

Abstract
Background.
Donor brain death (BD) triggers a systemic inflammatory response that reduces organ quality and increases immunogenicity of the graft. We characterized the early innate immune response induced by BD in the liver and peripheral blood of hemodinamically stable non-human primates (NHP).
Methods.
Rhesus macaques were assigned to either brain death or control group. BD was induced by inflation of a subdurally placed catheter and confirmed clinically and by cerebral angiography. Animals were monitored for 6 h after BD and managed to maintain hemodynamic stability.
Results.
Cortisol, epinephrine, nor-epinephrine, and IL-6 levels were elevated immediately after BD induction. Neutrophils and monocytes significantly increased in circulation following BD induction, while dendritic cells were decreased at 6 h post-induction. Flow cytometry revealed increased expression of chemokine receptors CxCR1, CxCR2, CCR2, and CCR5 in peripheral blood leukocytes from NHP subjected to BD. Microarray analysis demonstrated a significant up-regulation of genes related to innate inflammatory responses, toll-like receptor signaling, stress pathways, and apoptosis/cell death in BD subjects. Conversely, pathways related to glucose, lipid, and protein metabolism were down-regulated. In addition, increased expression of SOCS3, S100A8/A9, ICAM-1, MHC class II, neutrophil accumulation, and oxidative stress markers (carboxy-methyl-lysine and hydroxynonenal) were detected by immunoblot and immunohistochemistry.
Conclusions.
Activation of the innate immune response after BD in association with a down-regulation of genes associated with cell metabolism pathways in the liver. These findings may provide a potential explanation for the reduced post-transplant function of organs from brain dead donors. In addition, this work suggests potential novel targets to improve donor management strategies.
Keywords: inflammation, innate immunity, brain-death, transplantation, organ donor, liver, toll-like receptors
INTRODUCTION
Brain death (BD) remains the main clinical condition leading to organ donation for transplantation in the United States [1]. However, studies have shown that livers from a living donor (LD) have superior post-transplant survival compared with livers from deceased donors [2, 3]. Although increased cold ischemia time is a major determinant of organ quality between living and deceased donors, recent studies indicate that inflammation and innate immune activation prior to organ recovery are critical contributing factors [4–7]. This generalized inflammatory response is believed to follow the multiple hemodynamic and neurohormonal disturbances associated with severe irreversible neurologic damage that occurs throughout the BD period. Clinical studies have shown an association between BD and inflammatory response in the liver [5, 8, 9]; however, appropriate prospective investigation of these events in the clinical setting is very limited and highly controversial. Experimental data from rodent models has shown that acute traumatic neurologic injury is characterized by a catecholamine surge that is followed by the release of pro-inflammatory cytokines and chemokines to peripheral circulation [10–12]. Large animal models of BD have confirmed the hemodynamic and metabolic changes associated with BD in humans and rodents [13–15]. However, the molecular and cellular components of the early inflammatory cascade have not been consistently documented in large animal studies with appropriate hemodynamic resuscitation. Moreover, the role of inflammation in liver damage is still unclear [8, 16–20] and a comprehensive description of the early changes in hemodynamic status, metabolism, and the relationship to inflammation in the liver using a clinically relevant large animal model is lacking.
We developed a non-human primate (NHP) model of brain death to allow for the characterization of early molecular and cellular inflammatory pathway activation. Our study demonstrates that as early as 6 h after brain death, inflammation can be observed at the gene, protein, and cellular levels. These data have been integrated into a descriptive model that characterizes the early events and pathways leading to BD induced inflammation, which may provide guidance for future therapeutic targets to improve quality of organs for transplantation.
MATERIALS AND METHODS
Animal Care
Rhesus macaque (Macaca mulatta) were obtained from the University of Wisconsin-Madison (UW) Primate Center and housed in accordance with Institutional Animal Care and Use Committee guidelines. All NHP were pre-screened for common viral, bacterial, and parasitic infections prior to assignment into groups: 6 brain-dead (BD) and 6 non-brain-dead (NBD) controls. Complete description of the procedures for brain death induction and organ recovery are provided in Supplemental Methods.
