

Abstract
The discovery and validation of protein–protein interactions provides a knowledge base that is critical for defining protein networks and how they underpin the biology of the cell. Identification of protein interactions that are highly transient, or sensitive to biochemical disruption, can be very difficult. This challenge has been met by proximity labeling methods which generate reactive species that chemically modify neighboring proteins. The most widely used proximity labeling method is BioID, which features a mutant biotin ligase BirA(Arg118Gly), termed BirA*, fused to a protein of interest. Here, we explore how amino acid substitutions at Arg118 affect the biochemical properties of BirA. We found that relative to wild-type BirA, the Arg118Lys substitution both slightly reduced biotin affinity and increased the release of reactive biotinyl-5′-AMP. BioID using a BirA(Arg118Lys)-Lamin A fusion enabled identification of PCNA as a lamina-proximal protein in HEK293T cells, a finding that was validated by immunofluorescence microscopy. Our data expand on the concept that proximity labeling by BirA fused to proteins of interest can be modulated by amino acid substitutions that affect biotin affinity and the release of biotinyl-5′-AMP.
Keywords: Protein–protein interactions, Proximity ligation, BirA, BioID: biotinylation, Nuclear lamina
1. Introduction
1.1. Enzyme-based methods for identifying protein–protein interactions in cells
The identification of protein–protein interactions (PPIs) in eukaryotic cells is critical for understanding basic protein function as well as the organization and regulation of cellular pathways. A major challenge in this regard is that many protein–protein interactions are transient and/or characterized as low affinity. Widely-used biochemical techniques such as immunoprecipitation and in vitro binding assays, which can provide protein–protein interaction data, rely on interactions that are sufficiently stable or can be reconstituted outside of the environment of the cell. In recent years, methods that provide a snapshot of protein–protein interactions in the cell have been developed. Proximity-dependent labeling techniques utilize enzymes fused to a protein of interest (POI) that can transfer a molecular tag to proteins situated within a suitable labeling radius in the cell [1–3]. The molecular tag facilitates enrichment and identification of the labeled species, the latter usually by mass spectrometry (MS). Proximity-dependent labeling techniques can therefore overcome some of the difficulties with studying transient and/or weak protein–protein interactions in the cell, as well as the challenges associated with dissecting macromolecular complexes since the labeling occurs when cellular architecture is intact (Fig. 1A).
Fig. 1.
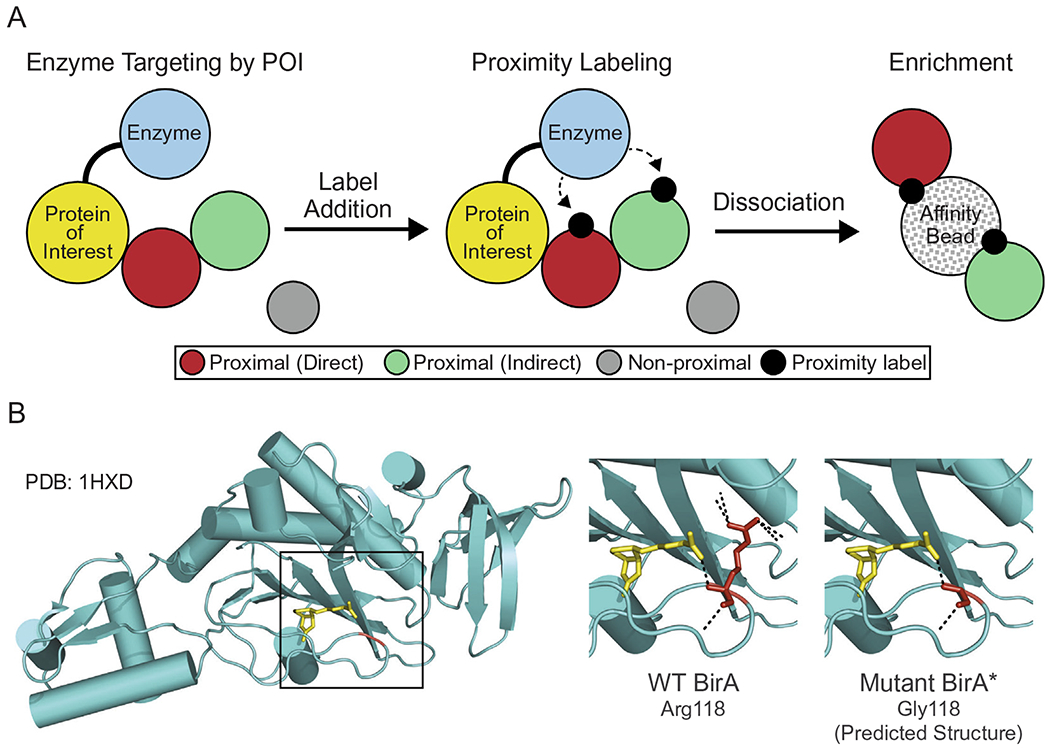
Proximity ligation protein labeling. Overview of enzyme-based proximity labeling. An enzyme capable of generating a reactive intermediate is expressed as a fusion with a protein of interest (yellow) that results in appropriate subcellular targeting. Label addition results in proximal labeling of proteins bound directly (red) and indirectly (green) to the protein of interest. Proteins are subsequently solubilized, and the proximity-modified proteins are enriched on beads by virtue of the label. The proteins labeled in this manner are identified by mass spectrometry. (B) Crystal structure of BirA (PDB: 1HXD) with biotin (yellow) bound to the active site. Residue 118 is highlighted in red with the biotin binding site boxed in black. The panels to the right show the biotin binding site of BirA and the hydrogen bonds (black dotted lines) associated with Arg118 and Gly118. Modeling performed in PyMol. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Most proximity-dependent labeling approaches utilize biotin, or biotin conjugates, as the tag, and streptavidin for detection and enrichment of labeled species. To date, two types of enzymes have been used, peroxidases and biotin-ligases. The peroxidases employed are horseradish peroxidase (HRP) and ascorbate peroxidase (APEX), while the biotin-protein ligase used for proximity labeling is the BirA enzyme from E. coli. HRP and APEX can, in the presence of hydrogen peroxide (H2O2), convert a phenolic substrate into a highly reactive radical. Phenolic substrates such as biotin-arylazides [4], biotin-tyramides [5], and biotin-phenols [6] can be used for proteomic applications, and 3,3′-diaminobenzidine can be used to generate an electron-dense product for visualization by electron microscopy [7]. Radicals released by both HRP and APEX are short lived (< 1 ms), but react with electron-rich amino acids including Tyr, Trp, His and Cys with a predicted labeling radius of 20 nm [8]. HRP-based labeling has been used to identify protein–protein interactions on the cell surface [9], but HRP is inactive in the reducing environment of the cytosol. The development of APEX circumvented this problem. The engineered form of APEX is functional over a broad pH range, and shows enzymatic activity in several cellular compartments of mammalian cells including the mitochondrial matrix, endoplasmic reticulum, cytoplasm, and nucleus [10].
The most widely used proximity-dependent labeling technique utilizes a mutant form of the biotin-protein ligase BirA. In E. coli and other bacteria, BirA regulates the enzyme activity of acetyl-CoA carboxylase through biotinylation of the carboxyl carrier protein (BCCP) subunit [11]. BirA also helps coordinate biotin synthesis by regulating transcription of the biotin biosynthetic operon [11–13]. The mechanism of BirA biotinylation has been studied in detail. Briefly, BirA binds biotin and ATP and generates the intermediate, biotinoyl-5′-AMP (bioAMP) [14]. At low biotin concentrations, bioAMP is transferred to the amine group of a lysine in BCCP. Under conditions of high biotin concentration, where all BCCP subunits are biotinylated, bioAMP remains bound to BirA; in vitro the BirA-bioAMP complex has a half-life of ~ 30 min [15]. In bacteria, the BirA-bioAMP complex binds to a 40-base pair sequence in the biotin biosynthetic operon, resulting in the transcriptional repression of genes encoding enzymes required for biotin biosynthesis [13].
The premise of identifying protein interactions through proximity labeling by BirA is based on structure–function work from Cronan and others. Amino acid substitutions in the biotin-binding site of BirA, notably Arg118Gly, were shown to dramatically increase the bioAMP dissociation rate [16,17]. Cronan suggested bioAMP release could provide the basis for promiscuous chemical biotinylation of lysine residues within proteins in the solvent [18,19]. Proof-of-concept came with the observations that recombinant BirA(Arg118Gly) could mediate biotinylation of purified proteins in vitro, and that multiple proteins in E. coli underwent promiscuous biotinylation in the presence of this mutant enzyme [18]. The BirA(Arg118Gly) mutant was designated BirA*. The Roux laboratory published an elegant implementation of the method in mammalian cells where they showed that BirA fused to Lamin A mediates proximal labeling of proteins within the nuclear lamina. These authors coined the term BioID to describe the technology [20]. When tethered to a nuclear pore complex protein, BirA* was shown to have an effective labeling radius of 10 nm [21]. This is an approximation based on high resolution mapping of nucleoporin positions within the nuclear pore complex. The effective labeling radius is presumably generated by a high local concentration of lysine-reactive bioAMP, but the labeling is likely influenced by multiple factors including how the POI is anchored within a macromolecular structure, the flexibility afforded by BirA* tethering, and the density of solvent-exposed lysine residues in proximity. BirA* fused to a POI can generate compartment-specific targeting and biotinylation of proteins, which could include proteins that bind directly or indirectly to the POI (Fig. 1A). Enrichment of the biotinylated species and MS analysis provides an inventory of proteins labeled by bioAMP released from BirA* in the compartment or structure of interest.
To date, BioID has been used successfully to study a variety of macromolecular structures. Examples include BirA* targeting to the nuclear lamina [20,22,23], the nuclear pore complex [21], cell junctions [24–26], and centrosomes [27–30]. BirA* has also been targeted to membrane-bound compartments such as the mitochondrial matrix [31], and it has also been used in contexts where the POI fusion is not tethered to a cellular structure [32–35].
1.2. Dependence on and limitations of the biotin-avidin interaction
The selective, high affinity interaction between biotin and avidin (and its derivatives; Kd = 10−14M; [36]) is a critical feature of BioID. Enrichment for biotinylated species is specific and tolerates the stringent wash conditions necessary to dissociate unlabeled proteins and contaminating macromolecular structures including cellular membranes. The wash stringency is important because in most published datasets, identification of proteins enriched on streptavidin beads by MS does not reveal a biotin mass addition (+226.08; biotinyl-lysine). In other words, MS typically scores peptides (masses) derived from the protein enriched without identifying the biotin-containing site responsible for enrichment. The fact that biotinylated peptides are not usually detected might reflect (i) the inefficient release of biotinylated peptides from streptavidin; (ii) biotin modification of lysine residues interfering with trypsin recognition of the cleavage site; and (iii) a low stoichiometry of biotin labeling. Thus, the proximity assessment of proteins is usually inferred from counting the number of peptides derived from a protein enriched on streptavidin beads. It is worth noting that one study succeeded in direct identification of biotinylated peptides using a strategy termed Biotin Site Identification Technology (BioSITe) [37]. In this method biotinylated proteins are digested with trypsin prior to enrichment with an immobilized anti-biotin antibody, which has a lower affinity for biotin than streptavidin. This approach helps eliminate non-biotinylated peptides as contaminates from the pipeline, it allows for more efficient elution of biotinylated peptides for MS analysis, and it enables MS detection of the biotin modification on peptides. A similar strategy involves using Strept-Tactin for enrichment, elution, and MS identification of biotinylated peptides [38].
