

Abstract
Background
Metastatic progression of breast cancer is still a challenge in clinical oncology. Therefore, an elucidation how carcinoma cells belonging to different breast cancer subtypes realize their metastatic capacities is needed. The aim of this study was to elucidate a similarity of activated molecular pathways underlying an enhancement of invasiveness of carcinoma cells belonging to different breast carcinoma subtypes.
Materials and methods
In order to reach this aim, parental and invasive (INV) MDA-MB-231 (triple-negative), T47D (hormone receptor-positive), and Au565 (Her2-positive) breast carcinoma cells were used and their molecular phenotypes were compared using a proteomic approach.
Results
Independently from breast cancer subtypes, INV cells have demonstrated fibroblast-like morphology accompanied by enhancement of invasive and migratory capacities, increased expression of cancer stem cell markers, and delayed tumor growth in in vivo animal models. However, the global proteomic analysis has highlighted that INV cells were different in protein expressions from the parental cells, and Her2-positive Au565-INV cells showed the most pronounced molecular differences compared to the triple-negative MDA-MB-231-INV and hormone receptor-positive T47D-INV cells. Although Au565-INV breast carcinoma cells possessed the highest number of deregulated proteins, they had the lowest overlapping in proteins commonly expressed in MDA-MB-231-INV and T47D-INV cells.
Conclusions
We can conclude that hormone receptor-positive cells with increased invasiveness acquire the molecular characteristics of triple-negative breast cancer cells, whereas Her2-positive INV cells specifically changed their own molecular phenotype with very limited partaking in the involved pathways found in the MDA-MB-231-INV and T47D-INV cells. Since hormone receptor-positive invasive cells share their molecular properties with triple-negative breast cancer cells, we assume that these types of metastatic disease can be treated rather equally with an option to add anti-hormonal agents. In contrast, Her2-positive metastasis should be carefully evaluated for more effective therapeutic approaches which are distinct from the triple-negative and hormone-positive metastatic breast cancers.
Key words: breast cancer cells, cancer stem cells, invasiveness, migration, metastasis, molecular profiling
Introduction
According to the World Health Organization (WHO) reports, 2.1 million women worldwide are diagnosed with breast cancer every year. In 2018, it was estimated that 627000 deaths were counted for breast cancer and it was approximately 15% of all cancer-related deaths among women (www.who.int). It is believed that metastatic progression is responsible for most breast cancer deaths.1 Therefore, elucidation of molecular properties of carcinoma cells responsible for metastatic spread can help in the development of biomarkers or therapeutic targets to predict or improve clinical outcome in breast cancer patients.
Metastasis cascade contains a number of steps such as cancer cell invasion in surrounding tissues followed by cell intravasation into the blood and/or lymphatic vessels, extravasation into the tissue of distant organs, and, finally, colonization and growth in the targeted organs.2 Since metastatic spread begins from cancer cell invasion through the biological membranes and extracellular matrix (ECM), it is logical to suggest that breast carcinoma cells with enhanced metastatic capacities should possess an augmented invasiveness.
It is currently known that different breast cancer phenotypes reveal different propensities for cancer cell dissemination. Thus, hormone-dependent breast carcinomas demonstrate lower metastatic potential in comparison with Her2-positive and triple-negative breast cancers.3 However, it is not fully understood whether carcinoma cells belonging to different breast cancer subtypes have an activation of similar or different pathways involved in metastatic progression. There is a limited number of reports on the molecular diversities of primary tumors and metastatic lesions. Therefore, it is difficult to study the molecular perturbations that occur in breast cancer cells during metastatic progression.
Since triple-negative breast cancer possesses the most aggressive metastatic potential, other types of breast carcinomas (hormone receptor-positive and Her2-positive) are considered as metastatically active if they demonstrate an overexpression of molecules upregulated in triple-negative tumors (EGFR, cytokeratines 5/6, 14, 17, etc.).4 Therefore, it is assumed that different subtypes of metastatic breast cancer cells can have the same therapeutic targets.5 This hypothesis leads to the current therapeutic strategy which does not really stratify the patients depending on the molecular patterns of the secondary tumors. Nowadays, a chemotherapeutic approach contains an arsenal of cytotoxic compounds nonspecifically killing the rapidly dividing carcinoma cells.6 There are only few additional options to use anti-hormonal and anti-Her2 agents in the treatment of patients with appropriate tumor subtypes.7 This strategy could be logical and acceptable, if metastatic breast cancer cells of different histopathological subtypes reveal the unified molecular properties and common molecular pathways are involved in metastatic cascade.
This study aimed to elucidate a similarity of activated molecular pathways underlying an enhancement of invasiveness in carcinoma cells belonging to different breast carcinoma subtypes.
Materials and methods
Cells and cell cultures
MDA-MB-231 (triple-negative type (TNBC): estrogen, progesterone and HER2/neu receptor negative (ER-, PR-, HER2/neu-), T47D (luminal A type: ER+, PR+, HER2/neu-), and Au565 (Her2/neu-positive cells: ER-, PR-, HER2/neu+) cells were purchased from the American Type Culture Collection. All cell lines were grown in RPMI1640 medium supplemented with 2 mM L-glutamine, 50 U/mL penicillin, 50 μg/mL streptomycin, and 10% fetal calf serum (FCS) (Thermo Fisher Scientific, Vienna, Austria). Cell cultures were maintained in a 5% CO2 humidified atmosphere.
Breast carcinoma cells with increased invasive abilities (INV cells) were obtained from parental breast carcinoma cells after repetitive cell migration through the uncoated 8 μm-pore membrane in the Boyden chamber (Corning Life Sciences, Berlin, Germany) toward the cell culture medium containing an attractant (10% FCS). Cell migration was repeated ten times every 2 weeks. In order to support and control the invasive abilities of the INV cells, cell migration through the membrane with 8 μm pores was repeated and evaluated every month.
3D tomographic microscopy
Parental and invasive MDA-MB-231, T47D, and Au565 cells were seeded into the glass-bottom dishes with diameter of 35 mm (Ibidi, Switzerland). Seeded cells were incubated for 24 hours at 37°C and 5% CO2 humidified atmosphere. Next, cells were analysed for their morphology using 3D tomographic microscope (3D Cell Exlplorer, Nanolive SA, Switzerland), and 3D tomographic images (z-stacks) were collected for further computational analysis using STEVE software (Nanolive SA, Switzerland).
Migration assay
Migratory capacities of the investigated breast cancer cells were assessed using the Boyden chambers and automated live cell imager Lionheart FX (BioTek, Vermont, USA). Parental and INV breast carcinoma cells were serum-starved for 24 hours, next harvested and seeded (5x105 cells in 2.5 mL RPMI 1640 serum-free medium) into the upper insert with an 8-μm pore size polycarbonate membrane. The lower chamber contained the same volume of cell culture medium with chemoattractant (10% FCS). After 24 hours of cell incubation at 37°C in a 5% CO2 humidified atmosphere, the inserts were removed and the migrated cells were stained with the Hoechst 33342 (1,5 μg). Using the Gen 5 3.0 software, the images of the stained cells were captured and a number of migrated cells were counted. All cell migration assays were performed in triplicates, and repeated at least three times.
Invasive abilities of breast carcinoma cells
Cell Biolabs CytoSelect TM Laminin and Collagen I Cell Invasion Assay Kits (Cell Biolabs, Inc., San Diego, USA) were used to study the invasive abilities of the investigated parental and newly obtained INV breast carcinoma cells. Briefly, breast carcinoma cells harvested in the RPMI1640 serum-free quenching medium were placed in the upper insert with the laminin- or collagen I-coated membranes. The lower chamber contained RPMI1640 medium with chemoattractant (10% FCS). Prepared plates were incubated for 24 hours at 37°C in a 5% CO2 humidified atmosphere. Cells migrated through the laminin- or collagen I-coated membranes were detached using the cell detachment solution, and then lysed in the lysis buffer containing CyQuant® GR dye solution. Fluorescence of the investigated samples was measured with a fluorescence microplate reader Synergy H1M (BioTek, Vermont, USA) at 480nm/520nm. Background readings from buffers were subtracted from the readings of each sample. Invasion assay was repeated at least three times for each investigated cell line.
Cell growth
Cell proliferation rate was determined as previously described.8 Parental MDA-MB-231, T47D and Au565 and their invasive pairs MDA-MB-231-INV, T47D-INV and Au565-INV breast cancer cells were seeded (1 x 104) in triplicates in 4.0-mL in 6-well plates. Cells were incubated for 24 hours at 37°C and then trypsinized and counted using Beckman Coulter Vi-CELL AS cell viability analyzer (Beckman Coulter, Fullerton, CA, USA). Cell population doubling time (DT) in hours was determined using the computerized tool (http://www.doubling-time.com/compute.php) and following equation:
DT (hours) = duration (24 hours) * log(2) / log (N1) – log (N0), where N1 – final cell number; N0 – initial cell number. Each experiment was repeated at least three times to ensure the reproducibility of the results.
Flow cytometry
For analysis of CD44 and CD24 expression in parental and invasive MDA-MB-231, T47D, and Au565 cells by flow cytometry (BD FACS Canto II, Becton Dickinson, San Jose, USA) 1x106 cells per tube were pelleted at 200xg, resuspended and stained with 4 μg/mL human monoclonal anti-CD44-PE and anti-CD24-FITC antibodies (Miltenyi Biotec, Bergisch Gladbach, Germany). An equivalent amount of PE- and FITC-conjugated IgG1k (Miltenyi Biotec, Bergisch Gladbach, Germany) were used as isotype controls. The labeled cells were washed in the FACS buffer and then analyzed by flow cytometry, and results were evaluated by the subsequent FACS DIVA Software 7.0. All measurements were repeated at least three times.
