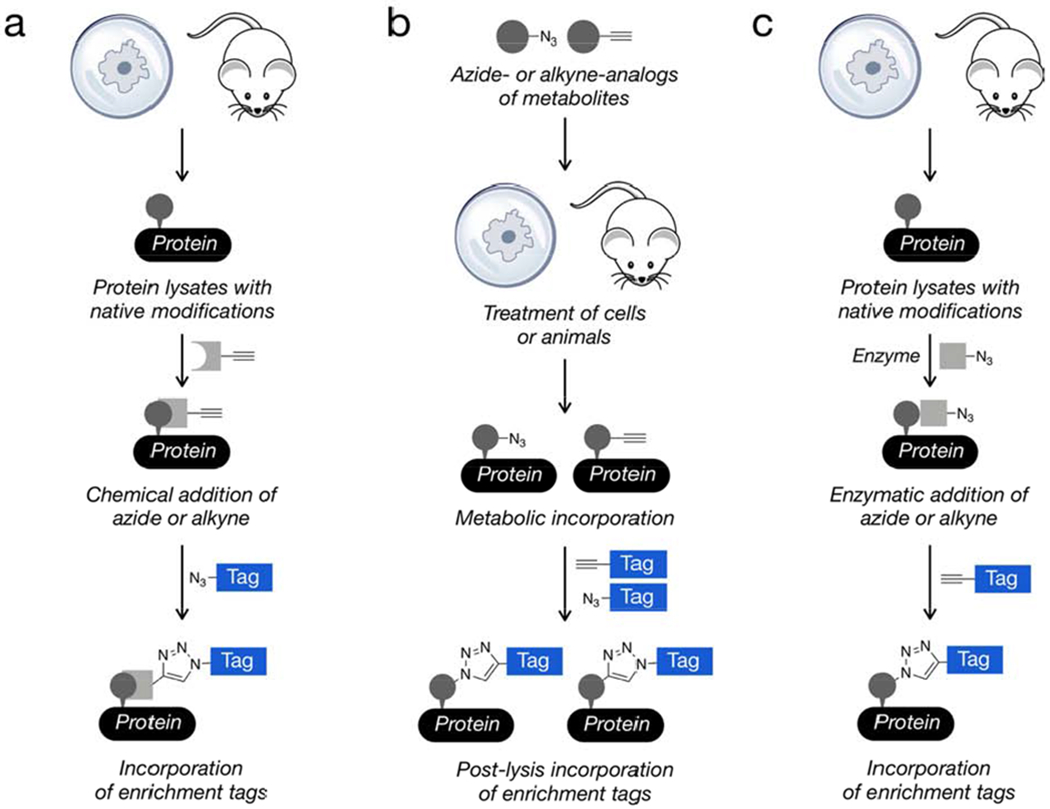
General classes of click-chemistry methods for the identification of post-translational modifications. a) The chemical reactivity of certain PTMs allows them to be reacted directly and selectively with bioorthogonal probes. b) The metabolism of living cells or animals can be exploited for the introduction of chemical probes with click-chemistry handles. c) Other PTMs can be modified enzymatically to install click-chemistry handles.
Glycosylation
Glycosylation is one of the most common protein PTMs, involving the covalent addition of carbohydrates with various sizes and compositions (Varki et al., 2015). In metazoans, proteins destined for the lumen of the secretory pathway, the cell surface, or the extracellular space can be subjected to a variety of different types of glycosylation. By far the most common of these forms are N-linked (Aebi, 2013; Schwarz and Aebi, 2011; Weerapana and Imperiali, 2006) and mucin O-linked glycosylation (Hang, 2005 145;Jensen, 2010 163;Tran, 2013 131). N-linked glycans are first biosynthesized as an oligosaccharide on a dolichol lipid before an en bloc transfer to the protein substrate in the endoplasmic reticulum (ER) lumen. Specifically, the carbohydrate-protein linkage can be made to the side chain of asparagine in the sequence motif Asn-Xxx-Ser/Thr, where Xxx is any amino acid except proline. As the glycoprotein moves through the ER and Golgi apparatus, the oligosaccharide can be trimmed and then elaborated with additional carbohydrates to generate a range of large and heterogeneous structures. N-linked glycans are critical for protein folding and intracellular protein trafficking, as well as mediating various interactions at the cell surface and the function and stability of circulating proteins. Mucin O-linked glycosylation first involves the addition of an N-acetylgalactosamine through an α-linkage to the side chains of serine or threonine residues of proteins transiting the Golgi. Like N-linked glycans, this core modification can then be elaborated through the addition of more carbohydrates. Mucin O-linked glycosylation plays key roles in cell-cell contacts, protective barrier formation, and the biophysics of the cell surface. Intracellular proteins in animals can be subjected to O-GlcNAc modification, the addition of N-acetylglucosamine through a β-linkage to serine or threonine residues (Yang and Qian, 2017). Unlike the major forms of glycosylation on secreted proteins, O-GlcNAc is not further elaborated. However, it is dynamically cycled on and off proteins and therefore acts as a signaling modification. Thousands of protein substrates of O-GlcNAc have been identified and this PTM can affect protein-protein interactions, enzymatic activity, and compete with other modifications like phosphorylation.
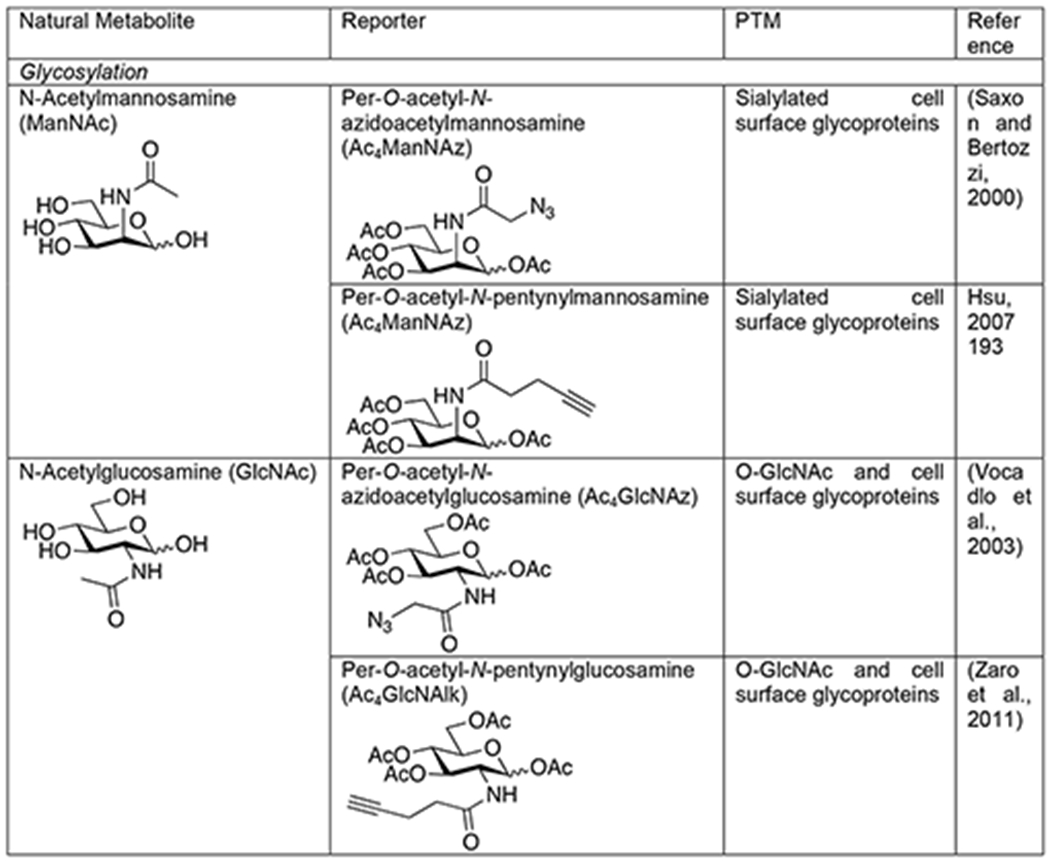
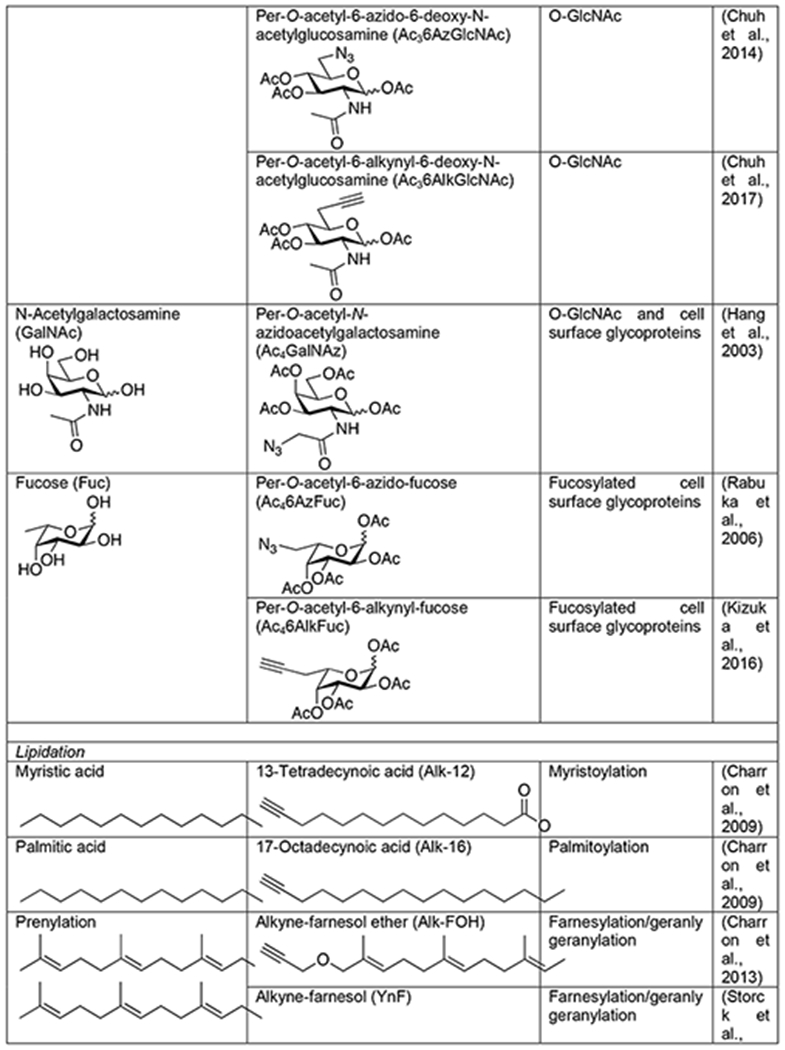
Traditional methods for studying glycoproteins relied on lectins and antibodies. Lectins are natural carbohydrate-binding proteins found in many organisms. Unfortunately, they suffer from a lack of complete specificity for particular carbohydrate but rather recognize structural “themes” that can be found in several different types of glycans. Additionally, they typically bind glycan structures with relatively low affinity. Some carbohydrate antibodies have been generated, but some of them display preference for the underlying peptide sequence, preventing them from being truly pan-selective reagents. Therefore, the field of glycobiology has benefited greatly from bioorthogonal approaches to the visualization and enrichment of glycoproteins. The first such method was pioneered by the Bertozzi lab, termed metabolic oligosaccharide engineering, involves the use of metabolic chemical reporters (MCRs) - analogs of naturally occurring monosaccharides bearing bioorthogonal functional groups (Mahal et al., 1997; Saxon and Bertozzi, 2000). These MCRs, can be accepted by living cells or even animals where they are biosynthetically converted to high-energy sugar donors and then directly incorporated into glycans by endogenous enzymes. For example, one of the first MCRs was an azide-containing analog of N-acetylmannosamine, Ac4ManNAz, with the hydroxyl groups masked as acetate esters to allow for passive diffusion into cells (Table 1) (Saxon and Bertozzi, 2000). Once internalized, the O- acetates are removed by esterases and the resulting ManNAz can be biosynthetically transformed into an azide-containing sialic acid monosaccharide that is then added to the termini of N-linked and mucin O-linked glycans. It is important to point out that in the case of the MCR method, the use of bioorthogonal chemistry is absolutely critical. Unlike some reactive probes that can be directly conjugated to tags without largely compromising their ability to covalently label their targets, direct tagging of monosaccharides would result in a large modification that would not be tolerated by the cellular biosynthetic enzymes, resulting in little or no labeling. Since this initial study using Ac4ManNAz, several other azide- or alkyne-containing MCRs have been developed, including analogs of N-acetylglucosamine (Vocadlo et al., 2003; Zaro et al., 2011), N-acetylgalactosamine (Hang et al., 2003), and fucose (Kizuka et al., 2016; Rabuka et al., 2006) (Table 1). Additionally, other bioorthogonal groups like alkenes (Niederwieser et al., 2013; Späte et al., 2014) and cyclopropenes (Cole et al., 2013; Patterson et al., 2014a; Patterson et al., 2012) that can undergo inverse-demand Diels-Alder reactions have been incorporated into monosaccharides.