1.3. Refining BirA-based proximity labeling
While BioID with BirA* has been highly successful, there have been efforts to improve the technology. A second-generation technique termed BioID2 was developed by Roux and colleagues based on a lower molecular weight biotin ligase from Aquifex aeolicus [39]. The smaller ligase (26 vs. 35 kD) was shown to improve targeting of protein fusions as well as mediate labeling with a lower biotin concentration in the culture media. Very recently, the Ting laboratory has used yeast display-based directed evolution to generate two BirA mutants, TurboID and miniTurbo, which rapidly release bioAMP and can be used for proximity dependent biotinylation in as little as 10 min in mammalian cells [40].
We took a highly focused approach that involved generating amino acid substitutions in Arg118 in the biotin-binding site of BirA. We focused on Arg118 in BirA, which in the crystal structure (PDB: 1HXD) is positioned to make polar contacts with the carboxyl group of biotin (yellow) and with Arg121 in the biotin binding loop (Fig. 1B; [16]). The Gly substitution for Arg118 in BirA* likely alters the loop position since this reduces biotin affinity by 200-fold [17]. We reasoned that other amino acid substitutions at this position might alter bioAMP dissociation (positively or negatively) and impact biotinylation. We found that BirA(Arg118Lys) can mediate proximity labeling in cells under low biotin concentrations, and in vitro, can be inactivated by automodification at a high biotin concentration.
2. Materials and methods
2.1. Expression, purification, and assay of BirA
BirA proteins were expressed and purified as Maltose Binding Protein (MBP) fusions. The MBP tag was selected as the fusion partner because it affords high levels of expression and solubility in E. coli, and it contains an abundance of lysine residues (33) that could potentially serve as promiscuous biotinylation sites. The BirA coding region was cloned into the pMal-p2x vector (NEB) and site-directed mutagenesis (QuickChange II; Agilent Technologies #200523) was performed to generate plasmids encoding 19 different amino acids at position 118 (primer sequences in Table S1).
BirA proteins were expressed in BL21 E. coli in LB broth and purified with amylose resin (NEB; E8021S). In brief, MBP-BirA plasmids were transformed into chemically competent BL21 E. coli (NEB; C2530), plated on LB-ampicillin plates, and incubated at 37 °C for 16 h. Starter cultures were made by scraping ~ 10 colonies into LB medium (25 ml) containing ampicillin (100 μg/ml) and shaken at 37 °C for 2h. The starter culture was then added to LB amp (1 L) with 0.2% glucose and shaken at 37 °C to an OD600 ~ 0.6. Cultures were cooled on ice for 10 min, induced with IPTG (1 mM), and shaken at 18 °C for 18 h. Prior to harvest, the cultures were supplemented with PMSF (1 mM) and shaken briefly. Cultures were collected by centrifugation (4300g for 15 min at 4 °C). Pellets were resuspended in 12.5 ml of amylose column buffer (20 mM Tris pH 8, 200 mM NaCl, 1 mM EDTA, 1 mM DTT, and 1 μg/ml each of leupeptin, pepstatin, and aprotinin). Cells were lysed by passage through a French press (3X). After clarification by centrifugation (30,000g for 30 min at 4 °C), the soluble extract (~ 15 ml) was combined with amylose resin (1 ml packed beads) in a chromatography column (27 ml; Kimble, 420400-1515). The column was rotated end-over-end (4 °C, 2 h), the resin allowed to settle, and flow-through was collected. Beads were then washed with amylose column buffer (50 ml), and MBP fusions subsequently eluted with 10 mM maltose in amylose column buffer. Peak protein fractions were determined by A280 measurement, and fractions were pooled and dialyzed into PBS containing DTT (1 mM). The dialyzed MBP-BirA proteins were then supplemented with protease inhibitors (1 μg/ml each of leupeptin, pepstatin, and aprotinin, 1 mM PMSF). The purified proteins (0.4–2 mg/ml, depending on the mutant) were dispensed into small aliquots, snap frozen in liquid nitrogen, and stored at −80 °C. Most experiments were performed using single-use aliquots, though there was no apparent loss of activity when BirA proteins were stored on ice for several days.
It has been previously shown that BirA* undergoes auto-biotinylation in E. coli [18]. This provides a simple readout for activity of BirA mutants. To determine the auto-modification state, each BirA protein (1 μg) was resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and detected with fluorescent-neutravidin (fl-neutra; ThermoFisher, 22853) by LI-COR Odyssey Clx infrared imager. A duplicate gel was stained with Coomassie Blue to ensure equal loading of the BirA proteins (Fig. 2A). From this analysis, we learned that different amino acid substitutions for Arg at position 118 can modestly increase or decrease biotin conjugation to MBP-BirA when expressed in E. coli.
Fig. 2.
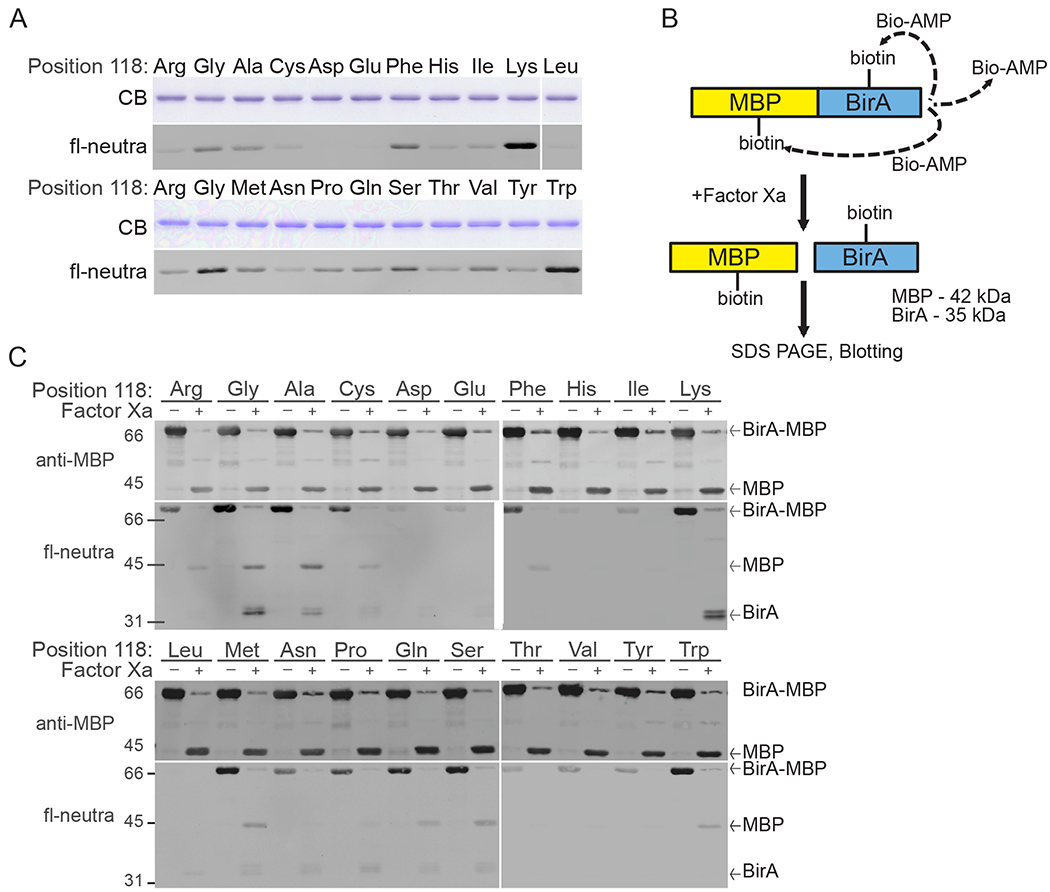
Evaluation of auto-biotinylation by mutants of BirA at position 118. (A) Purification and biotinylation state (fl-neutra detection) of wild-type and mutant BirA proteins expressed as MBP fusions in E. coli. Each mutant protein contains a single amino acid substitution at position 118, as indicated. Equal amounts (1 μg) of each MBP-BirA mutant were analyzed by Coomassie blue staining and by blotting. (B) Scheme for Factor Xa mediated cleavage and analysis of auto-biotinylation. (C) Biotin detection within the MBP and BirA catalytic fragments of each BirA mutant (2.5 μg) before and after cleavage with Factor Xa. Blots were also probed with anti-MBP antibody. Bands corresponding to MBP-BirA, MBP, and BirA are indicated to the right of each panel. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The biotinylation signal from the MBP-BirA fusion protein could reflect BirA auto-modification, or BirA labeling of MBP. BirA and MBP contain 18 and 33 Lys residues, respectively, any of which could be reactive with bioAMP. To distinguish between BirA auto-modification and MBP biotinylation, we subjected the recombinant fusion proteins to proteolytic cleavage using the Factor Xa cleavage site positioned between MBP and BirA. Following cleavage, the biotinylation state was assessed by probing with fl-neutra (Fig. 2B).
For cleavage reactions, MBP-BirA proteins (5 μg) were each dialyzed into Factor Xa cleavage buffer (20 mM Tris-HCl pH 8, 100 mM NaCl, 2mM CaCl2). The dialyzed proteins were then incubated with Factor Xa (0.5 μg; NEB; P8010S) at room temperature for 16 h on a benchtop shaker. Following this reaction, cleaved (2.5 μg) and un-cleaved (2.5 μg) mutant enzyme was separated on an 8% SDS-PAGE gel, transferred to nitrocellulose membrane and detected with fl-neutra and anti-MBP antibody (NEB; E8032S) with anti-mouse AlexaFluor680 secondary antibody (ThermoFisher, A28183) (Fig. 2C). We found differences in the relative levels of labeling of MBP and BirA that were dependent on the amino acid substitution. BirA(WT) displayed a very low level of biotinylation that was restricted to MBP. BirA* (Arg118Gly substitution) showed labeling on both MBP and BirA, but was biased towards modification of BirA itself. We observed that Arg118Gly and Arg118Lys showed the strongest level of automodification (Fig. 2C), and in both mutants BirA appeared as a doublet with slightly higher labeling of the faster migrating species. We also noted the Arg118Trp mutant biotinylated MBP but this occurred without obvious automodification. Based on these data, we set out to study the Arg118Lys and Arg118Trp mutants. We subsequently determined from transfection experiments that Lamin A-BirA (Arg118Trp) mutant has very low biotinylation activity in cells even with biotin supplementation (unpublished observations). These data led us to a comparative analysis of BirA(Arg118Lys), BirA(Arg118Gly) and BirA(WT).