ALDEFLUOR Assay for detection of the ALDH1 activity
To detect ALDH1 enzymatic activities in the investigated parental and INV breast carcinoma cells, ALDEFLUOR assay kit was used according to the manufacturer instructions (STEMCELL Technologies, Koeln, Germany). Briefly, 106 cells/mL were harvested from cell cultures, centrifuged at 200xg, and resuspended in 1 mL of ALDEFLUOR Buffer. For ALDEFLUOR staining, 5 μL/mL of the activated ALDEFLUOR™ Reagent was added into each investigated sample. Each experimental sample was accompanied by the negative control after adding 5 μL of diethylaminobenzaldehyde (DEAB), the ALDH1 inhibitor. All prepared test and negative control samples were incubated at 37°C for 30 minutes. Following incubation, the samples were centrifuged for 5 minutes at 250 xg. Supernatants were discarded, and cells were resuspended in 0.5 mL of ALDEFLUOR™ Assay Buffer and stored on for 30 minutes. Next fluorescence intensity of the stained cells was analyzed by the flow cytometry, and a percentage of breast carcinoma cells with ALDH1 activity was estimated by the subsequent FACS DIVA Software 7.0. All experiments were repeated three times.
Western blot analysis
Western blot was performed as published previously9 using ALDH1A3 rabbit polyclonal antibody (ab129815, Abcam, Cambridge, UK) or GAPDH rabbit monoclonal antibody (Cell Signaling Technologies, Frankfurt am Mein, Germany) as a loading control. For evaluation of protein expression, X-ray films (GE Healthcare, Chicago, IL, USA) were scanned and analyzed by the Image StudioTM Lite 5.0 (LI-COR Biotechnology, Lincoln, NB, USA). The Integrated Density Value (IDV) was obtained as a ratio of normalized protein band densities in parental and INV cells after background correction.
Proteomic analysis
Materials used for the analysis of protein profiling
TMT 6 plex™ Isobaric Label Reagent Set (6-plex TMT) was obtained from Life Technologies (Grand Island, NY, USA). Trypsin (Sequencing grade modified porcine) was obtained from Promega. Acetonitrile was purchased from JT Baker, and formic acid was obtained from EMD Millipore (Billerica, MA, USA). MyProt-Buffer 1, MyProt-Buffer 2, MyProt-Buffer 3, MyProt-PhosphoWASH Buffer, and MyProt-PhosphoELU Buffer were utilized by MyOmicsDx, Inc (Towson, MD, USA).
Protein preparation
Protein samples were processed by MyOmicsDx, Inc (Towson, MD, USA) using “Filter Assisted Sample Preparation” (FASP) method. Briefly, protein samples in 9M UREA were reduced with 5 mM TCEP at 37°C for 45 min and reduced cysteines were blocked using 50 mM iodoacetamide at 25°C for 15 min. Protein samples (100 μg each) were then cleaned using 10 kDa Amicon Filter (UFC501096, Millipore, USA) three times using 9M urea and two times using MyProt-Buffer 1 (MyOmicsDx, Inc). Samples were then proteolyzed with trypsin (V5111, Promega, USA) for 12 hrs at 37°C. The peptide solution then was acidified by adding 1% trifluoroacetic acid (TFA) and was incubated at room temperature for 15 min. A Sep-Pak light C18 cartridge (Waters Corporation, USA) was activated by loading 5 mL 100% (vol/vol) acetonitrile and was washed by 3.5 mL 0.1% TFA solution two times. Acidified digested peptide solution was centrifuged at 1,800 × g for 5 min, and the supernatant was loaded into the cartridge. To desalt the peptides bound to the cartridge, 1 mL, 3 mL, and 4 mL of 0.1% TFA were used sequentially. To elute the peptides from the cartridge, 2 mL of 40% (vol/vol) acetonitrile with 0.1% TFA was used, and this elution was repeated two more times (for a total of 6 mL of eluate). It was important to ensure that the cartridge had stopped dripping before each sequential wash and elution solution was applied. The eluted peptides were lyophilized overnight and reconstituted in 100 μL of MyProt-Buffer 2 (MyOmicsDx, Inc, MD, USA).
Multiplexed TMT labeling
Digested peptides from samples in a volume of 100 μl MyProt-Buffer 2 were labeled using 6-plex TMT reagents. After 2 hours, 8 μL of 5% hydroxylamine were added to the sample and incubated for 15 minutes to quench the reaction. Labeled peptides were dried to remove organic solvents and reconstituted in 500ul MyProt-Buffer 3 (MyOmicsDx, Inc), combined and fractionated on a bRPLC (basic Reverse Phase Liquid Chromatography) column (XBridge BEH C18 Column, 5 μm, 2.1 x 100 mm) via XBridge BEH C18 Guard Column (Waters Corporation, USA) using an Agilent 1260 HPLC system. Peptides in each fraction were dried.
Strong cation exchange chromatography and TiO2-based phosphopeptide enrichment
Peptides were fractionated by strong cation exchange (SCX) chromatography. Briefly, 10 mg of lyophilized peptides mixture was resuspended in 1 ml of SCX solvent A (5 mM KH2PO4 pH 2.7, 30% ACN) and fractionated by SCX chromatography on a PolySULPHOETHYL A (5 μm, 200 Å) column (200 × 9.4 mm; PolyLC Inc., Columbia, MD) by employing an increasing gradient of SCX solvent B (5 mM KH2PO4 pH 2.7, 30% ACN, 350 mM KCl) on an Agilent 1100 LC system. For each experiment, a total of 90 fractions were initially collected, which were pooled into fractions of various sizes (13, 16, or 22 fractions for each respective experiment), and dried using speedvac (Eppendorf, USA). Each fraction was subjected to TiO2-based phosphopeptide enrichment. Briefly, TiO2 beads were incubated with DHB solution (80% ACN, 1% TFA, 3% 2,5-dihydroxybenzoic acid (DHB)) for 2–4 hours at room temperature. Each fraction was resuspended in DHB and incubated with pretreated TiO2 beads (5 mg) overnight at room temperature. Phosphopeptide-bound TiO2 beads were washed three times with DHB solution and twice with MyProt-PhosphoWASH buffer. Peptides were eluted three times with 40 μl of MyProt-PhosphoELU buffer.
Nanoflow electrospray ionization tandem mass spectrometry analysis
Data dependent MS/MS analyses of the TMT labeled peptides were carried out on a Thermo Scientific™ EASY-nLC 1000™ HPLC system and Thermo Scientific EASYSpray™ source with analytical nanoflow column system includes 2 cm trap column and 75 μm x 20 cm analytical column both packed with Magic C18 AQ, 5 μm, 100 Å (Michrom Bioresources, USA). Samples were analyzed on a Thermo Scientific™ Q Exactive Mass Spectrometer using FT HCD MS2 fragmentation mode. Peptides were electrosprayed through a 15 μm emitter (PF3360-75-15-N-5, New Objective) at a voltage of 2.0 kV spray voltage. Reversed phase solvent gradient consisted of 0.1% formic acid with increasing levels of 90% acetonitrile in 0.1% formic acid over a period of 90 minutes. The Q Exactive instrument was operated to automatically switch between full scan MS and MS/MS acquisition. Survey full scan MS spectra (m/z 350−1800) was acquired in the Orbitrap with 35,000 resolution after accumulation of ions to a 3 × 106 target value based on predictive AGC from the previous full scan. Dynamic exclusion was set to 15 s. The 10 most intense multiply-charged ions (z ≥ 2) were sequentially isolated and fragmented in the Axial Higher energy Collision-induced Dissociation (HCD) cell using normalized HCD collision energy at 30% with an AGC target 1e5 and a maxima injection time of 400 ms at 35,000 resolution.
MS data analysis
Mass spectrometry raw files were automatically processed through Proteome Discoverer 2.1 software using Xtract in addition to default spectrum selector node. The searches were performed using Mascot search engine together with Sequest HT interfaced with different processing nodes of Proteome Discoverer 2.1 SP1. Search parameter included oxidation on methionine, deamidation on residues N and Q as different variable modifications and TMT 6-plex on N-terminus and lysine residue, carbamidomethyl on cysteine residue as different fixed modifications. Mass tolerances on precursor and fragment masses were set to 15 ppm and 0.03 Da, respectively. Peptide validator node was used for peptide validation with a stringent cutoff of 0.01 and relaxed cutoff of 0.05 (false discover rate, FDR) and 1% FDR cutoff was used to filter the data. High confidence (0.1% FDR) and top-ranked peptide were considered with protein grouping option. Protein ratios were normalized through MyProt-QuantiR (MyOmicDx, Inc) software package and peptides with >30% isolation interference were excluded from the protein quantification to avoid potential interference of reporter ions from contaminant peaks.