Table 1.
Chemical Reporters of PTMs
 |
 |
 |
 |
 |
Not surprisingly, MCRs of glycans have been extensively used for the proteomic identification of glycoproteins in a variety of contexts. To facilitate the identification of glycosylation sites, the Bertozzi lab recently developed isotope-targeted glycoproteomic (IsoTaG) reagents (Woo et al., 2017). These biotinylated tags generate a unique isotopic signature on glycosylated peptides post bioorthogonal tagging, facilitating the detection of low-abundance glycopeptides. For example, combining an IsoTag with Ac4GalNAz, an azide-containing analog of N-acetylgalactosamine, enabled the identification of over 2,000 potential O-GlcNAcylated peptides from primary human T-cells (Woo et al., 2018). Unfortunately, several glycan MCRs suffer from a lack of glycan selectivity. In retrospect this is not surprising, as individual monosaccharides are found in multiple types of glycans. Additionally, cells have evolved to interconvert different monosaccharides, resulting in potentially undesirable interconversion of MCRs as well (Boyce et al., 2011). In an attempt to address these selectivity issues, we and others have synthesized various structural analogs of MCRs that could bias the utilization of these reporters by different glycosylation pathways (Chuh et al., 2017; Chuh et al., 2014; Li et al., 2016; Shen et al., 2017; Zaro et al., 2017). Notably, several MCRs that are selective for O-GlcNAcylated proteins, suggesting that this strategy may be applicable for other classes of glycans. It is also important to note that because MCRs compete with natural monosaccharides, their incorporation levels do not necessarily reflect the natural amounts of protein glycosylation. Finally, recent evidence has suggested that some amount of signal generated by MCRs is likely due to either direct chemical labeling of proteins or their breakdown into intrinsically reactive metabolites (Qin et al., 2018); however, other data from the same labs indicates that this “background” labeling may be a minor component (Qin et al., 2017).
Complementary to these metabolic strategies, chemoenzymatic methods have been developed. The most well-established of these approaches for proteomics is in the area of O-GlcNAcylation and was inspired by early work on the enzymatic radiolabeling of native O-GlcNAc modifications in cell or tissue lysates. Specifically, terminal GlcNAc moieties can be labeled using a combination of radioactive UDP-galactose (UDP-Gal) and a β1,4-galactosyltransferase (GalT), which generates a Gal-GlcNAc disaccharide on O-GlcNAcylated proteins that can be detected by autoradiography (Torres and Hart, 1984). Replacing the native GalT with a mutant (Y289L) enabled the enzymatic transfer of N-azidoacetylgalactosamine (GalNAz) from UDP-GalNAz to generate a disaccharide bearing an azide for subsequent bioorthogonal chemistry (Table 1) (Clark et al., 2008). This technology has been transformative for the confirmation of potential O- GlcNAcylated proteins identified using MCRs, as well as the direct identification of modified proteins. For example, when combined with a photo-cleavable biotin enrichment-tag, this technique enabled the identification of 458 O-GlcNAc sites on 278 proteins from mouse cerebrocortical brain tissue (Alfaro et al., 2012) and subsequently 1094 sites on 530 proteins from human brain samples (Wang et al., 2017). Building upon this idea, several other UDP-azido-sugars and enzymes have been developed for the incorporation of bioorthogonal handles onto different cell surface glycans (Wen et al., 2018; Wu et al., 2018). However, to our knowledge, they have not yet been applied to proteomics analysis. Since these methods modify native glycosylation, they necessarily enrich endogenous modifications and can enable overall quantification.
Lipidation
Lipidation, the attachment of a variety of fatty acids, is a PTM that controls the localization and thereby function of proteins (Jiang et al., 2018). More specifically, this modification dynamically regulates the position of proteins throughout a sophisticated network of membranes and organelles that are critical to cellular function. Two of the most common types of lipidation are the addition of saturated fatty acids. Myristoylation typically occurs co-translationally on the N-terminus of proteins through the formation of an amide bond to the 14-carbon myristic acid. Likewise, palmitoylation is the reversible and often highly dynamic addition of the 16-carbon palmitic acid to cysteine side chains, resulting in fatty acid thioesters. Unsaturated lipids can also be irreversibly added as thioethers to cysteines through a PTM termed S-prenylation. More specifically, two different isoprenoids, a 15-carbon farnesyl or 20-carbon geranylgeranyl, can be found at one or two cysteines typically at the extreme C-terminus of certain proteins. Together these PTMs control the subcellular localization of proteins with a variety of biological consequences. For example, the combination of S-prenylation and dynamic palmitoylation control the positioning of activated Ras to the cellular membrane and therefore the subcellular localization of the associated signaling pathways.
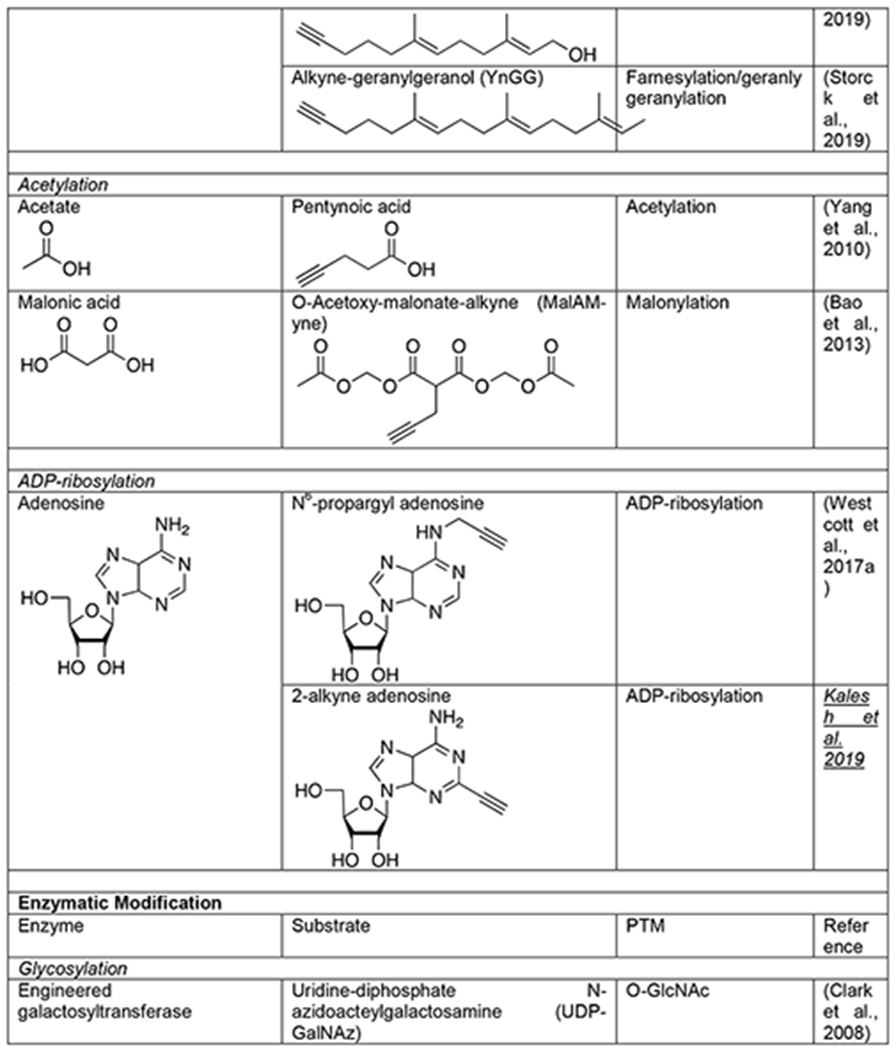
In contrast to their complex biology, lipids have chemically uniform structures and lack essentially any functional groups. This makes the creation of traditional enrichment tools like antibodies extremely difficult. Therefore, visualization of protein lipidation largely relied on the metabolic incorporation of radioactive lipids. This method suggested that, like the carbohydrate pathways discussed above, lipid MCRs bearing small bioorthogonal functionalities might also be tolerated and incorporated onto proteins. Again, the use of small bioorthogonal handles was absolutely required for this technology, as these relatively small modifications can be tolerated by the cellular metabolic and transferase enzymes. This possibility was first confirmed through the metabolic labeling of lipidated proteins with myrisitc- and palmitic-acid analogs bearing azides at the omega end of the lipid chain (Hang et al., 2007). These original lipid MCRs were quickly followed by the development of analogous alkyne-lipids that take advantage of the better CuAAC orientation described above (Table 1) (Charron et al., 2009). These alkynyl-lipids have enabled a variety of transformative proteomics experiments. For example, 13-tetradecynoic acid (Alk-12) was used as a myristic acid analog to identify myristoylated proteins from the Plasmodium falciparum, the malaria pathogen (Wright et al., 2014). This same reporter was also combined with SILAC to identify myristoylation events that were sensitive to an N-myristoyltransferase inhibitor (Thinon et al., 2014). Likewise, the palmitic acid analog 17-octadecyonic acid, or Alk-16, was used to identify critical palmitoylation events that control the antiviral activity of the protein IFITM3 (Yount et al., 2010). The unique nature of metabolic incorporation of Alk-16 was subsequently exploited in combination with SILAC to perform a pulse-chase experiment and catalog the dynamics of different palmitoylation events across the cell (Martin et al., 2012). And, this probe has been successfully applied to profile palmitoylated proteins in pathogens, like Toxoplasma gondii (Foe et al., 2015). Similarly to the saturated fatty acids, azide and alkyne analogs of isoprenoids have also been developed that can be metabolically incorporated into S-prenylation by cells (Table 1) (Charron et al., 2013; DeGraw et al., 2010; Storck et al., 2019). Again, when combined with proteomic strategies, these reporters have enabled the discovery of key S-prenylation events that control antiviral activity of proteins and the profiling of S- prenylation changes in disease.