Quantification of Serum Hormone and Cytokine Levels
Serum cortisol (General Medical Laboratories, Madison, WI), epinephrine, nor-epinephrine (Alpco Diagnostics, Salem, NH), and IL-1β (Cellsciences, Canton, MA) values were determined by ELISA. Serum levels of IL-2, IL-4, IL-5, IL-6, TNF-α, and IFN-γ were determined using a non-human primate Th1/Th2 cytokine cytometric bead array (BD Biosciences, San Diego, CA) and FACSCalibur flow cytometer (BD Biosciences).
RNA Isolation
Liver was homogenized in Trizol (Invitrogen, Carlsbad, CA) and RNA extracted. RNA purification was performed by Qiagen RNeasy column (Qiagen, Germantown, MD) including the on-column DNase digestion step. RNA concentration was measured using a NanoDrop ND-1000 UV-Vis spectrophotometer and integrity confirmed using the Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA).
Microarray
RNA (1 μg) was labeled and fragmented using the MessageAmp Biotin II-Enhanced IVT kit following manufacturer’s instructions. aRNA (10 μg) was applied to the Genechip Rhesus macaque Genome Array (Affymetrix, Santa Clara, CA) and hybridized for 16 h at 45°C according to manufacturer’s instructions. Genechips were processed on the Affymetrix Fluidics450 Station and data extracted from GC3000 G7 scanned image using the Affymetrix command console. Data has been deposited in the Gene Expression Omnibus with accession number GSE22624.
Microarray Data Analysis
Data (CHP files) were imported into the Genesifter software package (VixX labs, Seattle, WA) and analyzed using pairwise analysis of BD verses NBD liver using the following parameters: t-test with and without Benjamini and Hochberg correction (P ≤ 0.05), fold change ≥ 1.5, and log data transformation. KEGG pathway analysis in the Genesifter software package and EASE analysis using the DAVID Bioinformatics Resource was performed on genes determined to be differentially expressed using the following criteria: t-test (P ≤ 0.05, no correction), fold change ≥ 1.5, and log data transformation. Pathways were considered over-represented if the z-score was greater than 2 (Genesifter) [21]. In DAVID, KEGG pathway and Gene Ontology Biological functions were considered over-represented with a Benjamini and Hochberg P value less than 0.05 and 0.01, respectively [22, 23].
Western Blotting
Liver homogenates were made using the M-PER protein extraction kit (Pierce, Rockford, IL) and protein concentrations determined by Bradford assay (Pierce). Protein was denatured for 5 min at 90°C in SDS-PAGE buffer prior to loading on Ready Gels (Bio-Rad, Hercules, CA). The protein was transferred to nitrocellulose membranes and probed with antibodies to SOCS3 (rabbit polyclonal; Abcam, Cambridge, MA,) and S100A9 (goat polyclonal; Abcam). Isotype-specific horseradish peroxidase conjugated secondary antibodies were used [donkey anti-rabbit or anti-mouse IgG (Pierce)] and bands visualized using ECL (Pierce) and captured using a LAS 4000 gel imager (Fuji Film, Tokyo, Japan). Membranes were stripped with Restore Western Blot Stripping Buffer (Pierce) and re-probed with anti-GAPDH-HRP (Abcam).
Histological Analysis
Liver was fixed in 10% formalin, embedded in paraffin, and mounted onto slides for staining: myeloperoxidase (MPO) (rabbit polyclonal, Abcam), CD68 (clone KP1, DAKO North America, Carpinteria, CA), HLA-DR (clone LN3; Biolegend, San Diego, CA), 4-hydroxynonenal (HNE) (rabbit antiserum; Diagnostic Intl. Inc., San Antonio, TX), and carboxy methyl lysine (CML) (clone NF1G; Trans Genic, Inc., Minami Kumamoto, Japan). The HIER method was used for antigen retrieval (BioGenex, San Ramon, CA). Endogenous peroxidase activity was blocked with 3% hydrogen peroxide and non-specific staining with Sniper reagent (Biocare Medical, Concord, CA). Staining for ICAM-1 (rabbit polyclonal; Abcam) was performed on OCT embedded cryosections. Isotype-specific peroxidase-conjugated secondary antibodies (Sigma-Aldrich, St. Louis, MO, USA) were used and chromogen diaminobenzidine was applied for visualization. Images from 12 random fields within each slide were acquired at 400× magnification using an Olympus BX51 microscope (Olympus America Inc., Center Valley, PA) and processed using ImageJ software (NIH, Bethesda, MD). Positive-staining and cell counts for each image were quantified using color-separation and background-subtraction, automatic thresholding (e.g., Maximum Entropy, Triangle, Huang), and particle-analysis algorithms.
