

Abstract
Introduction
PRI‐002 is an orally available anti–amyloid beta (Aβ) prionic compound developed for direct disassembly of toxic Aβ oligomers relevant to Alzheimer's disease.
Methods
Two placebo‐controlled clinical phase I trials with oral dosing of PRI‐002 were conducted in healthy young subjects: A single ascending dose trial (4, 12, 36, 108, or 320 mg PRI‐002 or placebo) in 40 participants followed by a multiple ascending dose study with daily 160 mg PRI‐002 for 14 days or 320 mg for 28 days in 24 participants. The main objectives were safety, tolerability, and evaluation of pharmacokinetic (PK) parameters.
Results
PRI‐002 was safe and well tolerated after single and multiple oral administration up to the highest doses. PRI‐002 was absorbed rapidly and drug exposure increased proportional to dose. During repeated daily administration, the drug accumulated by a factor of about three. Steady‐state conditions were reached after 1 to 2 weeks.
Conclusions
The safety and PK results encourage further clinical development of PRI‐002.
Keywords: Alzheimer´s disease (AD), anti‐Aβ‐prionic, first‐in‐human (FIH), MAD, multiple ascending dose, pharmacokinetics, phase I, PRI‐002, SAD, safety, single ascending dose
1. INTRODUCTION
PRI‐002 (alias “RD2” in earlier publications, and alias “Contraloid acetate” in regulatory documents) is an all‐d‐enantiomeric peptide, which was developed to directly destroy toxic and replicating amyloid beta (Aβ) oligomer prions, by disassembling aggregates into non‐toxic Aβ monomers. 1 , 2 The investigational drug is specifically designed for the curative and/or disease‐modifying treatment of cognitive, memory deficits in patients with Alzheimer´s disease (AD). PRI‐002 is reaching its target organ, the brain 3 and has demonstrated target engagement in vitro and in vivo. 2 , 4 Preclinical proof‐of‐concept studies in three different transgenic mouse models (APPSL, APPswe/PS1∆E9, and TBA2.1) of AD in three different laboratories successfully proved therapeutic efficacy of PRI‐002 by improving cognition and decelerating neurodegeneration. Therapeutic efficacy could be shown even under truly non‐preventive treatment conditions, and when applied orally 2, , 4 , 5 , 6 normalized the cognitive abilities and behavior of transgenic mice up to levels of healthy wild‐type littermates. 4 , 5
2. MATERIALS AND METHODS
2.1. Study design
Two consecutive phase I, randomized, double‐blind, placebo‐controlled clinical studies with PRI‐002 were conducted in healthy young subjects. First, a single ascending dose (SAD) study—“A Randomized, Double‐blind and Placebo‐controlled Single Ascending‐dose Phase I Study (First‐in‐human) to Investigate the Safety, Tolerability and Pharmacokinetics of Contraloid Acetate (in Healthy Subjects).”; EUDRA‐CT: 2017‐000396‐93—and second, a multiple ascending dose (MAD) study—“Single‐center, Randomized, Prospective, Double‐blind, Placebo Controlled Phase Ib Study With an Adaptive Multiple Ascending Dose (MAD) Design to Investigate the Safety, Tolerability, Pharmacokinetics of Contraloid Acetate (Healthy Subjects)”; EUDRA‐CT: 2018‐002500‐14—were conducted. Both studies were approved by the ethics committee of the Medical University Vienna, Austria. All subjects signed an informed consent form prior to any study‐related activity. Both studies were conducted at the Medical University of Vienna, Department of Clinical Pharmacology, Vienna, Austria, under the EU regulations (Directive 2001/20/EC, 2003/94/EC, and 2017/1572/EC), defining Good Clinical Practices (GCP) and Good Manufacturing Practices (GMP) of human drugs as well as whose implementation in national Austrian law.
RESEARCH IN CONTEXT
Systematic review: Recent press release on the re‐evaluation of the aducanumab phase III study validated amyloid beta (Aβ) oligomers as the relevant target in Alzheimer's disease (AD) treatment. These results will re‐activate the long‐lasting discussion about the most favorable target against AD, and new evaluation is expected. So far, a curative treatment is missing.
Interpretation: PRI‐002 was developed to directly destroy toxic and replicating Aβ oligomer prions, by disassembling aggregates into non‐toxic Aβ monomers. PRI‐002 was safe and well tolerated in humans after single and multiple oral administrations up to the highest doses. Plasma levels achieved in humans already after single oral dosing are in the same range of those observed in the highest dosed transgenic mice in successful Proof of Concept (PoC) studies.