Acetylation
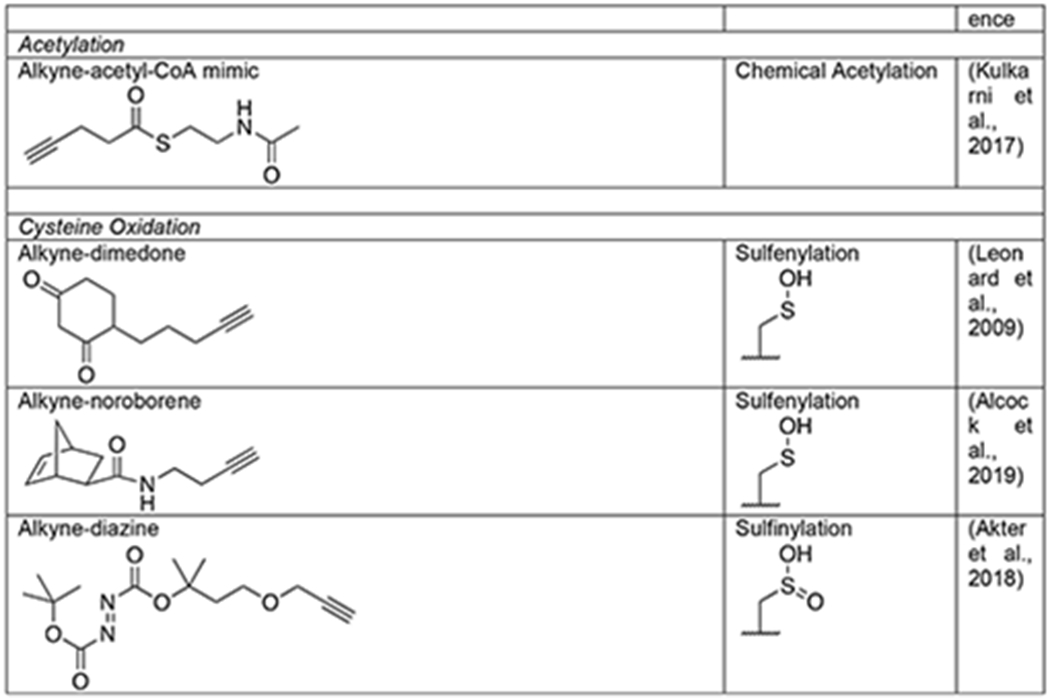
Acetylation of the ε-amine of lysine is a PTM best known for critical regulation of gene transcription through specific modification patterns on chromatin (Shahbazian and Grunstein, 2007) but can also play other important roles throughout the cell (Yang and Seto, 2008). Through capping of lysine, acetylation can directly alter the charge state of a protein and directly compete with other modifications like methylation, ubiquitination, and SUMOylation. Acetate is added to proteins by a family of acetyltransferases that use the high-energy acetyl-CoA cofactor, and the modification can be removed by another family of deacetylases. Anti-acetyl lysine antibodies have been particularly powerful for the identification of almost 5,000 modification sites across the mammalian proteome. However, these antibody tools cannot easily read out on the dynamics of protein acetylation or distinguish between the enzymatic modification of proteins by acetyltransferases versus the direct chemical modification by the reactive acetyl-CoA thioester. For example, a metabolic chemical reporter of acetylation can be used in a similar fashion to radioactive or bioorthogonal amino acids to perform pulse and/or pulse-chase experiments. As a first step towards using bioorthogonal chemistry to address these issues, 4-pentynoic acid was tested as an MCR and was shown to label a range of proteins in living cells, including known acetylated proteins (Table 1) (Yang et al., 2010). The utility of this short alkyne acid as an MCR for lysine acetylation was then bolstered by the fact that purified p300 acetyltransferase can utilize synthetic 4- pentynonyl-CoA to label proteins in cell lysates (Yang et al., 2011). In addition to acetylation, longer fatty acids and modified acetate groups, including propinylation, butryrylation, malonylation, and succinylation have been found on lysine side chains (Lin et al., 2012). This encouraged the more recent development of an MCR for protein malonylation, 2-propargyl malonate (Mal-yne) (Table 1) (Bao et al., 2013). Much like the monosaccharide MCRs described above, the carboxylic acids of Mal-yne were protected as acetoxymethyl esters to enable diffusion into living cells, enabling the identification of almost 400 proteins from HeLa cells, including 14 known malonylated proteins. As mentioned directly above, it is unclear how many lysine acetylation events occur enzymatically versus chemically, but bioorthogonal reactions have also provided an approach to answer this question through the development of a direct-modifying probe. More specifically, an alkyne-containing thioester was synthesized that mimics acetyl-CoA but cannot be used by acetyltransferases (Table 1) (Kulkarni et al., 2017). Profiling of cells using this reporter allowed for the identification of ~100 potential protein targets of direct chemical acylation and enabled the characterization of malonyl-CoA as a particularly reactive thioester for this type of reaction.
Methylation
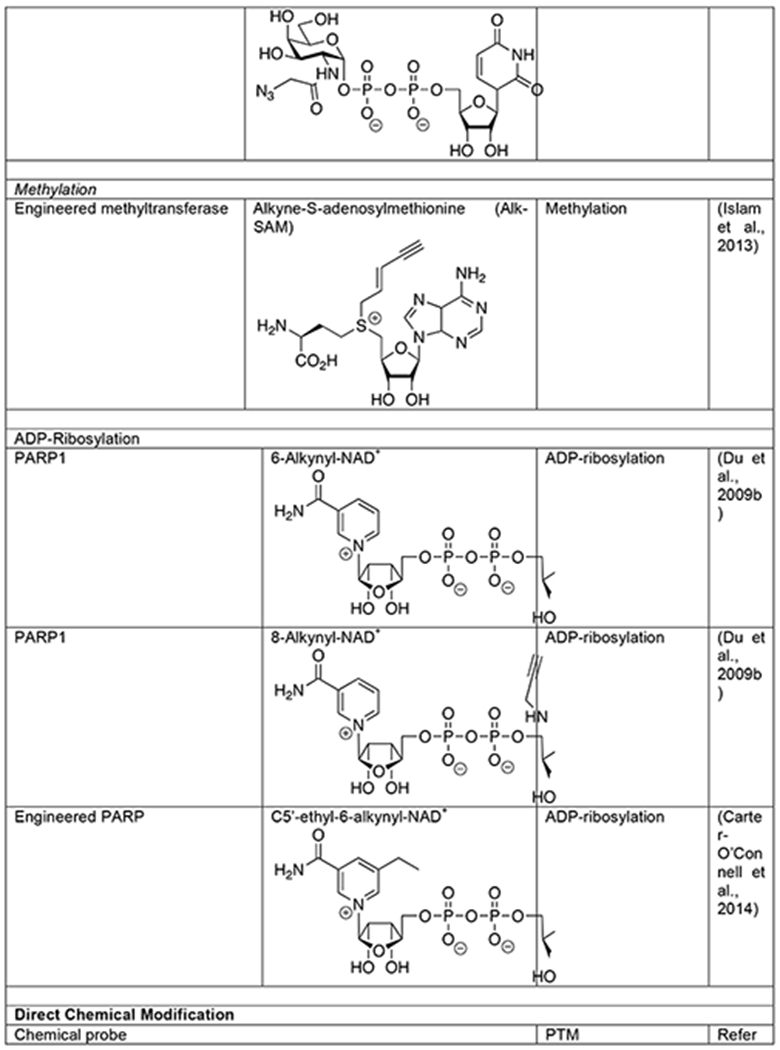
Similar to acetylation, lysine side chains can also be subjected to methylation, which also plays important epigenetic roles through chromatin biology and can more broadly affect the function of a variety of proteins (Bedford and Clarke, 2009; Biggar and Li, 2015; Greer and Shi, 2012). However, this PTM is somewhat more versatile by virtue of its ability to add one, two, or three methyl groups to the same lysine, as well as methylation of arginine side chains in three different patterns. Methylation is added to proteins by distinct lysine- and arginine-methyltransferases that all use S-adenoyl-L- methionine (SAM) as a cofactor. The modification can then be removed by two families of demethylases that are defined by their differential enzymatic mechanisms. Methylation affects proteins by altering the size of lysine and arginine side chains and antagonizing other modifications. Antibodies that recognize methylated lysines and arginines are available, but bioorthogonal chemistry has made important contributions to the characterization of this PTM. As a first step, two different analogs of SAM with short alkyne modifications were synthesized (Binda et al., 2011; Peters et al., 2010). In vitro analysis demonstrated that these SAM analogs were accepted by a subset of the methyltransferases tested. These results suggested that pairs of bioorthogonal SAM reporters and methyltransferase mutants could be identified using a “bump-hole” strategy. In other words, larger alkyne- or azide-modifications of SAM or “bumps” might be tolerated by specific enzymes that have had their active site mutated to generate “holes” that would accommodate the modifications. This goal was subsequently realized through rational engineering of the methyltransferase active site, enabling the labeling and identification of methyltransferase-specific substrates (Table 1) (Islam et al., 2012; Islam et al., 2013). Unfortunately, these SAM analogs are not cell permeable and therefore can only be used in cell lysates. To address this shortcoming, the SAM biosynthetic pathway was altered to accept alkyne-modifications in living mammalian cells (Wang and Luo, 2013). In this case, cells can be treated with the alkyne-modified analog of methionine that will subsequently act as an MCR in the engineered pathway to generate the corresponding alkyne-SAM. While a success, this engineering effort highlights both the fundamental limitation of the MCR-strategy, the acceptance of a bioorthogonal reporter by multiple enzymes, and the utility of the minimal bioorthogonal azide and alkyne handles that are best capable of sneaking through these pathways.
ADP-Ribosylation
Protein ADP-ribosylation results from the enzymatic transfer of ADP-ribose to a range of amino acid side chains, including lysine, asparagine, glutamine, serine and cysteine in mammals (Cohen and Chang, 2018; Palazzo et al., 2019). In humans, a family of 17 ADP-ribosyltransferases, termed poly-ADP-ribose polymerases (PARPs), can add this modification. Despite their name, the majority of PARPs appear to be mono-ADP-ribosyltransferases while a subset can generate poly-ADP-ribose through the extension of the ADP-ribose chain. This modification can also be rendered dynamic through removal by ADP-ribose hydrolases. ADP-ribosylation has been shown to play important roles in a variety of cellular processes, in particular stress response and DNA damage repair, making this PTM an intriguing therapeutic target in a variety of diseases (Curtin and Szabo, 2013). The structurally complex and chemically labile nature of poly- ADP-ribosylation has made it challenging to study and bioorthogonal probes have made important contributions to the global and enzyme specific analysis of substrate proteins. The ability of PARPs to accept modified analogs of ADP-ribose was first explored using 6- or 8-alkyne-NAD+ molecules (Table 1), which enabled the labeling and identification of PARP1 substrates when incubated with cell lysates (Chang et al., 2009; Du et al., 2009a). A bump-hole strategy similar to that used for SAM analogs described above was also exploited to create probes for specific PARP family members. More specifically, a hole could be generated in PARPs that could accept bumps at the C5- position of the nicotinamide on NAD+, allowing for the identification of specific substrates of PARP1 and PARP2 (Carter-O’Connell et al., 2014). Although powerful, these approaches rely on synthetic NAD+ analogs that are not cell permeable and therefore must be performed in cell lysates. To address this limitation, an MCR of ADP- ribosylation was recently developed (Westcott et al., 2017a). More specifically, cells can biosynthesize alkyne-containing NAD+ from N6-propargyl adenosine, resulting in labeling of ADP-ribosylation in living cells and characterization of Hras modification. Again, the small size of the alkyne was key to transiting the NAD+ biosynthetic pathway. More recently, a similar reporter bearing the alkyne at the 2-position of adenosine was shown to display similar cellular labeling and improved detection of protein targets by proteomics (Kalesh et al., 2019).
