

Abstract
Pro-inflammatory cytokines in the tumor microenvironment (TME) can promote tumor growth, yet their value as therapeutic targets remains underexploited. We validated the functional significance of the CLCF1-CNTFR signaling axis in lung adenocarcinoma (LUAD) and generated a high affinity soluble receptor (eCNTFR-Fc) that sequesters CLCF1, thereby inhibiting its oncogenic effects. eCNTFR-Fc inhibits tumor growth in multiple xenograft models and an autochthonous, highly aggressive genetically-engineered mouse model (GEMM) of LUAD driven by activation of oncogenic Kras and loss of Trp53. Abrogation of CLCF1 through eCNTFR-Fc appears most effective in tumors driven by oncogenic KRAS. We observed a correlation between effectiveness of eCNTFR-Fc and the presence of KRAS mutations that retain the intrinsic capacity to hydrolyze GTP, suggesting that the mechanism of action may be related to altered GTP loading. Overall, we nominate blockade of CLCF1-CNTFR signaling as a novel therapeutic opportunity for LUAD and potentially for other tumor types in which CLCF1 is present in the TME.
Keywords: lung cancer, lung adenocarcinoma, KRAS, G12C, CLCF1, CNTFR, CAFs, receptor decoy, gp130, therapy
INTRODUCTION
Lung cancer is the leading cause of cancer-related death worldwide. Non-small cell lung cancer (NSCLC) accounts for 85–90% of cases, and lung adenocarcinoma (LUAD) is the most common subtype. While approximately 30% of LUAD patients harbor a mutation in KRAS, there are currently few targeted therapeutic options1–3. In LUAD subtypes characterized by EGFR or ALK alterations, small molecule inhibitors are advantageous, although rapid drug resistance remains a major limitation4–6. Immunotherapy also has proven beneficial for some patients7–10. Despite these advances, there is a continued clinical need for innovative approaches to lung cancer treatment, especially those directed at mechanisms of oncogenesis not targeted by available agents. A large body of literature supports the hypothesis that the tumor microenvironment (TME) supports tumor growth via paracrine signaling11,12. A major cell type within the TME is the cancer-associated fibroblast (CAF). Several studies have specifically evaluated the role of CAFs in lung cancer, although these have had only limited impact on the development of new therapeutic approaches13–17.
We previously described a pro-oncogenic mechanism through which CAFs promote tumor growth in a mouse model of LUAD18. Cardiotrophin-like cytokine factor 1 (CLCF1) expressed by mouse CAFs promotes tumor growth, whereas knockdown of its cognate receptor, ciliary neurotrophic factor receptor (CNTFR) in mouse lung tumor cells decreases tumor growth. CLCF1 (NNT-1/BSF-3) was initially identified as a cytokine of the IL-6 family expressed in T cell lymphomas19. The CLCF1 receptor, CNTFR, is a member of the IL-6 receptor family and forms a trimeric complex with LIFR and gp130. Ligand binding activates signaling through gp130, leading to activation of downstream signaling pathways20. Our prior work nominated CLCF1-CNTFR as a potential novel therapeutic target in lung cancer. As other cancers also express both CLCF1 and CNTFR21–24, we reasoned that therapeutic intervention to block this pathway could have relevance to multiple cancer types.
Monoclonal antibodies have been the most widely used strategy to block ligand-receptor interactions25. An alternative approach involves engineering an extracellular fragment of a soluble receptor as a ‘decoy’ that binds to and sequesters the cognate ligand from activating cell surface receptors26. Decoy receptors have been used to block VEGFR27 and TNF28 with other molecules still in development29–31. Here, we describe the development and preclinical testing of a first-in-class therapeutic candidate based on an engineered version of the soluble CNTFR extracellular domain. This receptor decoy specifically binds to CLCF1 and neutralizes its biological activity, leading to significant anti-tumor effects. We demonstrate that blockade of the CLCF1-CNTFR signaling axis is particularly effective in a subset of LUAD, including those driven by specific genotypes of oncogenic KRAS, identifying a potential biomarker for further therapeutic development and providing a novel therapeutic opportunity for a LUAD subtype with currently limited available therapies.
RESULTS
Functional effect of CLCF1-CNTFR signaling in human LUAD
CLCF1 is upregulated in lung adenocarcinoma (LUAD) compared to normal lung (Extended Data Fig. 1a) and high expression is associated with decreased survival in patients with KRAS mutation [Cox hazard ratio: 2.53 (95% CI 1.43–4.48); P-value: 0.001] but not those without KRAS mutation [Cox hazard ratio: 0.86 (95% CI 0.51–1.4); P-value: 0.56] (Extended Data Fig. 1b,c). This suggested a specific role of CLCF1 signaling in KRAS-driven oncogenesis, which is further explored below. In contrast, CNTFR, IL6ST (gp130), and LIFR expression were not associated with survival (Extended Data Fig. 1d–i).
Our prior work indicated that in mouse lung tumors, cancer-associated fibroblasts (CAFs) are the primary source of CLCF118. To determine if human CAFs also produce CLCF1, we isolated CAFs from human lung cancer patients and matched normal lung fibroblasts (NLFs; Supplementary Table 1). Expression of CLCF1 was significantly elevated in six of eight human CAFs compared to patient-matched NLFs (Extended Data Fig. 2a). However, the LUAD cell lines tested also secrete CLCF1, suggesting the existence of both paracrine and autocrine signaling for this cytokine in human LUAD (Extended Data Fig. 2b,c).
We next evaluated the functional role of CLCF1 in LUAD cell lines. Exposure to recombinant CLCF1 led to increased cell viability in all lines examined (Fig. 1a and Extended Data Fig. 3a–c). Ligand binding to the CNTFR/LIFR/gp130 complex leads to phosphorylation of gp130 and activation of downstream signals including STAT3 and ERK20. Thus, as expected, CLCF1 induced phosphorylation of STAT3 (Fig. 1b–d). To further probe the functional significance of CLCF1-CNTFR signaling in human lung cancer, we used RNA interference to decrease CNTFR at the cell surface. Knockdown of CNTFR decreased viability of five LUAD cell lines tested (Fig. 1e–g and Extended Data Fig. 4a and Supplementary Table 2), suppressed clonogenic growth (Fig. 1h,i and Extended Data Fig. 4b) and led to decreased size and number of spheres in 3D culture (Fig. 1j,k and Extended Data Fig. 4c).
Figure 1.

Recombinant CLCF1 increases and CNTFR knockdown decreases tumor growth in human LUAD. (a) CLCF1 treatment for 72 h increases cell viability after serum starvation in LUAD cell lines A549, H23, and H358 in a concentration-dependent manner compared to untreated control (n = 3 independent experiments). * P = 0.04; ** P = 0.001; *** P < 0.001 using two-tailed unpaired Student’s t-test. Data represented as mean ± S.D. (b, d) Recombinant human CLCF1 phosphorylates STAT3 (Y705) in both a concentration ([CLCF1] = 10 nM) and (c, d) time-dependent (15 min after treatment) manner in A549, H23, and H358 as shown in the representative cropped western blots. Quantification from n = 3 independent experiments shown in graph and compared to the untreated control. * P = 0.037; ** P = 0.002; *** P < 0.001 using two-tailed unpaired Student’s t-test. Data represented as mean ± S.D. (e) qRT-PCR measurements of CNTFR knockdown with shCNTFR or control shGFP (n = 3 independent experiments with three technical replicates per group). ** P < 0.01; *** P < 0.001 using one-way analysis of variance (ANOVA) and Dunnett’s multiple comparison test (DMCT). Data represented as mean ± S.D. (f) Proliferation of A549 after knockdown with indicated shRNAs (n = 3 independent experiments with three technical replicates per group). *** P < 0.001 using Two-way ANOVA and DMCT. Data represented as mean ± S.D. (g) Proliferation rates for LUAD cells after CNTFR knockdown at day 7 (n = 3 independent experiments shown). *** P < 0.001 using one-way ANOVA and DMCT. Data represented as mean ± S.D. (h) Representative images of colony-formation assay in A549 and H23. (i) Quantification of colony number from (h) (n = 3 independent experiments shown). *** P < 0.001 using one-way ANOVA and DMCT. Data are represented as mean ± S.D. (j) Representative images of spheres from cells grown in anchorage-independent conditions in A549 and H23. (k) Quantification of sphere number (n = 3 independent experiments shown). *** P < 0.001 using one-way ANOVA and DMCT. Data represented as mean ± S.D. (l) Tumor volume quantification of A549 xenografts with indicated shRNAs (n = 6 biologically independent samples). *** P < 0.001 using two-way ANOVA and DMCT. Data represented as mean ± S.E.M. (m) Tumor volume quantification of final time point in indicated LUAD cell line xenografts (A549: n = 6 biologically independent samples; H23: n = 6 biologically independent samples; H2009: n = 8 biologically independent samples). *** P < 0.001 using one-way ANOVA and DMCT. Whiskers identify the maximum and minimum values; boxes indicate the 75th and 25th percentile and line the median. (n) Representative hematoxylin and eosin (H&E) staining and immunohistochemistry (IHC) for phospho-histone H3 (PH3) and cleaved caspase-3 (CC3) in A549 xenografts. Scale bars, 50 μm. (o) Quantification of PH3- and CC3-positive foci (A549 and H23: n = 6 biologically independent samples; H2009: n = 8 biologically independent samples with 10 technical replicates in each group). * P < 0.05; *** P < 0.001 using one-way ANOVA and DMCT. Data represented as mean ± S.E.M.
We then evaluated whether CNTFR knockdown would influence tumor growth in vivo. CNTFR knockdown in three LUAD cell lines tested decreased xenograft formation (Fig. 1l,m and Extended Data Fig. 4d,e). Moreover, tumors formed from LUAD cells with CNTFR knockdown exhibited a lower proliferative index and higher levels of apoptosis compared to control tumors (Fig 1n,o and Extended Data Fig. 4f,g). To test whether the effect of CLCF1 was mostly paracrine or autocrine, we knocked down CLCF1 in H2009 and implanted these cells as xenografts (Extended Data Fig. 5a). We observed a decrease in tumor growth in both the CLCF1 and the CNTFR knockdown tumors, suggesting that at least in subcutaneous xenograft models the source of CLCF1 is primarily autocrine secretion from the tumor cells.
To determine the mechanism of action of CNTFR blockade in LUAD, we evaluated the effect of knockdown on the MAPK, AKT, and STAT3 signaling pathways, all previously identified as activated downstream of gp13032. Phosphorylation of ERK and S6 were decreased in tumors after CNTFR knockdown (Extended Data Fig. 5b–d), indicating effects on both MAPK/ERK and AKT signaling. Decreased phosphorylation of STAT3 was also observed (Extended Data Fig. 5b,d). Taken together, these results indicate that CLCF1-CNTFR signaling is active in LUAD, has a pro-oncogenic role, and suggest that the mechanism of CNTFR inhibition involves dampening the activity of several signaling cascades including ERK, AKT, and STAT3 signaling.
Engineering a soluble receptor decoy to inhibit the CLCF1-CNTFR signaling axis
The functional studies above support the notion that inhibition of CLCF1-CNTFR signaling could be a therapeutic opportunity in lung cancer. Therefore, we sought to identify an effective strategy to target this pathway. CNTFR is anchored to the cell surface via a glycosylphosphatidylinositol (GPI) linkage that forms following proteolytic cleavage of a C-terminal propeptide (Fig. 2a,i)33. When bound to CLCF1, CNTFR forms a complex with gp130 and LIFR (Fig. 2a,ii). CNTFR lacking this GPI linkage can also be secreted from the cell, but can still bind to CLCF1 and activate downstream signaling, even in cells that do not express CNTFR through what is termed trans-signaling (Fig. 2a,iii). Thus, effective blockade of CLCF1 requires both increasing binding of the decoy to CLCF1 and decreasing binding to gp130 and LIFR (Fig. 2a,iv)34.
Figure 2.
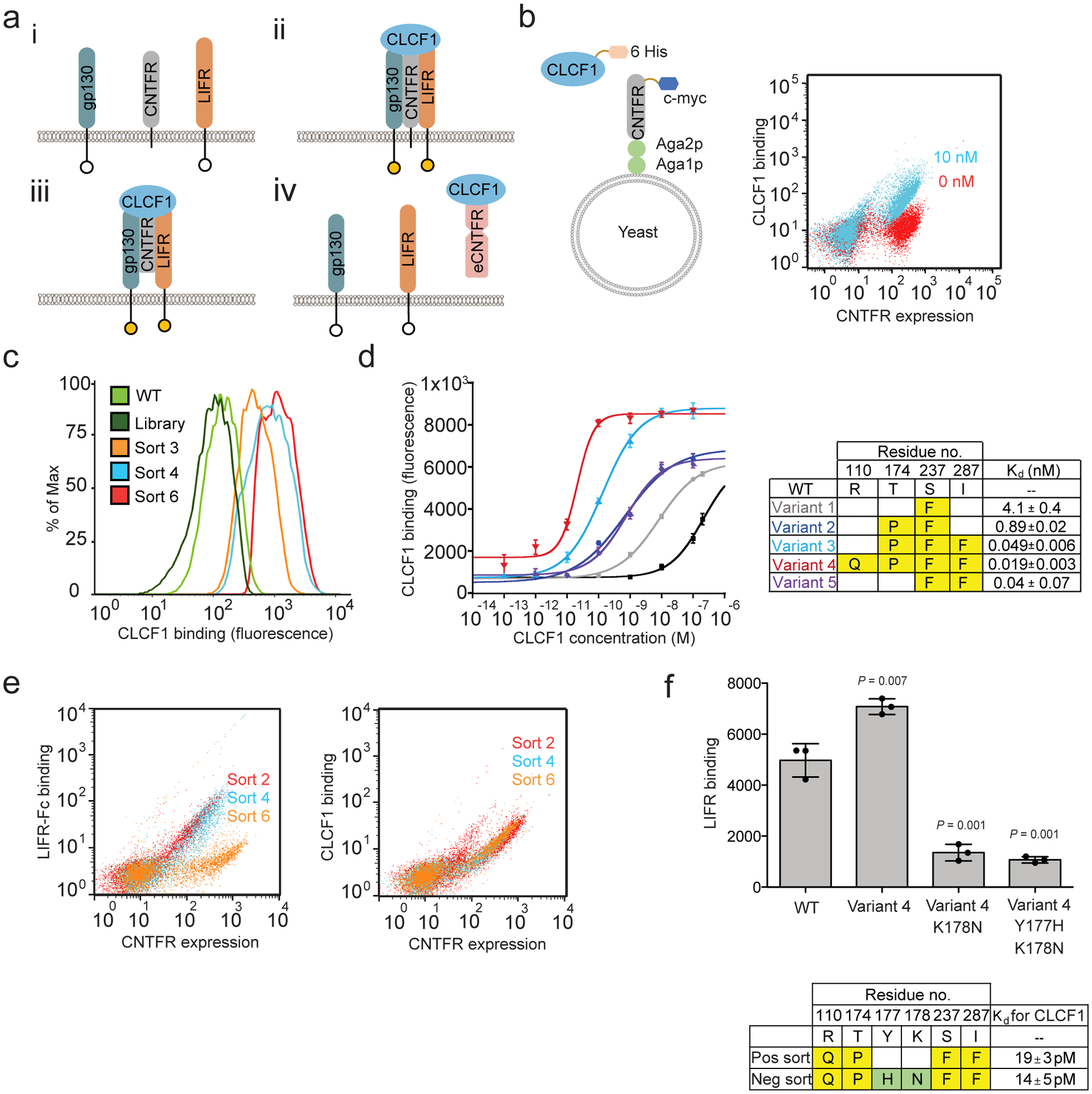
Engineering a CNTFR receptor decoy using yeast display. (a) i) CNTFR transmits signal through the β receptors, gp130 and LIFR. ii) The β receptors become activated when CLCF1 complexes with CNTFR. iii) Soluble CNTFR allows gp130 and LIFR to heterodimerize even in cells lacking CNTFR expression. iv) Engineered soluble CNTFR (eCNTFR) that does not bind to the β receptors can function as an antagonist. (b) Schematic representation of yeast-displayed CNTFR and overlaid flow cytometry dot plot representing binding of yeast-displayed wtCNTFR to 10 nM (cyan) and 0 nM (red) CLCF1-His. The experiment was repeated at least three times independently with similar results. (c) Flow cytometry histograms of the first CNTFR library and intermediate sorted population compared to wtCNTFR (WT), measuring binding to 0.5 nM CLCF1. Only the gated population of yeast expressing CNTFR is shown. The experiment was repeated at least three times independently with similar results. (d) Binding curves of affinity matured yeast-displayed CNTFR variants with various concentrations of CLCF1 and the measured apparent Kd values (n = 3 independent experiments). Data are represented as mean ± S.D. (e) Overlaid representative flow cytometry dot plots for sort 2 (red), sort 4 (blue), and sort 6 (orange) showing enrichment of non-LIFR binders. The experiment was repeated at least three time independently with similar results. (f) Y177H and K178N isolated from negative screening against LIFR-Fc additively decreases LIFR-Fc binding. The measured apparent Kd values represent binding affinity toward CLCF1. Error bars represent the standard deviation (n = 3 independent experiments). *** P < 0.001 using two-tailed unpaired Student’s t-test. Data represented as mean ± S.D.