2.2. Bead-based biotinylation assay with BirA
We designed a simple bead-based assay with the goal of testing whether BirA tethered to a surface could mediate promiscuous biotinylation of a substrate in trans (Fig. 3A). MBP-BirA fusion proteins (WT, Arg118Gly, and Arg118Lys; 5 μg each) and MBP (10 μg, corresponding to a 4-fold molar excess over MBP-BirA) were immobilized on amylose resin (10 μl packed beds) by rotation (4 h, 4 °C). We also immobilized MBP alone (10 μg) in the absence of BirA, which serves as a blotting control. Beads were then washed four times with in vitro biotinylation buffer (IVBB) (50 mM Tris pH8, 5 mM MgCl2, 1 mM DTT, 1 μg/ml each aprotinin, leupeptin, and pepstatin) and resuspended with IVBB (20 μl) and placed on ice. ATP (3 mM final) and biotin (10 μM) were added, and the reaction was supplemented with IVBB (50 μl total volume). Samples were then incubated at 37 °C for 30 min. The reactions were chilled briefly on ice, and then centrifuged to pellet the beads. The supernatant was aspirated, beads were washed four times with IVBB (300 μl), and 1X SDS-PAGE sample buffer (50 μl) was added to each tube. The sample (20%) was analyzed by SDS-PAGE and immunoblotting with anti-MBP antibody to confirm equivalent loading, and fl-neutra was used to detect biotin labeling (Fig. 3B). MBP and fl-neutra signals were quantified using Image Studio (LI-COR) or ImageJ software. Biotinylation was plotted by normalizing to the level of each protein (MBP signal) for MBP-BirA (cis-biotinylation) and MBP (trans-biotinylation) (Fig. 3C). The level of trans-biotinylation by the BirA(Arg118Lys) mutant was higher than that of BirA*. Surprisingly, BirA(WT) was also capable of labeling MBP in trans. This suggests that even the WT enzyme is capable of releasing sufficient bioAMP for biotinylation of a proximal protein. It should be noted that non-enzymatic (BirA-independent) biotinylation of MBP is not observed in this assay.
Fig. 3.
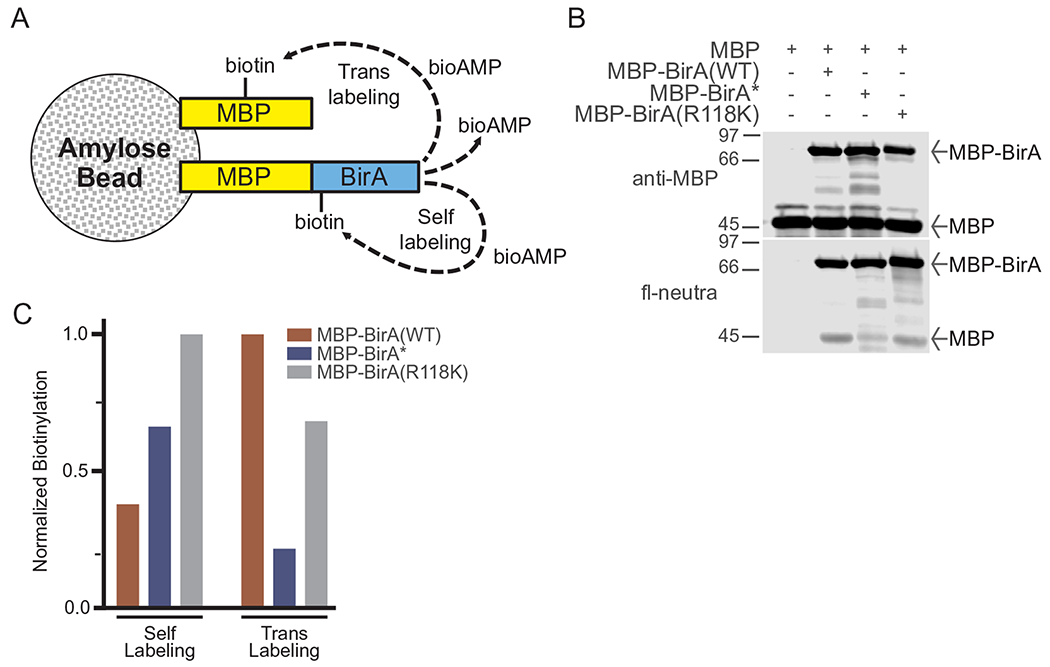
Bead-based analysis of proximal biotinylation by BirA mutants. (A) Scheme for proximal (self and trans) biotinylation by BirA immobilized on an amylose bead surface. (B) Biotinylation reactions were performed with MBP-BirA mutants (5 μg) and free MPB (10 μg) on amylose beads in the presence of biotin (50 μM). Samples were analyzed by fl-neutra. Normalized biotinylation of self and trans labeling was quantified and plotted in (C).
2.3. BirA activity measured using biotin acceptor peptides
To further characterize the biotin ligase activity of the BirA(Arg118Lys) mutant, we examined its ability to label a biotin acceptor peptide (BAP). We used a 23 amino acid BAP isolated by phage display [41] and expressed it as a GST fusion protein. The single lysine embedded in the BAP sequence (Fig. 4A, bold underlined) serves as the biotin acceptor in a reaction that requires addition of ATP and biotin. For controls, the acceptor lysine was changed to an alanine (K > A) or arginine (K > R), the latter to retain the positive charge (Fig. 4A). For the TLC experiments described below, synthetic peptides corresponding to the WT, Lys to Ala, and Lys to Arg sequences (Fig. 4A) were purchased (GenScript). Peptides were resuspended in water (1.5 mM) and frozen (−80 °C).
Fig. 4.
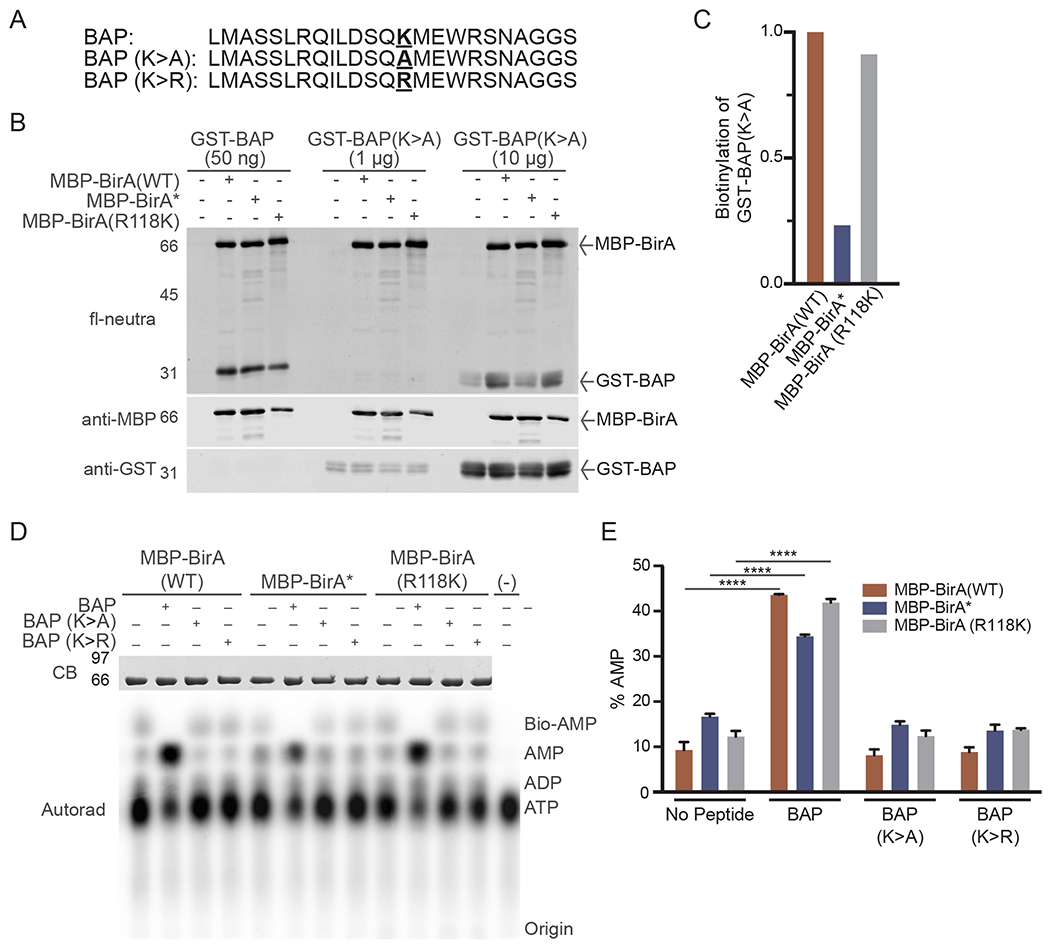
Utilization of Biotin Acceptor Peptide (BAPs) for analyzing BirA mutants. (A) BAP sequences showing the position of the acceptor lysine and amino acid substitutions (bold underline). (B) In vitro biotinylation of BAP substrates with purified BirA enzymes. Reactions contained MBP-BirA (1 μg) with the indicated amount of each BAP and biotin (50 μM). (C) Quantification of relative biotinylation of GST-BAP (K- > A) in (B) indicates proximity labeling of a site outside of the BAP acceptor lysine. (D) TLC analysis of basal and BAP-induced bioAMP and AMP generation by BirA proteins. Assays were performed in triplicate and analyzed by autoradiography, one set of which is shown in this panel. (E) Quantification of AMP generation by BirA mutants. The % AMP is the percentage of signal in each lane corresponding to [α-32P]-labeled AMP. (****p < 0.0001).
BAP amino acid sequences (WT, Lys to Ala) were reverse translated and codon optimized for expression in E. coli. Single strand oligonucleotides were purchased with overhangs to permit direct cloning into the pGEX-4 T1 plasmid after restriction digest with EcoRI and XhoI. Thus, the sense strand contained a 5′ overhang (5′-AATT), and the antisense strand contained a 5′ overhang (5′-TCGA) (Table S2). The pGEX-4 T1 plasmids were transformed into chemically competent BL21 E. coli, plated on LB-ampicillin plates, and grown at 37 °C for 16 h. Because E. coli contain endogenous BirA that biotinylates the GST-BAP substrate during growth, cultures are maintained in M9 minimal medium lacking biotin. M9 minimal medium is prepared as follows: (i) A 5X M9 minimal salts solution is prepared by dissolving NaHPO4 (30 g), KH2PO4 (15 g), NaCl (2.5 g) and NH4C1 (5 g) in H2O (1 L) followed by sterile filtration, (ii) Separately, glucose (4 g) is added to H2O (750 ml) and autoclaved, (iii) MgSO4 (1 mM final), CaCl2 (1 mM final) and 5X M9 salts (200 ml) are added to the glucose solution with sterile water for a final volume of 1 L.
Starter cultures were made by scraping ~ 10 colonies into M9 minimal medium (25 ml) containing ampicillin (100 μg/ml). The starter culture was shaken at 37 °C for 2 h, and then added to the growth culture in the same medium (1 L) and shaken at 37 °C until the OD600 reaches 0.6. The growth culture was induced with IPTG (1 mM) and EtOH (2%) and shaken for 3 h at 37 °C. Just prior to harvest, PMSF (1 mM) was added to the culture. The culture was collected by centrifugation (4300g for 15 min at 4 °C). The pellets were resuspended in PBS with DTT (1 mM), with leupeptin, pepstatin, and aprotinin (1 μg/ml each) and lysed by passing through a French press 3 times. The lysate was clarified by centrifugation (30,000g 30 min at 4 °C). Triton X-100 (using a 20% stock) was added to the soluble fraction to a final concentration of 1%, and the sample (12.5 ml) was combined with glutathione agarose (1 ml packed beads; Sigma; G4510). The sample was mixed by end-over-end rotation in a chromatography column (27 ml) at 4 °C (2 h). The beads were allowed to settle, the column flow-through was collected, and beads were washed sequentially with the following buffers: (1) PBS with 1% Triton X-100 (25 ml), (2) PBS with 500 mM NaCl (25 ml) and (3) PBS (25 ml). GST-BAP is eluted in glutathione (10 mM), 50 mM Tris, pH 8 (50 mM), DTT (1 mM), with leupeptin, pepstatin, and aprotinin (1 μg/ml each). The fractions were measured by A280, and peak fractions were selected, pooled, and dialyzed into PBS containing DTT (1 mM). Protease inhibitors (leupeptin, pepstatin, and aprotinin; 1 μg/ml each) were added after dialysis and proteins were aliquoted and stored at −80 °C.