Cysteine redox modifications
The electron rich thiol group of cysteine has unique nucleophilic properties as outlined above but can also undergo a broad range of oxidative reactions to generate unique functional groups, including S-sulfenylation (-SOH), S-sulfinylation (-SO2H), S-sulfonylation (-SO3H), S-nitrosylation (-SNO), S-sulfhydration (-SSH), and S- glutathionylation (-SSG). Most thiol redox modifications can be formed by the non-enzymatic reactions of reactive oxygen/nitrogen/sulfur species (ROS/RNS/RSS) with protein thiols, which has led to the widespread notion that these types of modifications are randomly distributed across the cysteine proteome. However, emerging evidence suggests that thiol redox modifications are well-controlled, site-specific cellular events, which play important roles in regulation of diverse protein functions, including catalytic or ligand binding activities, protein-protein interactions, and protein stability (Antelmann and Helmann, 2011). Identification of protein targets of thiol redox modifications is crucial to understanding their roles in biology and disease; however, proteome-wide analysis of thiol redox modifications is challenging due to the lack of selective biological reagents (e.g. antibodies) and is further complicated by the labile and highly dynamic nature of thiol redox forms. Not surprisingly, bioorthogonal chemistry has also provided solutions to these limitations (Yang et al., 2016). The first such method relied on the selective reaction between a sulfenylated cysteine and dimedone (5,5-dimethyl-1,3- cylcohexanedione) nucleophiles for the selective installation of a azide- or alkyne-tags as a stable thioethers that could then be subjected to bioorthogonal reaction with visualization or affinity tags (Table 1) (Leonard et al., 2009; Paulsen et al., 2011; Yang et al., 2014). Notably, this probe was used to identify a sulfenylated cysteine in the active site of the epidermal growth factor receptor (EGFR) that increases its kinase activity (Paulsen et al., 2011). More recently, sulfenylated cysteines were shown to selectively react with noroborene probes to give stable sulfoxide products (Table 1) (Alcock et al., 2019). The unique reactivity of electrophilic diazenes with -SO2H was likewise exploited to generate a alkyne-containing probe of sulfinylated cysteines termed DiaAlk (Table 1) (Akter et al., 2018). In the case of these PTMs, like some of the enzymatic activity profiling experiments we described above, the exploitation of bioorthogonal chemistry is not absolutely necessary but is rather a key feature that can be exploited for multiple types of analyses. The chemical mechanisms exploited to enable these selective labeling reactions are shown in Figure 8.
Figure 8.
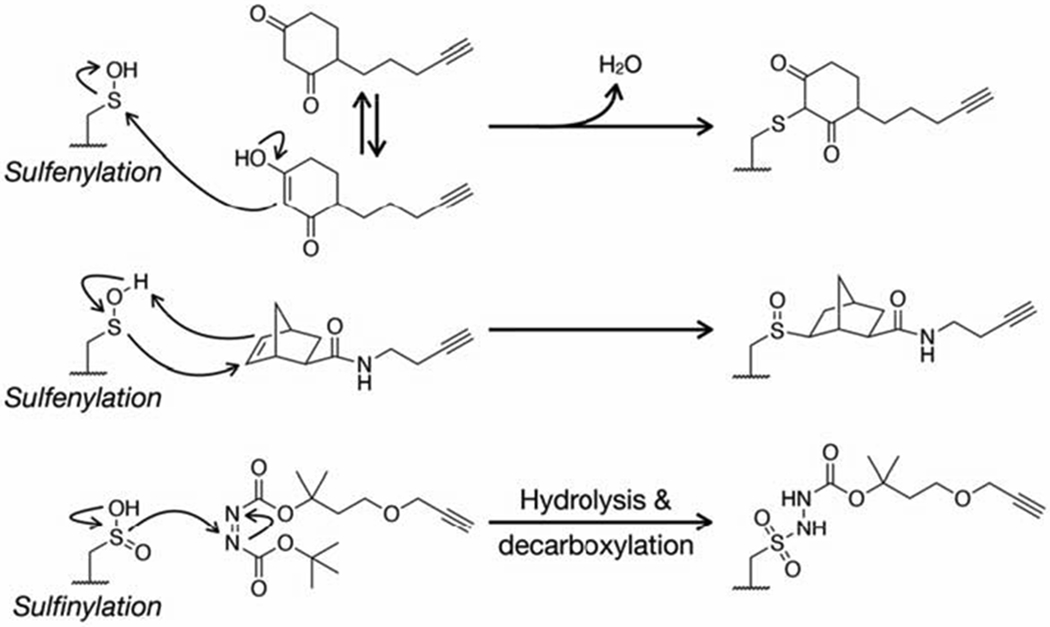
Chemical mechanisms responsible for the selective reaction of sulfenylated and sulfinylated cysteine residues with their respective probes.
Examples of biological insights into PTM function gained using click chemistry
Cataloging PTMs is in itself an important goal, reminiscent of RNA sequencing and global proteomics experiments. However, they have also enabled a range of important biological discoveries. For example, in the case of protein glycosylation, chemoenzymatic modification of mouse or human brain tissues followed by proteomics identified several O-GlcNAc modification sites on the Parkinson’s disease-causing protein α-synuclein (Alfaro et al., 2012; Wang et al., 2017). With this information, we subsequently used synthetic protein chemistry to generate site-specifically O- GlcNAcylated α-synuclein and showed that this modification inhibits the amyloid aggregation and toxicity of this protein (Levine et al., 2019). As another example, the Hang lab recently used a metabolic reporter of short-chain fatty acids to profile modified proteins in Salmonella (Zhang et al., 2020). They found that several virulence proteins are butyrylated and showed that site-specific modification of the transcription factor HilA inhibits its activity and attenuates bacterial entry into epithelial cells. Similarly, the same lab used the N6-propargyl adenosine metabolic reporter to discover that the critical signaling protein Hras is ADP-ribosylated under oxidative stress in cells and that this modification inhibits its activity (Westcott et al., 2017b). As a final example, the Caroll lab used their probe, alkyne-diazine, and quantitative proteomics to profile protein sulfinylation in wild-type and sulforedoxin-null cells, allowing them to directly identify numerous unknown substrates of this critical enzymatic regulator of oxidative stress (Akter et al., 2018). In all of these cases, these insights would have been impossible or at least difficult to obtain using traditional biological tools like enrichment with anti-PTM antibodies, which either do not exist or do not recognize the entire repertoire of modified proteins. For example, using our synthetic proteins we found that the two most commonly used pan-selective anti-O-GlcNAc antibodies only recognize one out of four modification sites on α-synuclein.
Extracting Biological Information from Click Chemistry Enabled Proteomic Investigations
At this stage, click chemistry-facilitated proteomics is robust, and proteomic lists of proteins captured or modified by small-molecules are routinely published. However, it is still challenging to identify the interactions and modifications that are most likely to affect protein biochemistry and thus mediate important biology. Therefore, it is important to narrow the list of potential proteins for follow-up biochemical or functional studies. Unfortunately, there are no silver bullets to accomplish this, and each investigation often necessitates unique solutions. In the case of understanding functional consequences of small molecule-protein interactions, the use of structurally similar but functionally inactive control probes are essential to prioritize biologically relevant protein targets, which can require additional medicinal chemistry effort. In addition, although click probes can reveal potential protein targets, often such experiments do not directly provide relative levels ligand-protein stoichiometry, thus competitive experiments with underivatized bioactive analogs can facilitate the identification of selective and high- stoichiometry targets. Furthermore, careful validation of identified small molecule-protein interactions is necessary to home in on proteins of functional relevance. Secondary target engagement assays should be used to confirm small-molecule target interactions and genetic perturbation strategies (knock out/down, overexpression, etc.) should be employed to assess whether genetic modulation of identified proteins mimic or alters the activity of the bioactive compound. In the case of PTMs, we recommend several approaches. All proteomics lists should be entered into biological pathway analysis programs that can provide clues to the overall biochemical pathways that might controlled by a PTM of interest. This algorithm guided analysis should be paired with a working knowledge of the literature surrounding a PTM, with an emphasis on the previously characterized phenotypes and biochemistry. If the specific site of a PTM is identified, genetics can also be a guide. For example, if a given modification site is conserved evolutionarily, it is more likely to be functionally relevant. Additionally, if a site is mutated in human disease (e.g., a mutation consistently found in cancer) the modification may also be playing an important role. Once these approaches have been used to narrow down the list of potentially interesting proteins, the modification stoichiometry of some PTMs can be determined on a case-by-case basis (Darabedian et al., 2018; Percher et al., 2016; Rexach et al., 2010), as it is more likely that proteins with a high fraction of modification will have altered biochemistry.
Considerations and Looking Forward
Bioorthogonal click chemistry has enabled the development of a wide range of chemical probes and reporters. In many cases, the use of small bioorthogonal reactive groups is essential for the application of probes to study living systems and, in all cases, increases probe versatility through the divergent attachment of a various enrichment and analysis tags. Despite the many successes of numerous clickable probes, some challenges still remain. Due in part to the diversity of biological applications, there is no standardized protocol covering the different aspects of probe treatment, click chemistry conditions, or analysis. This results in a choice of a variety of different methods that may seem overwhelming or arbitrary to the newcomer, thus the sharing of detailed protocols from expert labs remains an important goal. Additionally, while the commercial availability of both probes and tags has exploded in the past decade, some reagents and most chemical probes require synthetic chemistry skills to prepare. Therefore, there is an important need for the continued close collaboration between biology and chemistry labs to ensure the application of the best probes to the most important biological questions. Finally, the proteomic analyses used downstream of certain probe applications generates a large volume of data that needs to be analyzed and curated, which again requires some non-standard expertise, proper controls, and rigorous statistical evaluation.
In the case of illuminating small molecule interactions, despite the utility of protein-centric and chemocentric click probes, the vast majority of proteins still lack useful chemical tools to interrogate their function. ABPP and similar chemical proteomic platforms have aimed to identify chemical probes for functionally related protein families which can be used in complex native environments; however, there currently exists no general capability to identify chemical probes or small molecule ligands for all ligandable proteins (Schreiber, 2019). Proteome-wide fragment-based ligand discovery (Backus et al., 2016; Parker et al., 2017a; Wang et al., 2019) and other methods (Franzini and Randolph, 2016) represent promising strategies to systematically yet agnostically identify small molecule protein ligands. Furthermore, click handles enable the enrichment of protein targets; however, their presence can often perturb molecular interactions and biological activity. Recent advances in “label-free” proteomic methods, such as thermal proteome profiling (TPP) and drug affinity responsive target stability (DARTS), represent powerful complementary strategies to map small molecule-protein interactions, bypassing the requirement of enrichment handles such as click functional groups (Lomenick et al., 2009; Savitski et al., 2014). However, these methods rely on ligand-induced stabilization towards heat or proteolytic degradation, for example, which are irreversible processes that can be influenced by other complicating factors including PTMs and other interaction partners, and, furthermore, the lack of an enrichment step can lead to substantial false negative readouts.
In the case of PTMs, it still remains challenging to identify the exact site of modification on the protein and, in the case of heterogenous modifications like glycosylation, the exact structure of the modification. Improvements in mass spectrometry techniques, instrumentation, and data analysis will play key roles in the probe-related discoveries in the future. Additionally, many of the probes for PTMs require cellular metabolism to result in protein labeling. This reliance on interconnected biosynthetic pathways can result in the metabolic transformation of these reporters into “off target” pathways. Therefore, important future goals should be a more comprehensive understanding of the metabolism of probes and the generation selective reporters that are funneled down the PTM pathway of interest. Finally, continued chemical and biochemical insights are needed for the development of probes for currently untargeted PTMs, an ever expanding number in all kingdoms of life.