Future directions: PRI‐002 has demonstrated safety and comfortable pharmacokinetic properties in humans. A phase II study in AD patients aiming at the demonstration of efficacy will be the next step.
The primary end points were safety and tolerability. The secondary objective was the assessment of pharmacokinetic (PK) characteristics of PRI‐002.
For the single ascending dose (or SAD) study, capsules were filled with PRI‐002 acetate without any excipients. The study medication was prepared at the required dose for each treatment prior to administration at the hospital pharmacy. The placebo treatment matched the appearance and shape of PRI‐002 capsules. Each subject within the cohort received a single dose of PRI‐002 capsules or placebo capsules, under fasting conditions. Capsules were administered with 250 mL of tap water. Fluid intake was restricted during 1 hour before and 1 hour after drug administration. Food intake was prohibited for at least 8 hours pre‐dose (monitored by glucose testing) and 5 hours post‐dose followed by standardized meals.
In the multiple ascending dose (or MAD) study, PRI‐002 was administered as capsules filled with PRI‐002 acetate without any excipients or placebo. Aptuit, Verona, Italy, manufactured the study medication (with 40 mg or 100 mg PRI‐002). Capsules were administered with 250 mL of tap water. On days of PK sampling, fluid intake was restricted for 1 hour before and 1 hour after drug administration. At the first dosing, food intake was restricted for at least 8 hours pre‐dose (glucose testing) and 2 hours post‐dose, followed by a standardized meal.
In the SAD first‐in‐human study, 40 healthy male subjects (19‐45 years of age), randomly assigned to the treatment, received one oral dose of study drug in the morning after an overnight fast. Eight healthy subjects were assigned to each of the five sequential dose cohorts (4, 12, 36, 108, or 320 mg PRI‐002). For each cohort, the first two subjects (sentinels) were randomized in a 1:1 ratio to receive PRI‐002 or placebo, and the remaining six subjects in a ratio of 5:1 to receive PRI‐002 or placebo. All participants were admitted to the clinical facility on day 1. Sentinels remained at the clinical site through day 4; all other subjects were kept for 1 day after administration and returned thereafter to the facility once daily for visits. The last follow‐up visit was 1 week after study drug administration.
In the MAD study, 24 healthy male subjects (21‐43 years of age) were randomly assigned to the treatment and received the fixed dose daily for 14 or 28 days (cohort 1 and cohort 2, respectively). The study was open for the participation of female subjects, but probably due to the strict inclusion/exclusion criteria, no female subject was recruited.
The sequential cohorts were exposed to an increased dose of PRI‐002 in order to identify the maximal administered multiple dose. The study was planned with an adaptive design in order to be able to reduce the dose in case of severe side effects. The first cohort received 160 mg PRI‐002, 50% of the maximal administered single safe dose in the SAD study. In case of severe side effects, the dose for the second cohort would have been reduced to 80 mg. Because no severe side effects occurred in the first cohort, the second cohort was dosed with 320 mg.
For both cohorts, the first four subjects (sentinels) were randomized in a ratio of 2:2 to receive PRI‐002 or placebo, and the remaining eight subjects in a ratio of 6:2 to receive PRI‐002 or placebo. All participants were admitted to the clinical facility on day 1. Sentinels remained at the clinical site through day 8 and subsequently returned daily for dosing and study procedures to the clinical site until day 14/28 (cohort 1/cohort 2). All other subjects were allowed to leave the facility on day 2 after administration and returned for dosing and study procedures to the clinical site at a fixed morning time for 11/25 days (cohort 1/cohort 2). On day 14/28 (cohort 1/cohort 2), all participants were admitted to the clinical site for another 24 hours for PK sampling. A follow‐up was performed on days 16/30, 17/31, and at the end of study visit on day 21/35.
2.2. Participants
Healthy subjects (selected by medical history, physical examination, vital signs, and laboratory data) between 19 and 45/21 and 43 years of age (SAD/MAD) were enrolled. Any medical condition requiring chronic medication; any evidence of active infection requiring antibiotic therapy within 14 days prior to screening; or any medical history of vasculitis, autoimmune disease, or treatment for cancer within the past 2 years or of acute/chronic hepatitis B or C was exclusionary. All prescription, over‐the‐counter, and herbal medications were prohibited within 10 days of study dosing (with the exception of calcium/vitamin D supplements, nasal steroids, ocular medications, and paracetamol ≤1000 mg/day at the discretion of the investigator).