We used directed evolution to engineer a soluble CNTFR variant with stronger affinity for CLCF1 and lack of binding to gp130 and LIFR. We hypothesized that such a molecule would act as an efficient ligand trap and antagonize CLCF1-mediated oncogenic signaling. To develop a high-affinity receptor decoy, DNA encoding the extracellular domain of CNTFR was subjected to random mutagenesis via error-prone PCR. The corresponding protein library (~108 transformants) was displayed as fusions on the yeast cell surface (Fig. 2b)35. The library was screened to enrich for variants with increased CLCF1 binding using flow cytometry (Extended Data Fig. 6). After 3 rounds of screening, T174P and S237F appeared as consensus mutations, with substantial diversity observed at other amino acid positions (Supplementary Table 3a,b). To probe additive effects of these mutations, 20 randomly selected distinct clones from the sorted populations were shuffled using the Staggered Extension Process (StEP) method36 to create a second library. A combination of equilibrium binding and kinetic off-rate screens37 were used to sort this library to impose increased screening stringency (Fig. 2c). After three rounds of screening, combinations of four consensus mutations (R110Q, T174P, S237F, and I287F) emerged (Supplementary Table 4). Binding studies indicated that each of these mutations contributed to the higher binding affinity for CLCF1 (Fig. 2d), with the combination of all four mutations leading to an apparent Kd of 20 pM. This CNTFR variant (variant 4) was carried forward for further optimization.
As CLCF1-CNTFR binding activates downstream signaling through heterodimerization of LIFR and gp130, modifying CNTFR to reduce or prevent formation of this complex while sequestering CLCF1 is critical. We confirmed that yeast-displayed CNTFR does complex with gp130 and LIFR in a CLCF1-dependent manner (Extended Data Fig. 3c and Extended Data Fig. 7). Therefore, CNTFR variant 4 was further engineered to decrease its binding to the co-receptors. Random mutations were introduced into CNTFR variant 4 using error-prone PCR, and the resulting library was incubated with CLCF1 and screened for variants with decreased binding signal for LIFR by flow cytometry (Fig. 2e and Extended Data Fig. 8). Two consensus mutations (Y177H and K178N) were identified that reduced binding to LIFR (Fig. 2f and Extended Data Fig. 8). A final variant, eCNTFR, combined these two mutations, the four mutations that confer high affinity CLCF1 binding, and an additional two alanine substitutions (T268A and D269A) previously shown to weaken binding to gp13034.
Characterization of soluble eCNTFR
As structural information on full-length CNTFR is unavailable, wtCNTFR and eCNTFR were modeled using the Phyre2 server to predict the three-dimensional locations of eCNTFR mutations38. Three mutations (T174P, S237F, and I287F) were proximal to the aromatic cluster (F172, F199, and F238) and the conserved residues (E236 and E286) previously shown to be important for cytokine binding (Fig. 3a)39,40. Soluble eCNTFR was recombinantly expressed with a C-terminal hexahistidine tag (eCNTFR-His) or as an N-terminal fusion to an antibody Fc domain (eCNTFR-Fc), and affinity to CLCF1 was measured using a microtiter plate-based assay (Extended Data Fig. 9). Both eCNTFR-His and eCNTFR-Fc exhibited picomolar binding affinity to CLCF1 (Fig. 3b). In comparison, CLCF1 binding affinity was too weak to be quantified for soluble wild-type CNTFR constructs (wtCNTFR-His and wtCNTFR-Fc). A similar approach was used to characterize binding interactions with gp130 and LIFR. In these experiments, eCNTFR constructs showed no detectable binding to gp130 and LIFR, in contrast to wtCNTFR constructs, which bound to both receptors (Fig. 3c). Increasing the size of a protein to avoid glomerular filtration can significantly increase serum half-life, and the Fc domain can further increase half-life through FcRn-mediated recycling41. Therefore, the eCNTFR-Fc fusion was used to further evaluate the effect of eCNTFR in animal models of LUAD.
Figure 3.
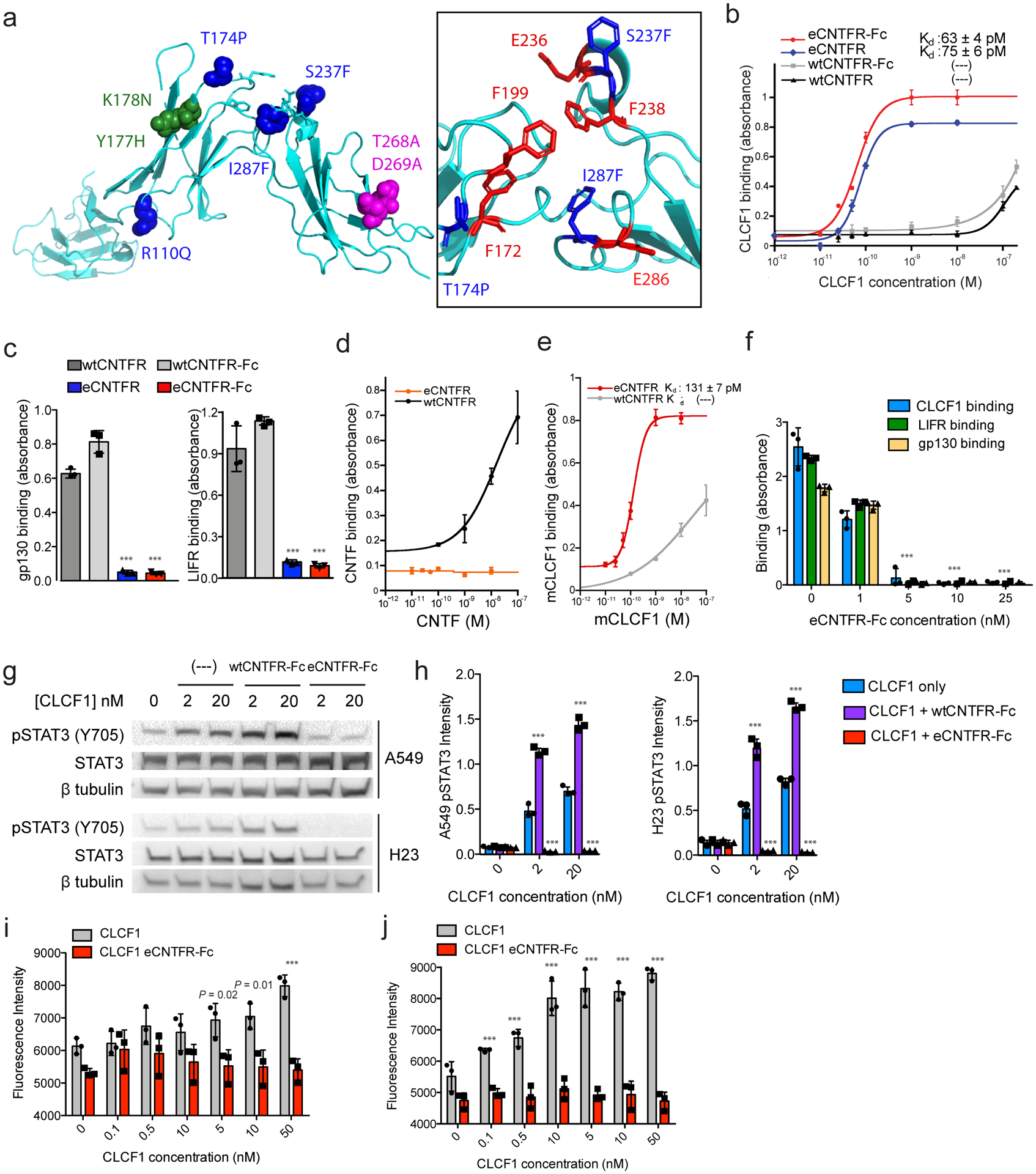
Characterization of eCNTFR constructs. (a) The 3D structure prediction of wtCNTFR (yellow) and eCNTFR (blue) was carried out with the Phyre 2 server (Protein Homology/analogY Recognition Engine V 2.0), showing locations of four mutations from affinity maturation (blue), two mutations to reduce LIFR binding (green), two mutations to reduce gp130 binding (magenta); the inset shows the aromatic cluster and conserved residues of CNTFR for cytokine binding (red) and mutations from affinity maturation (blue). Binding affinities of soluble wtCNTFR and eCNTFR constructs were compared for (b) CLCF1, (c) gp130-Fc and LIFR-Fc, (d) CNTF, and (e) mouse CLCF1. Kd values were calculated where appropriate. Error bars represent the standard deviation (n = 3 independent experiments). *** P < 0.001 compared to the corresponding wtCNTFR construct using two-tailed unpaired Student’s t-test. Data represented as mean ± S.D. (f) Competition assay using ELISA to measure the ability of eCNTFR-Fc to block binding between wtCNTFR-Fc and CLCF1-His, LIFR-His, and gp130-His. Where LIFR-His and gp130-His were included CLCF1 (10 nM) was also added to induce complex formation. Error bars represent the standard deviation (n = 3 independent experiments). *** P < 0.001 compared to the corresponding untreated control using two-tailed unpaired Student’s t-test. Data represented as mean ± S.D. (g, h) eCNTFR-Fc inhibits STAT3 phosphorylation (Y705) in A549 and H23 cells (cropped). Quantification from n = 3 independent experiments shown in graph and compared to the untreated control. *** P < 0.001 using two-tailed unpaired Student’s t-test. Data represented as mean ± S.D. (i, j) eCNTFR-Fc inhibits CLCF1 induced cell survival in serum starved A549 and H23 cells (n = 3 independent replicates). *** P < 0.001 compared to the corresponding non-eCNTFR-Fc treated control using two-tailed unpaired Student’s t-test. Data are represented as mean ± S.D.
CNTF also binds CNTFR, and CNTF-mediated signaling is important for neuronal cell survival42. Engineering binding selectivity of eCNTFR-Fc to CLCF1 over CNTF could help minimize any potential side effects from inhibiting CNTF43 signaling. Additionally, while CLCF1 is known to act only through CNTFR, CNTF also binds to the IL-6 receptor (IL-6R), suggesting that CLCF1 and CNTF have unique functional roles in regulating signaling pathways. wtCNTFR-Fc exhibited binding to recombinantly produced CNTF, while eCNTFR-Fc did not (Fig. 3d). These results are consistent with yeast-displayed binding data for wtCNTFR and eCNTFR (Extended Data Fig. 10a) and indicate that affinity maturation of CNTFR for CLCF1 led to increased specificity towards CLCF1 and decreased binding to CNTF. eCNTFR-Fc also bound to mouse CLCF1 with high affinity as compared to wtCNTFR-Fc, indicating its utility for in vivo experiments in which CLCF1 is of mouse origin (Fig. 3e and Extended Data Fig. 10b). Importantly, mouse CLCF1 can activate CNTFR in human cells (Extended Data Fig. 10c).
To assess whether eCNTFR-Fc could sequester CLCF1 and block receptor complex formation, we designed a competition binding assay to measure the effect of eCNTFR-Fc on the interaction between wtCNTFR and each of the other subunits of the receptor complex. Incubating eCNTFR-Fc in wtCNTFR-His-coated wells prevented CLCF1, LIFR, and gp130 constructs from interacting with wtCNTFR-His (Fig. 3f). To determine whether eCNTFR-Fc could effectively neutralize CLCF1 and inhibit gp130 signaling, LUAD cells were stimulated with CLCF1 in the presence and absence of the soluble CNTFR constructs. While wtCNTFR-Fc increased phosphorylation of STAT3 (Tyr705), eCNTFR-Fc decreased phosphorylation in both cell lines tested (Fig. 3g,h). Furthermore, incubation with eCNTFR-Fc inhibited CLCF1-mediated viability (Fig. 3i,j).
eCNTFR-Fc effect correlates with commonly seen KRAS mutations in lung cancer
The analysis of public gene expression data shown above suggest that CLCF1 expression is specifically prognostic of survival in patients with oncogenic KRAS-driven LUAD. As there are currently few therapeutic options for KRAS mutant tumors, developing new therapies for this subset is of particular clinical importance. To identify potential molecular determinants of response to eCNTFR-Fc, we assembled a panel of LUAD cell lines with a broad variety of genotypes and evaluated the effect of eCNTFR-Fc on cell viability (Fig. 4a). These cell lines exhibited a wide variety of sensitivities with the least sensitive (no effect) being normal lung cells (NL20) and the most sensitive being the LUAD cell line A549. Notably, sensitivity did not correlate with CNTFR expression (data not shown). The most sensitive cell lines all carried either KRAS or NRAS mutation. Cell lines with wild-type KRAS or an EGFR mutation exhibited intermediate sensitivity. Overall, sensitivity correlated with the ability to respond to upstream signals. H1755 and H1395 (both BRAFG469A) cells were completely insensitive to CNTFR blockade. The BRAFG469A mutation is a “Class 2” mutation that leads to signaling of constitutively active dimers and is expected to be independent of upstream KRAS signaling44,45. Similarly, the two KRAS mutant cell lines carrying the Q61H mutation (Fig. 4a) were completely insensitive to eCNTFR-Fc blockade. KRAS-GTP loading is the combined effect of intrinsic hydrolysis, which can vary widely among different mutations, and GAP-mediated hydrolysis, which is influenced both by the structure of specific mutations and the presence of GTPase activating proteins (GAPs) within the cell and their activation by upstream signals46. Q61H mutant KRAS lacks intrinsic GTPase activity46 and thus would also be expected to be largely insensitive to upstream signals that regulate GAPs. The three most common KRAS mutations in lung cancer, accounting for greater than seventy percent of the total, are G12C, G12D, and G12V47. Importantly, all cell lines carrying one of these three mutations were sensitive to eCNTFR-Fc. Taken together, these results are consistent with a model in which inhibition of CLCF1-CNTFR signaling regulates proteins involved in mediating KRAS-GTP hydrolysis and thereby regulating the output of oncogenic KRAS.
Figure 4.

Genotype specificity of eCNTFR-Fc in LUAD (a) Cell line viability 72 h after treatment with 2.5 μM eCNTFR-Fc or PBS (n = 3 independent experiments with four technical replicates per group). Exact P-values shown above respective cell line and *** P < 0.001 using two-tailed unpaired Student’s t-test. Data represented as mean ± S.D. (b) Representative cropped western blot of A549 treated with serum, CLCF1, eCNTFR-Fc, CLCF1 + eCNTFR-Fc, or eCNTFR-Fc + serum after 24 h serum starvation (n = 2 independent experiments with similar results). (c) Quantification of western blot from 4b and Extended Data Fig. 12a (n = 2 independent experiments with similar results). *** P < 0.001 using one-way analysis of variance (ANOVA) and Dunnett’s multiple comparison test (DMCT). Data represented as mean ± S.D. (d) RAS-GTP levels assessed by ELISA [Relative Light Units (RLU)] in cell lysates derived from A549 and H23 treated with serum, CLCF1, eCNTFR-Fc, CLCF1 + eCNTFR-Fc, or eCNTFR-Fc + serum after 24 h serum starvation (n = 2 independent experiments shown with three technical replicates per group). Data represented as mean.
After engagement with the ligand, CNTFR activates gp130, which leads to activation of SHP248. In turn, SHP2 functions as a key upstream regulator of both oncogenic and wild-type KRAS through regulation of GTP loading49–52. In both A549 and H23 LUAD cell lines, serum stimulation increased phosphorylation of SHP2, as well as STAT3 and ERK, as expected (Fig. 4b,c and Supplementary Fig. 1a). When cell lines were stimulated with recombinant CLCF1 in the absence of serum, phosphorylation levels of SHP2, STAT3, and ERK also increased, consistent with upstream signaling of CLCF1 serving to activate SHP2. Treatment with eCNTFR-Fc had a strong attenuation of the effect of CLCF1 but was less effective in inhibiting the effect of full serum, which is to be expected since serum has other effects independent of the CLCF1-CNTFR axis. Changes in levels of phosphorylated AKT were not observed (Supplementary Fig. 1b). To more directly establish the mechanistic link between the trimeric receptor complex and GTP loading of KRAS, we directly measured the levels of RAS-GTP in cells treated with recombinant CLCF1 and in the presence or absence of eCNTFR-Fc (Fig. 4d). Levels of RAS-GTP increased after CLCF1 treatment and this effect was attenuated by eCNTFR-Fc. These results point to a link between CLCF1-CNTFR signaling and oncogenic KRAS and may explain why CLCF1 inhibition appears to be more effective in some KRAS genotypes but not others. Taken together, these studies suggest that CLCF1 inhibition could be particularly effective in KRAS mutant tumors as further explored below.
eCNTFR-Fc sequesters CLCF1 and inhibits in vivo tumor growth
We next evaluated the role of eCNTFR-Fc as an anti-tumor therapeutic in vivo. To determine whether eCNTFR-Fc could effectively sequester mouse CLCF1, non-tumor bearing mice were treated with a single dose of eCNTFR-Fc. Serum levels of eCNTFR-Fc rapidly increased, along with a concomitant decrease in unbound CLCF1, which returned to baseline levels by 72 hours (Fig. 5a). These results indicate that eCNTFR-Fc effectively binds to mouse CLCF1 and reduces its availability in serum.
Figure 5.