Multiparametric Flow Cytometry
Peripheral blood leukocytes were isolated from whole blood by centrifugation on Ficoll gradients (LSM; Mediatech, Manassas, VA). Leukocytes were suspended in staining buffer (1X PBS, 1% BSA, 0.1% NaN3, pH 7.4) and incubated with fluorochrome conjugated antibodies. Leukocyte phenotype was identified by staining for CD11b (BD Biosciences, San Jose, CA), CD11c (Biolegend, San Diego, CA), CD14 (Biolegend), CD20 (Biolegend), CD31 (Biolegend), and MHC Class II DR (BD Biosciences). Leukocytes were stained with antibodies against chemokine receptors; CD181 (CxCR1; R&D Systems, Minneapolis, MN), CD182 (CxCR2; R&D Systems), CD192 (CCR2B; R&D Systems), CD195 (CCR5; BD Biosciences), and CD197 (CCR7; R&D Systems). Nonspecific binding was blocked with anti-CD16 (Biolegend) and human IgG (Sigma-Aldrich). Cells were stained with Live Dead Aqua (Invitrogen) to identify necrotic cells and fixed using the BD Cytofix/Cytoperm kit (BD Biosciences). Samples were analyzed using a BD LSR II flow cytometer (Becton Dickenson, Franklin Lakes, NJ). A minimum of 30,000 events were analyzed using Flo Jo software (Treestar Inc., Ashland, OR).
Data Analysis and Statistics
Descriptive statistics, Student’s t-test for comparison of means, and two-way analysis of variance (ANOVA) were performed using Graph-Pad Prism software (GraphPad Software, Inc., San Diego, CA). Statistical significance was set at a P value <0.05.
RESULTS
NHP Brain Death Model
NHP in the BD group showed increased blood pressure 10 min after inflation of the catheter (Mean Arterial pressure (MAP): 167.2 mmHg ± 41.9), followed by a period of severe hypotension and hemodynamic instability for 30–45 min (MAP: 44.5 ± 13.0) (Fig. 1A). Sustained sinus tachycardia (heart rate: 166.6 ± 27.8) (Fig. 1B) was documented