2.3. Safety assessments
Participants were closely monitored for adverse events (AEs) throughout the studies. Safety assessments including physical examination, vital sign measurements, electrocardiography (ECG) recordings including QTc measurement, and clinical laboratory tests were conducted at specified intervals throughout the studies.
The independent Data and Safety Monitoring Board (DSMB) assessed the safety and PK data of the sentinels and the entire cohort carefully and approved the continuation of the study.
2.4. Pharmacokinetic assessments
During the SAD study, blood samples for plasma PK analysis were collected pre‐dose and at 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 12, 18, 24, 36, 48, and 72 hours post‐dose. During the MAD study, blood samples for plasma PK analysis were collected pre‐dose and at 0.5, 1, 1.5, 2, 2.5, 4 6, 8, 12, and 24 hours post‐dose on day 1 for both cohorts and on day 14 (cohort 1) or day 28 (cohort 2). For the remaining study, the day 1 blood sample was collected pre‐dose. After the last administration, additional blood samples were collected at 36, 48, and 72 hours post‐dose and on day 21/35 (cohort 1/cohort 2). PRI‐002 plasma concentrations were quantified using a validated liquid chromatography method with tandem mass spectrometric detection. The method had been qualified in terms of specificity, reproducibility, precision, and robustness. The lower limit of quantification (LLOQ) had been set to 0.5 ng/mL. PRI‐002 levels of <0.5 ng/mL were set to zero for PK calculations. In the SAD study, plasma concentrations for each dose level following single oral doses of PRI‐002 were used to calculate the following PK parameters: Area under the plasma concentration versus time curve (AUC) between 0 and t, and from 0 to infinity as the sum of AUC0‐tlast + AUCtlast‐∞ (AUC0‐t, AUC0‐inf), maximum concentration (Cmax) and time to reach it (Tmax), terminal half‐life (t½), and total plasma clearance (CL/f = Dose/AUC). A non‐compartment analysis (NCA) was used to calculate visible disposition phases from semi‐log plots, their half‐lives (t½α, t½β), and respective elimination rate constants (ke).
In the MAD study, PRI‐002 levels found on day 1 of both cohorts were evaluated exclusively for Cmax, Tmax, and AUC0‐24h, but not for half‐lives because the dosing interval was too short for a reliable estimation of the half‐lives of the terminal disposition phase. Trough levels were inspected visually for the time of constant levels, and mean values were calculated from those levels representing steady‐state conditions (Css). For cohort 1, this interval was from days 8 to 12 and from days 20 to 28 for cohort 2. Individual mean values were assigned to Css. PK evaluation at the last study days included Cmax, Tmax, AUC0‐24h, Clast, AUC0‐tlast, AUC0‐inf, t½ α, and t½ ß.
AUC was calculated by the trapezoidal rule; Cmax, Tmax, and Clast were taken from actual data. For half‐life determination, the semi‐log plot of individual PRI‐002 plasma level profiles was inspected visually and then the time intervals were selected, for which the half‐lives (α, ß) were calculated by regression analysis. The calculation of the extrapolated AUC (to infinity) followed the equation AUCtlast‐inf = Clast/ke. Extrapolated AUC should account for <30% of total AUC0‐inf because extrapolation is highly dependent on the accuracy of ke. Total oral clearance is defined by CL/f = Dose/AUC0‐inf. Because the fraction (f) entering the systemic circulation unchanged is not known for PRI002, f was set to 1 for a rough estimate. Accordingly, CL data were not taken up in the Result section. The extent of accumulation (accumulation factor, AF) was estimated by the quotient of AUC0‐inf/AUC0‐24h at steady state.
In the MAD study in cohort 2 only one sentinel was available and passed the sentinel study plan including study day 7. In addition, one subject dropped out of the study after day 9. No samples were obtained on day 10 or later. Thus, a complete set of data was evaluated for six subjects receiving 320 mg/day of PRI‐002.
2.5. Statistical analysis
Both phase I studies were conducted with a sufficient number of volunteers to fulfill the study objectives of safety and PK. Descriptive statistics was used to evaluate the data. The PK and statistical analyses of the SAD and MAD studies were conducted by the clinical research team from the Clinical Unit of Hospital de La Princesa, Madrid, Spain, using the program WinNonlin 7 (Pharsight Corporation, Cary, NC). Further data evaluation and interpretation were conducted by KAIROSmetics UG (Berlin, Germany).