Bioinformatic analysis of proteomic data
Alterations in activation of intracellular molecular pathways relatively to normal samples were quantitatively assessed using the bioinformatical platform Oncobox.10 The structures of 3121 molecular pathways were extracted from the following public databases: Reactome,11 NCI Pathway Interaction Database,12 Kyoto Encyclopedia of Genes and Genomes,13 HumanCyc,14 Biocarta [www.biocarta.com] and Qiagen (www.qiagen.com/us/shop/genes-and-pathways/pathway-central/). Molecular pathways were visualized using Oncobox software.15
Pathway activation level (PAL) for each molecular pathway was calculated according to the formula:
where PALp - molecular pathway p activation level; CNRn (case-to-normal ratio) - ratio of protein n product concentrations in the test sample and in the norm (average value in the control group); ln – natural logarithm; discrete value ARRnp (activator/repressor role) for protein n in pathway p is defined as follows:
Visualization of the molecular pathways was performed using R “ggplot2” and “VennDiagram” packages. 1000 random permutations were made in order to test significance of overlaps for top deregulated proteins or molecular pathways.
Protein set enrichment analysis was performed as previously described.16 Top 10% of up- or down-regulated proteins were analyzed using “GSEAPreranked” module of the software using the following gene sets databases: c2.cp.kegg. v6.2.symbols.gmt, c2.cp.reactome.v6.2.symbols. gmt, c5.all.v6.2.symbols.gmt.
Animal experiments
Animal experiments were approved by the Ministry of Agriculture, Forestry and Food of the Republic of Slovenia No. 34401-15/2017/8 based on the approval of the National Ethics Committee for Experiments on Laboratory Animals, and were in compliance with the standards required by the EU Directive 2010/63/EU for animal experiments. Female NUDE (HSD: Athymic Nude-Foxn1NU, Envigo RMS Srl, San Pietro al Natisone, Italy) mice were maintained on a 12 h light–dark schedule under specific pathogen-free conditions at constant room temperature and humidity. Food and water were provided ad libitum. In order to estimate the tumorigenic capacities of the investigated cells, parental and INV breast carcinoma cells were injected at a concentration of 1×106 in 0.1 ml NaCl subcutaneously for the induction of subcutaneous tumors in 6 weeks old NUDE mice (6 animals per group). Tumor formation was monitored every day until tumors became palpable. Afterward tumors were measured every second day using Vernier Caliper in three perpendicular diameters (a, b, c) and tumor volumes were calculated according to formula V = (π × a × b × c)/6. The tumor doubling times were calculated as the time in which tumor reaches its double volume; i.e. from 40 mm3 to 80 mm3. When tumors reached 250 mm3 or significant palpable axillary or inguinal lymph nodes were detected, animals were sacrificed and autopsied. Lungs, liver, kidney, intestine, colon, ovarium, spleen, lymph nodes were visually inspected for macrometastases. Tumors and axillary and inguinal lymph nodes were excised for histological analysis. The tumors and lymph nodes were fixed in IHC zinc fixative (BD Biosciences, San Diego, CA, USA), embedded to paraffin blocks and cut into three consecutive 2-μm-thick sections. The first section of tumor and lymph nodes was H&E stained to evaluate the morphology. The second section of tumor was stained with Masson`s trichrome in order to determine collagen. To determine cell proliferation, we incubated the third tumor section with a monoclonal mouse anti-human antibody against Ki-67 (1:200; clone MIB-1 M7240; DAKO Agilent technologies inc., Denmark). The sections were stained on an automated slide stainer with an indirect, biotin-free system (Optiview DAB IHC Detection kits, 760-700; Ventana Medical Systems, Roche, USA). The other two sections of lymph nodes were stained with a monoclonal rabbit anti-human E-cadherin antibody (1:2500, ab40772, Abcam) and a polyclonal rabbit anti-human pan-cytokeratin antibody (1:3000, ab217916, Abcam). These primary antibodies were then detected with a peroxidase-conjugated streptavidin–biotin secondary antibody (Rabbit specific HRP/DAB detection IHC kit, ab64261, Abcam) and counterstained with hematoxylin. Ten images of each immunohistochemically stained sections were captured with a DP72 CCD camera (Olympus; Hamburg, Germany) connected to a BX-51 microscope (Olympus, Hamburg, Germany) under the 200x or 600x magnification. The number of Ki67-positive cells and the area of collagen were determined on the acquired images using CellSens Dimensions software (Olympus, Hamburg, Germany).
Results
Cellular morphology
As can be seen in Figure 1, INV breast carcinoma cells with enhanced invasive abilities have markedly changed their morphological phenotype toward the formation of fibroblast-like and spindle-shaped cells. INV carcinoma cells increased their cellular surfaces due to membrane ruffling accompanied by lamellipodia and filopodia formation. Independently from the breast cancer subtype, all INV cells were characterized by more pronounced mitochondria fission compared to parental breast carcinoma cells. Interesting to note that all INV cells were characterized by the lower number of lipid droplets in comparison with their parental counterparts. Additionally, T47D-INV and Au565-INV cells were characterized by the lipid droplets translocation to the filopodia tips, whereas a perinuclear lipid droplet localization was detected in parental cells only.
Figure 1.
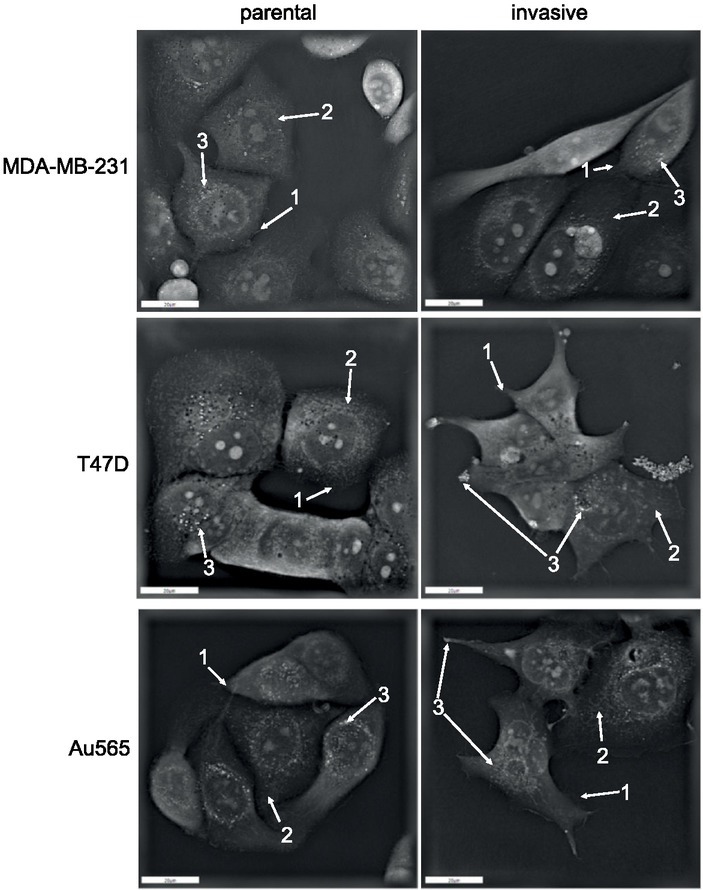
Holographic microscopy of breast cancer cells. Parental and invasive MDA-MB-231, T47D, and Au565 cells were analysed for their morphology using 3D tomographic microscope as described in Materials and Methods. Arrow 1: filopodia; Arrow 2: mitochondria; Arrow 3: lipid droplets.
Migratory and invasive abilities of the investigated breast carcinoma cells
To determine whether newly obtained breast carcinoma INV cells possess affected invasive and migratory abilities, appropriate assays were performed.
The investigated cells were evaluated for their migratory abilities through the uncoated 8-μm pore size polycarbonate membrane (Figure 2A). Among the parental MDA-MB-231, T47D, and Au565 breast carcinoma cells, triple-negative MDA-MB-231 and Her2-positive Au565 cells showed the more pronounced cell migration than hormone-dependent T47D cells. Thus, 901.7 + 109.4 MDA-MB-231 cells and 1089.7 + 360.8 Au565 cells migrated toward the RPMI 1640 medium containing attractant (10% FCS), whereas parental T47D cells demonstrated 176,3 + 54.4 migrated cells. All newly obtained MDA-MB-231-INV, T47D-INV Au565-INV invasive breast carcinoma cells were more migratory than their parental counterparts (5308.3 + 1513.5, 1261.7 + 776.6 and 19864.0 + 7830.6 migrated MDA-MB-231-INV, T47D-INV and Au565-INV cells, respectively).
Figure 2.
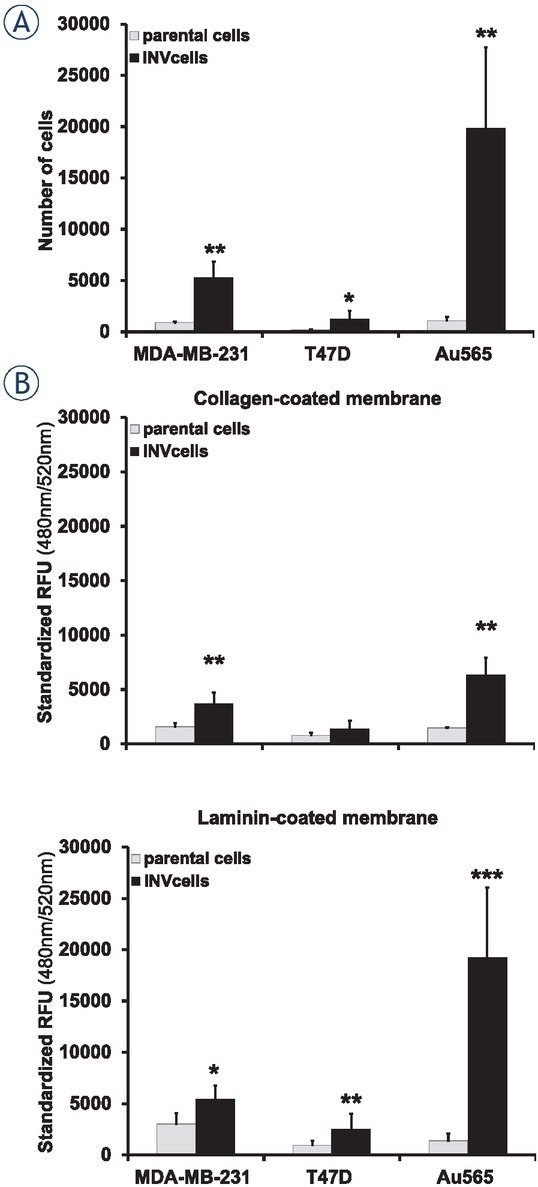
Breast cancer cell migration and invasion. (A) Differences in migration of parental and INV breast carcinoma cells. (B) Invasion of parental and invasive breast carcinoma cells through the collagen- and laminin coated membranes. Columns represent the mean value including standard deviation obtained from three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001).