Acknowledgments
M.R.P.’s research in this area is supported by the National Institutes of Health R01GM125939.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abegg D, Frei R, Cerato L, Hari DP, Wang C, Waser J, and Adibekian A (2015). Proteome-wide profiling of targets of cysteine reactive small molecules by using ethynyl benziodoxolone reagents. Angewandte Chemie-International Edition 54, 10852–10857. [DOI] [PubMed] [Google Scholar]
- Abo M, Bak DW, and Weerapana E (2017). Optimization of caged electrophiles for improved monitoring of cysteine reactivity in living cells. ChemBioChem 18, 81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abo M, and Weerapana E (2015). A caged electrophilic probe for global analysis of cysteine reactivity in living cells. J Am Chem Soc 137, 7087–7090. [DOI] [PubMed] [Google Scholar]
- Aebi M (2013). N-linked protein glycosylation in the er. Biochimica et biophysica acta 1833, 2430–2437. [DOI] [PubMed] [Google Scholar]
- Agard NJ, Prescher JA, and Bertozzi CR (2004). A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- Akter S, Fu L, Jung Y, Conte ML, Lawson JR, Lowther WT, Sun R, Liu K, Yang J, and Carroll KS (2018). Chemical proteomics reveals new targets of cysteine sulfinic acid reductase. Nat Chem Biol 14, 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcock LJ, Oliveira BL, Deery MJ, Pukala TL, Perkins MV, Bernardes GJL, and Chalker JM (2019). Norbornene probes for the detection of cysteine sulfenic acid in cells. ACS chemical biology 14, 594–598. [DOI] [PubMed] [Google Scholar]
- Alfaro JF, Gong CX, Monroe ME, Aldrich JT, Clauss TR, Purvine SO, Wang Z, Camp DG 2nd, Shabanowitz J, Stanley P, et al. (2012). Tandem mass spectrometry identifies many mouse brain o-glcnacylated proteins including egf domain-specific o-glcnac transferase targets. Proc Natl Acad Sci U S A 109, 7280–7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An H, and Statsyuk AV (2013). Development of an activity based probe for ubiquitin and ubiquitin-like protein signaling pathways. Abstracts of Papers of the American Chemical Society 246. [DOI] [PubMed] [Google Scholar]
- Antelmann H, and Helmann JD (2011). Thiol-based redox switches and gene regulation. Antioxidants & Redox Signaling 14, 1049–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, et al. (2015). The promise and peril of chemical probes. Nat Chem Biol 11, 536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backus KM (2019). Applications of reactive cysteine profiling. Curr Top Microbiol Immunol 420, 375–417. [DOI] [PubMed] [Google Scholar]
- Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, et al. (2016). Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard TE, Murrey HE, Geoghegan KF, Ende CWA, and Johnson DS (2014). Investigating gamma-secretase protein interactions in live cells using active site-directed clickable dual-photoaffinity probes. Medchemcomm 5, 321–327. [Google Scholar]
- Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, et al. (2007). Quantitative chemical proteomics reveals mechanisms of action of clinical abl kinase inhibitors. Nat Biotechnol 25, 1035–1044. [DOI] [PubMed] [Google Scholar]
- Bao X, Zhao Q, Yang T, Fung YME, and Li XD (2013). A chemical probe for lysine malonylation. Angewandte Chemie International Edition 52, 4883–4886. [DOI] [PubMed] [Google Scholar]
- Bar-Peled L, Kemper EK, Suciu RM, Vinogradova EV, Backus KM, Horning BD, Paul TA, Ichu TA, Svensson RU, Olucha J, et al. (2017). Chemical proteomics identifies druggable vulnerabilities in a genetically defined cancer. Cell 171, 696–709 e623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barglow KT, and Cravatt BF (2004). Discovering disease-associated enzymes by proteome reactivity profiling. Chem Biol 11, 1523–1531. [DOI] [PubMed] [Google Scholar]
- Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, and Bertozzi CR (2007). Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci U S A 104, 16793–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman LA, Nguyen TB, Roberts AM, Miyamoto DK, Ku WM, Huffman TR, Petri Y, Heslin MJ, Contreras CM, Skibola CF, et al. (2017). Chemoproteomics-enabled covalent ligand screen reveals a cysteine hotspot in reticulon 4 that impairs er morphology and cancer pathogenicity. Chemical Communications 53, 7234–7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman LA, Zaro BW, Miller SM, and Pratt MR (2013). An alkyne-aspirin chemical reporter for the detection of aspirin-dependent protein modification in living cells. J Am Chem Soc 135, 14568–14573. [DOI] [PubMed] [Google Scholar]
- Bedford MT, and Clarke SG (2009). Protein arginine methylation in mammals: Who, what, and why. Mol Cell 33, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshaw PJ, Ho SN, Crabtree GR, and Schreiber SL (1996). Controlling protein association and subcellular localization with a synthetic ligand that induces heterodimerization of proteins. Proceedings of the National Academy of Sciences of the United States of America 93, 4604–4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggar KK, and Li SSC (2015). Non-histone protein methylation as a regulator of cellular signalling and function. Nature Reviews Molecular Cell Biology 16, 5–17. [DOI] [PubMed] [Google Scholar]
- Binda O, Boyce M, Rush JS, Palaniappan KK, Bertozzi CR, and Gozani O (2011). A chemical method for labeling lysine methyltransferase substrates. ChemBioChem 12, 330–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagg J, and Workman P (2017). Choose and use your chemical probe wisely to explore cancer biology. Cancer Cell 32, 9–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blewett MM, Xie J, Zaro BW, Backus KM, Altman A, Teijaro JR, and Cravatt BF (2016). Chemical proteomic map of dimethyl fumarate-sensitive cysteines in primary human t cells. Sci Signal 9, rs10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce M, Carrico IS, Ganguli AS, Yu S-H, Hangauer MJ, Hubbard SC, Kohler JJ, and Bertozzi CR (2011). Metabolic cross-talk allows labeling of o-linked beta-n-acetylglucosamine-modified proteins via the n-acetylgalactosamine salvage pathway. Proceedings of the National Academy of Sciences of the United States of America 108, 3141–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-O’Connell I, Jin H, Morgan RK, David LL, and Cohen MS (2014). Engineering the substrate specificity of adp-ribosyltransferases for identifying direct protein targets. J Am Chem Soc 136, 5201–5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TR, Hilgraf R, Sharpless KB, and Fokin VV (2004). Polytriazoles as copper(i)-stabilizing ligands in catalysis. Organic Letters 6, 2853–2855. [DOI] [PubMed] [Google Scholar]
- Chang PV, Chen X, Smyrniotis C, Xenakis A, Hu T, Bertozzi CR, and Wu P (2009). Metabolic labeling of sialic acids in living animals with alkynyl sugars. Angew Chem Int Ed Engl 48, 4030–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron G, Li MMH, MacDonald MR, and Hang HC (2013). Prenylome profiling reveals s-farnesylation is crucial for membrane targeting and antiviral activity of zap long-isoform. Proceedings of the National Academy of Sciences of the United States of America 110, 11085–11090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron G, Zhang MM, Yount JS, Wilson J, Raghavan AS, Shamir E, and Hang HC (2009). Robust fluorescent detection of protein fatty-acylation with chemical reporters. J Am Chem Soc 131, 4967–4975. [DOI] [PubMed] [Google Scholar]
- Chen WT, Dong JJ, Plate L, Mortenson DE, Brighty GJ, Li SH, Liu Y, Galmozzi A, Lee PS, Hulce JJ, et al. (2016). Arylfluorosulfates inactivate intracellular lipid binding protein(s) through chemoselective sufex reaction with a binding site tyr residue. J Am Chem Soc 138, 7353–7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, and Mann M (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. [DOI] [PubMed] [Google Scholar]
- Chuh KN, Batt AR, and Pratt MR (2016). Chemical methods for encoding and decoding of posttranslational modifications. Cell Chem Biol 23, 86–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuh KN, Batt AR, Zaro BW, Darabedian N, Marotta NP, Brennan CK, Amirhekmat A, and Pratt MR (2017). The new chemical reporter 6-alkynyl-6-deoxy- glcnac reveals o-glcnac modification of the apoptotic caspases that can block the cleavage/activation of caspase-8. J Am Chem Soc 139, 7872–7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuh KN, and Pratt MR (2015). Chemical methods for the proteome-wide identification of posttranslationally modified proteins. Curr Opin Chem Biol 24, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuh KN, Zaro BW, Piller F, Piller V, and Pratt MR (2014). Changes in metabolic chemical reporter structure yield a selective probe of o-glcnac modification. J Am Chem Soc 136, 12283–12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisar EAG, Nguyen N, and Rosen H (2013). A gtp affinity probe for proteomics highlights flexibility in purine nucleotide selectivity. J Am Chem Soc 135, 4676–4679. [DOI] [PubMed] [Google Scholar]
- Cisar JS, Weber OD, Clapper JR, Blankman JL, Henry CL, Simon GM, Alexander JP, Jones TK, Ezekowitz RAB, O’Neill GP, et al. (2018). Identification of abx-1431, a selective inhibitor of monoacylglycerol lipase and clinical candidate for treatment of neurological disorders. J Med Chem 61, 9062–9084. [DOI] [PubMed] [Google Scholar]
- Clark PM, Dweck JF, Mason DE, Hart CR, Buck SB, Peters EC, Agnew BJ, and Hsieh-Wilson LC (2008). Direct in-gel fluorescence detection and cellular imaging of o-glcnac-modified proteins. J Am Chem Soc 130, 11576–11577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cognetta AB, Niphakis MJ, Lee HC, Martini ML, Hulce JJ, and Cravatt BF (2015). Selective n-hydroxyhydantoin carbamate inhibitors of mammalian serine hydrolases. Chem Biol 22, 928–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MS, and Chang P (2018). Insights into the biogenesis, function, and regulation of adp-ribosylation. Nat Chem Biol 14, 236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole CM, Yang J, Šečkutė J, and Devaraj NK (2013). Fluorescent live-cell imaging of metabolically incorporated unnatural cyclopropene-mannosamine derivatives. ChemBioChem 14, 205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, and Mann M (2014). Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed maxlfq. Mol Cell Proteomics 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Wright AT, and Kozarich JW (2008). Activity-based protein profiling: From enzyme chemistry to proteomic chemistry. Annu Rev Biochem 77, 383–414. [DOI] [PubMed] [Google Scholar]
- Curtin NJ, and Szabo C (2013). Therapeutic applications of parp inhibitors: Anticancer therapy and beyond. Molecular Aspects of Medicine 34, 1217–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darabedian N, Thompson JW, Chuh KN, Hsieh-Wilson LC, and Pratt MR (2018). Optimization of chemoenzymatic mass tagging by strain-promoted cycloaddition (spaac) for the determination of o-glcnac stoichiometry by western blotting. Biochemistry 57, 5769–5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayon L, Hainard A, Licker V, Turck N, Kuhn K, Hochstrasser DF, Burkhard PR, and Sanchez JC (2008). Relative quantification of proteins in human cerebrospinal fluids by ms/ms using 6-plex isobaric tags. Analytical Chemistry 80, 2921–2931. [DOI] [PubMed] [Google Scholar]
- DeGraw AJ, Palsuledesai C, Ochocki JD, Dozier JK, Lenevich S, Rashidian M, and Distefano MD (2010). Evaluation of alkyne-modified isoprenoids as chemical reporters of protein prenylation. Chemical Biology & Drug Design 76, 460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Amo DS, Wang W, Jiang H, Besanceney C, Yan AC, Levy M, Liu Y, Marlow FL, and Wu P (2010). Biocompatible copper(i) catalysts for in vivo imaging of glycans. J Am Chem Soc 132, 16893–16899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewes G, and Knapp S (2018). Chemoproteomics and chemical probes for target discovery. Trends Biotechnol 36, 1275–1286. [DOI] [PubMed] [Google Scholar]
- Du J, Jiang H, and Lin H (2009a). Investigating the adp-ribosyltransferase activity of sirtuins with nad analogues and 32p-nad. Biochemistry 48, 2878–2890. [DOI] [PubMed] [Google Scholar]
- Du J, Jiang H, and Lin H (2009b). Investigating the adp-ribosyltransferase activity of sirtuins with nad analogues and 32p-nad. Biochemistry 48, 2878–2890. [DOI] [PubMed] [Google Scholar]