3. RESULTS
3.1. Safety and tolerability results
3.1.1. Safety summary for the SAD study
There were no deaths, serious AEs, severe AEs, or discontinuations because of a treatment‐emergent AE reported in the SAD study. A total of six participants (20%) who were given PRI‐002 reported non‐serious AEs considered probably or possibly related to study treatment, compared with three participants (30%) who received placebo. A summary of these AEs is presented in Table 1. The severity of eight of these treatment‐emergent AEs was deemed mild and included EEG abnormalities only, whereas the severity of one AE (headache) was graded as moderate. No dose‐related trend in the incidence of AEs was observed with increasing doses of PRI‐002.
Table 1.
Summary of adverse events of the SAD study considered probably or possibly related to study treatment
| PRI‐002 | |||||||
|---|---|---|---|---|---|---|---|
| Cohort 1 (4 mg) | Cohort 2 (12 mg) | Cohort 3 (36 mg) | Cohort 4 (108 mg) | Cohort 5 (320 mg) | Placebo | Total | |
| System organ class, preferred term, n (%) | n = 6 | n = 6 | n = 6 | n = 6 | n = 6 | n = 10 | n = 40 |
| Nervous system disorders | |||||||
| EEG abnormal (grade 1) | 1 | 1 | 3 | 0 | 0 | 3 | 8 |
| (16.7%) | (16.7%) | (50.0%) | (30%) | (20%) | |||
| Headache (grade 2) | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| (16.7%) | (2.5%) | ||||||
| Total number of events | 1 | 1 | 4 | 0 | 0 | 3 | 9 |
| (16.7%) | (16.7%) | (66.7%) | (30%) | (22.5%) | |||
Overall, there were no major changes in the course of the blood chemistry and hematology attributable to study treatment. There were no clinically significant alterations in the physical exploration, vital signs, ECG, or EEG during the course of the study. No apparent treatment‐ or dose‐related trends in the 12‐lead ECG parameters were noted following any dose of PRI‐002 or placebo.
3.1.2. Safety summary for the MAD study
There were no deaths, serious AEs, severe AEs, or discontinuations because of a treatment‐emergent AE reported in the MAD study. One subject was withdrawn from the study due to an unrelated serious AE (broken lower jaw due to bicycle accident). A total of one participant (6.7%) given PRI‐002 reported non‐serious AEs compared with one participant (12.5%) who received placebo. A summary of these AEs is presented in Table 2.
Table 2.
Summary of adverse events of the MAD study considered probably or possibly related to study treatment
| PRI‐002 | ||||
|---|---|---|---|---|
| Cohort 1 (160 mg) | Cohort 2 (320 mg) | Placebo | Total | |
| System organ class, preferred term, n (%) | n = 8 | n = 7 | n = 8 | n = 23 |
| Nervous system disorders | ||||
| Disoriented three times | 1 | 0 | 0 | 1 |
| (12.5%) | (4.3%) | |||
| Headache (grade 2) | 0 | 0 | 1 | 1 |
| (12.5%) | (4.3%) | |||
| Total number of events | 1 | 0 | 1 | 2 |
| (12.5%) | (12.5%) | (8.7%) | ||
There were two non‐serious AEs assessed by the principal investigator to be possibly related to the study treatment (Table 2). One in the low dose and one in placebo; none in the high dose group. The severity of both AEs was deemed mild. The occurrence of these AEs does not appear to be dose‐related.
Overall, there were no major changes in the course of the blood chemistry and hematology attributable to study treatment. Clinically significant alterations in the physical exploration, vital signs, oral body temperature, or ECG were not found during the study. Although minor and transient changes in blood pressure and pulse rate were noted at isolated time points for some subjects, none of these findings were clinically relevant.
3.2. Pharmacokinetic results
3.2.1. Pharmacokinetic evaluation of the SAD study
Plasma levels of PRI‐002 at the two lower doses were below 0.5 ng/mL (LLOQ). At the dose level of 36 mg, levels were close to the determination limit and reliable PK parameters could not be calculated for all subjects. Therefore, no stable means were presentable. Mean PK parameters of PRI‐002 are presented in Table 3 and Figure 1.
Table 3.