2.4. BirA substrate recognition and promiscuity analyzed using BAPs
The purified GST-BBP fusions are used to examine the biotin acceptor site-specific and potentially promiscuous biotinylation by BirA proteins. In this experiment, the GST-BAP mutant (K > A) serves as a substrate for promiscuous biotinylation by BirA because the single lysine in the 23 amino acid BAP is changed to a non-biotinylated amino acid, alanine. Thus, any biotinylation observed with GST-BAP mutant (K > A), occurs as a result of promiscuous labeling on one or more of the 21 lysine residues in GST.
For each reaction, an MBP-BirA fusion (1 μg) was combined with either GST-BAP (50 ng), GST-BAP(K > A) (1 μg), or GST-BAP(K > A) (10 μg). Samples were prepared in a total volume of 50 μl in IVBB with ATP (3 mM) and biotin (50 μM) on ice. Reactions were incubated at 37 °C for 1 h and stopped with the addition of 5X SDS-PAGE sample buffer (10 ul). Samples (20% of total reaction) were analyzed by SDS-PAGE and immunoblotting with anti-MBP and anti-GST (Santa Cruz; sc-138) antibodies along with fl-neutra to detect biotinylation (Fig. 4B). Quantification was performed using Image Studio or ImageJ software where fl-neutra signal from samples with 10 μg of GST-BAP(K > A) were quantified by subtracting signal from the negative control and normalizing to each BirA enzyme (Fig. 4C). The results from this experiment indicated that all three forms of BirA biotinylate the WT BAP substrate when it is used at a low concentration (50 ng). Virtually no labeling of the mutant BAP(K > A) was detected when it is used at 20-fold higher concentration than the WT BAP. However, the mutant BAP did show a low level of labeling when used at the highest concentration (10 ug) by BirA(WT) and BirA(R118K). These data indicated that for this particular substrate, the WT and Arg118Lys mutant BirA mediate a higher level of promiscuous labeling of a mutant BAP substrate than the Arg118Gly mutant, BirA*.
To better characterize biotinylation mediated by the Arg118Gly and Arg118Lys mutants, we utilized thin layer chromatography (TLC) to monitor the chemical products of the reaction [42]. The ATP- and biotin-dependent generation of bioAMP by BirA is followed by AMP release and biotin conjugation to BAP. The use of [α-32P]-labeled ATP in biotinylation reactions with TLC enables the visualization of ATP, ADP, AMP, and bioAMP by autoradiography (autorad) because of the different mobilities of these chemical species. In these reactions, synthetic peptides encoding WT and mutant BAPs are added and the relative level of AMP production are determined in order to assess how well different BirA enzymes respond to substrate.
Biotinylation reactions for analysis by TLC were performed in a modified IVBB containing Tris, pH 8 (5 mM), TCEP (0.5 mM), MgCl2 (0.5 mM), KCl (10 mM), ATP (50 μM), and [α-32P] ATP (82.5 nM; Perkin Elmer). Each reaction (15 ul total) contained MBP-BirA (2 μM), BAP peptide (50 μM), and biotin (50 μM). Reactions were assembled on ice and incubated at 37 °C for 1 h. In duplicate, 2 μl of each reaction was spotted with 1 cm spacing exactly 2 cm from the base of a microcrystalline cellulose matrix plate (20 cm × 20 cm; Analtech, 05011) and placed in a chamber with ~1 cm depth of an isobutryic acid:NH4OH:water (66:1:33 by volume) solution. The buffer front was allowed to migrate ~15 cm (three quarters of the plate height) which typically takes between 1 and 2 h. The plate was removed from the chamber and allowed to air dry. The plate was then covered with clear plastic wrap and imaged by exposure to X-ray film and/or a Phosphor Imager (Molecular Dynamics). One-half of each reaction (7.5 μl) was also analyzed by SDS-PAGE and Coomassie blue staining to confirm equal loadings (Fig. 4D). Reactions were spotted on duplicate plates as technical replicates. Utilizing the plot lanes function in ImageJ, the signal from ATP, ADP, AMP, and bioAMP was quantified. The total signal in each lane was calculated and the percentage of signal corresponding to AMP was plotted (Fig. 4E). Percent AMP is a measure of turn-over of bioAMP to biotin and AMP in the active site of BirA. It was observed that all three BirA enzymes respond to WT BAP by generating AMP. BirA showed a slightly higher level of AMP production than BirA(WT) in the absence of BAP, which is to be expected since the fast off-rate of bioAMP from this mutant would be followed by a low level of spontaneous bioAMP hydrolysis. The BirA(R118K) mutant is virtually identical to BirA(WT) in terms of AMP production in response to WT BAP peptide.
2.5. Biotin affinity and bioAMP turnover of BirA analyzed with thin layer chromatography
To estimate the affinities of BirA proteins for biotin, we used TLC to measure the ratio of AMP to bioAMP as a function of biotin concentration. Because this reaction is performed in the absence of BAP, AMP generation reflects the spontaneous hydrolysis of bioAMP. Mutations in the biotin-binding loop of BirA, notably Arg118Gly, reduce the affinity for bioAMP and result in its dissociation from the enzyme active site [17]. This assay can be considered a partial reaction compared to what occurs in cells, where biotin would be added to a lysyl group on a proximal protein.
To analyze the affinity of BirA mutants for both biotin and bioAMP, we again utilized [α-32P]-labeled ATP, TLC, and autoradiography. Reactions were prepared and carried out in the same manner as the previously described TLC experiment (Section 2.4); however, the concentration of biotin in each reaction ranged from 0 to 250 μM. Each reaction (15 μl) was assembled on ice and contained MBP-BirA (2 μM). Reactions were incubated at 37 °C for 1 h. In duplicate, 2 μl of each reaction was spotted onto TLC plates as described above and analyzed by autoradiography and/or Phosphor Imager (Fig. 5A,C,E). Total signal in each lane was quantified using the plot lanes tool in ImageJ and the data plotted as a ratio of AMP to bioAMP (Fig. 5G). The AMP/bioAMP ratio reflects the basal level of dissociation of bioAMP from BirA and the subsequent hydrolysis of the biotinoyl-AMP linkage prior to TLC. BirA(WT) has an AMP/bioAMP ratio < 1 regardless of biotin concentration indicating minimal precocious release of bioAMP. At high biotin concentrations (> 50 μM), BirA* readily released bioAMP as indicated by the higher AMP/bioAMP ratio. BirA(Arg118Lys) demonstrated an AMP/bioAMP ratio greater than BirA(WT) (indicating increased precocious bioAMP release) and less than BirA*.
Fig. 5.
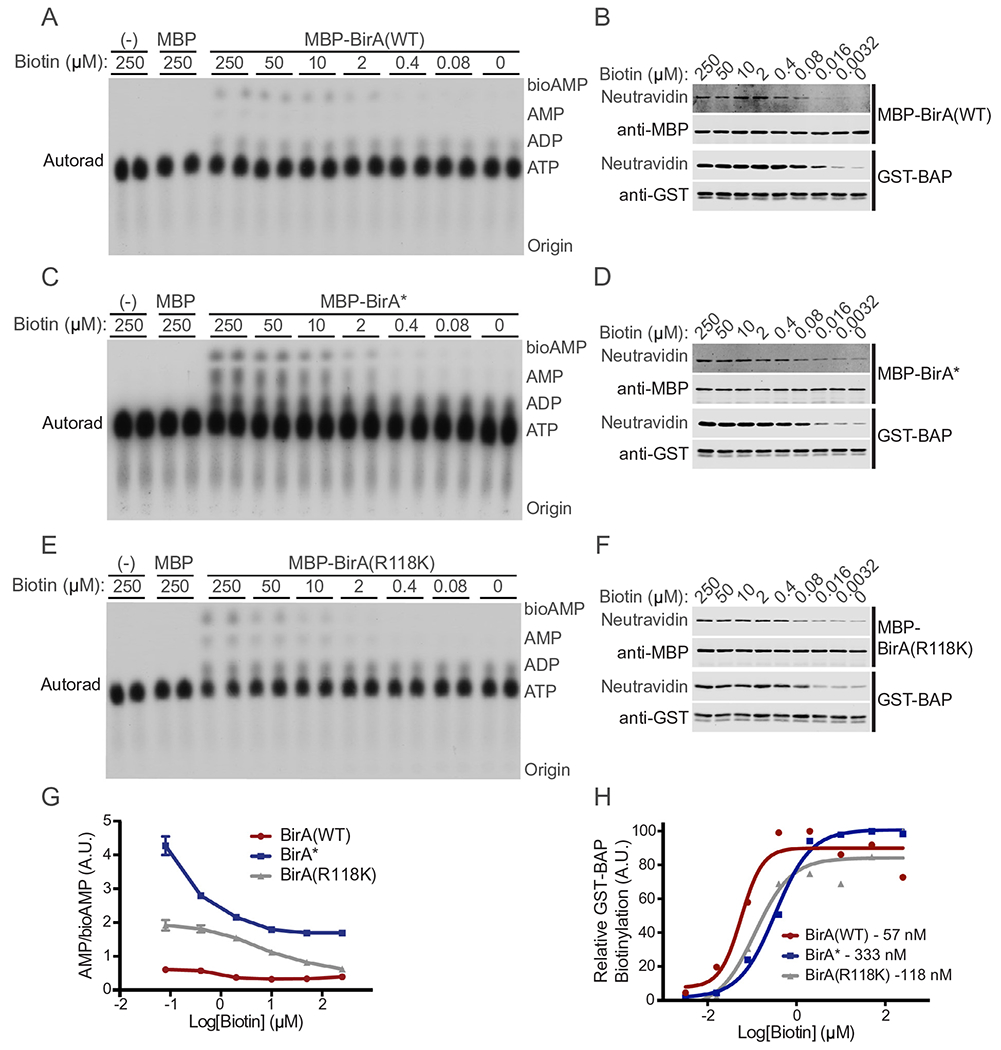
Evaluation of BirA biotin affinity and turn-over rate. TLC analysis of [α-32P]-labeled bioAMP and AMP generation as a function of biotin concentration by (A) BirA, (B) BirA*, and (C) BirA(R118K). Signal for each [α-32P]-labeled species was quantified using ImageJ and the data plotted as the ratio of AMP/bioAMP, a measure of the promiscuous release and hydrolysis of bioAMP to AMP in (G). (B, D, F) Biotinylation of BAP measured as a function of biotin concentration using fl-neutra. (H) BAP biotinylation is normalized to GST signal and plotted as a function of biotin concentration. Apparent KM values calculated in GraphPad Prism are shown.
2.6. Estimation of biotin affinity for BirA enzymes
To determine the approximate KM of each BirA enzyme for biotin we utilize the GST-BAP substrate. Reactions were prepared on ice in IVBB. Each reaction contained MBP-BirA (50 ng), GST-BAP (1 μg), and ATP (3 mM). Biotin was added to the desired concentration in a 30 μl total reaction volume before placing the sample at 37 °C. Each tube was incubated for exactly 20 min and then stopped by adding 5X SDS-PAGE sample buffer (6 μl). Half of each reaction was analyzed by SDS-PAGE and immunoblotted with anti-MBP and anti-GST antibodies as well as fl-neutra to detect biotinylation (Fig. 5B,D,F). Biotinylation of GST-BAP was normalized for protein loading and plotted on a log scale in GraphPad Prism. A non-linear regression was performed, and a sigmoidal fit applied to the data. From this analysis, approximate KM values were generated (BirA(WT), 57 nM; BirA(R118K), 118 nM; BirA*, 333 nM; Fig. 5H). These data indicate that BirA(R118K) has a lower affinity for biotin than BirA(WT), but a higher affinity than BirA*.