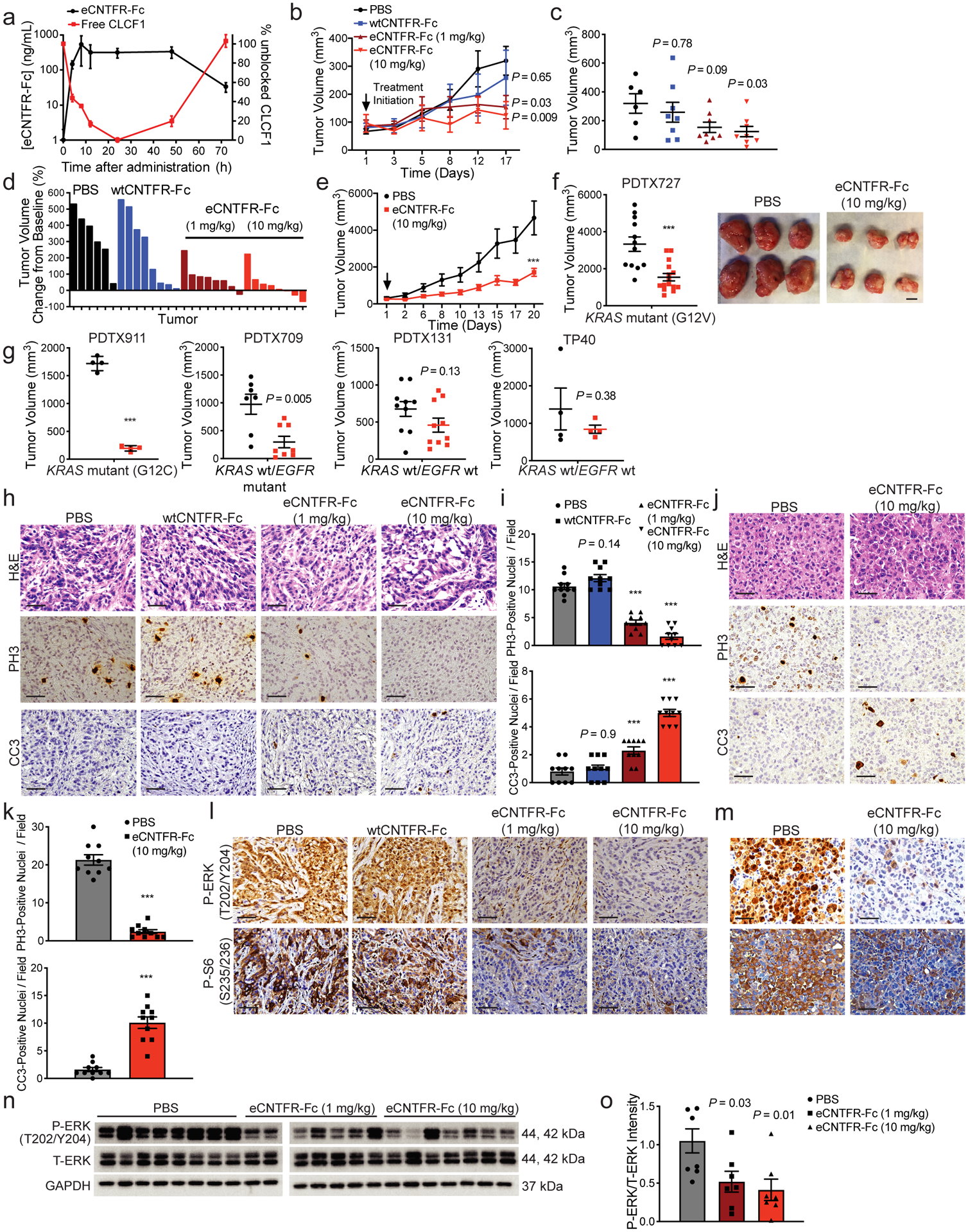
Effect of eCNTFR-Fc in preclinical xenograft models. (a) Blood clearance and CLCF1 sequestration after intraperitoneal (i.p.) dosing of 10 mg/kg eCNTFR-Fc in non-tumor bearing NOD scid gamma (NSG) mice. Serum samples were collected post injection and unbound CLCF1 was measured by ELISA using eCNTFR-Fc as the capture agent (n = 3 independent experiments). Vehicle-treated mice were used to determine baseline CLCF1 levels. Data represented as mean ± S.D. (b) Tumor volume quantification of A549 xenografts [n = 8 biologically independent samples except PBS (n = 6 at final time point)]. Two-way analysis of variance (ANOVA) and Dunnett’s multiple comparison test (DMCT). Data represented as mean ± S.E.M. (c) Tumor volume quantification of final time point of A549 xenografts [n = 8 biologically independent samples except PBS (n = 6)]. One-way ANOVA and DMCT. Data represented as mean ± S.E.M. (d) Waterfall plot showing tumor percent change from baseline for A549 xenografts. (e) Tumor volume quantification of patient-derived tumor xenograft 727 (PDTX727) model [PBS (n = 12) eCNTFR-Fc (n = 15) biologically independent samples]. *** P < 0.001 using two-way ANOVA and DMCT. Data represented as mean ± S.E.M. (f) Tumor volume quantification of final time point of PDTX727 [PBS: (n = 12) eCNTFR-Fc (n = 15) biologically independent samples] (left) and representative images of PDTX727 tumors (right). *** P < 0.001 using two-tailed unpaired Student’s t-test. Data represented as mean ± S.E.M. Scale bars, 10 mm. (g) Tumor volume quantification of final time point of PDTX models. [PDTX911 (n = 4), PDTX709 (PBS: n = 7 and eCNTFR-Fc: n = 8), PDTX131 (n = 10), TP40 (n = 4) biologically independent samples]. *** P < 0.001 using two-tailed unpaired Student’s t-test. Data represented as mean ± S.E.M. (h) Representative H&E staining and IHC for phospho-histone H3 (PH3) and cleaved caspase-3 (CC3) from A549 xenografts. Scale bars, 50 μm. (i) Quantification of PH3- and CC3-positive [n = 8 biologically independent samples except PBS (n = 6) with 10 technical replicates per group]. One-way ANOVA and DMCT. Data represented as mean ± S.E.M. (j) Representative H&E staining and IHC for PH3 and CC3 from PDTX xenografts. Scale bars, 50 μm. (k) Quantification of PH3- and CC3-positive foci. [PBS (n = 12), eCNTFR-Fc (n = 15) biologically independent samples with 10 technical replicates per group]. *** P < 0.001 using two-tailed unpaired Student’s t-test. Data represented as mean ± S.E.M. (l) Representative IHC for phospho-ERK (P-ERK) and Phospho-S6RP (P-S6) in A549 xenografts and (m) PDTX (n = 3 independent experiments with similar results). (n) Representative cropped western blot of A549 xenografts [n = 8 biologically independent samples except PBS (n = 6)]. (o) Quantification of western blot [depicting PBS (n = 8) and eCNTFR-Fc (n = 7 per group) biologically independent samples]. One-way ANOVA and DMCT. Data represented as mean ± S.E.M.
To test the therapeutic efficacy of eCNTFR-Fc, two LUAD cell lines were engrafted in immunodeficient mice and eCNTFR-Fc was dosed once tumors reached an average volume of 100 mm3. Treatment led to dose-dependent tumor inhibition in both xenograft models, whereas wtCNTFR-Fc had no effect (Fig. 5b–d and Supplementary Fig. 2a–d). Next, we evaluated the effect of eCNTFR-Fc on a panel of patient-derived tumor xenografts (PDTXs). Treatment with eCNTFR-Fc led to significant tumor growth inhibition in three of five LUAD PDTX models (Fig. 5e–g and Supplementary Fig. 2h,i and Supplementary Table 5). A significant decrease in proliferation markers and an increase in apoptosis were observed in both cell line xenografts and PDTX models (Fig. 5h–k and Supplementary Fig. 2e,f). The genotypes of the three PDTX models that responded to eCNTFR-Fc treatment were KRAS G12C, KRAS G12V, and KRAS wild-type (wt)/EGFR mutant, while the non-responders were KRAS and EGFR wt. We also noted that the CAFs with highest expression of CLCF1 were obtained from tumors with genotypes predicted to be most sensitive to eCNTFR-Fc (Supplementary Table 1).
As observed with CNTFR knockdown, treatment with eCNTFR-Fc also decreased activation of ERK (Fig. 5l–o and Supplementary Fig. 2g,j and Supplementary Fig. 3a,b), S6 Kinase (Fig. 5l,m and Supplementary Fig. 12g), and STAT3 (Supplementary Fig. 3c–f). To assess the time-dependent effect on signaling pathways, a short-term study was performed in which tumor-bearing mice were treated with eCNTFR-Fc and euthanized at different time points (Supplementary Fig. 3g–l). These results suggest that eCNTFR-Fc first leads to inhibition of STAT3, which is then followed by delayed inhibition of ERK and S6 signaling.
Next, we extended these studies to an autochthonous, highly aggressive genetically-engineered mouse (GEM) model of LUAD53. KrasG12D/Trp53f/f mice treated with eCNTFR-Fc demonstrated decreased tumor burden compared to vehicle-treated controls (Fig. 6a–e). Treatment with eCNTFR-Fc also led to decreased proliferation, increased apoptosis, and decreased activation of ERK, S6, and STAT3 signaling (Fig. 6f–h). We then performed a survival assay comparing eCNTFR-Fc treatment with cisplatin (Fig. 6i). We chose a platinum compound as a comparison as this is a commonly used standard chemotherapeutic therapy in human LUAD. Both cisplatin and eCNTFR-Fc treatment led to improved survival. However, cisplatin-treated mice had significant weight loss at the end of the study, whereas eCNTFR-Fc treated mice did not exhibit decreases in weight (Supplementary Fig. 4h). Extensive evaluation of mouse tissues post-mortem in eCNTFR-Fc-treated mice did not reveal any abnormalities, whereas platinum chemotherapy has been shown to induce significant adverse effects such as renal toxicity54 (Supplementary Fig. 4 and Supplementary Table 6). These results strongly support the potential therapeutic efficacy of eCNTFR-Fc in LUAD.
Figure 6.

Effect of eCNTFR-Fc in an autochthonous KRAS-driven genetically-engineered mouse model. (a) Representative 2D axial microCT (μCT) images, cross-section of mouse lungs at cervical vertebra 8 from KrasG12D/Trp53f/f mice treated 3 times per week with PBS or eCNTFR-Fc (10 mg/kg) by intraperitoneal (i.p.) injection for 4 weeks (Day 28) starting at 8 weeks post-delivery of 5 × 106 pfu of adenovirus-expressing Cre (Ad-Cre) (Day 0). Red outline surrounds the heart and red arrow identifies representative tumor nodule. (b) Quantification of μCT tumor burden using ImageJ software. Arbitrary units (A.U.) (n = 10 biologically independent samples with 12 technical replicates in each group). One-way analysis of variance (ANOVA) and Dunnett’s multiple comparison test (DMCT). Data represented as mean ± S.E.M. (c) Representative H&E images of lungs 28 days after treatment initiation. Scale bars, 1 mm. (d) Effect of treatment on tumor burden (%) and (e) tumor foci (n = 10 biologically independent samples). *** P < 0.001 using two-tailed unpaired Student’s t-test. Data represented as mean ± S.E.M. (f) Representative IHC for PH3 and CC3 from the GEM model. (g) Quantification of PH3- and CC3-positive foci. (n = 10 biologically independent samples with 12 technical replicates in each group). *** P < 0.001 using two-tailed unpaired Student’s t-test. Data represented as mean ± S.E.M. (h) Representative IHC for phospho-ERK (P-ERK), Phospho-S6RP (P-S6), and phopho-STAT3 (P-STAT3) 28 days after treatment initiation (n = 3 independent experiments with similar results). (i) Kaplan-Meier analysis of survival to ethical endpoint (n = 11 mice per group). Day 0 denotes the end of treatment and 12 weeks after administration of Ad-Cre. Two-sided log-rank test with Bonferroni correction. (j) CLCF1 ELISA performed on patient plasma samples (n = 25) and normal controls (n = 10). Mutation of interest = KRAS G12C, KRAS G12V, and KRAS wild-type / EGFR mutant. One-way ANOVA and DMCT. Data represented as mean ± S.E.M.
Further development of eCNTFR-Fc as a bona fide therapeutic agent will be specifically enhanced by identification of an appropriate biomarker for activity of this pathway. We noted a modest positive correlation between CLCF1 expression and decreased viability after treatment with eCNTFR-Fc in the cell line panel we tested in vitro (Supplementary Fig. 5a). While our data suggest that specific genotypes are more sensitive to eCNTFR-Fc, we sought to determine whether CLCF1 levels in the plasma could also serve as an indicator of activity of this pathway in individual patients. We developed a method to detect CLCF1 by ELISA, with eCNTFR-Fc acting as a capture agent, and used it to measure the levels of CLCF1 in the plasma of LUAD patients. We observed a trend towards higher levels of CLCF1 in LUAD patients relative to healthy controls. (Supplementary Fig. 5b and Supplementary Tables 7,8). Moreover, patients with genotypes sensitive to eCNTFR-Fc (with ‘mutation of interest’) had significantly higher levels of CLCF1 than those without the mutation of interest (Fig. 6j). We analyzed the data further using logistic regression (logit) to demonstrate whether CLCF1 in the blood can predict if a tumor has a particular mutation of interest (KRAS G12C, KRAS G12V, or KRAS wt/EGFR mutant) [Odds ratio: 8.346 (CI 95% 6.36–10.33); P-value: 0.0364] (Supplementary Fig. 5c–d). Our results demonstrate that when plasma CLCF1 (pg/mL) levels increase by one unit, it is 8.346 times more likely to be in the mutation group. Taken together, these results suggest that CLCF1 plasma concentration combined with genotypic analysis of the tumor could serve as potential biomarkers for selection of patients most likely to benefit therapeutically from eCNTFR-Fc.
DISCUSSION
Prior work in a genetically engineered mouse model of lung cancer revealed the CLCF1-CNTFR axis as a therapeutic target in LUAD. Here, we first demonstrated that CNTFR loss is deleterious in human LUAD cell lines. We then developed an approach for inhibiting CNTFR activity using a novel engineered decoy receptor using yeast phage display. This involved generating a molecule that has the capacity to bind CLCF1 with high affinity while simultaneously decreasing its affinity for LIFR and gp130. This new molecule, termed eCNTFR-Fc, was extensively tested in a wide panel of in vitro and in vivo models. Notably, in the course of these studies we found that CNTFR blockade is particularly effective in cell lines and PDTX models carrying a subset of oncogenic KRAS mutations, identifying a biomarker for further clinical evaluation using this decoy receptor. The effect on KRAS mutant tumors is particularly significant given that few therapeutic options currently exist for this subset of LUAD patients, despite the recent development of small molecules specifically targeting the G12C mutation55. Our results are consistent with the recently reported importance of upstream signaling molecules for both wild-type and oncogenic KRAS signaling when the capacity to cycle between a GTP bound and a GDP bound state is maintained, either by high intrinsic GTPase activity or low intrinsic GTPase activity paired with high GAP-mediated hydrolysis46,56,57. Recent work suggests that combined treatment with SHP2 and MEK inhibitors may be beneficial in LUAD. Our work provides another potential therapeutic option, which could involve combination of eCNTFR-Fc with MEK inhibitors49.
Monoclonal antibodies are a prominent class of therapeutic agents with well-established developmental and regulatory paths, yet a challenge for their therapeutic use is that many natural ligand/receptor interactions are of high affinity (low nM to pM range), and thus antibodies cannot effectively block them, which have typical antigen binding affinities of 1–10 nM58. While wild-type CNTFR has a weak binding affinity (Kd>100 nM) for its ligands, the CNTFR/LIFR/gp130 tripartite complex is expected to bind CLCF1 with an affinity in the low nanomolar range59. Hence, to effectively modulate the CLCF1/receptor interaction, a therapeutic candidate must bind CLCF1 with much greater affinity than the native quaternary complex or be dosed at concentrations far exceeding endogenous levels. However, such high doses can result in undesirable off-target effects. These features of CLCF1-CNTFR binding suggested that an engineered decoy would be an optimal strategy.
We describe the development of an engineered receptor decoy to effectively bind to and sequester CLCF1. We used directed evolution to screen libraries of 1 × 108 variants of soluble CNTFR to identify a candidate that bound with high affinity to the CLCF1 ligand. To add to the complexity, soluble wtCNTFR functions as an agonist by binding to co-receptors LIFR and gp130, thus requiring engineering of a variant that does not bind these co-receptors while retaining binding to CLCF1 with high affinity. The result of these protein engineering efforts was a soluble CNTFR variant termed eCNTFR, which contains 8 amino acid mutations and binds to CLCF1 with ~60 pM affinity while not binding to LIFR and gp130 at concentrations up to 100 nM. To increase serum half-life and aid in recombinant protein production and purification, eCNTFR was fused to an antibody Fc domain. This final variant, eCNTFR-Fc, retained its high binding affinity for CLCF1 and did not bind gp130 and LIFR.
Importantly, while eCNTFR-Fc also exhibited high affinity binding to mouse CLCF1, unlike wtCNTFR-Fc, it did not bind to the related cytokine CNTF. Given that both mouse CLCF1 and CNTF are known to share the ligand binding domain of CNTFR with human CLCF1, eCNTFR-Fc appears to have retained binding for mouse CLCF1 due to higher sequence homology (97% vs. 17%). As CNTF signaling is important for neuronal cell survival, it is possible that the selectivity of eCNTFR-Fc for CLCF1 over CNTF will minimize side effects. We have not observed adverse effects in animal experiments thus far, both in immunodeficient and immunocompetent mice. Moreover, our comprehensive studies monitoring mouse weight and examination of blood samples and internal organs by a veterinary pathologist, have shown no evidence of toxicity (Supplementary Fig. 4 and Supplementary Table 6).
We tested the therapeutic efficacy of eCNTFR-Fc in xenograft, PDTX, and GEM models of LUAD. In all three models, administration of eCNTFR-Fc significantly decreased tumor volume in a dose-dependent manner compared to the untreated control. When compared to wtCNTFR-Fc, the eCNTFR-Fc-treated group showed a substantial decrease in tumor size, demonstrating that the engineering efforts were necessary for therapeutic effect. We utilized an autochthonous model as the final assessment of efficacy of our decoy receptor and at the end of the study observed a significant decrease in tumor burden and an increase in survival, comparable to cisplatin, the standard of care. However, unlike cisplatin, treatment with eCNTFR-Fc showed no evidence of toxic side effects.