Mean pharmacokinetic parameters of PRI‐002 in subjects (SAD study)
| PRI‐002 dose (mg) | Study day | Cmax (ng/mL) | Tmax (hour) | AUC0‐24 (ng*hour/mL) | AUC0‐tlast (ng*hour/mL) | AUC0‐inf (ng*hour/mL) | t½ (hour) |
|---|---|---|---|---|---|---|---|
| Cohort 4 (108 mg) (n = 6) | Day 1 | 2.92 | 3.08 | 26.4 | 26.8 | 39.2 | 14.9 |
| Cohort 5 (320 mg) (n = 6) | Day 1 | 19.6 | 1.17 | 104 | 131 | 157 | 26.5 |
FIGURE 1.
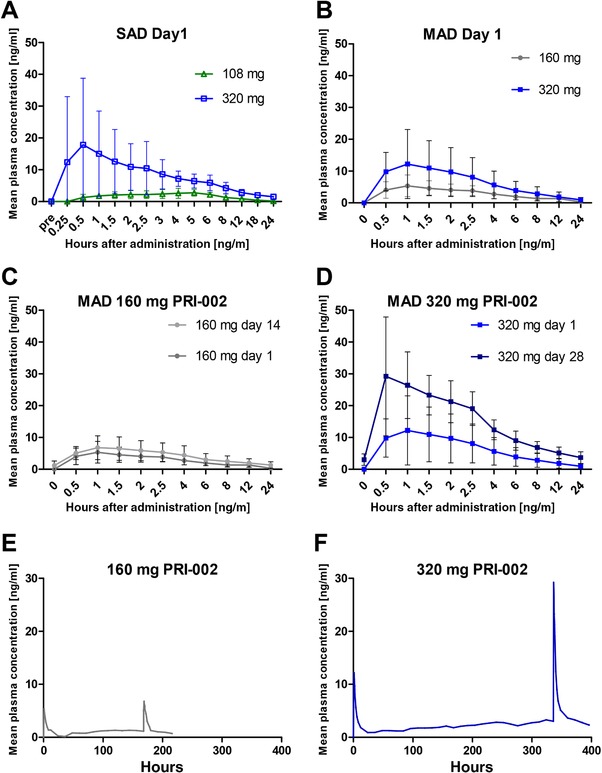
Mean plasma concentrations of PRI‐002 over time. (A) Pharmacokinetic (PK) profile of PRI‐002 after single oral doses. (B) Pharmacokinetic profile of PRI‐002 after a single oral dose of 160 mg compared to 320 mg. (C) and (D) Comparison of pharmacokinetic profiles of PRI‐002 between a single dose (day 1) and multiple oral doses (day 14/28) of 160/320 mg. (E) and (F) Mean plasma concentrations of PRI‐002 over time for the 160 and 320 mg cohorts. Please note that PK sampling was done only during the first 24 hours and after the last dosing for each cohort. On all other days, only one measurement has been done, pre‐dose. The lower limit of quantification (LLOQ) had been set to 0.5 ng/mL. PRI‐002 values of <0.5 ng/mL were set to zero for PK calculations
Following oral administration of the two higher doses (108 and 320 mg), PRI‐002 plasma levels were detectable after 0.25 to 0.5 hours and mean Cmax levels were 2.92 or 19.6 ng/mL reached at mean Tmax of 3.08 or 1.17 hours in cohorts 4 and 5, respectively. AUC0‐inf accounted for 39.2 ng*hour/mL (dose 108 mg) or 157 ng*hour/mL (dose 320 mg). Whereas the ratio of Cmax was 1:6.8 between both dose groups, the AUC0‐inf ratio of 1:4 was close to the dose ratio (1:3). According to the rather limited time interval of measurable plasma levels (about 18 hours after administration at the 108 mg dose), the calculation of the half‐lives of the terminal disposition phase was not sufficiently accurate. In the case of the 320 mg cohort, plasma levels were above the LLOQ for up to 48 hours. The estimated plasma half‐life of PRI‐002 of 26.5 hours in subjects receiving the 320 mg dose can be regarded as fairly reliable, because the documented time interval of this phase was roughly at least twice the half‐life. The limited data for the SAD study do not allow a statement about dose linearity of PRI‐002 PK over the investigated dose range.
3.2.2. Pharmacokinetic evaluation of the MAD study
3.2.2.1 Mean plasma levels
Table 4 shows the mean plasma PK parameters of cohort 1 and cohort 2. Figure 1 shows the mean PRI‐002 plasma concentrations at certain time points (full PK profile at day 1 and day 14/28) (A‐D) and over the total treatment period (E‐F).
Table 4.