2.7. BirA biotinylation visualized by confocal microscopy
The primary application of the characterized BirA mutants is proximity-dependent labeling in cells based on a fusion to a bait protein. For this analysis it is important to confirm that BirA fusion to the bait protein does not interfere with proper localization. We next explored the utility of BirA(R118K) for proximity labeling in cells using Lamin A as the fusion partner. We selected Lamin A for this purpose since BirA*-Lamin A was used in the original BioID method [20]. The coding region for human Lamin A was PCR amplified and cloned into the pcDNA3.1 (–) backbone using XhoI and AflII restriction sites. Subsequently, the gene encoding BirA(WT) was PCR amplified and cloned into the pcDNA-Lamin A construct using NheI and XhoI restriction sites. This creates an N-terminal BirA-Lamin A fusion. Finally, site-directed mutagenesis was performed to introduce the desired mutations at position 118, in this case R118G and R118K. For cell-based experiments, HEK293T cells were cultured in DMEM/F12 (Gibco; 11320082) supplemented with FBS (5%; Atlanta Biologicals, S11150), Sodium Pyruvate (1%; Gibco, 11360070), MEM Non-Essential Amino Acids (1%; Gibco, 11140076), and Penicillin/Streptomycin (1%; Gibco, 15070063). Transfections were performed using the Fugene6 transfection reagent (Promega, E2691).
Prior to transfection, HEK293T cells were seeded on sterile glass coverslips in 6-well plates (2.0 × 105 per well). The next day, cells (~ 40% confluency) were transfected with BirA-Lamin A fusion plasmids in duplicate wells. The medium for one coverslip of each transfection was supplemented with biotin (50 μM). DMEM/F12 medium contains 14 nM biotin while FBS addition to the medium may contribute up to 10 nM biotin [43]. This means that medium without biotin supplementation contains a low nanomolar concentration of biotin. After transfection, cells were grown for 24 h, and then fixed with formaldehyde (3.75%) in PBS for 10 min. Coverslips were washed 3x in PBS, permeabilized with Triton X-100 (0.2%) for 5 min and washed again with PBS (3x). Blocking was performed with BSA and FBS (2% each) in PBS for 1 h. Cells were then stained with anti-Lamin A antibody (Sigma, L1293) for 1 h, washed in PBS, and incubated with Streptavidin488 (Invitrogen; S11223) and Cy3 anti-rabbit secondary antibody (Jackson ImmunoReseach, 115–165-144) for 1 h. Coverslips were washed in PBS (3x) and stained with DAPI and mounted on slides using VectaShield (Vector Laboratories; H-1000).
Coverslips were imaged on a Zeiss LSM-880 confocal microscope. The Lamin A signal was used to focus and acquire a slice (1 μm thick) through the “middle” of individual nuclei. Image analysis was performed using Zeiss Zen software. Individual nuclei from different experimental conditions but with similar intensities of Lamin A signal were chosen for analysis (Fig. S1). Traces (indicated by white lines) were drawn across each nucleus and the fluorescence intensity of Lamin A and streptavidin signal was measured and plotted as a function of distance (Fig. 6A). Measuring the fluorescence intensities generated from detection of biotin and BirA-Lamin A permits a semi-quantitative comparison of the extent of coincidence. It should be noted that while most Lamin A is stably localized at the nuclear periphery in association with the nuclear lamina, there is also a nucleoplasmic pool of Lamin A. Additionally, since the nuclear lamina is disassembled during mitosis, soluble BirA-Lamin A fusion proteins would have the opportunity to biotinylate non-lamina proteins during part of the cell cycle.
Fig. 6.
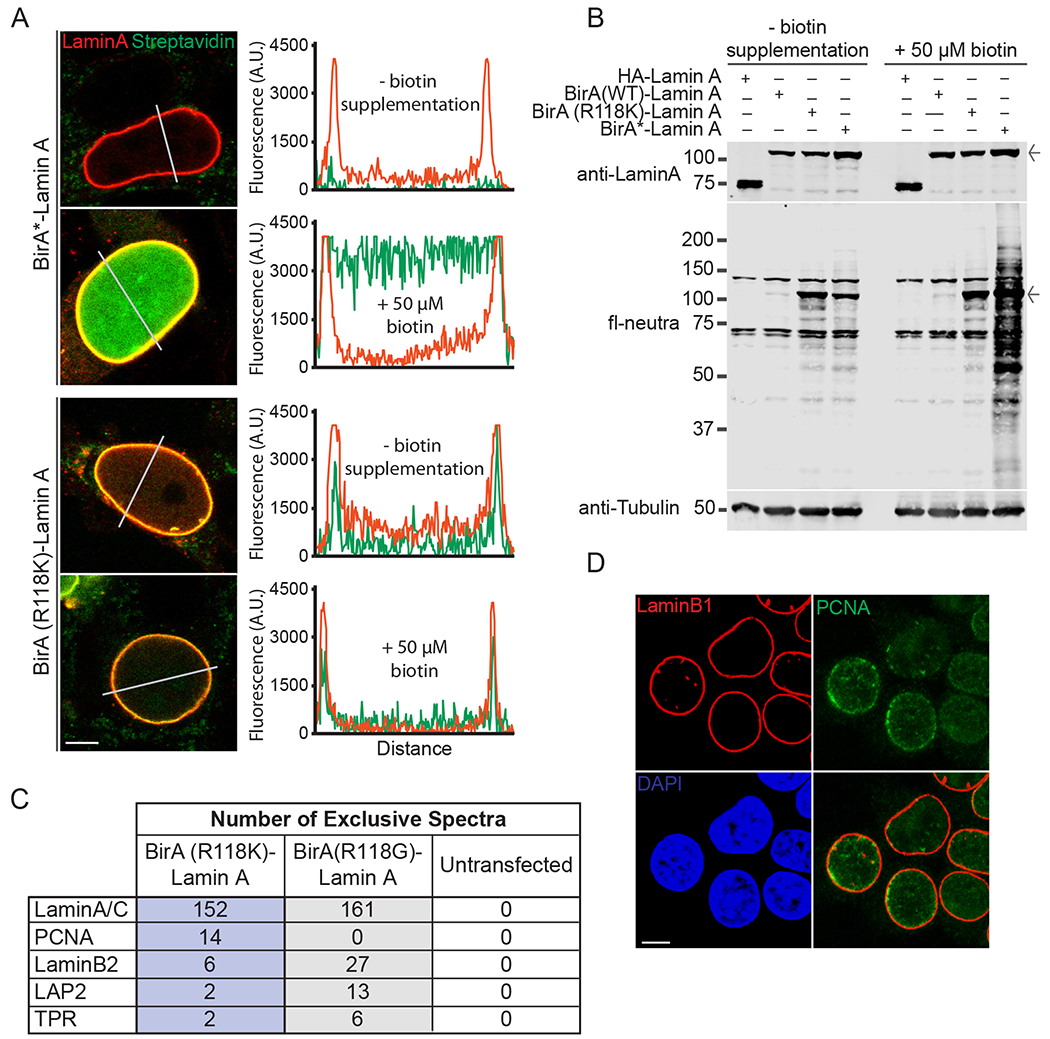
Cell-based experiments with BirA-Lamin A fusions. (A) Confocal microscopy showing the localization (red) and biotinylation (green) of BirA*-Lamin A and BirA(R118K)-Lamin A. HEK293T cells grown in the absence and presence of exogenous biotin (50 μM). Fluorescence intensities were measured across nuclei (white lines) and quantified (middle panels) (Scale bar 5 μm) (B) Whole cell lysate from HEK293T cells transfected with HA-Lamin A or BirA-Lamin A fusions with and without biotin supplementation (50 μM). Immunoblot of fl-neutra detection (arrow denotes BirA-Lamin A band) (C) Mass spectrometry results obtained from streptavidin pulldown of HEK293T lysate from cells expressing Lamin A-BirA(R118K) and Lamin A-BirA* in the absence of additional biotin supplementation. Shown are selected peptides derived from proteins known to be proximal to Lamin A in cells. A complete list of peptides from this analysis is provided (Table S3). (D) Localization of endogenous PCNA to the nuclear lamina by confocal fluorescence microscopy (Scale bar 5 μm). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Representative images of nuclei from transfected cells were obtained by confocal microscopy (Fig. 6A). The BirA(R118K)-Lamin A and BirA*-Lamin A fusions, revealed by staining for Lamin A (red), showed proper localization to the nuclear lamina. Both fusions displayed a low nucleoplasmic signal. Without biotin supplementation, cells expressing BirA*-Lamin A showed a low level of biotinylation by microscopy and blotting, as shown by Roux and co-workers [20]. Biotinylation by BirA*-Lamin A was greatly enhanced by biotin addition (Fig. 6A, B). BirA(R118K)-Lamin A showed similar levels of biotinylation under both culture conditions (Fig. 6A, B).
2.8. Identification of lamina-proximal proteins in cells
The next step was to test if the new BirA mutant, BirA(R118K), is capable of proximity labeling in cells. We used BirA(R118K)-Lamin A together with BirA*-Lamin A and untransfected cells as controls. The cell culture component of the experiment was performed without biotin supplemention since the KM of biotin for the R118K mutant is only two-fold greater than BirA(WT). We acknowledge that this condition might limit the number of protein partners labeled by BirA*-Lamin A given its reduced affinity for biotin [17]. BirA*-Lamin A can be viewed as an additional control for the biotin-selective enrichment of proximally labeled proteins. In other words, proteins identified by MS by virtue of enrichment on streptavidin beads in the BirA(R118K)-Lamin A sample cannot be explained by Lamin A binding interactions that are resistant to the stringent buffer conditions if they are not similarly enriched in the BirA*-Lamin A sample.
Plasmids encoding the BirA-Lamin A fusion proteins were each transfected into two 10 cm dishes of HEK293T cells using the Fugene6 transfection reagent. After 24 h, plates were washed with ice cold TBS and scraped into 1.5 ml cold TBS with PMSF (1 mM). Duplicate plates were combined and pelleted (800 × g, 5 min, 4 °C). The supernatant was aspirated, and pellets were resuspended in 500 μl lysis buffer (20 mM Tris pH 7.6, 0.65 M NaCl, 0.5% Triton X-100, 0.03% SDS, 2.5 mM EDTA, 2 mM DTT, 1 μM PMSF, 5 μg/mL aprotinin, and 5 μg/ml leupeptin/pepstatin). Tubes were incubated with agitation (4 °C for 20 min) followed by tip sonication (10 pulses at 0.4 s/pulse; Branson Sonifier 250). Lysates were clarified by centrifugation (16,000 × g, 20 min, 4 °C). For each condition, 5 μl (1%) was saved as input for the streptavidin pull-down. The lysate was added to streptavidin agarose (150 μl packed; GE Healthcare, 17-5113-01) in a siliconized 1.5 ml tube and incubated with end-over-end rotation (4 °C, 4 h). After incubation, the supernatant was collected, and beads were washed five times with 500 μl wash buffer (20 mM Tris pH 7.6, 0.25 M NaCl, 0.5% Triton X-100, 0.02% SDS, 1 mM EDTA, 2 mM DTT, 2 μg/mL aprotinin, and 2 μg/ml leupeptin/pepstatin) followed by TBS (500 μl, 5 times). For analysis by SDS-PAGE 1.5 μl (1%) of each sample was removed as input prior to further processing.