CLCF1 belongs to the IL-6 cytokine family. IL-6 itself has been extensively evaluated as a pro-oncogenic cytokine in breast60, lung61, and pancreatic cancer62. LIF, another cytokine belonging to the same family, was recently identified as a key paracrine factor in pancreatic cancer63. However, the pro-oncogenic effects of CLCF1-CNTFR signaling have received much less attention. Here, we demonstrate that CLCF1 activation plays an important pro-oncogenic role and that inhibition of this pathway has significant therapeutic effects across a wide range of genetic subtypes of LUAD. Indeed, the results described above indicate that eCNTFR-Fc is effective in tumors carrying various KRAS mutations, a subset of LUAD that has proven difficult to treat. The engineered decoy receptor was also effective against EGFR mutant PDTX models. In addition to LUAD, CLCF1-CNTFR signaling may also be relevant to other cancers including hepatocellular carcinoma, colorectal cancer, low-grade gliomas, and pancreatic cancer21–24. Collectively, our data indicate that eCNTFR-Fc is a promising therapeutic candidate for treating LUAD, and we have identified specific biomarkers that may portend increased sensitivity. These biomarkers could be used to identify patients with tumors most likely to be inhibited by this agent in clinical trials.
METHODS
Cell lines.
All cell lines were obtained from ATCC and maintained in RPMI-1640 supplemented with 10% Bovine Growth Serum (BGS), 4 mM glutamine, and 1% Penicillin and Streptomycin (P/S). Experiments were performed in DMEM supplemented with 10% BGS and P/S. Cells were incubated at 37°C in 5% CO2. Cells were routinely tested to exclude Mycoplasma contamination. Comparison of STR testing (Promega GenPrint10) on genomic DNA from our cell lines to ATCC data confirmed the identity of the cell lines.
Ethics statement.
Mice were maintained and animal experiments performed in accordance with policies approved by the Stanford University Administrative Panel on Laboratory Animal Care (Protocol #14625) and by the Institutional Animal Care and Use Committee at the University of California San Francisco (AN157619–01G). Primary human tissues were obtained with informed consent under a protocol reviewed by the Stanford University Institutional Review Board.
Mice
Lung adenocarcinoma mouse model.
Lox-stop-Lox-KrasG12D (129 Sv/Jae)64, Trp53fl/fl (FVB)65, (C57BL/6J) mice were maintained in a virus-free environment. Mice were intra-nasally infected with 5 × 106 pfu of adenovirus expressing Cre (University of Iowa) at 10- to 14-weeks of age64. Mice were dosed with eCNTFR-Fc (10 mg/kg) or PBS (vehicle) by intraperitoneal injection for four weeks three times per week beginning 8-weeks post-infection. Mice were weighed at the beginning of study and periodically throughout drug treatment. Equal number of male and female mice were utilized.
NSG Mice.
NOD-scid IL2Rgnull (NSG) mice (Jackson Laboratory Strain #005557) were obtained from Jackson Laboratory, Bar Harbor, Maine, USA, and bred in the barrier facility at Stanford University and UCSF. Both male and female mice were used for xenograft studies. Xenograft tumors were injected when mice were approximately 8–10 weeks of age. Mice were weighed at the beginning of study and periodically throughout drug treatment. Both male and female mice were used for xenograft studies.
Huma lung adenocarcinoma (LUAD) survival and gene expression analysis.
CLCF1 TPM log2 expression for the cohorts (LUAD; LUSC) were downloaded directly from the Broad Institute with R programming language using the package FirebrowseR (1.1.35)1. We used only expression data categorized as either TP (Primary Tumor) or NT (Normal). The full LUAD expected counts (RSEM level 3) were downloaded directly from the FIREHOSE Broad GDAC website. Somatic mutation for the LUAD data set was acquired from the UCSC Xena public repository. Only samples with a non-silent KRAS mutation(s) were associated with the KRAS mutation group; samples with KRAS silent mutations were not included as the KRAS wild-type group and were excluded from the analysis. Clinical data for LUAD survival analysis including censored data such as overall survival was acquired from published clinical aggregation of the TCGA dataset2. Survival analysis curves and multivariate Cox hazard regression were completed in R programming language using the survminer (0.4.3.999) and survival packages (2.44–1.1), respectively. For the multivariate Cox regression analysis, we adjusted for age of diagnosis, gender, and cancer stage. We grouped samples (Normal vs High) based on the quantile of the respective gene expression: normal is < 75th percentile and high is > 75th percentile.
| Resource | Source | Indentifier |
|---|---|---|
| CNTFR log2 expression (TPM) | FirebrowseR1 | https://github.com/mariodeng/FirebrowseR |
| Clinical data | Liu et al, 2018 | Supplemental Table S1 |
| LUAD RSEM | Direct download | http://gdac.broadinstitute.org/ |
| Mutation (LUAD) | Direct download | https://xenabrowser.net |
Generation of patient-derived tumor xenografts (PDTXs).
Fresh patient samples were cut into 1 × 1 mm fragments and either implanted fresh or frozen in 90% BGS and 10% DMSO for later use. Tumor fragments were dipped in Matrigel (Corning Matrigel #356234) and implanted in the subrenal capsule of NOD scid gamma (NSG) mice. Tumor growth was monitored for up to 1 year after implantation. Successfully implanted tumors were harvested at approximately 1 to 2 cm. A fragment was kept for histology, and the remainder was digested with collagenase for 45 min at 37°C and filtered through 100 μm and 40 μm cell strainers and centrifuged for 1,200 rpm for 8 min. Cells were incubated in RBC lysis buffer and resuspended in 6 mL of media and spun through 0.5 mL of serum (BGS) layered on the bottom of the tube to remove cellular debris. For subsequent passages and drug studies, cells were implanted subcutaneously in flanks of NSG mice (5 × 105 cells per flank) in 100 μL α-MEM and 20 μL Matrigel (Corning #356234). Xenograft tumor fragments were stored at −80°C until use. Tumor volume was measured at the times indicated and calculated using the ellipsoid formula [0.5(length × width2)].
Serum analysis and toxicity studies.
Blood samples from individual mice were collected at the end of the experiment under terminal anesthesia using cardiac puncture. Serum was separated from blood within 1 h by centrifugation at 500 g for 10 min. Samples were aliquoted and stored at −80°C for subsequent testing. Comprehensive Metabolic Panel (CMP) and Complete Blood Count (CBC) analyses were done by the Animal Diagnostic Laboratory at Stanford Veterinary Service Center. Toxicity studies including necropsy and comprehensive histopathological analysis of each organ were performed by a veterinary pathologist.
Blood specimen collection.
Blood samples were drawn from patients with lung adenocarcinoma (LUAD) before surgical resection of localized tumors at Stanford Hospital under IRB-approved protocol. Blood specimens were also drawn from healthy donors after obtaining informed consent. All specimens were collected into BD Vacutainer® tubes (#367841) containing the anticoagulant EDTA and were processed within 24 h. Between sample collection and sample processing, whole blood specimens were stored at 4°C on a rocking platform to prevent cell settling. Samples were centrifuged at 3,000 rpm for 10 min at 4°C. Following centrifugation, the supernatant (plasma) was removed and samples were stored at −80°C.
MicroCT imaging (μCT).
Scans were performed using the Xstrahl Small Animal Radiation Research Platform (SARRP, Xstrahl Inc., Suwanee, GA). The SARRP operates at a source-to-isocenter distance of 35 cm with a dual x-ray tube focal spot of 0.4mm (small) for the 40–80 kVp imaging beams66. μCT imaging is acquired in pancake geometry utilizing a stationary x-ray tube and detector with a 360° rotating couch, and is performed with 40–80 kVp imaging beams with 0.8 mm Be internal filtration and 1 mm Al added filtration67. The standard imaging protocol was used consisting of 60 kVp tube voltage, 0.8 mA beam current, and 1 min scanning time with a 0.275 × 0.275 × 0.275 mm3 voxel size. Maximum mean organ imaging doses to boney and non-boney structures were 10.5 cGy and 3.5 cGy, respectively, as reported previously68. Images were analyzed using 3D Slicer and axial images with lung tumor nodules were quantified using ImageJ.
Treatment of mice with eCNTFR-Fc.
When tumors reached an average size of 100 mm3 per tumor, mice were stratified into treatment arms based on average tumor size per group. Mice were then dosed with eCNTFR-Fc (10 mg/kg) or PBS (vehicle) by intraperitoneal (i.p.) injection for two to four weeks three times per week. Mice were weighed at the beginning of study and periodically throughout drug treatment. Tumor volume was measured with digital calipers three to four times per week.
Quantitative reverse transcriptase-PCR (qRT-PCR).
RNA was isolated using TRIZOL reagent (Invitrogen) and further purified using Qiagen miniRNA columns (Qiagen) following manufacturers’ specifications. cDNA was synthesized with a Maxima kit (Thermo Scientific), and quantitative reverse transcriptase-PCR (qRT-PCR) was performed using SYBRGreen (Applied Biosystems) also following manufacturers’ specifications. qRT-PCR was performed as follows using primer sequences in Supplementary Table 2: 95°C for 10 min, 35 cycles of 95°C for 15 s and 60°C for 1 min.
Knockdown studies in xenografts.
pLKO shRNA constructs were purchased from Thermo Fisher Scientific. Lentivirus for each construct was generated by transfecting HEK293 cells with polyethylenimine (PEI). Viral supernatants were collected on days 1 and 2 after transfection and pooled on day 2. Viral supernatants were then filtered through 0.45 μM PES filters. Viral pellets were re-suspended on a platform rocker for 2 h with approximately 500 μL fresh media. Cells were dissociated into a single cell suspension using Collagenase (Sigma) digestion buffer and filtered through 100 μm and 40 μm cell strainers as above. The resulting cell suspension was then plated at approximately 5 × 106 cells per well of a 6-well plate and spin infected with polybrene (Sigma) and virus in media at 1500 rpm at room temperature for 30 min (Sorvall XRT centrifuge) followed by incubation at 37°C. After selection with puromycin (2 μg/mL), cells were trypsinized, filtered, and counted for viable cells. Cells were then implanted (as above) keeping the viable cell count consistent between study groups. Remaining cells were kept for confirmation of gene knockdown.
Cell extracts and western blot analysis.
For total cell extracts, cells were lysed using NP-40 lysis buffer (20 mM Tris-HCl, pH 8.0, 137 mM NaCl, 10% Glycerol, 1% NP-40, dH20), 1x protease inhibitors (Sigma P8349–1ML) and 1x phosphatase inhibitor cocktail (Sigma P5726–1ML) for 15 minutes, sonicated and lysed for 30 minutes. Tumors were thawed and mechanically disrupted using the Bio-Gen PRO200 Homogenizer (PRO Scientific) on ice prior to lysis. Protein concentration was determined by BCA assay (Pierce). Samples were heat denatured at 95°C for 10 minutes. Proteins were resolved by SDS-PAGE, transferred to a PVDF membrane, and blocked in 5% non-fat dry milk. Primary antibodies were incubated overnight at 4°C (at indicated dilutions): P-AKT (#4060, Cell Signaling, 1:1000), T-AKT (#75692, Cell Signaling, 1:1000), P-ERK1/2 (#4370, Cell Signaling, 1:1000), T-ERK1/2 (#4695, Cell Signaling, 1:1000), P-MEK1/2 (#9121, Cell Signaling, 1:1000), T-MEK1/2 (#9122, Cell Signaling, 1:1000), P-SHP2 (#ab62322, Abcam, 1:1000), T-SHP2 (#3397, Cell Signaling, 1:1000), P-STAT3 (#9145, Cell Signaling, 1:1000), T-STAT3 (#9139, Cell Signaling, 1:1000), and GAPDH (#9485, Abcam, 1:1000). Proteins were analyzed by ChemiDoc XRS System (Bio-Rad), and, when necessary, quantification was performed using ImageJ software.
Histology and immunohistochemistry (IHC).
Tissue specimens were fixed in 10% buffered formalin for 24 h and stored in 70% ethanol until paraffin embedding. 5 μm sections were stained with hematoxylin and eosin (H&E) or used for immunohistochemical studies. Immunohistochemistry was performed on formalin-fixed, paraffin embedded mouse and human tissue sections using a biotin-avidin method per standard protocol. Briefly, sections were first deparaffinized in xylene and rehydrated in graded ethanols. Standard sodium citrate, pH 6.0, or 1 mM EDTA, pH 8.0 (for P-STAT3 antibody), buffered antigen retrieval was performed for 15 min prior to 3% H2O2 quenching for 10 min of endogenous peroxidase activity. Blocking and antibody dilutions were made in 5% normal goat serum (Vector Labs) in TBS containing 0.1% Triton X-100 (Sigma) and incubated overnight at 4°C in a humidified chamber. The following antibodies were used (at indicated dilutions): P-ERK1/2 (#4370, Cell Signaling, 1:400), P-Histone H3 (#9701, Cell Signaling, 1:400), Cleaved Caspase 3 (#9661, Cell Signaling, 1:400), P-STAT3 (#9145, Cell Signaling, 1:100), and P-S6 (#4858, Cell Signaling, 1:400). Biotinylated secondary goat anti-rabbit antibody (Vector Labs) and avidin-biotin (Vectastain Elite ABC kit) were incubated for 30 min at room temperature. Signal was developed with DAB peroxide substrate (Abcam #642238) per manufacturer’s instructions and counterstained with hematoxylin. Analysis of the tumor area and immunohistochemical analysis were done using ImageJ software by measuring pixel units. All quantification analyses of IHC staining were performed on three tumor specimens per treatment condition in three independent experiments, unless otherwise specified. Representative 40X fields of view (FOV) were imaged, avoiding areas of necrosis and poor cellularity. Ten to 12 FOVs were analyzed per condition. Data are displayed as mean per 40X FOV and error bars as standard error of the mean (S.E.M.). All quantification was done in a blinded fashion.
Cell assays.
Cell viability.
Cells were seeded in 96-well plates at 1,000 cells per well (optimal density for growth) in a total volume of 100 μL media containing 10% Bovine Growth Serum (BGS). After 24 h incubation, cell viability was assessed by AlamarBlue® assay (Thermo Fisher) for 7 days according to the manufacturer’s instructions. All data were compared to the first reading.
Colony-formation assay.
For long-term colony-formation assay, 10,000–50,000 cells per well were seeded in 6-well plates. After 12 days, cells were fixed with methanol, stained with crystal violet, photographed, and quantified.
3D Spheroid methylcellulose assay.
For anchorage-independent sphere growth, the cells were seeded into 24-well ultra-low attachment plates (20,000 viable cells per well) in 2 mL of complete medium supplemented with 0.5% methylcellulose. The spheres were allowed to form for 9–20 days (depending on the cell line). Spheres were imaged with Leica DMi8 microscope (brightfield). Sphere size and number were quantified using ImageJ.
Analysis of RAS-GTP Levels.
Levels of activated RAS-GTPase were determined using the RAS GTPase ELISA Kit (Abcam #134640) per the manufacturer’s instructions, similar to a previously published method69. Briefly, 1 × 106 cells were seeded in RPMI media supplemented with 10% Bovine Growth Serum (BGS), 4 mM glutamine, and Penicillin and Streptomycin (P/S) in 10-cm tissue culture dishes and incubated at 37°C in 5% CO2 until cells reached 60% confluence. Cells were then serum starved with RPMI and 1% penicillin/streptomycin for 24 h. Cells were subsequently incubated in CLCF1 (10 nM) and eCNTFR-Fc (2.5 μM) for 20 min at 37°C in 5% CO2. Media was then removed and cells were washed once in ice-cold PBS and processed following the manufacturer’s protocol.
Logistic regression (logit) model.
Logit regression was performed with R programming language using the glm function. Odds ratio (O.R.) was calculated using Wald test as part of the glm output. Our independent variable was a continuous value of blood plasma CLCF1 (pg/mL) and no other covariates were used in our model. Our dependent groups were samples with either ‘mutation of interest’ defined as KRAS G12C, KRAS G12V, or KRAS wild-type (wt) / EGFR mutant (dummy coded as 1, n = 17) or KRAS wt (dummy coded as 0, n = 8). The estimated probability was generated with the statistics package (predict function) in R using the results from our logit model. The conditions were set to blood plasma CLCF1 (pg/mL) levels for each quantile level (in 0.1 increments) for all samples (n = 25): range between 1.76 to 28.50 CLCF1 (pg/mL).
Recombinant CLCF1 production.
cDNA encoding for CLCF1 without the signal peptide sequence (28–225) was cloned into pET28b plasmid with inducible lac promoter using BsaI and XhoI restriction sites and amplified in DH10B cells. For expression, purified plasmids were transformed into Rosetta gami cells. Inclusion bodies were solubilized in 60% ddH2O, 40% acetonitrile, 0.1% TFA containing 5 mM DTT. Reversed-phase high-performance liquid chromatography (RP-HPLC) was used to purify CLCF1. Protein purity was further analyzed using SDS-PAGE and quantified using a Nanodrop 2000 (Thermo Scientific). A value of 39,549 M−1cm−1 was used as the extinction coefficient to quantify protein concentration.
Soluble CNTFR, LIFR, and gp130 production.
cDNA corresponding to the extracellular domains of CNTFR (1–342), LIFR (1–534), and gp130 (1–619) was cloned into the pAdd2 plasmid and amplified in DH10B cells. For expression, purified plasmids were transfected into human HEK 293 cells using PEI (#23966–2, Polysciences). Briefly, PEI was dissolved in dH2O to 1 g/L. For 500 mL transfection volume, 0.5 mg of purified DNA and 1 mL of PEI was dissolved in 10 mL of OptiPro Serum-Free Media (#12309–019, Thermo Fisher Scientific) each, then mixed immediately. After 15 min the solution was added dropwise to 500 mL of cells. The cells were incubated on a rotary shaker at 120 RPM in a humidified incubator at 37°C and 5% CO2. Fc fusion proteins were purified using a protein A (#101142, Fisher Scientific) affinity column; proteins containing a hexahistidine tag were purified using a nickel-NTA (#30210, Qiagen) affinity column. Proteins were then further purified using size exclusion chromatography. The following extinction coefficients were used for protein quantification: CNTFR variants: 70,275 M−1cm−1; CNTFR-Fc variants: 206,410 M−1cm−1; gp130: 130,470 M−1cm−1; gp130-Fc: 326,800 M−1cm−1; LIFR: 98,610 M−1cm−1; and LIFR-Fc: 263,080 M−1cm−1.