Mean pharmacokinetic parameters of PRI‐002 in subjects (MAD study)
| PRI‐002 dose (mg) | Study day | Cmax (ng/mL) (CV %) | Tmax (hour) (CV %) | AUC0‐24 (ng*hour/m) (CV %) | AUC0‐tlast (ng*hour/m) (CV %) | AUC0‐inf (ng*hour/mL) (CV %) | t1/2α (hour) (CV %) | t1/2β (hour) (CV %) | Clast | Css trough |
|---|---|---|---|---|---|---|---|---|---|---|
| Cohort 1 (160 mg) (n = 8) | Day 1 | 5.6 | 1.3 | 35.6 | ||||||
| (58%) | (50%) | (38%) | ||||||||
| Days 8‐12 | 1.28 | |||||||||
| (99%) | ||||||||||
| Day 14 | 7.2 | 1.3 | 62.7 | 147 | 181 | 4.6 | 38.5 | 0.6 | ||
| (53%) | (37%) | (64%) | (92%) | (64%) | (20%) | (34%) | (34%) | |||
| Cohort 2 (320 mg) (n = 6) | Day 1 | 13.8 | 1.3 | 76.2 | ||||||
| (76%) | (54%) | (75%) | ||||||||
| Days 20‐28 | 2.76 | |||||||||
| (57%) | ||||||||||
| Day 28 | 33.5 | 0.9 | 193 | 513 | 656 | 3.7 | 74.2 | 1.4 | ||
| (45%) | (64%) | (28%) | (40%) | (51%) | (25%) | (39%) | (47%) |
CV = coefficient of variation.
Mean plasma levels of the cohort 1 show a sharp peak on both full study days, a steady state of pre‐treatment levels after about 8 days, and a slow decrease of plasma levels at the end of treatment (Figure 1E). The coefficients of variation (CVs) of Cmax, Tmax, and AUC0‐24h were 58%, 50%, and 38%, respectively. Mean PK parameters of day 14 showed a similar variation as on day 1, with CVs of 53%, 37%, 64%, and 34% for Cmax, Tmax, AUC0‐24h, and Clast, respectively. The mean value of t½α was 4.6 hours, showing a low CV of 20%. The mean t½ß was 38.5 hours (CV = 34%), which is in agreement with the observation that steady‐state conditions were reached after 8 days (corresponding to four to five half‐lives). The extrapolated part of the total AUC was 20% on average and thereby met the rule of not being ≥30%. The accumulation factor at steady state was estimated by the quotient of AUC0‐inf/AUC0‐24h at day 14. The mean accumulation factor was 2.9 for the daily treatment interval.
Mean plasma levels of cohort 2 again show a sharp peak on both full study days, a steady state of pre‐treatment levels after about 15 to 20 days, and a slow decrease of plasma levels at the end of treatment (Figure 1F). The CVs of Cmax, Tmax, and AUC0‐24h were 76%, 54%, and 75%, respectively. Mean PK parameters of day 28 showed a somewhat lower variation compared to day 1, with CVs of 45%, 64%, 28%, and 47% for Cmax, Tmax, AUC0‐24h, and Clast, respectively. The mean value of t½α was 3.7 hours, showing a low variance of CV = 25%. The mean t½ß was 74.2 hours (CV = 39%), which again is in agreement with the observation that steady‐state conditions were reached after 15 days (corresponding to four to five half‐lives). The extrapolated part of the total AUC was 21% on average. The accumulation at steady state was 3.0 (using the quotient of Css (2.76 ng/mL) and C24h,day1 (0.92 ng/mL) or 3.4 using the quotient of day 28 AUC0‐inf (656 ng*hour/mL)/AUC0‐24h(193 ng*hour/mL) Thus, as in cohort 1, the mean accumulation factor of PRI‐002 found in cohort 2 is about three for a daily treatment interval.
When the dose‐dependent parameters in both treatment groups were compared, there was a dose‐proportional increase from 160 to 320 mg for day 1 and Cmax, AUC0‐24h, and Css, with Cohort2/Cohort1‐quotients of 2.5, 2.1, and 2.2. Comparing days 14 and 28, there was, however, a dose over‐proportional increase in Cmax, AUC0‐24h, AUC0‐tlast, and total AUC0‐inf, with quotients of 4.6, 3.1, 3.5, and 3.6. Differences are generated by the slower elimination of PRI‐002 in subjects of cohort 2. The mean terminal half‐life calculated for cohort 2 was twofold higher than for cohort 1. Because of small group sizes, half‐life differences are regarded as chance results.
Oral clearance rates in humans have been estimated to 2500 to 3500 L/hour, and in the present study mean oral clearance of cohort 5 (SAD study) was calculated to be 566 L/hour.