Samples, for MS were reduced with DTT (10 mM, 1 h, RT) followed by alkylation with iodoacetamide (50 mM, 1 h, RT). Samples were then digested overnight with alkylated trypsin (1 μg; Promega, V5111). The digestion was quenched with 3 μl acetic acid. Samples were clarified by centrifugation prior to loading of the extract (45%) on the liquid chromatography mass spectrometry system (LC-MS). The LC-MS system consisted of a Thermo Electron Velos Orbitrap ETD mass spectrometer system with a Protana nanospray ion source interfaced to a self-packed 8 cm × 75 μm ID Phenomenex Jupiter 10 μm C18 reversed-phase capillary column. Peptides were eluted from the column with an acetonitrile/0.1 M acetic acid gradient at a flow rate of 0.5 μl/min over 1.3 h. The nanospray ion source was operated at 2.4 kV and 265C. Instrument settings were as follows: 1 MS scan (FT, 60 K resolution, 1 microscan, AGC 9E5, Max IT 500 ms) followed by 20 MS/MS (IT, 1 microscan, AGC 8E3, Max IT 25 ms) with dynamic exclusion enabled (Repeat count 1, Repeat Duration 30 s, List Size 400, Exclusion Duration 60 s). This mode of analysis produced approximately 40,000 MS/MS spectra of ions ranging in abundance over several orders of magnitude. These data were analyzed by database searching using the Sequest search algorithm (10 ppm parent, 1 Da fragments, cys – carbamidomethyl fixed, met – oxidized variable) contained within Proteome Discoverer 1.4.1 against the UniProt Human database (03/2015, 89,663 entries) followed by loading raw search results into Scaffold v4.4.6 (2 peptides, xcorr – +1 > 1.8, +2 > 2.0, +3 > 2.3, + 4 > 2.5, peptide prophet > 60%, protein prophet > 90%) (Table S3). Proteins with exclusive spectra identified in the untransfected sample were removed and any protein with 2 or greater exclusive spectra were considered positive identifications (Table S4). All MS and analysis was done in the W.M. Keck Biomedical Mass Spectrometry Laboratory at the University of Virginia.
2.9. Biotin ligase activity of BirA-Lamin A fusions prepared from mammalian cells
We set out to measure the activity of the BirA-Lamin A fusions by biochemical isolation and in vitro biotinylation of the BAP substrate. Given that a small number of peptides derived from lamina-associated proteins were identified by MS using the BirA (R118K)-Lamin A, we considered it possible that this mutant has a lower activity when expressed in cells. Plasmids encoding HA-Lamin A and each BirA-Lamin A fusion were transiently expressed in HEK293T cells by transfection with Fugene6. After 24 h without biotin supplementation, the cells were washed with ice cold TBS and scraped into 1.5 ml cold TBS with PMSF (1 mM). Cells were pelleted by centrifugation (800 × g, 5 min, 4 °C). The supernatant was aspirated, and pellets were resuspended in 220 μl lysis buffer (20 mM Tris pH 7.6, 0.5% Triton X-100, 2.5 mM EDTA, 2mM DTT, 1 μM PMSF, 5 μg/mL aprotinin, and 5 μg/ml leupeptin/pepstatin). Tubes were incubated with agitation (4 °C for 20 min) before clarification by centrifugation (16,000×g, 20 min, 4 °C). The supernatant was aspirated and set aside. The triton resistant pellet was then resuspended in IVBB without biotin (150 ul). The pellets were solubilized by sonication (10 pulses at 0.2 s/pulse; Branson Sonifier 250) and 35 ul was separated into 4 tubes for each transfection. One tube was placed directly on ice. GST-BAP (5 μg) was added to each of the other tubes. Biotin (50 μM) was added to two tubes before incubation at 37 °C for either 30 min or 4 h. Reactions were stopped by the addition of 5X SDS sample buffer. Samples (20%) were analyzed by SDS-PAGE and immunoblot (Fig. 7A). The biotinylation state of GST-BAP for each condition was quantified by normalization of the fl-neutra signal to anti-GST signal and plotted (Fig. 7B).
Fig. 7.
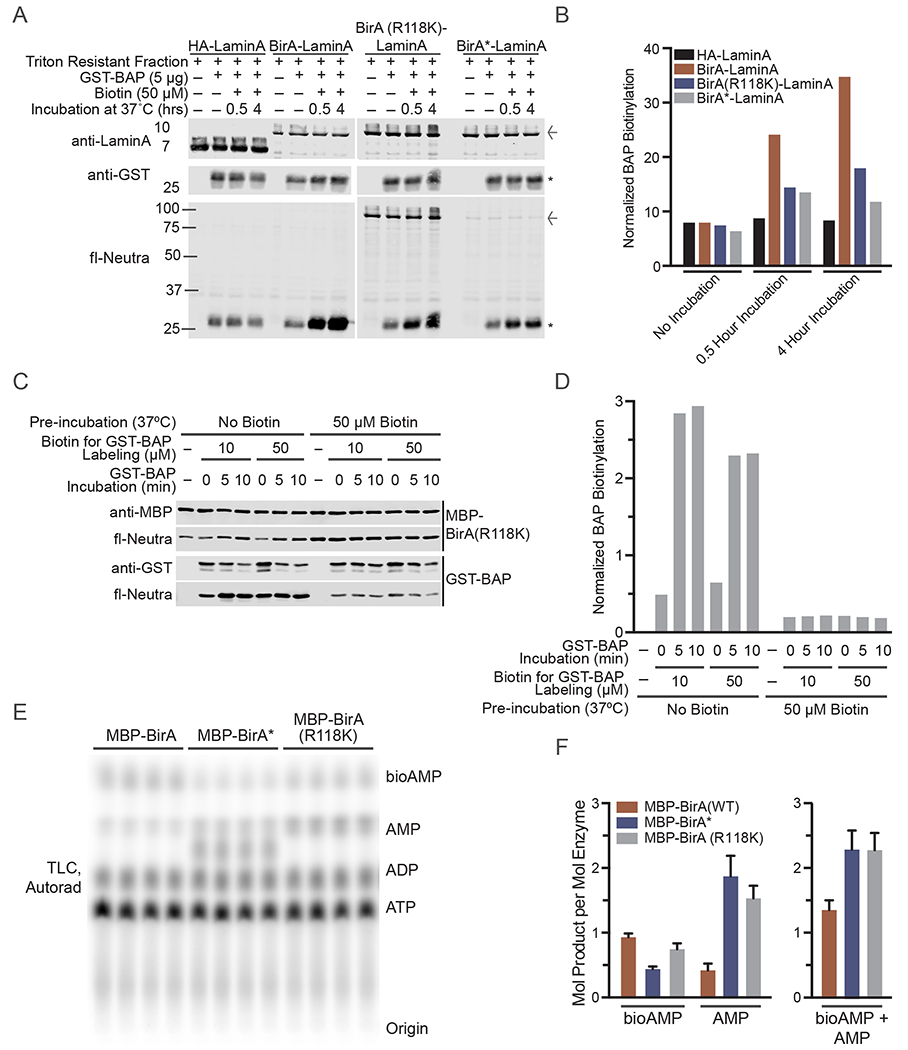
Evaluation of BirA(R118K) activity after auto-modification. (A) Triton resistant fraction from HEK293T cells transfected, in the absence of biotin supplementation, with HA-Lamin A or BirA-Lamin A fusions were harvested and subsequently incubated with biotin for the indicated times. Immunoblot of fl-neutra detection as well (arrow denotes BirA-Lamin A band; asterisk denotes GST-BAP). Signal from fl-neutra on GST-BAP was normalized to GST for each lane and plotted in (B). (C) In vitro biotinylation of GST-BAP substrate (1 μg) with BirA(R118K) (0.2 μg) with and without a pre-incubation with 50 μM biotin for 2 h at 37 °C. Signal from fl-neutra on GST-BAP was normalized to GST for each lane and plotted in (D). (E) TLC based assay to evaluate production of [α-32P]-labeled bioAMP and AMP by BirA, BirA* and BirA(R118K) after 90 min of in vitro biotinylation in the presence 50 μM biotin and 50 μM ATP (82.5 nM [α-32P]-ATP) at 37 °C. (F) Quantification of the mols of product generated per mol of BirA enzyme based on a standard curve quantifying [α-32P]-ATP signal per mol quantified by PhosphorImager (Fig. S2).
2.10. Test for autoinhibition and quantification of bioAMP production
To test whether the BirA(R118K) mutant is capable of undergoing auto-modification, potentially via biotinylation of the Lys residue engineered into the biotin binding site, we subjected the recombinant MBP-BirA(R118K) to a preincubation without and with biotin (50 μM) to allow for self-biotinylation (Fig. 7C, D). Samples were then incubated with BAP substrate in a second reaction (0, 5, 10 min). BAP biotinylation was measured and normalized to the GST signal from blotting. To examine the amount of bioAMP and AMP generated by BirA proteins, we mixed a defined amount of each MBP-BirA protein with biotin and [α-32P]-labeled ATP and examined the bioAMP and AMP production by TLC. To estimate amount of bioAMP and AMP produced per mole of enzyme, we generated a standard curve by spotting 0.003–0.01 pmol of radiolabeled ATP on a TLC plate, exposing the TLC plate on a Phoshor Imager, and plotting the signal as a function of [α-32P]-ATP concentration (Fig. S2).
3. Results and discussion
3.1. Mutagenesis of position 118 alters BirA biotinylation capabilities
The mutagenesis of position 118 in BirA (shown in Fig. 2A) revealed that changing the residue at this position can either enhance or reduce auto-biotinylation of the BirA protein. BirA(WT), with an arginine at position 118, displayed low level auto-biotinylation, a result of the sequence specific characteristics of the enzyme and therefore low promiscuity. Substitution with an acidic residue such as aspartate or glutamate resulted in an enzyme which appeared to be catalytically dead, while glycine (the residue in BirA*), lysine, and tryptophan greatly increased native biotinylation levels. Separation of the MBP tag from the BirA enzyme (Fig. 2C) demonstrated there also were differences in auto-biotinylation of the two species. The high levels of self-modification by BirA(R118K) were almost entirely restricted to BirA, which may partially be a result of biotinylation of the new lysine residue. This high level of specificity as well as the chemical similarity of lysine to arginine (in BirA(WT)) led us to use BirA(R118K) as our model mutant.
3.2. BirA(R118K) has properties that are similar to the WT enzyme and mediates proximity labeling in cells
The bead-based assay designed to measure promiscuous self- and trans- biotinylation by BirA mutants underscores the high levels of self-biotinylation by BirA(R118K) (Fig. 3C). Interestingly, BirA* also demonstrated higher self- than trans-biotinylation. This result is surprising and suggests that bioAMP release from BirA* is more effective for self-labeling than trans-modification of MBP in this assay. In contrast, BirA(WT) actually demonstrated greater trans-biotinylation activity than either BirA* or BirA(R118K) suggesting that in this assay BirA(WT) can release bioAMP, which mediates a low level of promiscuous biotinylation.