- Eirich J, Orth R, and Sieber SA (2011). Unraveling the protein targets of vancomycin in living s. Aureus and e. Faecalis cells. J Am Chem Soc 133, 12144–12153. [DOI] [PubMed] [Google Scholar]
- Foe IT, Child MA, Majmudar JD, Krishnamurthy S, van der Linden WA, Ward GE, Martin BR, and Bogyo M (2015). Global analysis of palmitoylated proteins in toxoplasma gondii. Cell Host & Microbe 18, 501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford B, Bateman LA, Gutierrez-Palominos L, Park R, and Nomura DK (2017). Mapping proteome-wide targets of glyphosate in mice. Cell Chemical Biology 24, 133–140. [DOI] [PubMed] [Google Scholar]
- Franzini RM, and Randolph C (2016). Chemical space of DNA-encoded libraries. J Med Chem 59, 6629–6644. [DOI] [PubMed] [Google Scholar]
- Galmozzi A, Kok BP, Kim AS, Montenegro-Burke JR, Lee JY, Spreafico R, Mosure S, Albert V, Cintron-Colon R, Godio C, et al. (2019). Pgrmc2 is an intracellular haem chaperone critical for adipocyte function. Nature 576, 138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Mfuh A, Amako Y, Woo CM (2018). Small Molecule Interactome Mapping by Photoaffinity Labeling Reveals Binding Site Hotspots for NSAIDs. J Am Chem Soc 140, 4259–4268. [DOI] [PubMed] [Google Scholar]
- Garbaccio RM, and Parmee ER (2016). The impact of chemical probes in drug discovery: A pharmaceutical industry perspective. Cell Chem Biol 23, 10–17. [DOI] [PubMed] [Google Scholar]
- Geurink PP, Prely LM, van der Marel GA, Bischoff R, and Overkleeft HS (2012). Photoaffinity labeling in activity-based protein profiling. Activity-Based Protein Profiling 324, 85–113. [DOI] [PubMed] [Google Scholar]
- Gillet LC, Namoto K, Ruchti A, Hoving S, Boesch D, Inverardi B, Mueller D, Coulot M, Schindler P, Schweigler P, et al. (2008). In-cell selectivity profiling of serine protease inhibitors by activity-based proteomics. Mol Cell Proteomics 7, 1241–1253. [DOI] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, and Aebersold R (2012). Targeted data extraction of the ms/ms spectra generated by data- independent acquisition: A new concept for consistent and accurate proteome analysis. Molecular & Cellular Proteomics 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammel M, and Hang HC (2013). Chemical reporters for biological discovery. Nat Chem Biol 9, 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, and Shi Y (2012). Histone methylation: A dynamic mark in health, disease and inheritance. Nature reviews Genetics 13, 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman EA, Ward CC, Spradlin JN, Bateman LA, Huffman TR, Miyamoto DK, Kleinman JI, and Nomura DK (2017). Covalent ligand discovery against druggable hotspots targeted by anti-cancer natural products. Cell Chemical Biology 24, 1368–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Shannon DA, Colby T, Wang ZM, Shabab M, Kumari S, Villamor JG, McLaughlin CJ, Weerapana E, Kaiser M, et al. (2013). Chemical proteomics with sulfonyl fluoride probes reveals selective labeling of functional tyrosines in glutathione transferases. Chem Biol 20, 541–548. [DOI] [PubMed] [Google Scholar]
- Gushwa NN, Kang SM, Chen J, and Taunton J (2012). Selective targeting of distinct active site nucleophiles by irreversible src-family kinase inhibitors. J Am Chem Soc 134, 20214–20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, and Aebersold R (1999). Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol 17, 994–999. [DOI] [PubMed] [Google Scholar]
- Hacker SM, Backus KM, Lazear MR, Forli S, Correia BE, and Cravatt BF (2017). Global profiling of lysine reactivity and ligandability in the human proteome. Nat Chem 9, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang H, Geutjes E, Grotenbreg G, Pollington A, Bijlmakers M, and Ploegh H (2007). Chemical probes for the rapid detection of fatty-acylated proteins in mammalian cells. J Am Chem Soc 129, 2744–2745. [DOI] [PubMed] [Google Scholar]
- Hang HC, Yu C, Kato DL, and Bertozzi CR (2003). A metabolic labeling approach toward proteomic analysis of mucin-type o-linked glycosylation. Proceedings of the National Academy of Sciences of the United States of America 100, 14846–14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding MW, Galat A, Uehling DE, and Schreiber SL (1989). A receptor for the immunosuppressant fk506 is a cis-trans peptidyl-prolyl isomerase. Nature 341, 758–760. [DOI] [PubMed] [Google Scholar]
- Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, et al. (2015). A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163, 712–723. [DOI] [PubMed] [Google Scholar]
- Hewings DS, Heideker J, Ma TP, AhYoung AP, El Oualid F, Amore A, Costakes GT, Kirchhofer D, Brasher B, Pillow T, et al. (2018). Reactive-site-centric chemoproteomics identifies a distinct class of deubiquitinase enzymes. Nat Commun 9, 1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horning BD, Suciu RM, Ghadiri DA, Ulanovskaya OA, Matthews ML, Lum KM, Backus KM, Brown SJ, Rosen H, and Cravatt BF (2016). Chemical proteomic profiling of human methyltransferases. J Am Chem Soc 138, 13335–13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Ogasawara D, Seneviratne UI, Cognetta AB 3rd, Am Ende CW, Nason DM, Lapham K, Litchfield J, Johnson DS, and Cravatt BF (2019). Global portrait of protein targets of metabolites of the neurotoxic compound bia 10–2474. ACS Chem Biol 14, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisgen R (1963). 1,3-dipolar cycloadditions. Past and future. 2, 565–598. [Google Scholar]
- Hulce JJ, Cognetta AB, Niphakis MJ, Tully SE, and Cravatt BF (2013). Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat Methods 10, 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, et al. (2015). The bioplex network: A systematic exploration of the human interactome. Cell 162, 425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam K, Bothwell I, Chen Y, Sengelaub C, Wang R, Deng H, and Luo M (2012). Bioorthogonal profiling of protein methylation using azido derivative of s-adenosyl- l-methionine. J Am Chem Soc 134, 5909–5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam K, Chen Y, Wu H, Bothwell IR, Blum GJ, Zeng H, Dong A, Zheng W, Min J, Deng H, et al. (2013). Defining efficient enzyme-cofactor pairs for bioorthogonal profiling of protein methylation. Proceedings of the National Academy of Sciences of the United States of America 110, 16778–16783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessani N, Liu Y, Humphrey M, and Cravatt BF (2002). Enzyme activity profiles of the secreted and membrane proteome that depict cancer invasiveness. Proc Natl Acad Sci USA 99, 10335–10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessani N, Niessen S, Wei BQ, Nicolau M, Humphrey M, Ji Y, Han W, Noh DY, Yates JR 3rd, Jeffrey SS, et al. (2005). A streamlined platform for high-content functional proteomics of primary human specimens. Nat Methods 2, 691–697. [DOI] [PubMed] [Google Scholar]
- Jiang H, Zhang X, Chen X, Aramsangtienchai P, Tong Z, and Lin H (2018). Protein lipidation: Occurrence, mechanisms, biological functions, and enabling technologies. Chemical reviews 118, 919–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalesh K, Lukauskas S, Borg AJ, Snijders AP, Ayyappan V, Leung AKL, Haskard DO, and DiMaggio PA (2019). An integrated chemical proteomics approach for quantitative profiling of intracellular adp-ribosylation. Sci Rep 9, 6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambe T, Correia BE, Niphakis MJ, and Cravatt BF (2014). Mapping the protein interaction landscape for fully functionalized small-molecule probes in human cells. J Am Chem Soc 136, 10777–10782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato D, Boatright KM, Berger AB, Nazif T, Blum G, Ryan C, Chehade KAH, Salvesen GS, and Bogyo M (2005). Activity-based probes that target diverse cysteine protease families. Nat Chem Biol 1, 33–38. [DOI] [PubMed] [Google Scholar]
- Kidd D, Liu Y, and Cravatt BF (2001). Profiling serine hydrolase activities in complex proteomes. Biochemistry 40, 4005–4015. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Kang DW, Leucke HF, Bond MR, Ghosh S, Love DC, Ahn JS, Kang DO, and Hanover JA (2013). Optimizing the selectivity of difo-based reagents for intracellular bioorthogonal applications. Carbohydr Res 377, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizuka Y, Funayama S, Shogomori H, Nakano M, Nakajima K, Oka R, Kitazume S, Yamaguchi Y, Sano M, Korekane H, et al. (2016). High-sensitivity and low-toxicity fucose probe for glycan imaging and biomarker discovery. Cell Chemical Biology 23, 782–792. [DOI] [PubMed] [Google Scholar]
- Kolb HC, Finn MG, and Sharpless KB (2001). Click chemistry: Diverse chemical function from a few good reactions. Angew Chem Int Ed Engl 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- Kulkarni RA, Worth AJ, Zengeya TT, Shrimp JH, Garlick JM, Roberts AM, Montgomery DC, Sourbier C, Gibbs BK, Mesaros C, et al. (2017). Discovering targets of non-enzymatic acylation by thioester reactivity profiling. Cell Chemical Biology 24, 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AC, and Crews CM (2017). Induced protein degradation: An emerging drug discovery paradigm. Nat Rev Drug Discov 16, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang K, and Chin JW (2014a). Bioorthogonal reactions for labeling proteins. ACS Chem Biol 9, 16–20. [DOI] [PubMed] [Google Scholar]
- Lang K, and Chin JW (2014b). Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem Rev 114, 4764–4806. [DOI] [PubMed] [Google Scholar]
- Lannine BR, Whitby LR, Dix MM, Douhan J, Gilbert AM, Hett EC, Johnson T, Joslynl C, Kath JC, Niessen S, et al. (2014). A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat Chem Biol 10, 760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapinsky DJ, and Johnson DS (2015). Recent developments and applications of clickable photoprobes in medicinal chemistry and chemical biology. Future Medicinal Chemistry 7, 2143–2171. [DOI] [PubMed] [Google Scholar]
- Laughlin ST, Baskin JM, Amacher SL, and Bertozzi CR (2008). In vivo imaging of membrane-associated glycans in developing zebrafish. Science 320, 664–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughlin ST, and Bertozzi CR (2009). In vivo imaging of caenorhabditis elegans glycans. Acs Chemical Biology 4, 1068–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard SE, Reddie KG, and Carroll KS (2009). Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS chemical biology 4, 783–799. [DOI] [PubMed] [Google Scholar]
- Leung D, Hardouin C, Boger DL, and Cravatt BF (2003). Discovering potent and selective reversible inhibitors of enzymes in complex proteomes. Nat Biotechnol 21, 687–691. [DOI] [PubMed] [Google Scholar]
- Levine PM, Galesic A, Balana AT, Mahul-Mellier AL, Navarro MX, De Leon CA, Lashuel HA, and Pratt MR (2019). Alpha-synuclein o-glcnacylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America 116, 1511–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang J, Wen L, Zhu H, Li S, Huang K, Jiang K, Li X, Ma C, Qu J, et al. (2016). An oga-resistant probe allows specific visualization and accurate identification of o-glcnac-modified proteins in cells. ACS chemical biology 11, 3002–3006. [DOI] [PubMed] [Google Scholar]
- Li ZQ, Hao PL, Li L, Tan CYJ, Cheng XM, Chen GYJ, Sze SK, Shen HM, and Yao SQ (2013). Design and synthesis of minimalist terminal alkyne-containing diazirine photo-crosslinkers and their incorporation into kinase inhibitors for cell- and tissue-based proteome profiling. Angewandte Chemie-International Edition 52, 8551–8556. [DOI] [PubMed] [Google Scholar]
- Li ZQ, Wang DY, Li L, Pan SJ, Na ZK, Tan CYJ, and Yao SQ (2014). “Minimalist” cyclopropene-containing photo-cross-linkers suitable for live-cell imaging and affinity-based protein labeling. J Am Chem Soc 136, 9990–9998. [DOI] [PubMed] [Google Scholar]
- Lin HN, Su XY, and He B (2012). Protein lysine acylation and cysteine succination by intermediates of energy metabolism. Acs Chemical Biology 7, 947–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SX, Yang XY, Jia S, Weeks AM, Hornsby M, Lee PS, Nichiporuk RV, Iavarone AT, Wells JA, Toste FD, et al. (2017). Redox-based reagents for chemoselective methionine bioconjugation. Science 355, 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Farmer JD, Lane WS, Friedman J, Weissman I, and Schreiber SL (1991). Calcineurin is a common target of cyclophilin-cyclosporine-a and fkbp-fk506 complexes. Cell 66, 807–815. [DOI] [PubMed] [Google Scholar]