As it was detected, all INV breast carcinoma cells demonstrated an enhancement of their capacities to invade through the collagen 1- and laminin-coated membranes (Figure 2B). Triple-negative MDA-MB-231-INV and hormone-dependent T47D-INV cells showed a ~ 2.38- and ~ 1.72-fold and ~ 1.8- and ~ 2.67-fold enhancement of invasiveness over that in parental cells through the collagen 1- and laminin-coated membranes, respectively. Invasive capacities of Her2-positive Au565-INV breast carcinoma cells through the collagen 1- and laminin-based matrix was estimated as a ~4.29- and ~13.85-fold higher than in their parental counterparts Au565 cells, respectively. Among all investigated INV cells, hormone-positive T47D-INV cells revealed the lowest invasive potential compared to triple-negative MDA-MB-231-INV and Her2-positive Au565-INV breast carcinoma cells. Thus, the invasiveness of T47D-INV cells through the collagen 1-coated membranes was ~2.7- and ~4.6-fold lower than of MDA-MB-231-INV and Au565-INV, respectively. Similarly, T47D-INV cells were ~2.2-fold less invasive through the laminin-coated membrane than MDA-MB-231-INV cells. In contrast, T47D-INV cells demonstrated ~7.6-fold lower invasiveness through the laminin-coated membrane than Au565-INV breast carcinoma cells.
Breast cancer stem cell (CSC) markers CD24, CD44 and ALDH1
Since an enhancement of the invasive abilities of carcinoma cells could be associated with altered expression of CD24 and CD44 markers17,18, we have evaluated their expressions in the INV breast cancer cells in comparison with parental cells (Figure 3). It was found that only MDA-MB-231-INV and T47D-INV cells changed their phenotype toward the loss CD44-/CD24+ subpopulation and increased number of CD44+/CD24-/low cells. Thus, ~ 90% and ~ 80% of MDA-MB-231-INV and T47D-INV cells possessed CD44+/CD24- phenotype, whereas parental MDA-MB-231 showed CD44+/CD24 expression in ~ 50% cells and parental T47D cell line did not contain CD44+/CD24- cells. Interesting to note that Her2-positive Au565-INV cells did not markedly differ in CD44+/CD24- expression from their parental counterparts (~70% in parental Au565 versus ~ 80% in Au565-INV cells).
Figure 3.
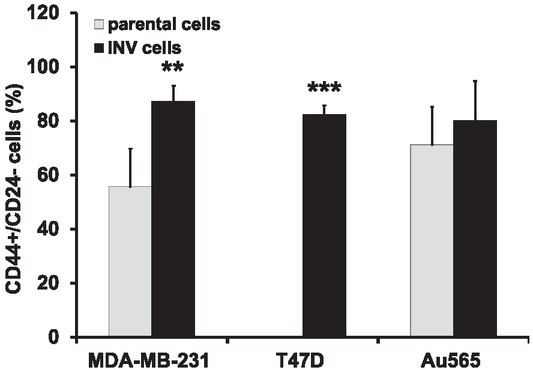
Expression of CD44+/CD24- cancer stem cell markers in breast carcinoma cells. Columns represent the mean value including standard deviation obtained from three independent experiments (**p < 0.01; ***p < 0.001).
It is known that enhanced ALDH1 expression and activity can be associated with breast carcinoma cells aggressiveness, invasiveness and metastasis.19 Therefore, we next determined the ALDH1A1 and ALDH1A3 expressions and ALDH1 activity in all investigated breast carcinoma cells (Figure 4). Although ALDH1A1 was expressed neither in parental nor in INV breast carcinoma cells (data not shown), ALDH1A3 was differently presented in parental and INV cells (Figure 4A).
Figure 4.
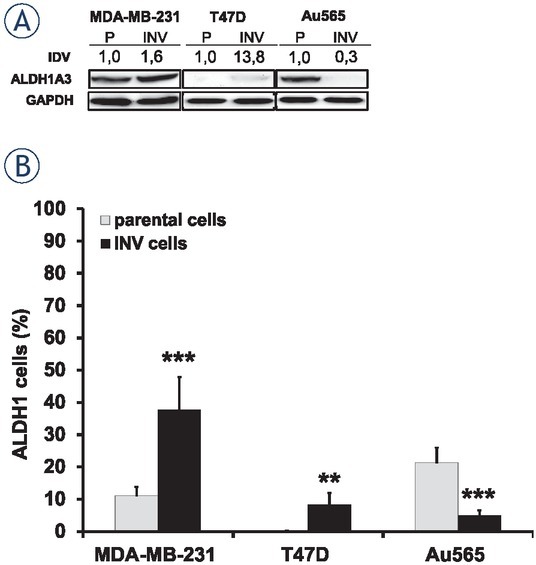
ALDH1 expression and activity in breast cancer cells. (A) Exponentially growing parental and INV breast carcinoma cells were collected for Western blot analysis. Total protein extracts were prepared from the cells and then processed for immunoblotting using antibodies to detect ALDH1A3. GAPDH was used as a loading control. IDV was determined as described in the section Materials and Methods. (B) ALDH1 activities were determined in the investigated parental and INV cells. Columns represent the mean value including standard deviation obtained from three independent experiments (**p < 0.01; ***p < 0.001).
Parental MDA-MB-231 and Au565 cells demonstrated equal ALDH1A3 expression, whereas parental T47D possessed very weak ALDH1A3 expression. Both invasive MDA-MB-231-INV and T47D-INV cells revealed an upregulation of ALDH1A3. In contrast, Au565-INV cells showed significant downregulation of this protein. ALDH1 activity was detected in parental MDA-MB-231 and Au565 cells only (11.13 + 2.65 % and 21.42 + 4.59 %, respectively) (Figure 4B), whereas parental T47D cell line did not contain cells with ALDH1 activity. Similarly to ALDH1A3 expression, ALDH1 activity was increased in MDA-MB-231-INV and T47D-INV cells (37.70 + 10.2 % and 8.31 + 3.62 %, respectively). Again, Au565-INV cells showed a repression of the ALDH1 activity (4.92 + 1.61 %) compared to parental Au565 cells.
Cell proliferation
To estimate the proliferation rates of the parental (MDA-MB-231, T47D, Au565) and INV cells (MDA-MB-231-INV, T47D-INV, Au565-INV), cell growth was evaluated and the doubling time of each investigated cell line was determined (Figure 5). Hormone-dependent T47D cells had the slowest proliferation capacities with a doubling time of 36.6 + 1.8 hours. MDA-MB-231 and Au565 breast carcinoma cells revealed shorter doubling times of 18.9 + 3.9 hours and 20.6 + 3.7 hours, respectively. Although invasive triple-negative MDA-MB-231-INV cells did not demonstrate the significant differences with their parental counterparts, hormone receptor-positive T47D-INV and Her2-positive Au565-INV cells showed an affected doubling time (~ 1.5-fold shorter time for T47D-INV and ~1.6-fold longer doubling time for Au565-INV compared to their parental counterparts).
Figure 5.
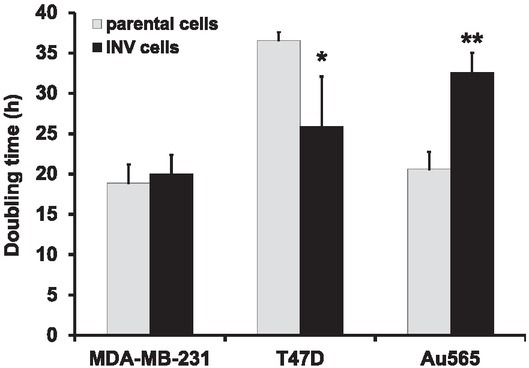
Cell proliferation rate in breast carcinoma cells. Parental and INV cells were evaluated for their doubling time as described in the Materials and Methods. For statistical evaluation, mean values and SD were calculated using at least three independent experiments; significance was determined by paired Student’s t-test (*p < 0.05; **p < 0.01).
Protein expression analysis and molecular pathway activation
To investigate molecular features linked to breast cancer cells invasiveness, we intersected top 10% differentially regulated proteins in all three cell lines, up- and downregulated proteins were intersected separately. Only six proteins were common for top upregulated ones in all the cell lines (Figure 6A), and three – for downregulated proteins (Figure 6C).
Figure 6.