4. DISCUSSION
A disease‐modifying therapy against AD is of fundamental interest for aging societies, health care systems, as well as for each single current and future AD patient. Because Aβ oligomers are the major neurotoxic agent responsible for disease development and progression 7 , 8 , 9 , 10 , 11 and growing evidence suggests that Aβ oligomers, or at least sub‐fractions of them, can replicate in a prion‐like fashion, 12 , 13 , 14 anti‐Aβ‐prionic compounds eliminating toxic Aβ oligomers are a promising new treatment strategy for AD. 1 PRI‐002 was designed specifically to eliminate these toxic oligomers and already demonstrated its pre‐clinical in vivo efficacy, including in vivo target engagement. 4 In this first‐in‐human study we could show that single and multiple oral doses of PRI‐002 were safe and well tolerated by healthy subjects when administered at single doses ranging from 4 to 320 mg (SAD study) and in the MAD study at daily oral doses of 320 mg for up to 28 days. In both studies (SAD and MAD), AEs were generally mild and balanced between the treatment groups (PRI‐002 vs placebo), indicating that they were potentially not drug and dose related. The only serious AE, a broken mandible due to a bicycle accident, was unrelated to the study medication and led to withdrawal of the subject from the study.
Pharmacokinetic assessment revealed a rapid liberation and absorption of PRI‐002 acetate from the formulation (lyophilized powder in hydroxypropyl methylcellulose (HPMC) capsules without excipients). Tmax was found as early as 0.5 hours after administration in fasted condition, and in the MAD study, mean Tmax values ranged between 0.9 and 1.3 hours.
Values for oral clearance in humans in the SAD study are unphysiologically high (10‐ to 70‐fold the physiological plasma flow through the liver of 48 L/hour 15 ). The reason is that within the equation defining the oral clearance (CL/f = Dose/AUC) the fraction of the dose that is systemically available (f) is not known. In principal, f can become <1 by two mechanisms. The first is incomplete enteral absorption and the second is pre‐systemic elimination by biotransformation before absorption, during absorption, and first liver passage (first pass effect [FPE]). The metabolic stability of PRI‐002 was found to be high in gastric and intestinal simulated fluid as well as in liver microsomes. 16 It is therefore rather unlikely that PRI‐002 is rapidly degraded before (gastric or intestinal fluid), during (gut wall) or shortly after absorption (FPE). Therefore, f has most probably been lowered mainly by incomplete absorption. This is not in contrast to the fact that absorption was rapid and occurred shortly after treatment. Of note, never a second peak or a late Tmax occurred.
The terminal disposition half‐life of 34 hours (cohort 1) or 78 hours (cohort 2) might reflect the relatively high protease‐resistance of d‐amino acid peptides. 16 , 17 Due to the limited data of the studies presented here, it is not possible to make a statement regarding the excretion of PRI‐002. The present data clearly demonstrate that there is a dose‐proportional increase of dose‐dependent PK parameters like Cmax, AUCs, and Css. It is suggested that the overproportional increase of AUCs in cohort 2 compared to cohort 1 is subject to chance and based on the longer t½ß of subjects in cohort 2.
When administered daily, the accumulation factor is about 3. Steady‐state plasma levels were reached after about 5 t½β half‐lives. They increased proportional to dose and reached 2.8 ng/mL (1.8 nM) at an oral dose of 320 mg PRI‐002/day. When compared to plasma levels yielded in successful proof‐of‐concept studies with transgenic AD mice, plasma levels achieved in humans already after single oral dosing are in the same range of those observed in transgenic mice that received a high therapeutic dose 4 . Therefore, it can be assumed that the necessary plasma levels required for the treatment of AD patients with PRI‐002 can be accomplished. The presented data have been obtained in healthy young volunteers and does not allow a sample size calculation of a phase II proof‐of‐concept trial in AD patients. In addition, metabolization and blood–brain barrier (BBB) penetration in the AD study population can be different to young volunteers. An allometric scaling should be considered for getting a more precise estimation on the clearance in the intended study population and calculating the dosing for a proof‐of‐concept trial. In any case, the presented plasma levels of the SAD and MAD studies offer an adequate range for the desired treatment of AD in patients. In conclusion, safety and pharmacokinetic data support further clinical development of PRI‐002.
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.