Generation and screening of a CNTFR library created via error-prone PCR.
CNTFR was expressed in yeast as a genetic fusion to the agglutinin mating protein Aga2p. cDNA encoding the human CNTFR extracellular domain (residues 18–342) was cloned into the pCTCON2 yeast display plasmid using NheI and BamHI restriction sites. An error-prone library was created using the CNTFR extracellular domain as a template, and mutations were introduced by using Taq polymerase (#50-811-694, Fisher Scientific) and 55 mM MgCl2. Separate PCR reactions were performed using different concentrations of MnCl2 (0, 0.01, 0.05, 0.1, and 015 mM). Products from these reactions were purified using gel electrophoresis. Purified mutant cDNA and linearized plasmid were electroporated into EBY100 yeast, where they were assembled in vivo through homologous recombination. Library size was estimated to 8.1 × 107 by dilution plating and colony counting.
Yeast displaying high-affinity CNTFR variants were isolated using fluorescence-activated cell sorting (FACS) using a BD Aria II flow cytometer (Stanford FACS Core Facility) and analyzed using a BD FACSCalibur. Screens were carried out using equilibrium binding conditions where yeast were incubated at room temperature in phosphate-buffered saline containing 1 mg/mL BSA (PBSA) with the following CLCF1 concentrations: for sort 1, 20 nM CLCF1 for 3 h; for sort 2, 2 nM CLCF1 for 6 h; for sort 3, 0.5 nM CLCF1 for 12 h. After incubation with CLCF1, yeast were pelleted, washed and resuspended in PBSA with 1:500 ratio of chicken anti-c-Myc (#A21281, Invitrogen) for 30 min at 4°C. Yeast were then washed and pelleted, and secondary labeling was performed on ice for 30 min using PBSA with 1:100 dilution of goat anti-chicken PE (#sc-3730, Santa Cruz Biotech) and mouse anti-HIS Hilyte Fluor 488 (#61250-H488, Anaspec).
Sorted clones were propagated and subjected to further rounds of FACS. After the last round of screening plasmid DNA was recovered using a Zymoprep kit (#50-444-107, Zymo Research Corp), transformed into DH10B electrocompetent cells, and isolated using plasmid miniprep kit. Sequencing was performed by Molecular Cloning Laboratories. Samples were analyzed on a FACSCalibur (BD Biosciences), and data were analyzed using FlowJo software (Treestar Inc).
Generation and screening of a CNTFR library created via Staggered Extension Process (StEP).
The StEP method was performed as described previously36 and the resulting library was displayed on yeast. Briefly, 20 unique sequences were selected randomly from the yeast population isolated from the final sort round of the error-prone PCR library. 1 ng of each of the templates was combined and 20 ng total template was mixed with the final concentrations of 0.15 μM each primer, 1X PCR buffer, 200 μM dNTP mix, 1.5 mM MgCl2, and 2.5 U Taq polymerase in sterile dH2O to 50 μL. The extension protocol was run for 100 cycles using the following parameters: 94°C for 30s (denaturation) and 55°C for 10 s. Products from these reactions were purified using gel electrophoresis. Purified mutant cDNA and linearized plasmid were electroporated in EBY100 yeast, where they were assembled in vivo through homologous recombination. Library size was estimated to 7.9 × 107 by dilution plating.
Screens were performed using a single round of equilibrium binding sorting followed by two rounds of kinetic off-rate sorts. For kinetic off-rate sorts, yeast were incubated with 2 nM CLCF1 for 2 h at room temperature, after which cells were washed twice to remove excess unbound CLCF1 and resuspended in PBSA containing 20 nM wtCNTFR-Fc to prevent rebinding of dissociated CLCF1. For the length of the unbinding steps, 10 h was used for sort 2, and 24 h was used for sort 3. Libraries were stained to detect CLCF1 binding and c-myc expression as described above and sorts were conducted such that the 0.5–1% of clones with the highest CLCF1 binding/c-Myc expression ratio were collected by FACS, enriching the library for clones with the highest binding affinity to CLCF1. Plasmid DNA was isolated and sequenced as described above.
Library generation and screening for CNTFR variants that do not bind LIFR.
To engineer CNTFR variants with decreased binding for LIFR, error-prone PCR was used to introduce random mutations into CNTFR variant 4, creating a library with an estimated diversity of about 1 × 108 transformants. The resulting library was displayed as fusion proteins on the yeast cell surface and screened to isolate the population with decreased binding signal for LIFR-Fc in the presence of CLCF1. To retain the binding affinity for CLCF1, screening was performed by alternating between positive selection for 0.5 nM CLCF1 and negative selection for increasing concentrations of LIFR-Fc. After six rounds of sorting, two consensus mutations emerged (Y177H and K178N). These mutations additively contributed to decreased LIFR binding.
Yeast-displayed CNTFR binding assays.
Yeast displaying the CNTFR constructs were incubated with varying concentrations of CLCF1 for 12 h at room temperature to reach equilibrium binding. This was followed by washing with PBSA and resuspension in PBSA with 1:500 ratio of chicken anti-c-Myc antibody for 30 min at 4°C. Yeast were then washed and pelleted, and secondary labeling was performed on ice for 30 min using PBSA with 1:100 dilution of goat anti-chicken PE antibody and mouse anti-HIS Hilyte Fluor 488 antibody. Then samples were washed and analyzed by flow cytometry using BD Accuri flow cytometer. Samples were analyzed on BD Bioscience software, and data were analyzed using FlowJo software (Treestar Inc).
For assays to detect binding with the β receptors, varying concentrations of LIFR constructs and/or gp130 constructs with 10 nM CLCF1 were added to yeast-displayed CNTFR. For His-tagged constructs, mouse anti-HIS Hilyte Fluor 488 antibody was used to detect binding. For detecting Fc-fusion constructs, anti-mouse-Fc Alexa 488 antibody (#A11029, Thermo Fisher) was used.
Cell-free binding assays.
96-well plates were coated with 10 μg/mL of anti-HIS antibody or anti-mouse-Fc antibody overnight and blocked with 5% milk for 1 h. The plates were then washed twice with PBSA. Varying concentrations of soluble CNTFR-HIS or CNTFR-Fc fusion constructs were incubated with 2 nM CLCF1 in PBSA for 12 h at room temperature. The mixture was then added to 96-well plates coated with anti-HIS antibody or anti-mouse-Fc antibody respectively for 1 h followed by washing twice with BPBS. Subsequently, the wells were incubated with 1:1000 diluted anti-CLCF1 rabbit antibody (#ab26125, Abcam) for 2 h at room temperature then washed four times with PBS. The wells were incubated with 1:1000 diluted HRP conjugated anti-rabbit antibodies (#111-035-144, Jackson ImmunoResearch) for 2 h at room temperature, washed four times with PBS. 1-Step Ultra TMB ELISA (#34029, Thermo Fisher Scientific) was used for the readout.
Phosphorylation assays.
A549 or H23 cells were grown until 50% confluence in 6-well plates. The cells were incubated in CLCF1 (10 nM) and CNTFR constructs (10 nM) for 20 min at 37°C in 5% CO2, then lysed with NP-40 buffer containing protease inhibitor (#P8340, Sigma Aldrich) and phosphatase inhibitor (#P5726, Sigma Aldrich). Equal amounts of lysate were loaded on Bis-Tris gels and transferred onto nitrocellulose membrane. Western Blot analysis was performed with the reagents above. Chemiluminescence was detected using the ChemiDoc XRS System (Bio-Rad). NP-40 buffer was composed of 20 mM Tris pH 8.0, 137 mM NaCl, 10% glycerol, and 1% IGEPAL/NP40.
CLCF1 cell proliferation assay.
5 × 103 A549 and H23 cells were seeded and grown for 24 h, and then serum starved by incubating for 24 h in DMEM with 0.1% BSA. CLCF1 and CNTFR constructs were then added and incubated for 72 h at 37°C in 5% CO2. Next, AlamarBlue® reagent (#DAL1025, Fisher Scientific) was added to each well and incubated for 1 h at 37°C in 5% CO2. The cell metabolic activity was detected by measuring fluorescence using 560EX nm/590EM nm. Error bars represent the standard deviation of triplicate wells. Data were measured against negative control with only media.
Analysis of in vivo CLCF1 sequestration by eCNTFR-Fc.
Non-tumor bearing NSG mice were administered a single dose of eCNTFR-Fc at 10 mg/kg body weight via intraperitoneal injection. The doses were formulated in 200 μL volume. Two mice were analyzed per condition, and untreated mice were used to determine baseline CLCF1 levels. Terminal blood collection was done at euthanasia by cardiac puncture at 6 h, 12 h, 24 h, 36 h, 48 h, and 72 h after injection, and serum was isolated for analysis. CLCF1 levels were measured using a sandwich ELISA. In this assay, eCNTFR-Fc was used as a capturing agent to ensure the detection of free, unbound CLCF1. 96-well plates were coated with 10 μg/mL of eCNTFR-Fc overnight at room temperature and blocked with 5% milk. After the coated plates were washed twice with PBSA, the plates were incubated with the collected serum at room temperature for 2 hours. After the plates were washed with BPBS twice, detection of CLCF1 was carried out using polyclonal anti-CLCF1 antibody and anti-rabbit HRP. After washing the plates 4 times with BPBS, ELISAs were developed using the 1-Step Ultra TMB ELISA.
Statistical analysis.
The specific statistical tests utilized are indicated in the figure legends. In sum, for comparison between two groups, statistical significance was assayed by two-tailed unpaired Student’s t-test with Prism GraphPad software. For comparison of more than two groups, variance was first systematically examined using an F-test for both one-way and two-way Analysis of Variance (ANOVA), depending on the experiment, combined with Dunnett’s multiple correction test (DMCT) for post-hoc analysis with Prism GraphPad software, * P < 0.05; ** P < 0.01; *** P < 0.001. Data are represented as mean ± S.D. for in vitro experiments and mean ± S.E.M. for in vivo experiments. In boxplots, box represents 25th and 75th percentiles with midline indicating the median; whiskers extend to the lowest/highest value within 1.5 times the interquartile range (IQR). Kaplan-Meier survival curves were calculated using the survival time for each mouse. The two-sided log-rank test with Bonferroni correction was used for post-hoc pairwise analysis with Prism GraphPad software. Unless otherwise specified, each in vitro experiment was subjected to at least three independent experiments (n biological replicates), and the average of the independent experiments was recorded and used in corresponding statistical analysis. For the in vivo animal studies, n is provided for each biologically independent sample. For human lung adenocarcinoma (LUAD) survival and gene expression data, analyses were performed with R programming language using the package FirebrowserR. Kaplan-Meier survival analysis curves and multivariate Cox hazard regression were completed in R programming language using the survminer and survival packages, respectively. For the multivariate Cox regression analysis, adjustments were made for age of diagnosis, gender, and cancer stage. Logistic regression (logit) analysis was performed with R programming language using the glm function.
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this published article and its supplementary information files or are available from the corresponding author upon reasonable request. Statistical Source Data underlying all Figures are provided as a separate file with a tab for each panel and the unprocessed western blots as Source Data. A Life Sciences Reporting Summary for this article is available as a Supplementary Information file.
Extended Data
Extended Data Figure 1:

High levels of CLCF1 in KRAS mutant lung adenocarcinoma (LUAD) are associated with decreased patient survival. (a) CLCF1 expression is plotted as log2 transcripts per million (TPM) on the y-axis. Each cohort and group are plotted on the x-axis as LUAD (normal; tumor) and lung squamous cell carcinoma (LUSC) (normal; tumor), respectively. Each violin plot is overlaid with a boxplot depicting the maxima, upper quartile, median, lower quartile, and minima for each group. The sample sizes are shown below each plot as n = 59, 515, 511, and 501, respectively. P-values were calculated with two-tailed unpaired Student’s t-test and corrected for multiple comparisons with Bonferroni adjustment. (b-e) Kaplan-Meier survival curves for LUAD samples. Samples sizes are listed on each plot. Hazard ratio and its respective P-values were calculated with a multivariate Cox hazard regression correcting for age of diagnosis, gender, and cancer stage. Blue solid line represents the normal group (gene expression < 75th percentile) and red solid line represents the high group (gene expression > 75th percentile). (b) CLCF1 expression from samples with KRAS mutation. (c) CLCF1 expression from samples with no KRAS mutations. (d) CNTFR expression from samples with KRAS mutation. (e) CNTFR expression from samples with no KRAS mutations. (f) IL6ST (gp130) expression from samples with KRAS mutation. (g) IL6ST (gp130) expression from samples with no KRAS mutations. (h) LIFR expression from samples with KRAS mutation. (i) LIFR expression from samples with no KRAS mutations. HR = adjusted hazard ratio; CI = confidence interval for the adjusted hazard ratio.
Extended Data Figure 2:
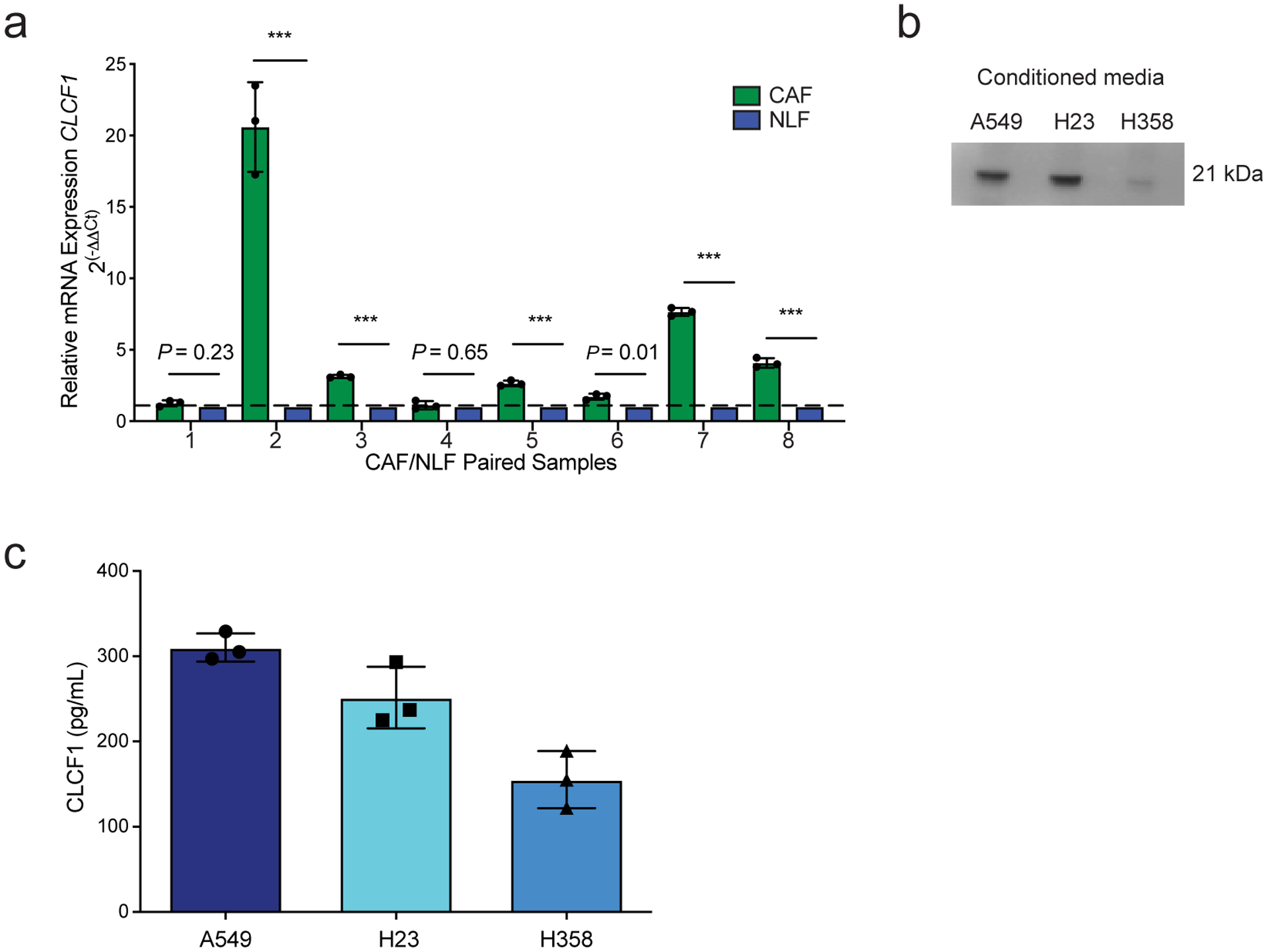
CLCF1 secreted by cancer-associated fibroblasts and LUAD cell lines. (a) Quantitative real-time PCR (qRT-PCR) of CLCF1 mRNA expression in human paired cancer-associated fibroblasts (CAFs) relative to normal lung fibroblasts (NLFs) from the same patient. qRT-PCR data are representative of n = 3 independent experiments. GAPDH and HPRT expression were used for normalization. *** P < 0.001 using two-tailed unpaired Student’s t-test. Data represented as mean ± S.D. (b) Representative cropped western blot analysis of conditioned media CLCF1 levels of indicated LUAD cell lines (representative of n = 2 independent experiments). (c) ELISA results showing CLCF1 levels (pg/mL) in conditioned media from indicated cell lines (n = 3 independent experiments). Data represented as mean ± S.D.