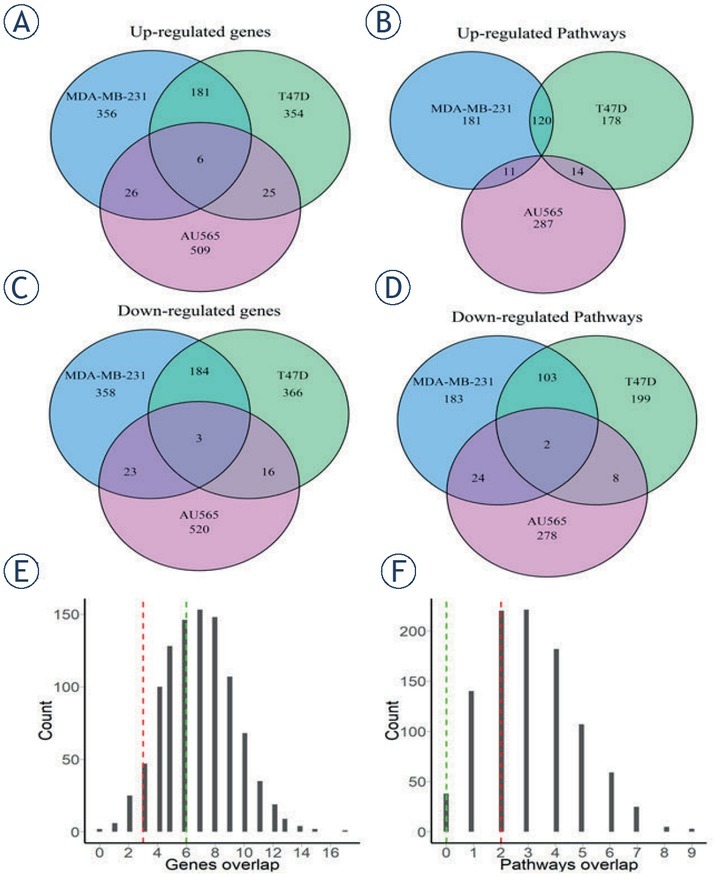
Bioinformatic analysis of proteins and pathways dysregulated in the INV compared to parental breast carcinoma cells. Overlap of up-regulated (A, B) or down-regulated (C, D) genes (A, C) and molecular pathways (B, D) between the investigated INV breast carcinoma cells. Top 10% features (proteins or pathways) were selected for the analysis. (E, F) Distribution of overlapped proteins for randomly selected groups (1000 random permutations).
We then tested whether this intersection is statistically significant by intersecting three random sets (576 out of 5755 proteins) 1000 times, the resulting histogram is shown in Figure 6E. The observed real intersection of top differential proteins didn’t exceed values obtained during random permutations, thus suggesting that the intersections observed may be random. This suggested also that the differential protein profiles were strongly different between the cell lines investigated.
Similarly, we calculated activation levels of 3118 molecular pathways using Oncobox algorithm that quantitatively estimates activation of molecular pathways.10,20 Pathway activation level (PAL) values were calculated for three cell lines tested in comparison with the control samples using quantitative proteomics expression data (supplementary Table 1). Positive values of PAL score indicate down-regulation of a pathway, and vice versa, negative values mean downregulation of a pathway.21 We then separately intersected top 10% of up- and downregulated pathways and found no common upregulated pathways in the three cell lines tested (Figure 6B), whereas two pathways were found to be strongly downregulated in all three cell lines (Figure 6D). As in the above protein intersection significance test, we intersected three random sets of pathways (312 out of 3118) for 1000 times, obtained histogram is shown on Figure 6F. We found that the real intersection of top deregulated pathways between the cell lines tested didn‘t exceed values for these random permutations. Again, this suggested that the intersections observed may be random and that the differential pathway profiles shared little identity for the cell lines investigated.
Table 1.
Proliferation abilities and collagen content in breast tumor xenografts
| Tumor cell line | % of proliferating | % of collagen |
|---|---|---|
| cells (mean ±se) | (mean ±se) | |
| MDA-MB-231 | 46.1 ± 3.1 | 18.7 ± 2.0 |
| MDA-MB-231-INV | 69.8 ± 4.7* | 22.9 ± 3.5 |
| T47D | 43.3 ± 2.3 | 3.3 ± 0.7 |
| T47D-INV | 22.9 ± 2.4*** | 21.6 ±1 .9*** |
| Au565 | 24.3 ± 1.7 | 32.3 ± 1.8 |
| Au565-INV | 57.7 ± 3.5*** | 6.1 ± 1.4*** |
p < 0.5; *** P < 0.001
In order to study the mechanisms of different cell line invasiveness, we focused on top differentially regulated pathways specific for each cell line. For this analysis we selected only pathways with >9 genes and used the following criteria: the pathway should be up-regulated in one cell line but down-regulated or at least 5-fold less activated in other lines. The most activated and satisfying this requirement pathway in T47D-INV cell line was “NCI Syndecan 3 mediated signaling events Main Pathway” (Figure 7A).
Figure 7.

Visualization of pathways involved in breast cancer cell invasiveness. (A) “NCI Syndecan 3 mediated signaling events Main Pathway” (B) “PECAM1_interactions_Main_Pathway”; (C) “reactome Regulation of innate immune responses to cytosolic DNA Main Pathway”; (D) “ILK Signaling Pathway Actin Polymerization Cytoskeletal Reorganization”. The pathway is shown as an interacting network, where green arrows indicate activation, red arrows indicate inhibition. Color depth of each node of the network corresponds to the logarithms of the case-to-normal (CNR) expression rate for each node, where “normal” is expression level in the control group, the scale represents extent of up/downregulation.
It was not up-regulated in MDA-MB-231-INV but instead was down-regulated in AU565-INV cells. Syndecans are membrane proteins controlling cell proliferation, differentiation, adhesion and migration 22 and were recently shown to promote breast cancer metastasis.23 Thus, invasiveness of T47D-INV cells may potentially be linked with activation of this pathway.
The top up-regulated pathway in AU565-INV cells was “PECAM1_interactions_Main_Pathway” (Figure 7B), but it was not upregulated in MDA-MB-231-INV and T47D-INV cells. This pathway appeared to be activated mostly due to over-expression of Src family tyrosine kinases (SFKs). These kinases are known to be deregulated in many cancer types and their inhibitors were tested in clinical trials.24 Indeed, inhibiting Src was previously associated with inhibited breast cancer cell migration and invasion and therefore may be linked with Au565-INV cell invasiveness in our case.25
The most up-regulated pathway in MDA-MB-231-INV cell line was “reactome Regulation of innate immune responses to cytosolic DNA Main Pathway” (Figure 7C), mostly due to over-expression of TRIM32. TRIM32 expression was previously found to be up-regulated in breast cancer tissues and cell lines, promoting cell proliferation and colony formation.26 High TRIM32 expression was also positively associated with lymph node metastasis.26
As MDA-MB-231-INV and T47D-INV cells had much more common up-regulated pathways, when compared to AU565-INV, we also studied which pathway is most up-regulated in both cell lines. It appeared that top pathway was “ILK Signaling Pathway Actin Polymerization Cytoskeletal Reorganization” (Myosin and actin were over-expressed, Figure 7D), which indicates that cytoskeletal re-arrangements may be common in these cell lines.
In addition we performed gene set enrichment analysis (GSEA) for top 10% of up- or down-regulated proteins. It appeared that, there no common gene sets at nominal p-value cut-off of 0.05 for up-regulated genes (Figure S1A), and 1 common gene set for down-regulated genes (Figure S1B), namely „GO_Neurological_system_process“. Detailed results of GSEA can be found supplementary Table S2.
In vivo
Next, we have evaluated the tumorigenic capacities of INV cells compared to their parental counterparts. It was observed that tumors originated from the INV cells demonstrated significantly delayed growth rates. Thus, parental and invasive Her2-positive Au565 had the slowest growth with tumor doubling times of 11.4 ± 1.6 days and 19.7 ± 0.9 days, respectively. The tumor doubling times of MDA-MD-231 and MDA-MB-231-INV tumors were 2.9 ± 0.2 and 10.7 ± 1.0 days, respectively.
Surprisingly, hormone receptor-positive T47D and T47D-INV tumors revealed the fastest growth rates compared to other tumor xenografts, with a tumor doubling times of 1.5 ± 0.3 and 3.1 ± 0.4 days, respectively (Figure 8).
Figure 8.