D.W. is co‐inventor of patents covering the composition of matter of RD2. D.W., D.J, K.A., and A.W. are co‐founders and shareholders of the company “Priavoid GmbH,” which is further developing PRI‐002 for clinical use. J.K., C.F., M.W., M.H., W.R., and M.W. declare no competing interests.
ACKNOWLEDGMENTS
The German Helmholtz Association and the Forschungszentrum Jülich funded the phase I study and the necessary pre‐clinical research. The MAD study was funded additionally in part by Part the Cloud: Translational Research Funding for Alzheimer's Disease (PTC) Award Number 18PTC‐19‐605853 from the Alzheimer's Association.
Kutzsche J, Jürgens D, Willuweit A, et al. Safety and pharmacokinetics of the orally available antiprionic compound PRI‐002: A single and multiple ascending dose phase I study. 2020;6:e12001 10.1002/trc2.12001
REFERENCES
- 1. Willbold D, Kutzsche J. Do we need anti‐prion compounds to treat Alzheimer's disease. Molecules. 2019;24(12):2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Groen T, Schemmert S, Brener O, et al. The Abeta oligomer eliminating D‐enantiomeric peptide RD2 improves cognition without changing plaque pathology. Sci Rep. 2017;7(1):16275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Leithold LH, Jiang N, Post J, et al. Pharmacokinetic properties of a novel D‐peptide developed to be therapeutically active against toxic beta‐amyloid oligomers. Pharm. Res. 2016;33(2):328‐336. [DOI] [PubMed] [Google Scholar]
- 4. Schemmert S, Schartmann E, Zafiu C, et al. Abeta oligomer elimination restores cognition in transgenic Alzheimer's mice with full‐blown pathology. Mol. Neurobiol. 2019;56(3):2211‐2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kutzsche J, Schemmert S, Tusche M, et al. Large‐scale oral treatment study with the four most promising D3‐derivatives for the treatment of Alzheimer's disease. Molecules. 2017;22(10). 10.3390/molecules22101693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schemmert S, Schartmann E, Honold D, et al. Deceleration of the neurodegenerative phenotype in pyroglutamate‐Abeta accumulating transgenic mice by oral treatment with the Abeta oligomer eliminating compound RD2. Neurobiol. Dis. 2019;124:36‐45. [DOI] [PubMed] [Google Scholar]
- 7. Forny‐Germano L, Lyra e Silva NM, Batista AF, et al. Alzheimer's disease‐like pathology induced by amyloid‐beta oligomers in nonhuman primates. J Neurosci. 2014;34(41):13629‐13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gandy S, Simon AJ, Steele JW, et al. Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid‐beta oligomers. Ann Neurol. 2010;68(2):220‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sengupta U, Nilson AN, Kayed R. The role of amyloid‐β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine. 2016;6:42‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long‐term potentiation in vivo. Nature. 2002;416(6880):535‐539. [DOI] [PubMed] [Google Scholar]
- 11. Walsh DM, Klyubin I, Shankar GM, et al. The role of cell‐derived oligomers of Abeta in Alzheimer's disease and avenues for therapeutic intervention. Biochem Soc Trans. 2005;33(Pt 5):1087‐1090. [DOI] [PubMed] [Google Scholar]
- 12. Olsson TT, Klementieva O, Gouras GK. Prion‐like seeding and nucleation of intracellular amyloid‐β. Neurobiol Dis. 2018;113:1‐10. [DOI] [PubMed] [Google Scholar]
- 13. Cohen M, Appleby B, Safar JG. Distinct prion‐like strains of amyloid beta implicated in phenotypic diversity of Alzheimer's disease. Prion. 2016;10(1):9‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Walker LC, Schelle J, Jucker M. The prion‐like properties of amyloid‐beta assemblies: implications for Alzheimer's disease. Cold Spring Harb Perspect Med. 2016;6(7). 10.1101/cshperspect.a024398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Greenway CV, Stark RD. Hepatic vascular bed. Physiol Rev. 1971;51(1):23‐65. [DOI] [PubMed] [Google Scholar]
- 16. Elfgen A, Hupert M, Bochinsky K, et al. Metabolic resistance of the D‐peptide RD2 developed for direct elimination of amyloid‐beta oligomers. Sci. Rep. 2019;9(1):5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elfgen A, Santiago‐Schubel B, Gremer L, Kutzsche J, Willbold D. Surprisingly high stability of the Abeta oligomer eliminating all‐d‐enantiomeric peptide D3 in media simulating the route of orally administered drugs. Eur J Pharm Sci. 2017;107:203‐207. [DOI] [PubMed] [Google Scholar]