- Liu Y, Patricelli MP, and Cravatt BF (1999). Activity-based protein profiling: The serine hydrolases. Proc Natl Acad Sci U S A 96, 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S, Wang JN, Wu RP, Gomez F, Loo JA, et al. (2009). Target identification using drug affinity responsive target stability (darts). Proceedings of the National Academy of Sciences of the United States of America 106, 21984–21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong KK, Bradner JE, and Kaelin WG Jr. (2014). The myeloma drug lenalidomide promotes the cereblon-dependent destruction of ikaros proteins. Science 343, 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahal LK, Yarema KJ, and Bertozzi CR (1997). Engineering chemical reactivity on cell surfaces through oligosaccharide biosynthesis. Science 276, 1125–1128. [DOI] [PubMed] [Google Scholar]
- Martin BR, Wang C, Adibekian A, Tully SE, and Cravatt BF (2012). Global profiling of dynamic protein palmitoylation. Nat Methods 9, 84–U205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Gago P, and Olsen CA (2019). Arylfluorosulfate-based electrophiles for covalent protein labeling: A new addition to the arsenal. Angewandte Chemie-International Edition 58, 957–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay CS, and Finn MG (2014). Click chemistry in complex mixtures: Bioorthogonal bioconjugation. Chem Biol 21, 1075–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery DC, Sorum AW, and Meier JL (2014). Chemoproteomic profiling of lysine acetyltransferases highlights an expanded landscape of catalytic acetylation. J Am Chem Soc 136, 8669–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrey HE, Judkins JC, Am Ende CW, Ballard TE, Fang Y, Riccardi K, Di L, Guilmette ER, Schwartz JW, Fox JM, et al. (2015). Systematic evaluation of bioorthogonal reactions in live cells with clickable halotag ligands: Implications for intracellular imaging. J Am Chem Soc 137, 11461–11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan A, and Jones LH (2015). Sulfonyl fluorides as privileged warheads in chemical biology. Chemical Science 6, 2650–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemmara VV, Subramanian V, Muth A, Mondal S, Salinger AJ, Maurais AJ, Tilvawala R, Weerapana E, and Thompson PR (2018). The development of benzimidazole-based clickable probes for the efficient labeling of cellular protein arginine deiminases (pads). ACS Chem Biol 13, 712–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederwieser A, Späte A-K, Nguyen LD, Jüngst C, Reutter W, and Wittmann V (2013). Two-color glycan labeling of live cells by a combination of diels-alder and click chemistry. Angewandte Chemie International Edition 52, 4265–4268. [DOI] [PubMed] [Google Scholar]
- Niessen S, Dix MM, Barbas S, Potter ZE, Lu S, Brodsky O, Planken S, Behenna D, Almaden C, Gajiwala KS, et al. (2017). Proteome-wide map of targets of t790m-egfr-directed covalent inhibitors. Cell Chem Biol 24, 1388–1400 e1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niphakis MJ, and Cravatt BF (2014). Enzyme inhibitor discovery by activity-based protein profiling. Annu Rev Biochem 83, 341–377. [DOI] [PubMed] [Google Scholar]
- Niphakis MJ, Lum KM, Cognetta AB, Correia BE, Ichu TA, Olucha J, Brown SJ, Kundu S, Piscitelli F, Rosen H, et al. (2015). A global map of lipid-binding proteins and their ligandability in cells. Cell 161, 1668–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, and Cravatt BF (2010). Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira BL, Guo Z, and Bernardes GJL (2017). Inverse electron demand diels-alder reactions in chemical biology. Chem Soc Rev 46, 4895–4950. [DOI] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, and Mann M (2002). Stable isotope labeling by amino acids in cell culture, silac, as a simple and accurate approach to expression proteomics. Molecular & Cellular Proteomics 1, 376–386. [DOI] [PubMed] [Google Scholar]
- Palazzo L, Mikolčević P, Mikoč A, and Ahel I (2019). Adp-ribosylation signalling and human disease. Open Biology 9, 190041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CG, Galmozzi A, Wang Y, Correia BE, Sasaki K, Joslyn CM, Kim AS, Cavallaro CL, Lawrence RM, Johnson SR, et al. (2017a). Ligand and target discovery by fragment-based screening in human cells. Cell 168, 527–541 e529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CG, Kuttruff CA, Galmozzi A, Jorgensen L, Yeh CH, Hermanson DJ, Wang YJ, Artola M, McKerrall SJ, Josyln CM, et al. (2017b). Chemical proteomics identifies slc25a20 as a functional target of the ingenol class of actinic keratosis drugs. Acs Central Science 3, 1276–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, and Kozarich JW (2007). Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry 46, 350–358. [DOI] [PubMed] [Google Scholar]
- Patterson DM, Jones KA, and Prescher JA (2014a). Improved cyclopropene reporters for probing protein glycosylation. Molecular bioSystems 10, 1693. [DOI] [PubMed] [Google Scholar]
- Patterson DM, Nazarova LA, and Prescher JA (2014b). Finding the right (bioorthogonal) chemistry. ACS Chem Biol 9, 592–605. [DOI] [PubMed] [Google Scholar]
- Patterson DM, Nazarova LA, Xie B, Kamber DN, and Prescher JA (2012). Functionalized cyclopropenes as bioorthogonal chemical reporters. J Am Chem Soc 134, 18638–18643. [DOI] [PubMed] [Google Scholar]
- Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, and Carroll KS (2011). Peroxide-dependent sulfenylation of the egfr catalytic site enhances kinase activity. Nat Chem Biol 8, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percher A, Ramakrishnan S, Thinon E, Yuan XQ, Yount JS, and Hang HC (2016). Mass-tag labeling reveals site-specific and endogenous levels of protein s-fatty acylation. Proceedings of the National Academy of Sciences of the United States of America 113, 4302–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters W, Willnow S, Duisken M, Kleine H, Macherey T, Duncan KE, Litchfield DW, Luscher B, and Weinhold E (2010). Enzymatic site-specific functionalization of protein methyltransferase substrates with alkynes for click labeling. Angewandte Chemie-International Edition 49, 5170–5173. [DOI] [PubMed] [Google Scholar]
- Prescher JA, and Bertozzi CR (2005). Chemistry in living systems. Nat Chem Biol 1, 13–21. [DOI] [PubMed] [Google Scholar]
- Qin W, Lv P, Fan X, Quan B, Zhu Y, Qin K, Chen Y, Wang C, and Chen X (2017). Quantitative time-resolved chemoproteomics reveals that stable o-glcnac regulates box c/d snornp biogenesis. Proceedings of the National Academy of Sciences of the United States of America 114, E6749–E6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Qin K, Fan X, Peng L, Hong W, Zhu Y, Lv P, Du Y, Huang R, Han M, et al. (2018). Artificial cysteine s-glycosylation induced by per-o-acetylated unnatural monosaccharides during metabolic glycan labeling. Angewandte Chemie (International ed in English) 57, 1817–1820. [DOI] [PubMed] [Google Scholar]
- Rabuka D, Hubbard S, Laughlin S, Argade S, and Bertozzi C (2006). A chemical reporter strategy to probe glycoprotein fucosylation. J Am Chem Soc 128, 12078–12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexach JE, Rogers CJ, Yu SH, Tao JF, Sun YE, and Hsieh-Wilson LC (2010). Quantification of o-glycosylation stoichiometry and dynamics using resolvable mass tags. Nat Chem Biol 6, 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rix U, and Superti-Furga G (2009). Target profiling of small molecules by chemical proteomics. Nat Chem Biol 5, 616–624. [DOI] [PubMed] [Google Scholar]
- Roberts AM, Miyamoto DK, Huffman TR, Bateman LA, Ives AN, Akopian D, Heslin MJ, Contreras CM, Rape M, Skibola CF, et al. (2017a). Chemoproteomic screening of covalent ligands reveals uba5 as a novel pancreatic cancer target. Acs Chemical Biology 12, 899–904. [DOI] [PubMed] [Google Scholar]
- Roberts AM, Ward CC, and Nomura DK (2017b). Activity-based protein profiling for mapping and pharmacologically interrogating proteome-wide ligandable hotspots. Curr Opin Biotechnol 43, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross PL, Huang YLN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, et al. (2004). Multiplexed protein quantitation in saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Molecular & Cellular Proteomics 3, 1154–1169. [DOI] [PubMed] [Google Scholar]
- Rostovtsev VV, Green LG, Fokin VV, and Sharpless KB (2002). A stepwise huisgen cycloaddition process: Copper(i)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- Rutkowska A, Thomson DW, Vappiani J, Werner T, Mueller KM, Dittus L, Krause J, Muelbaier M, Bergamini G, and Bantscheff M (2016). A modular probe strategy for drug localization, target identification and target occupancy measurement on single cell level. ACS Chem Biol 11, 2541–2550. [DOI] [PubMed] [Google Scholar]
- Saghatelian A, Jessani N, Joseph A, Humphrey M, and Cravatt BF (2004). Activity-based probes for the proteomic profiling of metalloproteases. Proc Natl Acad Sci U S A 101, 10000–10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salisbury CM, and Cravatt BF (2007). Activity-based probes for proteomic profiling of histone deacetylase complexes. Proceedings of the National Academy of Sciences of the United States of America 104, 1171–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salisbury CM, and Cravatt BF (2008). Optimization of activity-based probes for proteomic profiling of histone deacetylase complexes. J Am Chem Soc 130, 2184–2194. [DOI] [PubMed] [Google Scholar]
- Sanman LE, and Bogyo M (2014). Activity-based profiling of proteases. Annu Rev Biochem 83, 249–273. [DOI] [PubMed] [Google Scholar]
- Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, et al. (2017). A comprehensive map of molecular drug targets. Nature Reviews Drug Discovery 16, 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski MM, Reinhard FBM, Franken H, Werner T, Savitski MF, Eberhard D, Molina DM, Jafari R, Dovega RB, Klaeger S, et al. (2014). Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346, 1255784. [DOI] [PubMed] [Google Scholar]
- Saxon E, and Bertozzi C (2000). Cell surface engineering by a modified staudinger reaction. Science (New York, NY) 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- Schreiber S (2018). A chemical biology view of bioactive small molecules and a binder-based approach to connect biology to precision medicines. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SL (2019). A chemical biology view of bioactive small molecules and a binder-based approach to connect biology to precision medicines. Israel Journal of Chemistry 59, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz F, and Aebi M (2011). Mechanisms and principles of n-linked protein glycosylation. Curr Opin Struct Biol 21, 576–582. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, and Grunstein M (2007). Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem 76, 75–100. [DOI] [PubMed] [Google Scholar]
- Shannon DA, Gu C, McLaughlin CJ, Kaiser M, van der Hoorn RAL, and Weerapana E (2012). Sulfonyl fluoride analogues as activity-based probes for serine proteases. ChemBioChem 13, 2327–2330. [DOI] [PubMed] [Google Scholar]
- Sharma K, D’Souza RCJ, Tyanova S, Schaab C, Wiśniewski JR, Cox J, and Mann M (2014). Ultradeep human phosphoproteome reveals a distinct regulatory nature of tyr and ser/thr-based signaling. Cell Rep 8, 1583–1594. [DOI] [PubMed] [Google Scholar]
- Shen DL, Liu T-W, Zandberg W, Clark T, Eskandari R, Alteen MG, Tan HY, Zhu Y, Cecioni S, and Vocadlo D (2017). Catalytic promiscuity of o-glcnac transferase enables unexpected metabolic engineering of cytoplasmic proteins with 2-azido-2-deoxy-glucose. ACS chemical biology 12, 206–213. [DOI] [PubMed] [Google Scholar]
- Shi HB, Zhang CJ, Chen GYJ, and Yao SQ (2012). Cell-based proteome profiling of potential dasatinib targets by use of affinity-based probes. J Am Chem Soc 134, 3001–3014. [DOI] [PubMed] [Google Scholar]
- Sieber SA, Niessen S, Hoover HS, and Cravatt BF (2006). Proteomic profiling of metalloprotease activities with cocktails of active-site probes. Nat Chem Biol 2, 274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon GM, and Cravatt BF (2010). Activity-based proteomics of enzyme superfamilies: Serine hydrolases as a case study. J Biol Chem 285, 11051–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Thornton ER, and Westheimer FH (1962). The photolysis of diazoacetylchymotrypsin. J Biol Chem 237, 3006–3008. [PubMed] [Google Scholar]