in all BD animals along with increased urine output (5.1 ± 1.7 ml/kg/hr 6 h post-induction) indicating diabetes insipidus (Fig. 1C). NBD controls remained normotensive and did not show alterations in BP, heart rate, or urine output. Cerebral angiograms performed prior to catheter inflation demonstrated normal perfusion in both hemispheres (Suppl. Fig. 1A). Angiograms performed after BD induction showed incomplete diffusion of contrast media above the internal carotid arteries and abrogation of supratentorial blood flow in the circle of Willis and main branches while perfusion via extracranial arteries was preserved (Suppl. Fig. 1 B–E).
FIG. 1.
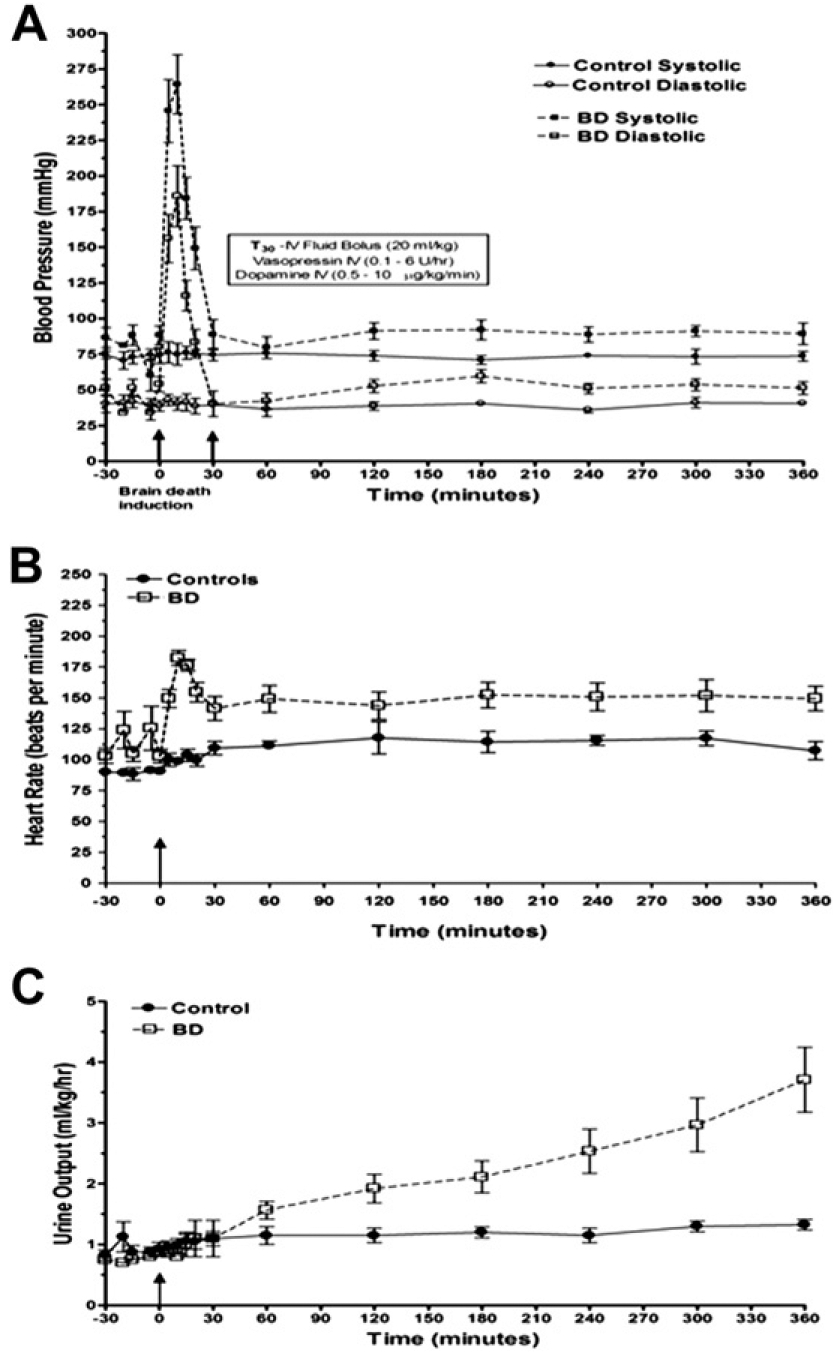
Brain death induction in NHP induces changes in physiologic parameters that model the human clinical scenario. Hemodynamic instability characterized by a sharp increase in arterial blood pressure after catheter inflation followed by hypotension (A), sustained tachycardia (B), and progressive increase in urinary output (C) was observed in all NHP subjected to BD induction (n = 5). A subset of NHP were anesthetized and mechanically ventilated without brain death induction to determine control physiologic values over the 6 h period (n = 6).
Serum epinephrine and nor-epinephrine levels increased significantly after 30 min of BD and remained elevated (Fig. 2A). Cortisol levels increased 30 min after induction and subsequently decreased below baseline levels six hours after BD (Fig. 2B). Serum levels of IL-6 were elevated in the BD group (281.4 pg/mL ± 88.2) relative to NBD control (13.3 pg/mL ± 11.2, P < 0.0001) or pre-BD samples (20.0 pg/mL ± 28.3, P < 0.01). The serum levels of IL-1β, IL-2, IL-4, IL-5, TNFα, and IFNγ were not different between BD and NBD control NHP (data not shown).
FIG. 2.