Extended Data Figure 3:
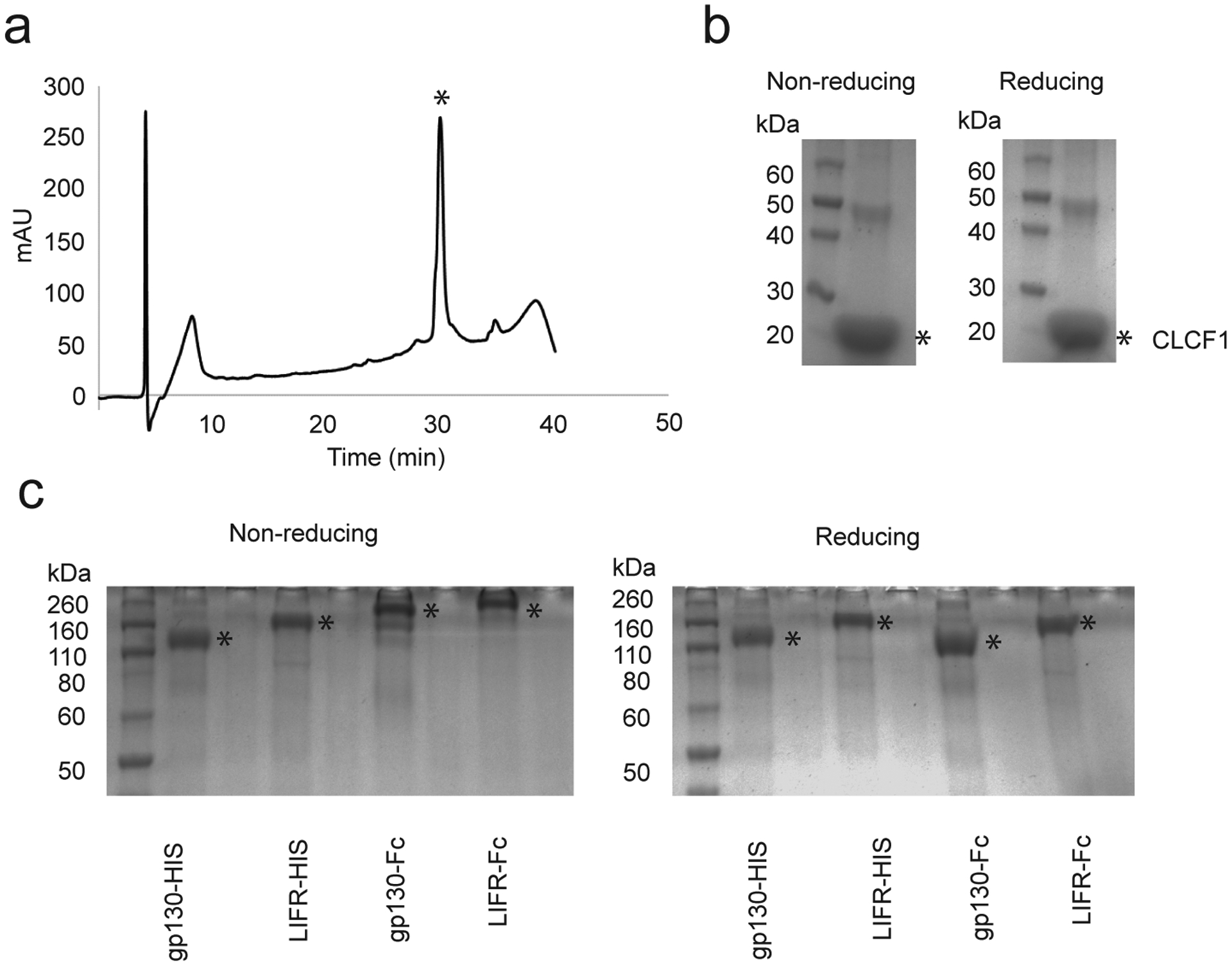
Recombinant production of CLCF1 and soluble β-receptors. (a) Representative analytical RP-HPLC chromatogram of purified CLCF1. A gradient of 10 – 60% solvent B (90% acetonitrile/10% water/0.1% trifluoroacetic acid) over 38 min was used. Asterisk (*) indicates peak corresponding to CLCF1. (b) SDS-PAGE of purified CLCF1 under non-reducing and reducing conditions. (c) SDS-PAGE of purified soluble β-receptor constructs under reducing and non-reducing conditions. Asterisk (*) indicates the corresponding band for each protein. All experiments were repeated at least three times with similar results.
Extended Data Figure 4:

CNTFR knockdown decreases proliferation in vitro and tumor growth in vivo. (a) Proliferation curves for H23, H358, H1975, and H2009, respectively, after CNTFR knockdown by the AlamarBlue® proliferation assay (n = 3 independent experiments with three technical replicates per group). *** P < 0.001 using two-way analysis of variance (ANOVA) and Dunnett’s multiple comparison test (DMCT). Data represented as mean ± S.D. (b) Representative images of colony-formation assay in indicated LUAD cell lines (n = 3 independent experiments with similar results). (c) Representative images of spheres from cells grown in anchorage-independent conditions in indicated LUAD cell lines (n = 3 independent experiments with similar results). (d) Tumor volume quantification of H23 xenografts with indicated shRNAs [shGFP (n = 8), shCNTFR 1 (n = 6), and shCNTFR 2 (n = 4) biologically independent samples]. *** P < 0.001 using two-way ANOVA and DMCT. Data represented as mean ± S.E.M. (e) Tumor volume quantification of H2009 xenografts with indicated shRNAs (n = 8 biologically independent samples). *** P < 0.001 using two-way ANOVA and DMCT. Data represented as mean ± S.E.M. (f-g) Representative H&E staining and IHC for phospho-histone H3 (PH3) and cleaved caspase-3 (CC3) from (f) H23 and (g) H2009 xenografts (n = 3 independent experiments with similar results). The quantification of PH3- and CC3-positive foci is presented in Fig. 1o. Scale bars, 50 μm.
Extended Data Figure 5:
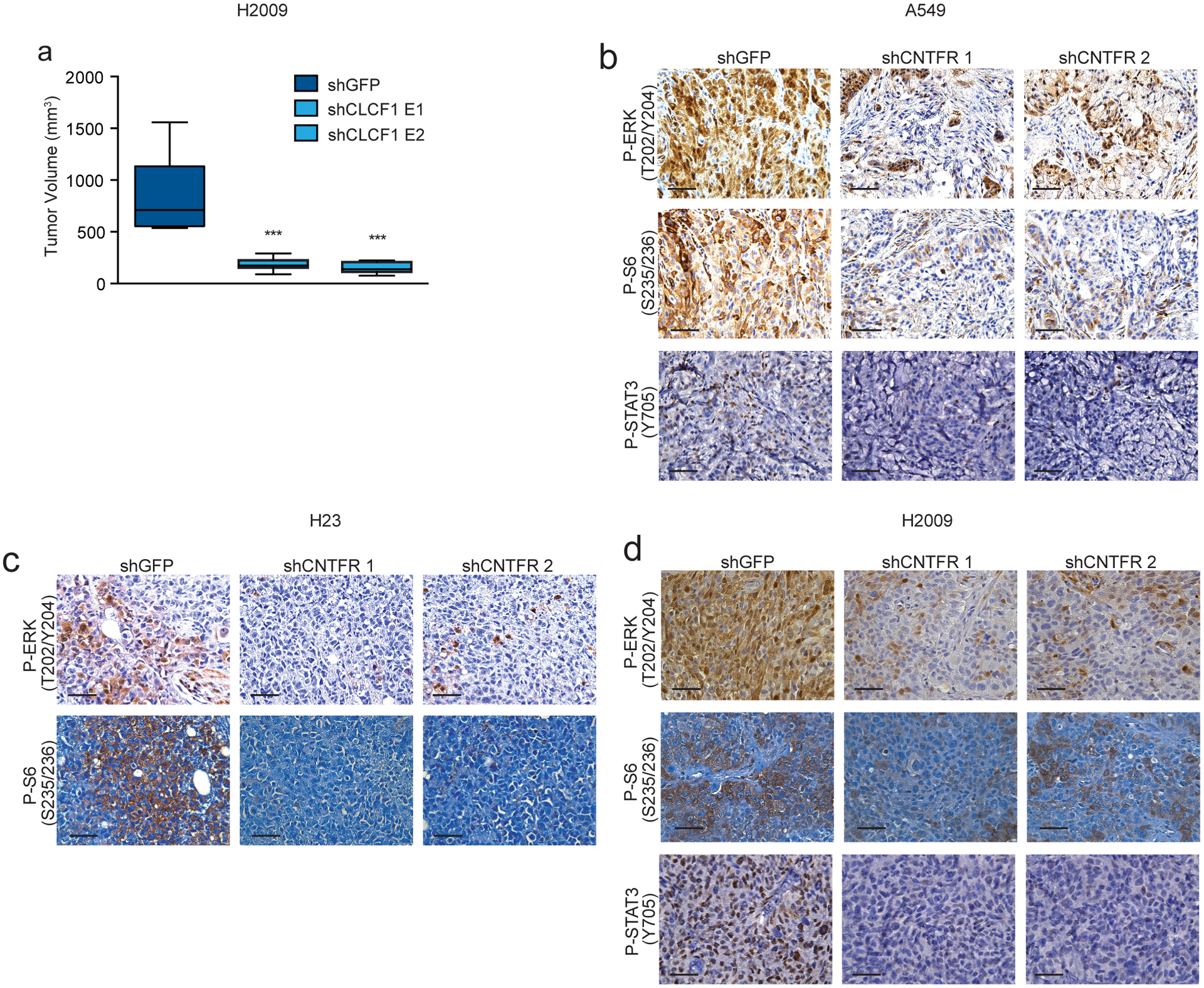
CLCF1 knockdown decreases tumor growth and CNTFR knockdown decreases ERK, S6, and STAT3 signaling in vivo. (a) Tumor volume quantification of final time point of H2009 xenografts with indicated shRNAs (n = 8 biologically independent samples). *** P < 0.001 using one-way ANOVA and Dunnett’s multiple comparison test (DMCT). Whiskers identify the maximum and minimum values; boxes indicate the 75th and 25th percentile and the line the median. (b-d) Representative IHC for phospho-ERK (P-ERK), Phospho-S6RP (P-S6), and phospho-STAT3 (P-STAT3) from A549, H23, and H2009 xenografts. Note: P-STAT3 baseline levels were below the level of detection for H23 (n = 3 independent experiments with similar results). Scale bars, 50 μm.
Extended Data Figure 6:
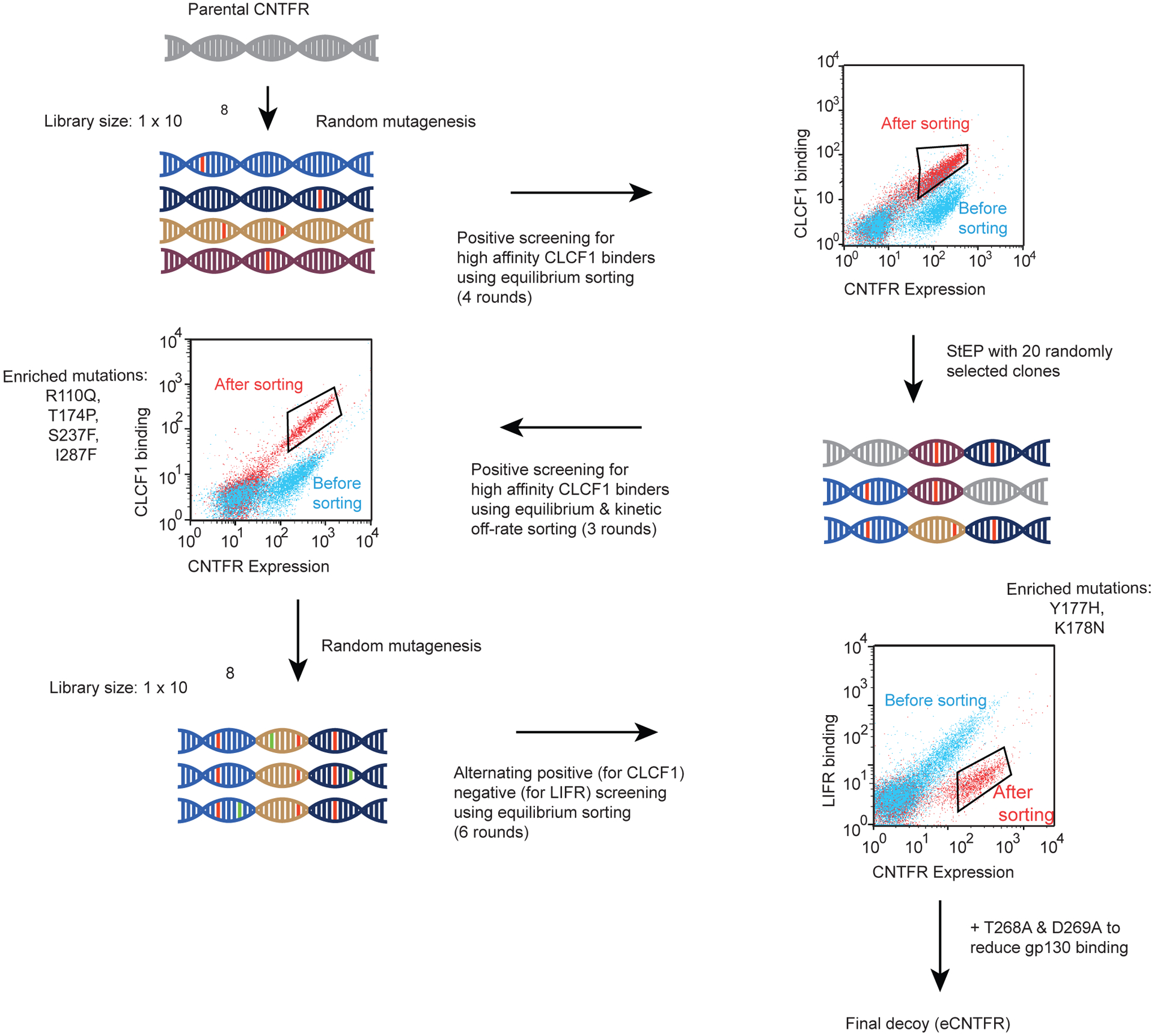
Schematic representation of the overall engineering strategy. 1) Parental CNTFR was randomly mutagenized via error-prone PCR and the resulting yeast-displayed library was screened to isolate high affinity CLCF1 binders using equilibrium sorting conditions. 2) Twenty clones were randomly selected from enriched yeast pools following sort 3 and sort 4, and the mutant CNTFR DNA was extracted and shuffled together using staggered extension process (StEP). The resulting library was screened against CLCF1 using equilibrium and kinetic off-rate sorting conditions to isolate yeast displaying high affinity binders. After 3 rounds of sorting, a combination of four consensus mutations was determined to comprise the highest affinity CLCF1 binder. 3) A further round of random mutagenesis was performed on this variant (variant 4) and the resulting yeast displayed library was screened to isolate a population that showed reduced binding to LIFR while retaining binding for CLCF1, as shown in Fig. 4e. From this screening approach, two mutations, Y177H and K178N, were identified (Fig. 4f) that conferred reduced LIFR binding. 4) The addition of point mutations T268A and D269A conferred reduced gp130 binding. 5) A combination of these 8 mutations comprised the final CNTFR variant (eCNTFR), an engineered protein that possessed high affinity binding to CLCF1 (R110Q, T174P, S237F, and I287F), and lack of binding to LIFR (Y177H and K178N) and gp130 (T268A and D269A).
Extended Data Figure 7:
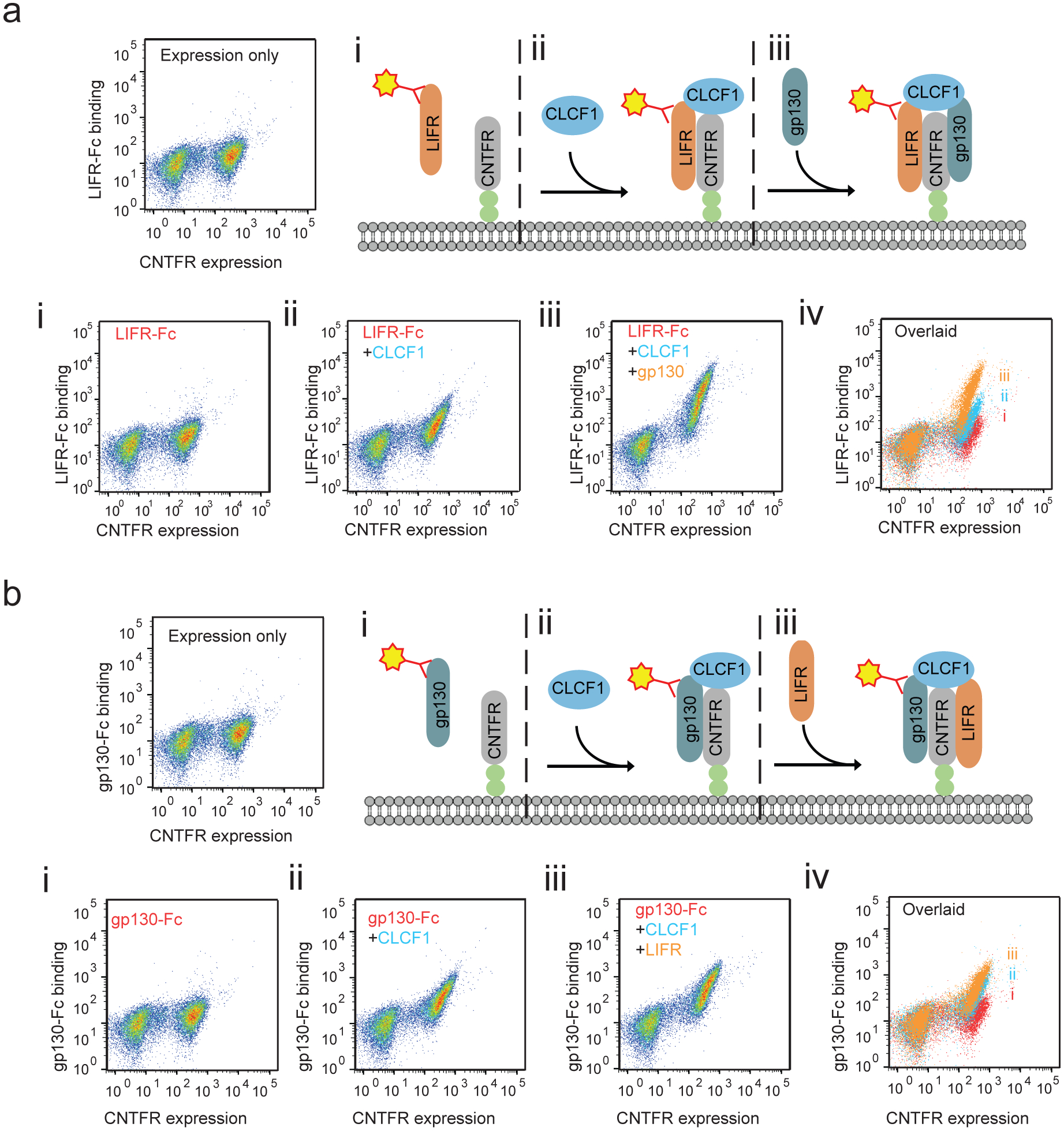
Two soluble constructs, one containing a hexahistidine tag and another fused to a mouse Fc domain, were prepared for each of the β subunits (LIFR-Fc, LIFR-His, gp130-Fc, and gp130-His). (a) As shown by the flow cytometry dot plots, i) when yeast-displayed CNTFR was incubated with LIFR-Fc (10 nM), no binding signal was detected. ii) Upon addition of CLCF1 (10 nM), LIFR-Fc binding signal increases indicating that in the presence of CLCF1, CNTFR interacts with LIFR. iii) When gp130-His was included in addition to CLCF1 and LIFR-Fc, LIFR-Fc binding signal to yeast-displayed CNTFR increased further. (b) i) Similarly, without CLCF1, gp130-Fc showed no detectable binding to yeast-displayed CNTFR. ii) Upon addition of CLCF1, gp130-Fc signal increased. iii) When LIFR-His was added gp130-Fc binding increased further, suggesting coordinated binding between LIFR and gp130 to the CLCF1-CNTFR complex. All experiments were repeated at least three times with similar results.