Analysis of the ability of each BirA mutant to recognize and label WT and mutant BAP sequences demonstrates BirA(WT), BirA*, and BirA(R118K) can each biotinylate the WT BAP (Fig. 4B). These data suggest that BirA with mutations in position 118 of the biotin-binding loop maintains its recognition and specific biotinylation of a BAP sequence. Biotinylation of the mutant BAP sequence (promiscuous labeling), required a substrate concentration 200-fold greater than WT BAP. This requirement likely reflects low level bioAMP release from the BirA enzymes and labeling of GST (Fig. 4C). Consistent with the results of the bead assay (Fig. 3) BirA(WT) is capable of a low level of promiscuous biotinylation. Examining the actual production and turnover of bioAMP by TLC supports the ability of each tested BirA to generate bioAMP in the absence of substrate and utilize it for biotinylation when substrate is present (Fig. 4D,E).
Examination of AMP/bioAMP ratios and biotinylation of BAP, each as a function of biotin concentration, revealed BirA(R118K) has characteristics intermediate to BirA(WT) and BirA*. With both the BirA(R118K) and BirA(WT), the AMP/bioAMP ratio appears to saturate at 50 μM biotin, whereas BirA* did not show signs of saturation at 250 μM biotin (Fig. 5A,C,E,G). The apparent KM for biotinylation of a WT BAP sequence is 57, 118, and 333 nM for BirA(WT), BirA(R118K), and BirA* respectively (Fig. 5B,D,F,H). Thus, relative to the glycine substitution in BirA*, substituting a lysine at position 118 in BirA is associated with higher affinity biotin binding and an intermediate level of bioAMP release.
3.3. BirA(R118K) demonstrates restricted labeling in cells
To determine if the BirA(R118K) mutant was capable of proximity labeling in cells, HEK293T cells were transfected with Lamin A fusions of BirA(R118K) and BirA*. The cells were grown for 24 h with and without biotin supplementation and were subsequently examined by confocal fluorescence microscopy as described in Section 2.7. We generated fluorescence intensity scans across the nuclei to help establish the location and extent of biotinylation. In BirA*-Lamin A transfected cells, minimal biotinylation was observed without biotin supplementation (Fig. 6A, green tracing), but biotinylation was increased dramatically by biotin supplementation (Fig. 6A, upper panels). BirA(R118K) labeling was mostly restricted to the nuclear lamina region with and without biotin supplementation (Fig. 6A, lower panels). These data suggest that BirA(R118K) may be suitable for proximity labeling of proteins without the necessity of biotin supplementation in culture.
To further evaluate biotinylation by BirA(R118K) in cells, HEK293T cells were transfected with HA-Lamin A, and BirA(WT), BirA(R118K), and BirA* Lamin A fusions in the presence and absence of biotin supplementation (50 μM). Whole cell lysate was assessed by western blot (Fig. 6B). Probing with fl-neutra again demonstrated increased biotinylation by BirA(R118K) in the absence of biotin supplementation compared to BirA-WT or BirA*. This biotinylation level was affected minimally by the addition of 50 μM biotin, suggesting a retention of proximity labeling.
3.4. BirA(R118K)-Lamin A mediates proximity labeling
We also determined the extent of proximity labeling by BirA(R118K) in cells by streptavidin pulldown and MS analysis (Section 2.8) using BirA(R118K)-Lamin A, BirA*-Lamin A, and untransfected cells (Fig. 6C) in the absence of biotin supplementation. We eliminated proteins from consideration if peptides from said proteins were identified in the control (no transfection sample). We focused on peptides derived from a few extensively characterized proteins of the nuclear lamina and nuclear pore (Fig. 6C). We found that BirA(R118K)-Lamin A can mediate proximal biotinylation of select nuclear proteins (Lamin A/C, Lamin B2, LAP2, TPR). These proteins were also identified by proximity labeling in the original BioID study that used BirA*-Lamin A [20]. We found that BirA(R118K)-Lamin A also enabled identification of PCNA as a proximal protein based on the 14 exclusive spectra from peptides from this protein. PCNA has functional links to the nuclear lamina [44,45], and is detected proximal to the nuclear lamina in untransfected HEK293T cells by confocal microscopy (Fig. 6D). This staining likely reflects centers of replication in late S-phase where both PCNA and Lamin A are known to associate [46,47], but it should be noted the signal could be indicative of centers of DNA damage as well [48].
The number of exclusive spectra based on Lamin A peptides identified (which probably reflect self-labeling) was slightly lower for BirA(R118K)-Lamin A compared to BirA* (152 versus 161) (Fig. 6C). BirA(R118K)-Lamin A also resulted in smaller numbers of exclusive spectra identified for LaminB2 and LAP2 peptides. These data are consistent with the fact that BirA(R118K) is more similar to BirA(WT) in terms of bioAMP release (Fig. 5G). The surprising finding was that BirA(R118K)-Lamin A was able to label PCNA, a nuclear protein with links to the nuclear lamina in the context of DNA replication [44,46,47]. PCNA was not identified by our BirA*-Lamin A sample, and it has not been identified by proximity labeling using Lamin A-BirA* or Lamin B-BirA* in other published studies [20,23]. Why a reduced rate of biotin release by a BirA mutant is apparently advantageous for labeling PCNA is not clear. A list of all proximity labeled proteins that yielded at least two exclusive spectra in either BirA-Lamin A sample is provided (Table S4). A total of 72 and 58 proteins were identified by BirA(R118K)-Lamin A and BirA*-Lamin A, respectively. The small number of proteins identified by BirA*-Lamin A in this experiment reflects the fact that biotin supplementation in culture was not performed. We found that 46 proteins were identified by both BirA-Lamin A fusions, indicating reasonable overlap under the conditions of this experiment.
3.5. BirA(R118K) is active in cells but can undergo auto-inhibition in vitro
The BirA(WT), BirA(R118K), and BirA(R118G) Lamin A fusions immunoprecipitated from cells were biochemically active, as each was able to biotinylate BAP (Fig. 7A,B). Not surprisingly, the WT enzyme showed the highest activity. We tested whether the BirA(R118K) enzyme can undergo auto-inhibition by preincubation with biotin, and assay for its biotin ligase activity toward BAP in a second incubation. This experiment revealed that when preincubated with a large excess of biotin and ATP, MBP-BirA(R118K) activity towards BAP can be significantly reduced. These data indicate that BirA(R118K) auto-modification is auto-inhibitory. The results from the TLC experiment indicate that comparable molar amounts of bioAMP and AMP are generated by the two mutants (Fig. 7E,F). Collectively, the data indicate that although automodification of BirA(R118K) certainly can occur, it reduces but does not eliminate activity in mammalian cells. BirA(R118K) and BirA* generated comparable levels of bioAMP and AMP, which under our assay conditions are in the range of 1–2 mol of product per mole of enzyme during a 90 min reaction in vitro (Fig. 7E,F). Thus, BirA(R118K) remains active in the time scale of a BioID experiment.
BioID with the mutant biotin-protein ligase BirA* has proven to be a powerful method for identifying proximal proteins in cells [20]). BioID has important advantages over traditional biochemical isolation methods. The promiscuous activity of BirA* is mediated by the Arg118Gly substitution within the flexible biotin binding loop in the active site of BirA. We explored the effects of other substitutions at the same position, and identified a substitution, Arg118Lys, that has biochemical properties that can be considered “intermediate” between the WT and Arg118Gly forms of BirA*. Our characterization of BirA (Arg118Lys) incorporated multiple biochemical methods developed by other groups working on BirA [22,49]. We also provided proof-of-principle that BirA (Arg118Lys), despite having a lower activity than BirA* [17,18,20], mediates proximal labeling in cells. Employing BirA mutants that show a range of biotin affinity and biotinoyl-5′-AMP release, including the Arg118Lys variant described here, might be helpful for the discovery of protein interactions by BioID.
Supplementary Material
Acknowledgments
Funding
This work was supported by the National Cancer Institute [CA214872].
Abbreviations:
- POI
protein of interest
- PPI
protein–protein interaction
- MS
mass spectrometry
- HRP
horseradish peroxidase
- APEX
engineered ascorbate peroxidase
- BCCP
biotin carboxyl carrier protein
- BioID
proximity dependent biotin identification
- BirA(WT)
BirA (Arg-118)
- BirA*
BirA (Arg118Gly)
- MBP
Maltose binding protein
- GST
Glutathione S-transferase
- fl-neutra
fluorescent neutravidin
- bioAMP
biotinoyl-5′-AMP
- IVBB
In vitro biotinylation buffer
- BAP
biotin acceptor peptide
- ATP
adenosine triphosphate
- ADP
adenosine diphosphate
- AMP
adenosine monophosphate
- TLC
thin layer chromatography
- LC-MS
liquid chromatography mass spectrometry
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- PBS
phosphate buffered saline
- TBS
tris buffered saline
- autorad
autoradiography
- BSA
bovine serum albumin
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ymeth.2018.11.003.