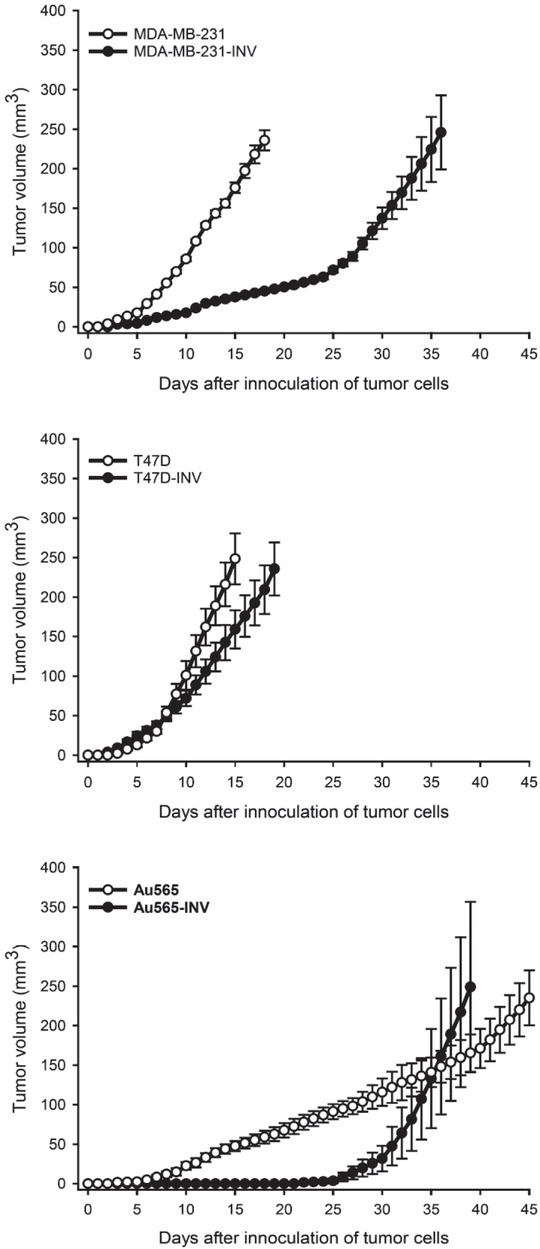
Tumorigenic abilities of parental and INV breast cancer cells. Tumor growth curves of breast cancer xenografts, parental (MDA-MB-231, T47D, Au565, ) and INV counterparts (MDA-MB-231-INV, T47D-INV, Au565-INV) are received as descibed in the section Materials and Methods.
Histological analysis of the investigated xenografts has shown that both parental and invasive T47D tumors were more differentiated (presence of nests) compared to parental and invasive Au565 and MDA-MB-565 tumors (Figure 9). Although MDA-MB-231-INV and Au565-INV xenografts were markedly delayed in their initial growth, they have demonstrated an exponential increase of tumor volume after day 25, and their proliferative potential was up to 70% (Figure 9, 10A, Table 1).
Figure 9.
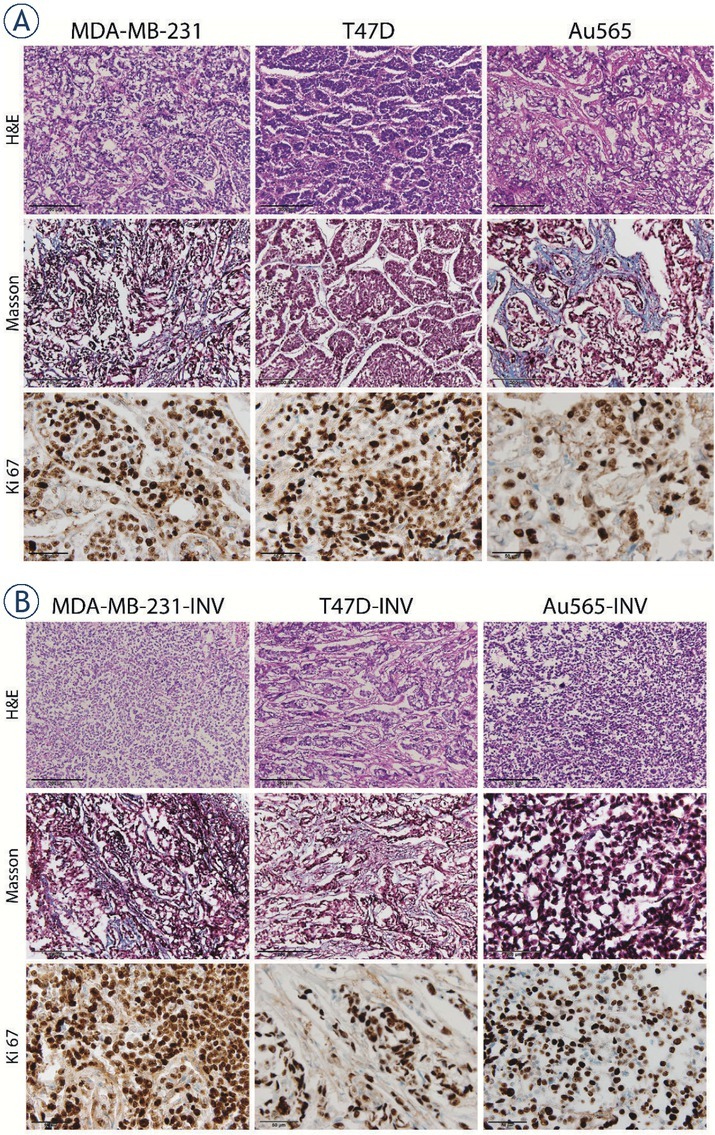
Representative histological images of breast cancer xenografts. Tumors originating from parental (A) and invasive (B) MDA-MB-231, T47D and Au565 breast carcinoma cells were stained with H&E for evaluation of tumor morphology (200× magnification). Collagen (blue) was stained using Masson`s staining (200× magnification). Ki-67 proliferative tumor cells are stained brown (600× magnification).
Figure 10.
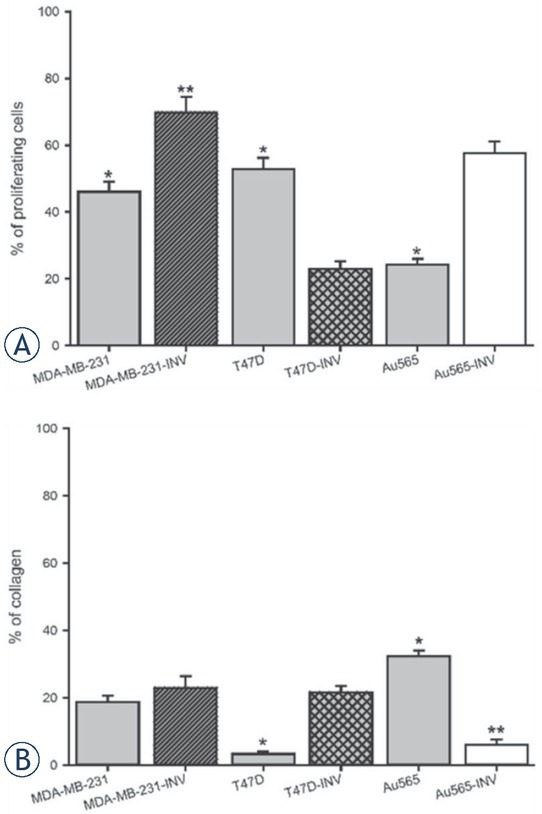
Histological analysis of parental and INV breast xenografts. Tumors originated from the parental (MDA-MB-231, T47D, Au565) and INV breast carcinoma cells (MDA-MB-231-INV, T47D-INV, Au565-INV) were evalueated for desmoplasia (intratumoral collagen content) and percentage of proliferative cells (Ki67 expression) (*p < 0.05; **p < 0.01; ***p < 0.001).
In contrast, in T47D-INV tumors demonstrated a lower number of proliferating cells (~ 20%) and increased desmoplasia compared to parental T47D tumors (Figure 9, 10B; Table 1). There was an opposite observation in parental and invasive Au565 xenografts: ~24% versus ~57% of proliferating parental Au565 and invasive Au565-INV cells, respectively; ~32% versus ~6% of stromal compartment with collagen in parental and invasive Au565 tumor, respectively (Figure 9, 10; Table 1).
In addition, the lymph nodes corresponding to the invasive MDA-MB-231, T47D, and Au565 xenografts were markedly but not significantly increased in sizes and weight compared to their parental counterparts (Figure 11). Histological analysis has shown that all investigated lymph nodes from both parental and invasive xenografts revealed medullar sinus histiocytosis and plasmacytosis (Figure 12). However, they were more pronounced in the lymph nodes obtained from animals bearing the tumors originated from INV breast carcinoma cells. Among the parental breast carcinoma xenografts, only Au565 tumors have given a growth of microscopic metastasis in the lymph node parenchyma in 8% of all investigated lymph nodes, whereas parental MDA-MB-231 and T47D tumors were not characterized with any suspicious lymph node metastasis. In contrast, growth of all tumors originated from the MDA-MB-231-INV, T47D-INV, and Au565-INV was accompanied by appearance of carcinoma cells in lymph nodes. Thus, few individual MDA-MB-231-INV, T47D-INV, and Au565-INV tumor cells were found in the subcapsular sinus in 17%, 8%, and 8% of all analyzed lymph nodes, preferentially inguinal, respectively.
Figure 11.
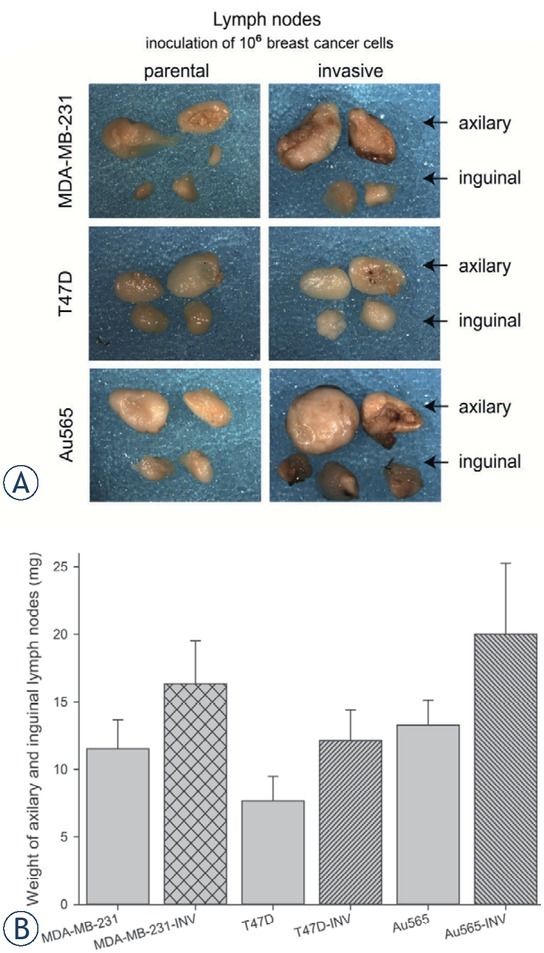
Lymph nodes of mice bearing breast cancer xenografts originated from parental (MDA-MB-231, T47D, Au565) and INV counterparts (Au565-INV, MDA-MB-231-INV, T47D-INV). (A) Representative images of macromorphology of the lymph nodes; (B) Cumulative weight of lymph nodes (AM+SE of 6 mice per group).