Extended Data Figure 8:

Library sorting process for non-LIFR binders. (a) Representative flow cytometry dot plot showing yeast library sorting process: isolation of non-LIFR binders (Sort 2 with 1 nM LIFR-Fc), isolation of CLCF1 binders (Sort 3 with 0.5 nM CLCF1-His), and isolation of non-LIFR binders (Sort 4 with 10 nM LIFR-Fc). To retain the binding affinity for CLCF1, strategy was alternated between positive screening for 0.5 nM CLCF1 and negative screening for increasing concentrations of LIFR-Fc. (b) Flow cytometry histogram representing yeast-displayed CNTFR variant containing T268A and D269A (CNTFR_AA) binding to CLCF1 (10 nM), gp130-Fc (10 nM), and LIFR-fc (10 nM) relative to untreated negative control (red). CNTFR_AA binds to CLCF1 and LIFR but not to gp130. For gp130-Fc and LIFR-Fc binding studies 10 nM of CLCF1 was added to induce receptor complex formation. All experiments were repeated at least three times with similar results.
Extended Data Figure 9:
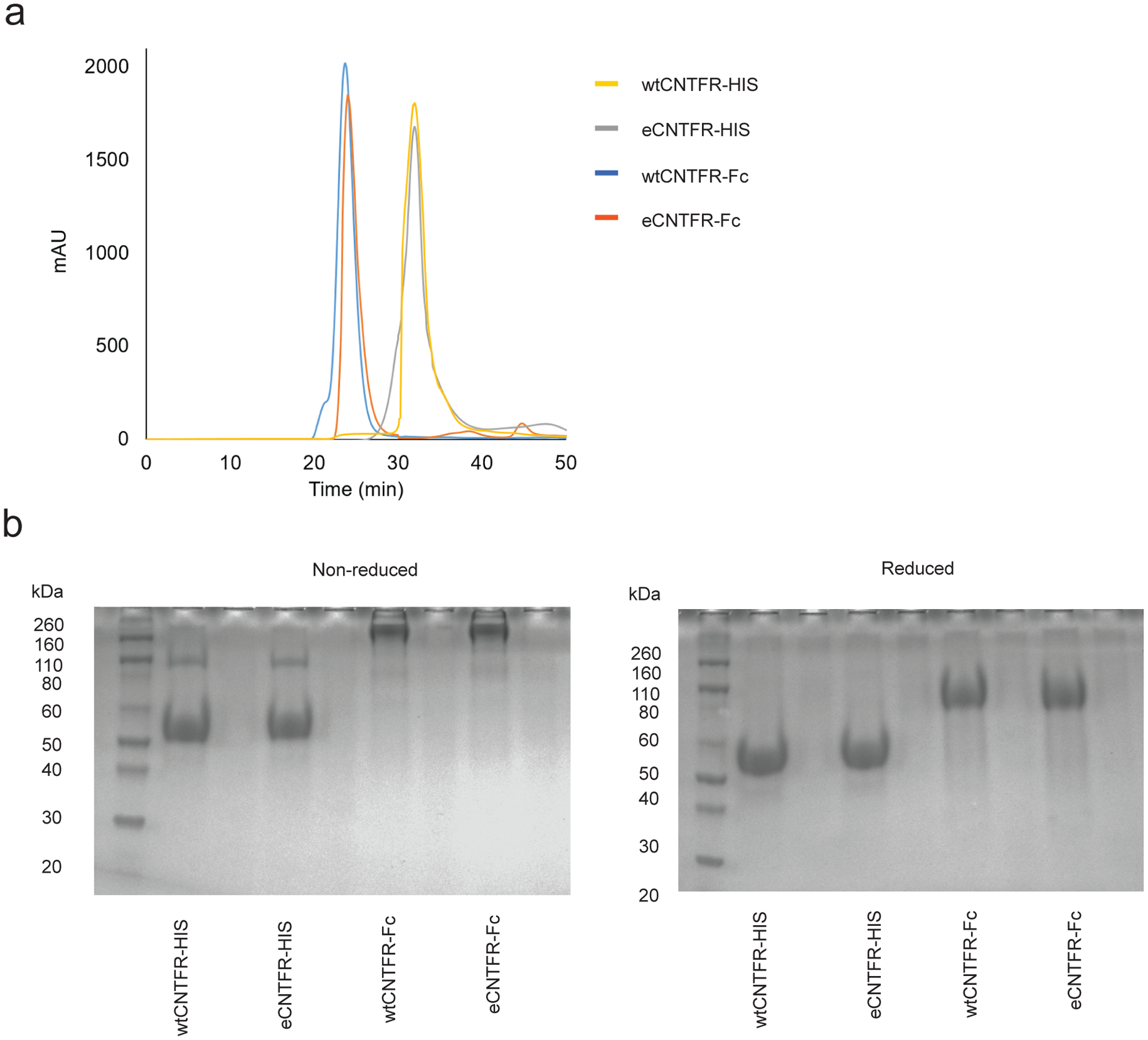
Recombinant production of soluble CNTFR proteins. (a) Overlaid analytical size exclusion chromatography chromatograms of purified wtCNTFR-HIS, wtCNTFR-Fc, eCNTFR-HIS, and eCNTFR-Fc. (b) SDS-PAGE of purified CNTFR constructs under reducing and non-reducing conditions. All experiments were repeated at least three times with similar results.
Extended Data Figure 10:

Yeast-displayed affinity matured eCNTFR does not bind to CNTF but retains high affinity binding to mCLCF1. (a) Binding curves representing yeast-displayed wild-type CNTFR (wtCNTFR) and engineered CNTFR (eCNTFR) binding to CNTF measured by flow cytometry (n = 3 independent experiments). Data represented as mean ± S.D. (b) Binding curves representing yeast-displayed wtCNTFR and eCNTFR binding to mCLCF1 measured by flow cytometry. While eCNTFR still binds to mCLCF1 with comparable affinity to hCLCF1, there was no detectable binding to CNTF (n = 3 independent experiments). Data represented as mean ± S.D. (c) Representative cropped western blot of two independent experiments showing A549 cells treated with mCLCF1 and phosphorylation of STAT3 (Y705) in a time-dependent manner.
Supplementary Material
Supplementary Figure 1. CLCF1-CNTFR downstream signaling pathways. (a) Representative cropped western blot of H23 treated with serum, CLCF1, eCNTFR-Fc, CLCF1 + eCNTFR-Fc, or eCNTFR-Fc + serum after 24 h serum starvation (n = 2 independent experiments with similar results). (b) Representative cropped western blot of A549 and H23 treated with serum, CLCF1, eCNTFR-Fc, CLCF1 + eCNTFR-Fc, or eCNTFR-Fc + serum after 24 h serum starvation (n = 2 independent experiments with similar results).
Supplementary Figure 2: eCNTFR-Fc decreases tumor growth in xenograft and PDTX models containing a KRAS or EGFR mutation. (a) Tumor volume quantification of H23 xenografts (n = 16 biologically independent samples). *** P < 0.001 using two-way analysis of variance (ANOVA) and Dunnett’s multiple comparison test (DCMT). Data represented as mean ± S.E.M. (b) Tumor volume quantification of final time point of H23 xenografts. Whiskers identify the maximum and minimum values; boxes indicate the 75th and 25th percentile and the line the median. *** P < 0.001 using one-way ANOVA and DCMT. (c) Waterfall plot showing tumor percent change from baseline for H23 xenografts. (d) Representative images of H23 xenograft tumors (n = 16 biologically independent samples). Scale bars, 10 mm. (e) Representative H&E staining and IHC for phospho-histone H3 (PH3) and cleaved caspase-3 (CC3) from H23 xenografts. Scale bars, 50 μm. (f) Quantification of PH3- and CC3-positive foci (n = 16 biologically independent samples with 10 technical replicates per group). *** P < 0.001 using one-way ANOVA and DCMT. Data are represented as mean ± S.E.M. (g) Representative IHC for phospho-ERK (P-ERK) and Phospho-S6RP (P-S6) in H23 xenografts (n = 3 independent experiments with similar results). (h) Waterfall plot showing tumor percent change from baseline for PDTX 727 model. (i) Tumor volume quantification of PDTXs [PDTX911 (n = 4), PDTX709 (PBS: n = 7 and eCNTFR-Fc: n = 8), PDTX131 (n = 10), and TP40 (n = 4) biologically independent samples]. *** P < 0.001 using using one-way ANOVA and DCMT. Data represented as mean ± S.E.M. (j) Representative cropped western blot of PDTX727 xenografts (n = 2 independent experiments with similar results). (k) Quantification of western blot (n = 2 independent experiments with similar results). One-way ANOVA and DMCT. Data represented as mean ± S.E.M.
Supplementary Figure 3: Signaling changes mediated in a time-dependent manner by ERK and STAT3 in vivo. (a) Quantification of P-ERK data in A549 xenografts presented in Fig. 5l (n = 3 independent experiments with 10 technical replicates per group). *** P < 0.001 using one-way analysis of variance (ANOVA) and Dunnett’s multiple comparison test (DMCT). Data represented as mean ± S.E.M. (b) Quantification of P-ERK data in PDTX727 presented in Fig. 5m. (n = 3 independent experiments with 10 technical replicates per group). *** P < 0.001 using two-tailed unpaired Student’s t-test. Data represented as mean ± S.E.M. (c) Representative IHC for phospho-STAT3 (P-STAT3). (d) Quantification of this P-STAT3 data in A549 xenografts (n = 3 independent experiments with 10 technical replicates per group). One-way ANOVA and DMCT. Data represented as mean ± S.E.M. (e) Representative IHC for P-STAT3 data in PDTX727. (f) Quantification of P-STAT3 data (n = 3 independent experiments with 10 technical replicates per group). *** P < 0.001 using two-tailed unpaired Student’s t-test. Data represented as mean ± S.E.M. (g) Representative IHC for P-ERK and P-STAT3 in A549 xenografts treated with eCNTFR-Fc (10 mg/kg) and harvested at time points shown. (h) Quantification of P-ERK and P-STAT3 data (n = 2 independent experiments with 10 technical replicates per group). *** P < 0.001 using one-way ANOVA and DMCT. Data represented as mean ± S.E.M. (i) Representative IHC for phospho-histone H3 (PH3) and cleaved caspase-3 (CC3) in A549 xenografts treated with eCNTFR-Fc (10 mg/kg) and harvested at time points shown. (j) Quantification of PH3 and CC3 data (n = 2 independent experiments with 10 technical replicates per group). *** P < 0.001 using one-way ANOVA and DMCT. Data represented as mean ± S.E.M. (k) Representative IHC for PH3 and CC3 in PDTX727 treated with eCNTFR-Fc (10 mg/kg) and harvested at time points shown. (l) Quantification of PH3 and CC3 data (n = 2 independent experiments with 10 technical replicates per group). One-way ANOVA and DMCT. Data represented as mean ± S.E.M. Scale bars, 50 μm.
Supplementary Figure 4: eCNTFR-Fc produces no observable adverse effects in PDTXs and GEMMs. Histologic sections and H&E staining of liver (a), kidney (b), left ventricle of the heart (c), retina (d), and proximal tibia (e) from a mouse that received the highest dose of CNTFR (10 mg/kg) for four weeks [n = 2 independent experiments (n = 3 mice per group) with similar results]. (f) Mouse weights following treatment course for PDTX911 and PDTX131 [PDTX911 (n = 2) and PDTX131 (n = 5) biologically independent samples]. (g) Mouse weights following treatment course for PDTX709 (n = 4 biologically independent samples). Two-way analysis of variance (ANOVA) and Dunnett’s multiple comparison test (DMCT). Data represented as mean ± S.E.M. (h) Endpoint mouse weights for KrasG12D/Trp53f/f mice treated 3 times per week with PBS, eCNTFR-Fc (10 mg/kg), or Cisplatin (7 mg/kg) for 4 weeks (Day 28) by intraperitoneal (i.p.) injection starting at 8 weeks post-delivery of 5 × 106 pfu of adenovirus-expressing Cre (Ad-Cre) (Day 0) (n = 10 biologically independent samples). *** P < 0.001 using one-way ANOVA and DMCT. Data represented as mean ± S.E.M.
Supplementary Figure 5: CLCF1 as a predictive biomarker in LUAD. (a) Scatter plot depicting Pearson’s correlation coefficient r (two-tailed) for CLCF1 mRNA expression obtained from the Cancer Cell Line Encyclopedia (CCLE) and the change in viability after treatment with 2.5 μM eCNTFR-Fc (in the cell line panel presented in Figure 4a) (n = 3 independent experiments). (b) CLCF1 ELISA performed on patient plasma samples and normal controls [Normal (n = 10) and Tumor (n = 26)]. (c) Odds ratio (O.R.) was calculated from a logistic regression (logit) model, and the P-value (two-tailed) was calculated using Wald test, P-value = 0.0364. Lung cancer samples were categorized as either having a ‘mutation of interest’ defined as KRAS G12C, KRAS G12V, or KRAS wild-type (wt) / EGFR mutant (dummy coded as 1, n = 17) or KRAS wt (dummy coded as 0, n = 8). (d) Graph showing the predictive probability of being in the mutation group as a function of blood plasma CLCF1 (pg/mL): y-axis is the predicted probability and the x-axis is the blood plasma CLCF1 (pg/mL).
ACKNOWLEDGMENTS
We thank Dana Gwinn especially and all of the other members of the Sweet-Cordero and Cochran labs for helpful suggestions as well as Sean Hunter, Tony Hunter, and Yu Shi for assistance with the STAT3 immunohistochemical assay. C.P.M. was supported by a fellowship from the Howard Hughes Medical Institute, by the National Cancer Institute of the National Institutes of Health under the Ruth L. Kirschstein National Service Research Award (NRSA) F31 (F31CA236324), and by the Stanford Medical Scientist Training Program T32 Grant. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. J.W.K was funded by a graduate fellowship from the Stanford Bio-X Program. K.K. was supported by a Postdoc Mobility grant (P300PB-174377) from the Swiss National Science Foundation. E.A.S.C and J.R.C. were funded by a grant from the Lungevity Foundation and Upstage Lung Cancer. J.R.C was funded by a Stanford Coulter Foundation Translational Partnership Award. E.A.S.C received funding from the American Lung Association. This work was also funded by a multi-investigator grant from the National Cancer Institute (R01CA225103) to E.A.S.-C and J.R.C.
Footnotes
COMPETING INTERESTS STATEMENT
J.W.K., C.P.M., J.R.C., and E.A.S.-C. are included as inventors on university-owned intellectual property related to the work described in this manuscript.