- Slack JL, Causey CP, Luo Y, and Thompson PR (2011). Development and use of clickable activity based protein profiling agents for protein arginine deiminase 4. Acs Chemical Biology 6, 466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten EM, and Bertozzi CR (2009). Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew Chem Int Ed Engl 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, and Collins I (2015). Photoaffinity labeling in target- and binding-site identification. Future Medicinal Chemistry 7, 159–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Späte A-K, Bußkamp H, Niederwieser A, Schart VF, Marx A, and Wittmann V (2014). Rapid labeling of metabolically engineered cell-surface glycoconjugates with a carbamate-linked cyclopropene reporter. Bioconj Chem 25, 147–154. [DOI] [PubMed] [Google Scholar]
- Speers AE, Adam GC, and Cravatt BF (2003). Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3+2] cycloaddition. J Am Chem Soc 125, 4686–4687. [DOI] [PubMed] [Google Scholar]
- Speers AE, and Cravatt BF (2004). Profiling enzyme activities in vivo using click chemistry methods. Chem Biol 11, 535–546. [DOI] [PubMed] [Google Scholar]
- Storck EM, Morales-Sanfrutos J, Serwa RA, Panyain N, Lanyon-Hogg T, Tolmachova T, Ventimiglia LN, Martin-Serrano J, Seabra MC, Wojciak-Stothard B, et al. (2019). Dual chemical probes enable quantitative system-wide analysis of protein prenylation and prenylation dynamics. Nature Chemistry 11, 552–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taunton J, Hassig CA, and Schreiber SL (1996). A mammalian histone deacetylase related to the yeast transcriptional regulator rpd3p. Science 272, 408–411. [DOI] [PubMed] [Google Scholar]
- Thinon E, Serwa RA, Broncel M, Brannigan JA, Brassat U, Wright MH, Heal WP, Wilkinson AJ, Mann DJ, and Tate EW (2014). Global profiling of co- and post-translationally n-myristoylated proteomes in human cells. Nat Commun 5, 4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornoe CW, Christensen C, and Meldal M (2002). Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem 67, 3057–3064. [DOI] [PubMed] [Google Scholar]
- Torres CR, and Hart GW (1984). Topography and polypeptide distribution of terminal n-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for o-linked glcnac. The Journal of biological chemistry 259, 3308–3317. [PubMed] [Google Scholar]
- Tsai CS, Yen HY, Lin MI, Tsai TI, Wang SY, Huang WI, Hsu TL, Cheng YS, Fang JM, and Wong CH (2013). Cell-permeable probe for identification and imaging of sialidases. Proc Natl Acad Sci U S A 110, 2466–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully SE, and Cravatt BF (2010). Activity-based probes that target functional subclasses of phospholipases in proteomes. J Am Chem Soc 132, 3264–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttamapinant C, Tangpeerachaikul A, Grecian S, Clarke S, Singh U, Slade P, Gee KR, and Ting AY (2012). Fast, cell-compatible click chemistry with copper-chelating azides for biomolecular labeling. Angew Chem Int Ed Engl 51, 5852–5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Esbroeck ACM, Janssen APA, Cognetta AB 3rd, Ogasawara D, Shpak G, van der Kroeg M, Kantae V, Baggelaar MP, de Vrij FMS, Deng H, et al. (2017). Activity-based protein profiling reveals off-target proteins of the faah inhibitor bia 10-2474. Science 356, 1084–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Geel R, Pruijn GJ, van Delft FL, and Boelens WC (2012). Preventing thiolyne addition improves the specificity of strain-promoted azide-alkyne cycloaddition. Bioconjug Chem 23, 392–398. [DOI] [PubMed] [Google Scholar]
- Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, et al. (2015). Essentials of glycobiology. [PubMed] [Google Scholar]
- Venable JD, Dong MQ, Wohlschlegel J, Dillin A, and Yates JR (2004). Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat Methods 1, 39–45. [DOI] [PubMed] [Google Scholar]
- Vocadlo D, Hang H, Kim E, Hanover J, and Bertozzi C (2003). A chemical approach for identifying o-glcnac-modified proteins in cells. Proceedings of the National Academy of Sciences of the United States of America 100, 9116–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Weerapana E, Blewett MM, and Cravatt BF (2014). A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat Methods 11, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, and Finn MG (2003). Bioconjugation by copper(i)-catalyzed azide-alkyne [3 + 2] cycloaddition. J Am Chem Soc 125, 3192–3193. [DOI] [PubMed] [Google Scholar]
- Wang R, and Luo M (2013). A journey toward bioorthogonal profiling of protein methylation inside living cells. Curr Opin Chem Biol 17, 729–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Yang F, Petyuk VA, Shukla AK, Monroe ME, Gritsenko MA, Rodland KD, Smith RD, Qian WJ, Gong CX, et al. (2017). Quantitative proteomics identifies altered o-glcnacylation of structural, synaptic and memory-associated proteins in alzheimer’s disease. J Pathol 243, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Dix MM, Bianco G, Remsberg JR, Lee HY, Kalocsay M, Gygi SP, Forli S, Vite G, Lawrence RM, et al. (2019). Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs. Nature Chemistry 11, 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CC, Kleinman JI, and Nomura DK (2017). Nhs-esters as versatile reactivity-based probes for mapping proteome-wide ligandable hotspots. Acs Chemical Biology 12, 1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JA, McLellan L, Stockley M, Gibson KR, Whitlock GA, Knights C, Harrigan JA, Jacq X, and Tate EW (2016). Quantitative chemical proteomic profiling of ubiquitin specific proteases in intact cancer cells. ACS Chem Biol 11, 3268–3272. [DOI] [PubMed] [Google Scholar]
- Weerapana E, and Imperiali B (2006). Asparagine-linked protein glycosylation: From eukaryotic to prokaryotic systems. Glycobiology 16, 91R–101R. [DOI] [PubMed] [Google Scholar]
- Weerapana E, Simon GM, and Cravatt BF (2008). Disparate proteome reactivity profiles of carbon electrophiles. Nat Chem Biol 4, 405–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E, Speers AE, and Cravatt BF (2007). Tandem orthogonal proteolysis-activity-based protein profiling (top-abpp) - a general method for mapping sites of probe modification in proteomes. Nature Protocols 2, 1414–1425. [DOI] [PubMed] [Google Scholar]
- Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, and Cravatt BF (2010). Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–U779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L, Gadi MR, Zheng Y, ACS CG, and 2018 (2018). Chemoenzymatic synthesis of unnatural nucleotide sugars for enzymatic bioorthogonal labeling. ACS Catalysis 8, 7659–7666. [Google Scholar]
- Westcott NP, Fernandez JP, Molina H, and Hang HC (2017a). Chemical proteomics reveals adp-ribosylation of small gtpases during oxidative stress. Nat Chem Biol 13, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westcott NP, Fernandez JP, Molina H, and Hang HC (2017b). Chemical proteomics reveals adp-ribosylation of small gtpases during oxidative stress. Nat Chem Biol 13, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitby LR, Obach RS, Simon GM, Hayward MM, and Cravatt BF (2017). Quantitative chemical proteomic profiling of the in vivo targets of reactive drug metabolites. Acs Chemical Biology 12, 2040–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolters DA, Washburn MP, and Yates JR 3rd (2001). An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem 73, 5683–5690. [DOI] [PubMed] [Google Scholar]
- Woo CM, Felix A, Byrd WE, Zuegel DK, Ishihara M, Azadi P, Iavarone AT, Pitteri SJ, and Bertozzi CR (2017). Development of isotag, a chemical glycoproteomics technique for profiling intact n- and o-glycopeptides from whole cell proteomes. Journal of proteome research 16, 1706–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo CM, Lund PJ, Huang AC, Davis MM, Bertozzi CR, and Pitteri SJ (2018). Mapping and quantification of over 2000 o-linked glycopeptides in activated human t cells with isotope-targeted glycoproteomics (isotag). Molecular & cellular proteomics : MCP 17, 764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AT, and Cravatt BF (2007). Chemical proteomic probes for profiling cytochrome p450 activities and drug interactions in vivo. Chem Biol 14, 1043–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright MH, Clough B, Rackham MD, Rangachari K, Brannigan JA, Grainger M, Moss DK, Bottrill AR, Heal WP, Broncel M, et al. (2014). Validation of n-myristoyltransferase as an antimalarial drug target using an integrated chemical biology approach. Nature Chemistry 6, 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright MH, and Sieber SA (2016). Chemical proteomics approaches for identifying the cellular targets of natural products. Natural Product Reports 33, 681–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZL, Person AD, Anderson M, Burroughs B, Tatge T, Khatri K, Zou Y, Wang L, Geders T, Zaia J, et al. (2018). Imaging specific cellular glycan structures using glycosyltransferases via click chemistry. Glycobiology 28, 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Carroll KS, and Liebler DC (2016). The expanding landscape of the thiol redox proteome. Molecular & cellular proteomics : MCP 15, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Gupta V, Carroll KS, and Liebler DC (2014). Site-specific mapping and quantification of protein s-sulphenylation in cells. Nature Communications 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, and Liu K (2015). Activity-based protein profiling: Recent advances in probe development and applications. ChemBioChem 16, 712–724. [DOI] [PubMed] [Google Scholar]
- Yang X, and Qian K (2017). Protein o-glcnacylation: Emerging mechanisms and functions. Nature Reviews Molecular Cell Biology 18, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X-J, and Seto E (2008). Lysine acetylation: Codified crosstalk with other posttranslational modifications. Mol Cell 31, 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y-Y, Grammel M, and Hang HC (2011). Identification of lysine acetyltransferase p300 substrates using 4-pentynoyl-coenzyme a and bioorthogonal proteomics. Bioorganic & medicinal chemistry letters 21, 4976–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YY, Ascano JM, and Hang HC (2010). Bioorthogonal chemical reporters for monitoring protein acetylation. J Am Chem Soc 132, 3640–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount JS, Moltedo B, Yang Y-Y, Charron G, Moran TM, López CB, and Hang HC (2010). Palmitoylome profiling reveals s-palmitoylation–dependent antiviral activity of ifitm3. Nat Chem Biol 6, 610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaro BW, Batt AR, Chuh KN, Navarro MX, and Pratt MR (2017). The small molecule 2-azido-2-deoxy-glucose is a metabolic chemical reporter of o-glcnac modifications in mammalian cells, revealing an unexpected promiscuity of o-glcnac transferase. ACS chemical biology 12, 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaro BW, Vinogradova EV, Lazar DC, Blewett MM, Suciu RM, Takaya J, Studer S, de la Torre JC, Casanova JL, Cravatt BF, et al. (2019). Dimethyl fumarate disrupts human innate immune signaling by targeting the irak4-myd88 complex. J Immunol 202, 2737–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaro BW, Yang Y-Y, Hang HC, and Pratt MR (2011). Chemical reporters for fluorescent detection and identification of o-glcnac-modified proteins reveal glycosylation of the ubiquitin ligase nedd4-1. Proceedings of the National Academy of Sciences of the United States of America 108, 8146–8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Fonslow BR, Shan B, Baek MC, and Yates JR 3rd (2013). Protein analysis by shotgun/bottom-up proteomics. Chem Rev 113, 2343–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZJ, Pedicord VA, Peng T, and Hang HC (2020). Site-specific acylation of a bacterial virulence regulator attenuates infection. Nat Chem Biol 16, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Ouyang X, Wan X, Gajiwala KS, Kath JC, Jones LH, Burlingame AL, and Taunton J (2017). Broad-spectrum kinase profiling in live cells with lysine-targeted sulfonyl fluoride probes. J Am Chem Soc 139, 680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuhl AM, Nolan CE, Brodney MA, Niessen S, Atchison K, Houle C, Karanian DA, Ambroise C, Brulet JW, Beck EM, et al. (2016). Chemoproteomic profiling reveals that cathepsin d off-target activity drives ocular toxicity of beta-secretase inhibitors. Nature Communications 7. [DOI] [PMC free article] [PubMed] [Google Scholar]