Figure 12.
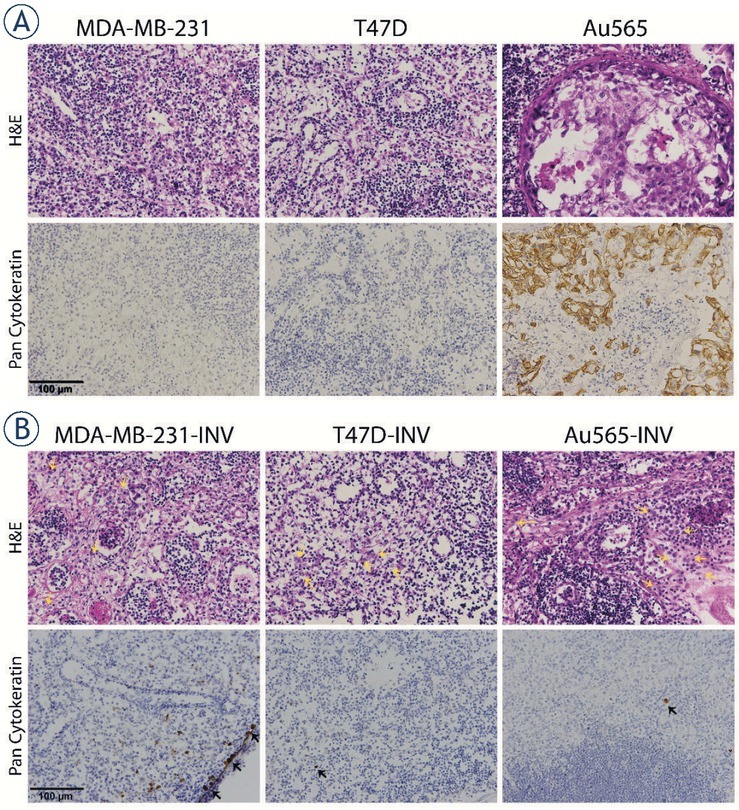
Histology of lymph nodes of mice bearing breast cancer xenografts. (A) HE staining in lymph nodes corresponding to the parental MDA-MB-231, T47D, Au565 and (B) invasive MDA-MB-231-INV, T47D-INV, Au565-INV xenografts. Representative images of lymph nodes taken under 400 × magnification. Brown staining represent pan-cytokeratin positive tumor cells in lymph node.
Discussion
Breast cancer metastases are still a challenge in oncology and one of the main causes of cancer-related deaths in women. Carcinoma cells entering the metastatic cascade should reveal an increased invasive potential to pass a number of biological barriers to reach the target organ. It seems that invasive abilities of carcinoma cells are required during the whole process of metastatic spread of carcinoma cells. Although there is a number of reports discussing an involvement of a variety of the molecules and pathways in cell invasiveness resulting in metastasis development, only limited data on the global molecular patterns in metastatic breast cancer cells are available.4,27 Furthermore, the majority of publications indicate that initiation of metastasis is uniformly regulated independently from the molecular subtypes of breast cancer. However, these reports demonstrated an up-regulation of the known metastasis-related pathways (EMT markers, TGF, VEGF, IL6, etc.) which were detected in the metastatic lesions.1,4,23,28 In our opinion, these pathways are the final “downstream” events in the chain of molecular perturbations resulting in metastasis development. Unfortunately, it is still not fully elucidated which molecules and pathways can be considered as initiators of metastatic cascade in breast cancer. Furthermore, it is still unclear, whether pathways-initiators are common for all breast cancer subtypes.
Hence, the main aim of our study was to determine how an enhancement of invasive and metastatic patterns in breast carcinoma cells is regulated in different breast cancer subtypes. It was assumed that breast cancer cell invasiveness should be accompanied by the acquiring of similar cellular morphology and molecular phenotype. Indeed, the newly obtained cells have demonstrated equal morphology which is specific for carcinoma cells possessing the augmented invasive and migratory capacities. Spindle-like form of the INV cells with more pronounced filopodia formation, diminished number of lipid droplets and their translocation to the filopodia tips result in cell abilities to pass the biological barriers and use the energy from the lipid droplets.29,30 As it was expected, all three INV cell lines belonging to different subtypes of breast cancer showed the enhanced migratory and invasive abilities. Although the parental breast carcinoma cells had equal invasive and migratory properties, and the procedure to obtain new cell lines with increased invasiveness was common for all investigated cells, Her2-positive Au565-INV cells revealed the most pronounced augmentation of the capacities to invade and migrate through the collagen- and laminin-coated membranes compared to other INV cells. Our experimental findings fully correlate with the clinical data describing the higher frequencies of positive lymph nodes in patients with Her2-positive tumors compared to triple-negative and hormone receptor-positive ones.31 Furthermore, Her2-positive breast cancers have a strong correlation with the margin positivity after operation.32 We suppose that Her2-positive cells with increased invasiveness can be found in the surgical margins due to enhanced capabilities to invade the surrounding tissues and migrate markedly more actively compared to the cells of other morphological subtypes of breast cancer. These differences in invasion and migration allowed us to assume that other molecular properties of invasive carcinoma cells belonging to different subtypes of breast cancer are also distinct.
It is assumed that tumors containing carcinoma cells with stemness features and mesenchymal phenotype demonstrate a higher probability for metastatic progression.17,18,19,28,33 Again, all investigated INV breast carcinoma cells have shown different enrichment for cells carrying stemness markers CD44+/CD24- and ALDH1. Despite an increased content of carcinoma cells with stemness capabilities, the tumorigenic capacities of all INV cells were comparable. Therefore, it is possible to conclude that neither CD44+/CD2- nor ALDH1 are the determinants for tumorigenic capacities of the investigated cells. However, the delayed tumor growth of the INV xenografts could be explained by a higher number of the injected CD44+/CD24-/ALDH1+ cells that are affected in their proliferation and can be dormant compared to the parental xenografts.17,34, 35, 36, 37 We would like to emphasize that the balance between CD44, CD24, and ALDH1 expression was differently affected in INV cells compared to the parental cells belonging to different subtypes of breast cancer. In accordance with the report by Sulaiman et al. 38, MDA-MB-231-INV and T47D-INV cells acquired a hybrid stemness phenotype, and Au565-INV cells switched their phenotype from hybrid to mesenchymal one. Differences in the acquired molecular phenotypes of INV cells were not limited to the observed distinctions in the invasiveness, migratory and stemness abilities, it was also found that INV cells not equally mobilized the molecular pathways potentially involved in the metastasis development. Taking into account the bioinformatic analysis of the proteomic data of the investigated INV cells, we can conclude that hormone receptor-positive T47D-INVcells with increased invasiveness acquire the molecular characteristics of triple-negative breast cancer cells, whereas Her2-positive Au565-INV cells specifically changed their own molecular phenotype with very limited partaking in the involved pathways found in the MDA-MB-231-INV and T47D-INV cells. These data demonstrate that triple-negative, hormone receptor-positive and Her2-positive breast carcinoma cells realize their invasive potential through activation or repression of different molecular pathways. To our knowledge, there are no reports on the differences in molecular patterns underlying an enhancement of breast cancer cell invasiveness depending on the type of breast cancer. Undoubtedly, further detailed investigation of the molecular pathways involved in breast cancer cell invasiveness is required and clinically important.
Taking together, we conclude that breast carcinoma cells having the same cellular morphology accompanied by the increased invasiveness and migratory capacities, reveal an activation of distinct pathways associated with increased metastatic properties. These pathways should be further elucidated to develop the biomarkers and potential therapeutic targets to predict, prevent or combat the metastatic spread of carcinoma cells belonging to different subtypes of breast cancer. Since hormone receptor-positive invasive cells share their molecular properties with triple-negative breast cancer cells, we assume that these types of metastatic disease can be treated rather equally with an option to add anti-hormonal agents. In contrast, Her2-positive metastasis should be carefully evaluated for more effective therapeutic approaches which are distinct from the triple-negative and hormone-positive metastatic cancers.
Acknowledgement
We are deeply grateful to the colleagues at OmicsTECH (USA) for their excellent technical support in proteomics performance and analysis.
This study was supported by Austrian Science Fund (FWF, P29457; I4140), Anniversary Fund of Austrian National Bank (ÖNB 17620), Ingrid Shaker-Nessmann Cancer Research Foundation, Austrian agency for international mobility and cooperation in education, science and research (OeAD-WTZ, IN05/2017; SI25/2018); ESMO Translational Research Fellowship (M.V.-S.).