REFERENCES
- 1.Ostrem JM & Shokat KM Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov 15, 771–785, doi: 10.1038/nrd.2016.139 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Cox AD, Fesik SW, Kimmelman AC, Luo J & Der CJ Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov 13, 828–851, doi: 10.1038/nrd4389 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herbst RS, Morgensztern D & Boshoff C The biology and management of non-small cell lung cancer. Nature 553, 446–454, doi: 10.1038/nature25183 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Lin JJ, Riely GJ & Shaw AT Targeting ALK: Precision Medicine Takes on Drug Resistance. Cancer Discov 7, 137–155, doi: 10.1158/2159-8290.CD-16-1123 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chong CR & Janne PA The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med 19, 1389–1400, doi: 10.1038/nm.3388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camidge DR, Pao W & Sequist LV Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol 11, 473–481, doi: 10.1038/nrclinonc.2014.104 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Forde PM et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N Engl J Med, doi: 10.1056/NEJMoa1716078 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Gandhi L et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med, doi: 10.1056/NEJMoa1801005 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Hellmann MD et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N Engl J Med, doi: 10.1056/NEJMoa1801946 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camidge DR, Doebele RC & Kerr KM Comparing and contrasting predictive biomarkers for immunotherapy and targeted therapy of NSCLC. Nat Rev Clin Oncol 16, 341–355, doi: 10.1038/s41571-019-0173-9 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Kalluri R The biology and function of fibroblasts in cancer. Nat Rev Cancer 16, 582–598, doi: 10.1038/nrc.2016.73 (2016). [DOI] [PubMed] [Google Scholar]
- 12.De Palma M, Biziato D & Petrova TV Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer 17, 457–474, doi: 10.1038/nrc.2017.51 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Su S et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 172, 841–856 e816, doi: 10.1016/j.cell.2018.01.009 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Kunita A et al. MicroRNA-21 in cancer-associated fibroblasts supports lung adenocarcinoma progression. Scientific reports 8, 8838, doi: 10.1038/s41598-018-27128-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Labernadie A et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol 19, 224–237, doi: 10.1038/ncb3478 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Albrengues J et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun 6, 10204, doi: 10.1038/ncomms10204 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen WJ et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat Commun 5, 3472, doi: 10.1038/ncomms4472 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Vicent S et al. Cross-Species Functional Analysis of Cancer-Associated Fibroblasts Identifies a Critical Role for CLCF1 and IL-6 in Non-Small Cell Lung Cancer In Vivo. Cancer Res 72, 5744–5756, doi: 10.1158/0008-5472.CAN-12-1097 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Senaldi G et al. Novel neurotrophin-1/B cell-stimulating factor-3: a cytokine of the IL-6 family. Proc Natl Acad Sci U S A 96, 11458–11463 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lelievre E et al. Signaling pathways recruited by the cardiotrophin-like cytokine/cytokine-like factor-1 composite cytokine: specific requirement of the membrane-bound form of ciliary neurotrophic factor receptor alpha component. J Biol Chem 276, 22476–22484, doi: 10.1074/jbc.M101681200 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Hu X et al. Ciliary neurotrophic factor receptor alpha subunit-modulated multiple downstream signaling pathways in hepatic cancer cell lines and their biological implications. Hepatology 47, 1298–1308, doi: 10.1002/hep.22163 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Kober P, Bujko M, Oledzki J, Tysarowski A & Siedlecki JA Methyl-CpG binding column-based identification of nine genes hypermethylated in colorectal cancer. Mol Carcinog 50, 846–856, doi: 10.1002/mc.20763 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Fan K et al. Hypomethylation of CNTFRalpha is associated with proliferation and poor prognosis in lower grade gliomas. Sci Rep 7, 7079, doi: 10.1038/s41598-017-07124-9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu J et al. CNTF receptor subunit alpha as a marker for glioma tumor-initiating cells and tumor grade: laboratory investigation. J Neurosurg 117, 1022–1031, doi: 10.3171/2012.9.JNS1212 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pento JT Monoclonal Antibodies for the Treatment of Cancer. Anticancer Res 37, 5935–5939, doi: 10.21873/anticanres.12040 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Chiu ML & Gilliland GL Engineering antibody therapeutics. Curr Opin Struct Biol 38, 163–173, doi: 10.1016/j.sbi.2016.07.012 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Ciombor KK & Berlin J Aflibercept--a decoy VEGF receptor. Curr Oncol Rep 16, 368, doi: 10.1007/s11912-013-0368-7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weinblatt ME et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med 340, 253–259, doi: 10.1056/NEJM199901283400401 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Economides AN et al. Cytokine traps: multi-component, high-affinity blockers of cytokine action. Nat Med 9, 47–52, doi: 10.1038/nm811 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Hunter SA & Cochran JR Cell-Binding Assays for Determining the Affinity of Protein-Protein Interactions: Technologies and Considerations. Methods Enzymol 580, 21–44, doi: 10.1016/bs.mie.2016.05.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim JW & Cochran JR Targeting ligand-receptor interactions for development of cancer therapeutics. Current opinion in chemical biology 38, 62–69, doi: 10.1016/j.cbpa.2017.03.010 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Silver JS & Hunter CA gp130 at the nexus of inflammation, autoimmunity, and cancer. J Leukoc Biol 88, 1145–1156, doi: 10.1189/jlb.0410217 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis S et al. Released Form of Cntf Receptor-Alpha Component as a Soluble Mediator of Cntf Responses. Science 259, 1736–1739, doi: 10.1126/science.7681218 (1993). [DOI] [PubMed] [Google Scholar]
- 34.Auguste P et al. Alanine substitution for Thr268 and Asp269 of soluble ciliary neurotrophic factor (CNTF) receptor alpha component defines a specific antagonist for the CNTF response. J Biol Chem 271, 26049–26056 (1996). [DOI] [PubMed] [Google Scholar]
- 35.Van Deventer JA & Wittrup KD Yeast surface display for antibody isolation: library construction, library screening, and affinity maturation. Methods Mol Biol 1131, 151–181, doi: 10.1007/978-1-62703-992-5_10 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Aguinaldo AM & Arnold F Staggered extension process (StEP) in vitro recombination. Methods Mol Biol 192, 235–239, doi: 10.1385/1-59259-177-9:235 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Boder ET, Midelfort KS & Wittrup KD Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc Natl Acad Sci U S A 97, 10701–10705, doi: 10.1073/pnas.170297297 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelley LA, Mezulis S, Yates CM, Wass MN & Sternberg MJE The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protocols 10, 845–858, doi: 10.1038/nprot.2015.053 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perret D et al. Two different contact sites are recruited by cardiotrophin-like cytokine (CLC) to generate the CLC/CLF and CLC/sCNTFR alpha composite cytokines. Journal of Biological Chemistry 279, 43961–43970, doi: 10.1074/jbc.M407686200 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Rousseau F et al. Ciliary Neurotrophic Factor, Cardiotrophin-like Cytokine, and Neuropoietin Share a Conserved Binding Site on the Ciliary Neurotrophic Factor Receptor alpha Chain. Journal of Biological Chemistry 283, 30341–30350, doi: 10.1074/jbc.M803239200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kintzing JR, Filsinger Interrante MV & Cochran JR Emerging Strategies for Developing Next-Generation Protein Therapeutics for Cancer Treatment. Trends Pharmacol Sci 37, 993–1008, doi: 10.1016/j.tips.2016.10.005 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ip NY et al. CNTF and LIF act on neuronal cells via shared signaling pathways that involve the IL-6 signal transducing receptor component gp130. Cell 69, 1121–1132 (1992). [DOI] [PubMed] [Google Scholar]
- 43.Leibinger M, Andreadaki A, Diekmann H & Fischer D Neuronal STAT3 activation is essential for CNTF- and inflammatory stimulation-induced CNS axon regeneration. Cell Death Dis 4, doi:ARTNe80510.1038/cddis.2013.310 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yao Z et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 28, 370–383, doi: 10.1016/j.ccell.2015.08.001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yao Z et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 548, 234–238, doi: 10.1038/nature23291 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hunter JC et al. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res 13, 1325–1335, doi: 10.1158/1541-7786.MCR-15-0203 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Kempf E, Rousseau B, Besse B & Paz-Ares L KRAS oncogene in lung cancer: focus on molecularly driven clinical trials. Eur Respir Rev 25, 71–76, doi: 10.1183/16000617.0071-2015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tebbutt NC et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat Med 8, 1089–1097, doi: 10.1038/nm763 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Mainardi S et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med 24, 961–967, doi: 10.1038/s41591-018-0023-9 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Ruess DA et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med 24, 954–960, doi: 10.1038/s41591-018-0024-8 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Nichols RJ et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol 20, 1064–1073, doi: 10.1038/s41556-018-0169-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wong GS et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med 24, 968–977, doi: 10.1038/s41591-018-0022-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jackson EL et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res 65, 10280–10288 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Marini KD et al. Inhibition of activin signaling in lung adenocarcinoma increases the therapeutic index of platinum chemotherapy. Sci Transl Med 10, doi: 10.1126/scitranslmed.aat3504 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Janes MR et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 172, 578–589 e517, doi: 10.1016/j.cell.2018.01.006 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Haigis KM KRAS Alleles: The Devil Is in the Detail. Trends Cancer 3, 686–697, doi: 10.1016/j.trecan.2017.08.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lito P, Solomon M, Li LS, Hansen R & Rosen N Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 351, 604–608, doi: 10.1126/science.aad6204 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim JW & Cochran JR Targeting ligand-receptor interactions for development of cancer therapeutics. Curr Opin Chem Biol 38, 62–69, doi: 10.1016/j.cbpa.2017.03.010 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Saggio I et al. Nonradioactive receptor binding assay for ciliary neurotrophic factor. Anal Biochem 221, 387–391 (1994). [DOI] [PubMed] [Google Scholar]
- 60.Sansone P et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest 117, 3988–4002, doi: 10.1172/JCI32533 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brooks GD et al. IL6 Trans-signaling Promotes KRAS-Driven Lung Carcinogenesis. Cancer Res 76, 866–876, doi: 10.1158/0008-5472.CAN-15-2388 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Wu X, Cao Y, Xiao H, Li C & Lin J Bazedoxifene as a Novel GP130 Inhibitor for Pancreatic Cancer Therapy. Mol Cancer Ther 15, 2609–2619, doi: 10.1158/1535-7163.MCT-15-0921 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shi Y et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 569, 131–135, doi: 10.1038/s41586-019-1130-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jackson EL et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15, 3243–3248 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jonkers J et al. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 29, 418–425 (2001). [DOI] [PubMed] [Google Scholar]
- 66.Wong J et al. High-resolution, small animal radiation research platform with x-ray tomographic guidance capabilities. Int J Radiat Oncol Biol Phys 71, 1591–1599, doi: 10.1016/j.ijrobp.2008.04.025 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnstone CD et al. Multi-institutional MicroCT image comparison of image-guided small animal irradiators. Phys Med Biol 62, 5760–5776, doi: 10.1088/1361-6560/aa76b4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnstone CD & Bazalova-Carter M MicroCT imaging dose to mouse organs using a validated Monte Carlo model of the small animal radiation research platform (SARRP). Phys Med Biol 63, 115012, doi: 10.1088/1361-6560/aac335 (2018). [DOI] [PubMed] [Google Scholar]
- 69.Lee SJ et al. Regulation of hypoxia-inducible factor 1alpha (HIF-1alpha) by lysophosphatidic acid is dependent on interplay between p53 and Kruppel-like factor 5. J Biol Chem 288, 25244–25253, doi: 10.1074/jbc.M113.489708 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. CLCF1-CNTFR downstream signaling pathways. (a) Representative cropped western blot of H23 treated with serum, CLCF1, eCNTFR-Fc, CLCF1 + eCNTFR-Fc, or eCNTFR-Fc + serum after 24 h serum starvation (n = 2 independent experiments with similar results). (b) Representative cropped western blot of A549 and H23 treated with serum, CLCF1, eCNTFR-Fc, CLCF1 + eCNTFR-Fc, or eCNTFR-Fc + serum after 24 h serum starvation (n = 2 independent experiments with similar results).
Supplementary Figure 2: eCNTFR-Fc decreases tumor growth in xenograft and PDTX models containing a KRAS or EGFR mutation. (a) Tumor volume quantification of H23 xenografts (n = 16 biologically independent samples). *** P < 0.001 using two-way analysis of variance (ANOVA) and Dunnett’s multiple comparison test (DCMT). Data represented as mean ± S.E.M. (b) Tumor volume quantification of final time point of H23 xenografts. Whiskers identify the maximum and minimum values; boxes indicate the 75th and 25th percentile and the line the median. *** P < 0.001 using one-way ANOVA and DCMT. (c) Waterfall plot showing tumor percent change from baseline for H23 xenografts. (d) Representative images of H23 xenograft tumors (n = 16 biologically independent samples). Scale bars, 10 mm. (e) Representative H&E staining and IHC for phospho-histone H3 (PH3) and cleaved caspase-3 (CC3) from H23 xenografts. Scale bars, 50 μm. (f) Quantification of PH3- and CC3-positive foci (n = 16 biologically independent samples with 10 technical replicates per group). *** P < 0.001 using one-way ANOVA and DCMT. Data are represented as mean ± S.E.M. (g) Representative IHC for phospho-ERK (P-ERK) and Phospho-S6RP (P-S6) in H23 xenografts (n = 3 independent experiments with similar results). (h) Waterfall plot showing tumor percent change from baseline for PDTX 727 model. (i) Tumor volume quantification of PDTXs [PDTX911 (n = 4), PDTX709 (PBS: n = 7 and eCNTFR-Fc: n = 8), PDTX131 (n = 10), and TP40 (n = 4) biologically independent samples]. *** P < 0.001 using using one-way ANOVA and DCMT. Data represented as mean ± S.E.M. (j) Representative cropped western blot of PDTX727 xenografts (n = 2 independent experiments with similar results). (k) Quantification of western blot (n = 2 independent experiments with similar results). One-way ANOVA and DMCT. Data represented as mean ± S.E.M.
Supplementary Figure 3: Signaling changes mediated in a time-dependent manner by ERK and STAT3 in vivo. (a) Quantification of P-ERK data in A549 xenografts presented in Fig. 5l (n = 3 independent experiments with 10 technical replicates per group). *** P < 0.001 using one-way analysis of variance (ANOVA) and Dunnett’s multiple comparison test (DMCT). Data represented as mean ± S.E.M. (b) Quantification of P-ERK data in PDTX727 presented in Fig. 5m. (n = 3 independent experiments with 10 technical replicates per group). *** P < 0.001 using two-tailed unpaired Student’s t-test. Data represented as mean ± S.E.M. (c) Representative IHC for phospho-STAT3 (P-STAT3). (d) Quantification of this P-STAT3 data in A549 xenografts (n = 3 independent experiments with 10 technical replicates per group). One-way ANOVA and DMCT. Data represented as mean ± S.E.M. (e) Representative IHC for P-STAT3 data in PDTX727. (f) Quantification of P-STAT3 data (n = 3 independent experiments with 10 technical replicates per group). *** P < 0.001 using two-tailed unpaired Student’s t-test. Data represented as mean ± S.E.M. (g) Representative IHC for P-ERK and P-STAT3 in A549 xenografts treated with eCNTFR-Fc (10 mg/kg) and harvested at time points shown. (h) Quantification of P-ERK and P-STAT3 data (n = 2 independent experiments with 10 technical replicates per group). *** P < 0.001 using one-way ANOVA and DMCT. Data represented as mean ± S.E.M. (i) Representative IHC for phospho-histone H3 (PH3) and cleaved caspase-3 (CC3) in A549 xenografts treated with eCNTFR-Fc (10 mg/kg) and harvested at time points shown. (j) Quantification of PH3 and CC3 data (n = 2 independent experiments with 10 technical replicates per group). *** P < 0.001 using one-way ANOVA and DMCT. Data represented as mean ± S.E.M. (k) Representative IHC for PH3 and CC3 in PDTX727 treated with eCNTFR-Fc (10 mg/kg) and harvested at time points shown. (l) Quantification of PH3 and CC3 data (n = 2 independent experiments with 10 technical replicates per group). One-way ANOVA and DMCT. Data represented as mean ± S.E.M. Scale bars, 50 μm.
Supplementary Figure 4: eCNTFR-Fc produces no observable adverse effects in PDTXs and GEMMs. Histologic sections and H&E staining of liver (a), kidney (b), left ventricle of the heart (c), retina (d), and proximal tibia (e) from a mouse that received the highest dose of CNTFR (10 mg/kg) for four weeks [n = 2 independent experiments (n = 3 mice per group) with similar results]. (f) Mouse weights following treatment course for PDTX911 and PDTX131 [PDTX911 (n = 2) and PDTX131 (n = 5) biologically independent samples]. (g) Mouse weights following treatment course for PDTX709 (n = 4 biologically independent samples). Two-way analysis of variance (ANOVA) and Dunnett’s multiple comparison test (DMCT). Data represented as mean ± S.E.M. (h) Endpoint mouse weights for KrasG12D/Trp53f/f mice treated 3 times per week with PBS, eCNTFR-Fc (10 mg/kg), or Cisplatin (7 mg/kg) for 4 weeks (Day 28) by intraperitoneal (i.p.) injection starting at 8 weeks post-delivery of 5 × 106 pfu of adenovirus-expressing Cre (Ad-Cre) (Day 0) (n = 10 biologically independent samples). *** P < 0.001 using one-way ANOVA and DMCT. Data represented as mean ± S.E.M.
Supplementary Figure 5: CLCF1 as a predictive biomarker in LUAD. (a) Scatter plot depicting Pearson’s correlation coefficient r (two-tailed) for CLCF1 mRNA expression obtained from the Cancer Cell Line Encyclopedia (CCLE) and the change in viability after treatment with 2.5 μM eCNTFR-Fc (in the cell line panel presented in Figure 4a) (n = 3 independent experiments). (b) CLCF1 ELISA performed on patient plasma samples and normal controls [Normal (n = 10) and Tumor (n = 26)]. (c) Odds ratio (O.R.) was calculated from a logistic regression (logit) model, and the P-value (two-tailed) was calculated using Wald test, P-value = 0.0364. Lung cancer samples were categorized as either having a ‘mutation of interest’ defined as KRAS G12C, KRAS G12V, or KRAS wild-type (wt) / EGFR mutant (dummy coded as 1, n = 17) or KRAS wt (dummy coded as 0, n = 8). (d) Graph showing the predictive probability of being in the mutation group as a function of blood plasma CLCF1 (pg/mL): y-axis is the predicted probability and the x-axis is the blood plasma CLCF1 (pg/mL).
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files or are available from the corresponding author upon reasonable request. Statistical Source Data underlying all Figures are provided as a separate file with a tab for each panel and the unprocessed western blots as Source Data. A Life Sciences Reporting Summary for this article is available as a Supplementary Information file.