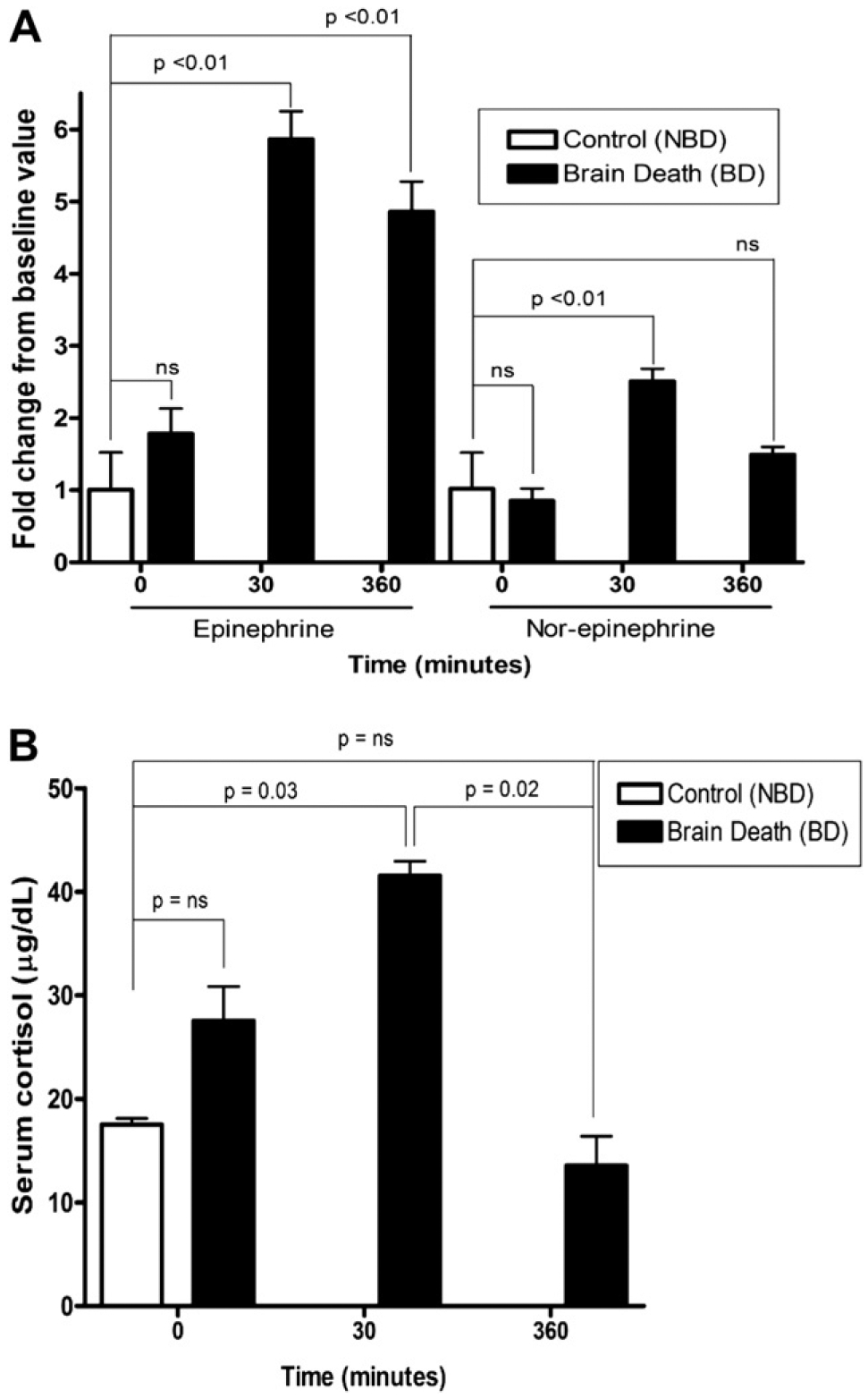
Epinephrine and Nor-epinephrine changes in brain-dead (n = 5) and Non-Brain Dead control (NBD) NHP (n = 6) (A). Bars represent fold change above baseline. Cortisol levels in NBD (n = 5) and brain-dead primates (n = 6) before and after brain death (B). Data are means ± SEM. ns = not-significant. Significance calculated by non-parametric, two-sided t-test.
Brain Death Induces Peripheral Blood Leukocyte Activation
To assess the effect of BD on systemic inflammation and peripheral blood leukocyte populations we performed multi-parametric flow cytometry analysis on leukocytes isolated prior to (T0) and 6 h after BD induction (T6). Comparisons were made with blood drawn from control animals sedated for the T0 blood draw and anesthetized and ventilated briefly 6 h later for the blood draw and tissue recovery. The percentage of peripheral blood neutrophils (CD11b+, CD11c−, CD14low, CD20−, MHC Class II DR−) and monocytes (CD11b+, CD11c−, CD14high, CD20−, MHC Class DR+) increased in both control and BD NHP (Fig. 3A). However, the increase (4-fold) in neutrophils was more pronounced in the BD group, although not reaching statistical significance. In contrast, peripheral blood myeloid dendritic cells (CD11blow, CD11c+, CD14low, CD20−, MHC Class II DRlow) were relatively unchanged in frequency in control animals, but decreased nearly 5-fold in BD (P < 0.05). Characterization of the activation states of peripheral blood leukocytes was performed by quantification of chemokine receptor expression and comparison of samples taken at T6 with those from T0. Neutrophils expressing CxCR1, CxCR2, and CCR7 increased in frequency in the BD animals, while decreasing in the controls (Fig. 3B). Monocytes expressing CCR2 and CCR5 increased in frequency in the BD animals, while remaining relatively unchanged in the controls (Fig. 3C). The small number of myeloid dendritic cells detected in the peripheral blood of BD NHP at T6 were found to be increased in the percentage of CxCR1, CxCR2, CCR2, CCR5, and CCR7 expressing cells (Fig. 3D).