The authors acknowledge the financial support of the Slovenian Research Agency (research program No. P3-003, No. BI-AT/18-19-003). The funder had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure
No potential conflicts of interest were disclosed.
Supplementary Material.
References
- 1.Scully OJ, Bay BH, Yip G, Yu Y. Breast cancer metastasis. Cancer Genomics Proteomics. 2012;9:311–20. PMID: 22990110. [PubMed] [Google Scholar]
- 2.Van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res. 2011;728:23–34. doi: 10.1016/j.mrrev.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiao W, Zheng S, Yang A, Zhang X, Zou Y, Tang H. Breast cancer subtypes and the risk of distant metastasis at initial diagnosis: a population-based study. Cancer Manag Res. 2018;10:5329–38. doi: 10.2147/CMAR.S176763. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuo WH, Chang YY, Lai LC, Tsai MH, Hsiao CK, Chuang EY. Molecular characteristics and metastasis predictor genes of triple-negative breast cancer: a clinical study of triple-negative breast carcinomas. PLoS One. 2012;7:e45831. doi: 10.1371/journal.pone.0045831. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weigelt B, Peterse JL, van ‚t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005;5:591–602. doi: 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- 6.Dwivedi AR, Thakur A, Kumar V, Skvortsova I, Kumar V. Targeting cancer stem cells pathways for the effective treatment of cancer. Curr Drug Targets. 2019 doi: 10.2174/1389450120666190821160730. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 7.Redig AJ, McAllister SS. Breast cancer as a systemic disease: a view of metastasis. J Intern Med. 2013;274:113–26. doi: 10.1111/joim.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skvortsova I, Skvortsov S, Stasyk T, Raju U, Popper BA, Schiestl B. Intracellular signaling pathways regulating radioresistance of human prostate carcinoma cells. Proteomics. 2008;8:4521–33. doi: 10.1002/pmic.200800113. et al. [DOI] [PubMed] [Google Scholar]
- 9.Skvortsova I, Skvortsov S, Haidenberger A, Devries A, Nevinny-Stickel M, Saurer M. Effects of paclitaxel and docetaxel on EGFR-expressing human carcinoma cells under normoxic versus hypoxic conditions in vitro. J Chemother. 2004;16:372–80. doi: 10.1179/joc.2004.16.4.372. et al. [DOI] [PubMed] [Google Scholar]
- 10.Buzdin A, Sorokin M, Garazha A, Sekacheva M, Kim E, Zhukov N. Molecular pathway activation - New type of biomarkers for tumor morphology and personalized selection of target drugs. Semin Cancer Biol. 2018;53:110–24. doi: 10.1016/j.semcancer.2018.06.003. et al. [DOI] [PubMed] [Google Scholar]
- 11.Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016;44:D481–7. doi: 10.1093/nar/gkv1351. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T. PID: the Pathway Interaction Database. Nucleic Acids Res. 2009;37:D674–9. doi: 10.1093/nar/gkn653. et al. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–60. doi: 10.1093/nar/gkp896. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romero P, Wagg J, Green ML, Kaiser D, Krummenacker M, Karp PD. Computational prediction of human metabolic pathways from the complete human genome. Genome Biol. 2005;6:R2. doi: 10.1186/gb-2004-6-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zolotovskaia MA, Sorokin MI, Roumiantsev SA, Borisov NM, Buzdin AA. Pathway instability is an effective new mutation-based type of cancer biomarkers. Front Oncol. 2018;8:658. doi: 10.3389/fonc.2018.00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinbichler TB, Savic D, Dudas J, Kvitsaridze I, Skvortsov S, Riechelmann H. Cancer stem cells and their unique role in metastatic spread. Semin Cancer Biol. 2019. et al. Epub ahead of print. [DOI] [PubMed]
- 18.Velasco-Velazquez MA, Popov VM, Lisanti MP, Pestell RG. The role of breast cancer stem cells in metastasis and therapeutic implications. Am J Pathol. 2011;179:2–11. doi: 10.1016/j.ajpath.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li W, Ma H, Zhang J, Zhu L, Wang C, Yang Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci Rep. 2017;7:13856. doi: 10.1038/s41598-017-14364-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sorokin M, Kholodenko R, Suntsova M, Malakhova G, Garazha A, Kholodenko I. Oncobox bioinformatical platform for selecting potentially effective combinations of target cancer drugs using high-throughput gene expression data. Cancers (Basel) 2018;10(10):E365. doi: 10.3390/cancers10100365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spirin PV, Lebedev TD, Orlova NN, Gornostaeva AS, Prokofjeva MM, Nikitenko NA. Silencing AML1-ETO gene expression leads to simultaneous activation of both pro-apoptotic and proliferation signaling. Leukemia. 2014;28:2222–8. doi: 10.1038/leu.2014.130. [DOI] [PubMed] [Google Scholar]
- 22.Lambaerts K, Wilcox-Adelman SA, Zimmermann P. The signaling mechanisms of syndecan heparan sulfate proteoglycans. Curr Opin Cell Biol. 2009;21:662–9. doi: 10.1016/j.ceb.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sayyad MR, Puchalapalli M, Vergara NG, Wangensteen SM, Moore M, Mu L. Syndecan-1 facilitates breast cancer metastasis to the brain. Breast Cancer Res Treat. 2019;178:35–49. doi: 10.1007/s10549-019-05347-0. et al. [DOI] [PubMed] [Google Scholar]
- 24.Zhang S, Yu D. Targeting Src family kinases in anti-cancer therapies: turning promise into triumph. Trends Pharmacol Sci. 2012;33:122–8. doi: 10.1016/j.tips.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ying X, Huang A, Xing Y, Lan L, Yi Z, He P. Lycorine inhibits breast cancer growth and metastasis via inducing apoptosis and blocking Src/FAK-involved pathway. Sci China Life Sci. 2017;60:417–28. doi: 10.1007/s11427-016-0368-y. [DOI] [PubMed] [Google Scholar]
- 26.Zhao TT, Jin F, Li JG, Xu YY, Dong HT, Liu Q. TRIM32 promotes proliferation and confers chemoresistance to breast cancer cells through activation of the NF-kappaB pathway. J Cancer. 2018;9:1349–56. doi: 10.7150/jca.22390. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanpaolo P, Barbieri V, Genovesi D. Prognostic value of breast cancer subtypes on breast cancer specific survival, distant metastases and local relapse rates in conservatively managed early stage breast cancer: a retrospective clinical study. Eur J Surg Oncol. 2011;37:876–82. doi: 10.1016/j.ejso.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 28.Geng SQ, Alexandrou AT, Li JJ. Breast cancer stem cells: multiple capacities in tumor metastasis. Cancer Lett. 2014;349:1–7. doi: 10.1016/j.canlet.2014.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antalis CJ, Uchida A, Buhman KK, Siddiqui RA. Migration of MDA-MB-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification. Clin Exp Metastasis. 2011;28:733–41. doi: 10.1007/s10585-011-9405-9. [DOI] [PubMed] [Google Scholar]
- 30.Chao H, Deng L, Xu F, Yu Z, Xu X, Huang J. MEX3C regulates lipid metabolism to promote bladder tumorigenesis through JNK pathway. Onco Targets Ther. 2019;12:3285–94. doi: 10.2147/OTT.S199667. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu N, Yang Z, Liu X, Niu Y. Lymph node status in different molecular subtype of breast cancer: triple negative tumours are more likely lymph node negative. Oncotarget. 2017;8:55534–43. doi: 10.18632/oncotarget.15022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jia H, Jia W, Yang Y, Li S, Feng H, Liu J. HER-2 positive breast cancer is associated with an increased risk of positive cavity margins after initial lumpectomy. World J Surg Oncol. 2014;12:289. doi: 10.1186/1477-781912-289. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rabinovich I, Sebastiao APM, Lima RS, Urban CA, Junior ES, Anselmi KF. Cancer stem cell markers ALDH1 and CD44+/CD24- phenotype and their prognosis impact in invasive ductal carcinoma. Eur J Histochem. 2018;62 doi: 10.4081/ejh.2018.2943. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou N, Wu X, Yang B, Yang X, Zhang D, Qing G. Stem cell characteristics of dormant cells and cisplatininduced effects on the stemness of epithelial ovarian cancer cells. Mol Med Rep. 2014;10:2495–504. doi: 10.3892/mmr.2014.2483. [DOI] [PubMed] [Google Scholar]
- 35.Chaterjee M, van Golen KL. Breast cancer stem cells survive periods of farnesyl-transferase inhibitor-induced dormancy by undergoing autophagy. Bone Marrow Res. 2011;2011:362938. doi: 10.1155/2011/362938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steinbichler TB, Dudas J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova II. Therapy resistance mediated by cancer stem cells. Semin Cancer Biol. 2018;53:156–67. doi: 10.1016/j.semcancer.2018.11.006. [DOI] [PubMed] [Google Scholar]
- 37.Skvortsov S, Debbage P, Skvortsova I. Proteomics of cancer stem cells. Int J Radiat Biol. 2014;90:653–8. doi: 10.3109/09553002.2013.873559. [DOI] [PubMed] [Google Scholar]
- 38.Sulaiman A, McGarry S, Han X, Liu S, Wang L. CSCs in breast cancer-one size does not fit all: Therapeutic advances in targeting heterogeneous epithelial and mesenchymal CSCs. Cancers (Basel) 2019;11 doi: 10.3390/cancers11081128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.