References
- [1].Rees JS, Li X-W, Perrett S, Lilley KS, Jackson AP, Protein neighbors and proximity proteomics, Mol. Cell. Proteomics 14 (2015) 2848–2856, 10.1074/mcp.R115.052902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chen C-L, Perrimon N, Proximity-dependent labeling methods for proteomic profiling in living cells, WIREs Dev. Biol (2017), 10.1002/wdev.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Li P, Li J, Wang L, Di L-J, Proximity labeling of interacting proteins: application of BioID as a discovery tool, Proteomics 17 (2017) 1700002, 10.1002/pmic.201700002. [DOI] [PubMed] [Google Scholar]
- [4].Honke K, Kotani N, Identification of cell-surface molecular interactions under living conditions by using the enzyme-mediated activation of radical sources (EMARS) method, Sensors 12 (2012) 16037–16045, 10.3390/s121216037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Li X-W, Rees JS, Xue P, Zhang H, Hamaia SW, Sanderson B, Funk PE, Farndale RW, Lilley KS, Perrett S, Jackson AP, New insights into the DT40 B cell receptor cluster using a proteomic proximity labeling assay, J. Biol. Chem 289 (2014) 14434–14447, 10.1074/jbc.M113.529578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hung V, Zou P, Rhee H-W, Udeshi ND, Cracan V, Svinkina T, Carr SA, Mootha VK, Ting AY, Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging, Mol. Cell 55 (2014) 332–341, 10.1016/J.MOLCEL.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hopkins C, Gibson A, Stinchcombe J, Futter C, Chimeric molecules employing horseradish peroxidase as reporter enzyme for protein localization in the electron microscope, Methods Enzymol. 327 (2000) 35–45, 10.1016/S0076-6879(00)27265-0. [DOI] [PubMed] [Google Scholar]
- [8].Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY, Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging, Science (80-) 339 (2013) 1328–1331, 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jiang S, Kotani N, Ohnishi T, Miyagawa-Yamguchi A, Tsuda M, Yamashita R, Ishiura Y, Honke K, A proteomics approach to the cell-surface interactome using the enzyme-mediated activation of radical sources reaction, Proteomics 12 (2012) 54–62, 10.1002/pmic.201100551. [DOI] [PubMed] [Google Scholar]
- [10].Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, Ellisman MH, Ting AY, Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy, Nat. Biotechnol 30 (2012) 1143–1148, 10.1038/nbt.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cronan JE, The E. coli bio operon: transcriptional repression by an essential protein modification enzyme, Cell 58 (1989) 427–429, 10.1016/0092-8674(89)90421-2. [DOI] [PubMed] [Google Scholar]
- [12].Nenortas E, Beckett D, Purification and characterization of intact and truncated forms of the Escherichia coli biotin carboxyl carrier subunit of acetyl-CoA carboxylase, J. Biol. Chem 271 (1996) 7559–7567, 10.1074/jbc.271.13.7559. [DOI] [PubMed] [Google Scholar]
- [13].Otsuka A, Abelson J, The regulatory region of the biotin operon in Escherichia coli, Nature 276 (1978) 689–694, 10.1038/276689a0. [DOI] [PubMed] [Google Scholar]
- [14].McAllister HC, Coon MJ, Further studies on the properties of liver propionyl coenzyme A holocarboxylase synthetase and the specificity of holocarboxylase formation, J. Biol. Chem 241 (1966) 2855–2861 http://www.jbc.org/content/241/12/2855.full.pdf (accessed 25.06.18). [PubMed] [Google Scholar]
- [15].Xu Y, Beckett D, Kinetics of biotinyl-5′-adenylate synthesis catalyzed by the Escherichia coli repressor of biotin biosynthesis and the stability of the enzymeproduct complex, Biochemistry 33 (1994) 7354–7360, 10.1021/bi00189a041. [DOI] [PubMed] [Google Scholar]
- [16].Weaver LH, Kwon K, Beckett D, Matthews BW, Corepressor-induced organization and assembly of the biotin repressor: a model for allosteric activation of a transcriptional regulator, Proc. Natl. Acad. Sci. U.S.A 98 (2001) 6045–6050, 10.1073/pnas.111128198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kwon K, Beckett D, Function of a conserved sequence motif in biotin holoenzyme synthetases, Protein Sci. 9 (2000) 1530–1539, 10.1110/ps.9.8.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Choi-Rhee E, Schulman H, Cronan JE, Promiscuous protein biotinylation by Escherichia coli biotin protein ligase, Protein Sci. 13 (2004) 3043–3050, 10.1110/ps.04911804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cronan JE, Targeted and proximity-dependent promiscuous protein biotinylation by a mutant Escherichia coli biotin protein ligase, J. Nutr. Biochem 16 (2005) 416–418, 10.1016/j.jnutbio.2005.03.017. [DOI] [PubMed] [Google Scholar]
- [20].Roux KJ, Kim DI, Raida M, Burke B, A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells, J. Cell Biol 196 (2012) 801–810, 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kim DI, Birendra KC, Zhu W, Motamedchaboki K, Doye V, Roux KJ, Probing nuclear pore complex architecture with proximity-dependent biotinylation, Proc. Natl. Acad. Sci. U.S.A 111 (2014) E2453–E2461, 10.1073/pnas.1406459111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mehus AA, Anderson RH, Roux KJ, BioID identification of lamin-associated proteins, Methods Enzymol. 569 (2016) 3–22, 10.1016/bs.mie.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fu Y, Lv P, Yan G, Fan H, Cheng L, Zhang F, Dang Y, Wu H, Wen B, MacroH2A1 associates with nuclear lamina and maintains chromatin architecture in mouse liver cells, Sci. Rep 5 (2015) 17186, 10.1038/srep17186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Van Itallie CM, Aponte A, Tietgens AJ, Gucek M, Fredriksson K, Anderson JM, The N and C termini of ZO-1 are surrounded by distinct proteins and functional protein networks, J. Biol. Chem 288 (2013) 13775–13788, 10.1074/jbc.M113.466193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fredriksson K, Van Itallie CM, Aponte A, Gucek M, Tietgens AJ, Anderson JM, Proteomic analysis of proteins surrounding occludin and claudin-4 reveals their proximity to signaling and trafficking networks, PLoS One 10 (2015) 0117074, , 10.1371/journal.pone.0117074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Van Itallie CM, Tietgens AJ, Aponte A, Fredriksson K, Fanning AS, Gucek M, Anderson JM, Mareel M, van Roy F, Biotin ligase tagging identifies proteins proximal to E-cadherin, including lipoma preferred partner, a regulator of epithelial cell-cell and cell-substrate adhesion, J. Cell Sci 127 (2014) 885–895, 10.1242/jcs.140475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gupta GD, Coyaud É, Gonçalves J, Mojarad BA, Liu Y, Wu Q, Gheiratmand L, Comartin D, Tkach JM, Cheung SWT, Bashkurov M, Hasegan M, Knight JD, Lin Z-Y, Schueler M, Hildebrandt F, Moffat J, Gingras A-C, Raught B, Pelletier L, A Dynamic protein interaction landscape of the human centrosomecilium interface, Cell 163 (2015) 1484–1499, 10.1016/J.CELL.2015.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Firat-Karalar EN, Rauniyar N, Yates JR, Stearns T, Proximity interactions among centrosome components identify regulators of centriole duplication, Curr. Biol 24 (2014) 664–670, 10.1016/J.CUB.2014.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Firat-Karalar EN, Stearns T, Probing mammalian centrosome structure using BioID proximity-dependent biotinylation, Methods Cell Biol. 129 (2015) 153–170, 10.1016/BS.MCB.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Comartin D, Gupta GD, Fussner E, Coyaud É, Hasegan M, Archinti M, Cheung SWT, Pinchev D, Lawo S, Raught B, Bazett-Jones DP, Lüders J, Pelletier L, CEP120 and SPICE1 cooperate with CPAP in centriole elongation, Curr. Biol 23 (2013) 1360–1366, 10.1016/J.CUB.2013.06.002. [DOI] [PubMed] [Google Scholar]
- [31].Cole A, Wang Z, Coyaud E, Voisin V, Gronda M, Jitkova Y, Mattson R, Hurren R, Babovic S, Maclean N, Restall I, Wang X, Jeyaraju DV, Sukhai MA, Prabha S, Bashir S, Ramakrishnan A, Leung E, Qia YH, Zhang N, Combes KR, Ketela T, Lin F, Houry WA, Aman A, Al-awar R, Zheng W, Wienholds E, Xu CJ, Dick J, Wang JCY, Moffat J, Minden MD, Eaves CJ, Bader GD, Hao Z, Kornblau SM, Raught B, Schimmer AD, Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia, Cancer Cell. 27 (2015) 864–876, 10.1016/J.CCELL.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhou Z, Rawnsley DR, Goddard LM, Pan W, Cao X-J, Jakus Z, Zheng H, Yang J, Arthur JSC, Whitehead KJ, Li D, Zhou B, Garcia BA, Zheng X, Kahn ML, The cerebral cavernous malformation pathway controls cardiac development via regulation of endocardial MEKK3 signaling and KLF expression, Dev. Cell 32 (2015) 168–180, 10.1016/J.DEVCEL.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Couzens AL, Knight JDR, Kean MJ, Teo G, Weiss A, Dunham WH, Lin Z-Y, Bagshaw RD, Sicheri F, Pawson T, Wrana JL, Choi H, Gingras A-C, Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions, Sci. Signal 6 (2013) rs15, 10.1126/scisignal.2004712. [DOI] [PubMed] [Google Scholar]
- [34].Cheng Y-S, Seibert O, Klöting N, Dietrich A, Straßburger K, Fernández-Veledo S, Vendrell JJ, Zorzano A, Blüher M, Herzig S, Berriel Diaz M, Teleman AA, PPP2R5C couples hepatic glucose and lipid homeostasis, PLOS Genet. 11 (2015) e1005561, 10.1371/journal.pgen.1005561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sun BK, Boxer LD, Ransohoff JD, Siprashvili Z, Qu K, Lopez-Pajares V, Hollmig ST, Khavari PA, CALML5 is a ZNF750- and TINCR-induced protein that binds stratifin to regulate epidermal differentiation, Genes Dev. 29 (2015) 2225–2230, 10.1101/gad.267708.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Green NM, Avidin, Adv. Protein Chem 29 (1975) 85–133, 10.1016/S0065-3233(08)60411-8. [DOI] [PubMed] [Google Scholar]
- [37].Kim DI, Cutler JA, Na CH, Reckel S, Renuse S, Madugundu AK, Tahir R, Goldschmidt HL, Reddy KL, Huganir RL, Wu X, Zachara NE, Hantschel O, Pandey A, BioSITe: a method for direct detection and quantitation of site-specific biotinylation, J. Proteome Res 17 (2018) 759–769, 10.1021/acs.jproteome.7b00775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Opitz N, Schmitt K, Hofer-Pretz V, Neumann B, Krebber H, Braus GH, Valerius O, Capturing the Asc1p/receptor for activated C kinase 1 (RACK1) microenvironment at the head region of the 40S ribosome with quantitative BioID in yeast, Mol. Cell. Proteomics 16 (2017) 2199–2218, 10.1074/mcp.M116.066654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kim DI, Jensen SC, Noble KA, Kc B, Roux KH, Motamedchaboki K, Roux KJ, An improved smaller biotin ligase for BioID proximity labeling, Mol. Biol. Cell 27 (2016) 1188–1196, 10.1091/mbc.E15-12-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, Carr SA, Feldman JL, Perrimon N, Ting AY, Efficient proximity labeling in living cells and organisms with TurboID, Nat. Biotechnol 36 (2018) 880–887, 10.1038/nbt.4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Schatz PJ, Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: a 13 residue consensus peptide specifies biotinylation in Escherichia coli, Bio/Technol. 11 (1993) 1138–1143, 10.1038/nbt1093-1138. [DOI] [PubMed] [Google Scholar]
- [42].Chakravartty V, Cronan JE, Altered regulation of escherichia coli biotin biosynthesis in bira superrepressor mutant strains, J. Bacteriol 194 (2012) 1113–1126, 10.1128/JB.06549-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dakshinamurti K, Chalifour LE, The biotin requirement of HeLa cells, J. Cell. Physiol 107 (1981) 427–438, 10.1002/jcp.1041070314. [DOI] [PubMed] [Google Scholar]
- [44].Dittmer TA, Sahni N, Kubben N, Hill DE, Vidal M, Burgess RC, Roukos V, Misteli T, Systematic identification of pathological lamin A interactors, Mol. Biol. Cell 25 (2014) 1493–1510, 10.1091/mbc.E14-02-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cobb AM, Murray TV, Warren DT, Liu Y, Shanahan CM, Disruption of PCNAlamin A interactions by prelamin A induces DNA replication fork stalling, Nucleus 7 (2016) 1–22, 10.1080/19491034.2016.1239685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Shumaker DK, Solimando L, Sengupta K, Shimi T, Adam SA, Grunwald A, Strelkov SV, Aebi U, Cardoso MC, Goldman RD, The highly conserved nuclear lamin Ig-fold binds to PCNA: its role in DNA replication, J. Cell Biol 181 (2008) 269–280, 10.1083/jcb.200708155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kennedy BK, Barbie DA, Classon M, Dyson N, Harlow E, Nuclear organization of DNA replication in primary mammalian cells, Genes Dev. 14 (2000) 2855–2868, 10.1101/gad.842600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hwang BJ, Toering S, Francke U, Chu G, Houtsmuller AB, Kanaar R, Vermeulen W, p48 Activates a UV-damaged-DNA binding factor and is defective in xeroderma pigmentosum group E cells that lack binding activity, Mol. Cell. Biol 18 (1998) 4391–4399, 10.1128/mcb.25.21.9350-9359.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Roux KJ, Kim DI, Burke B, BioID: a screen for protein-protein interactions, Curr. Protoc. Protein Sci 74 (2013) Unit 19.23, 10.1002/0471140864.ps1923s74. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.