FIG. 3.
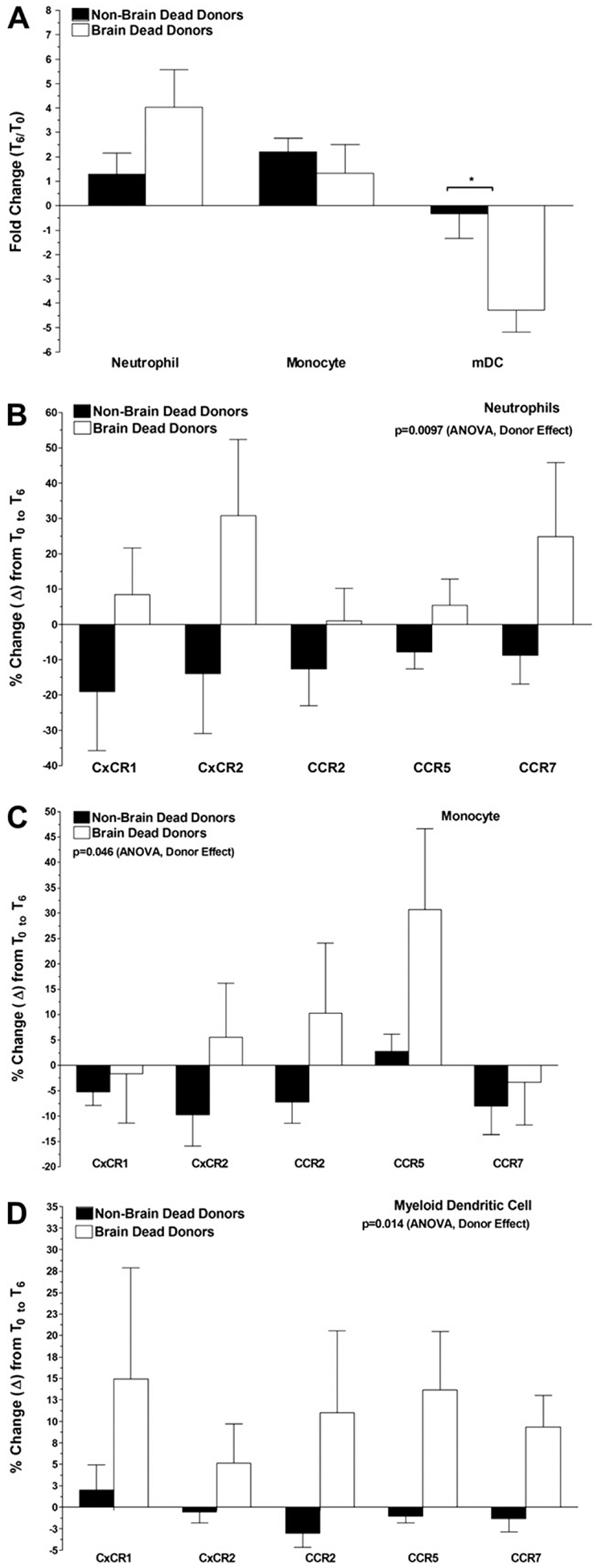
Brain death leads to changes in innate immune components of the peripheral blood leukocyte pool. Peripheral blood was drawn from control non-brain dead (NBD) and brain dead (BD) monkeys at T0 (prior to BD induction) and T6 (6 h post-BD induction) and leukocytes isolated by gradient centrifugation. Neutrophils, monocytes, and myeloid dendritic cells were quantified and changes in population frequency compared at T6 relative to T0 (A), chemokine receptor expression by neutrophils (B), monocytes (C), and myeloid dendritic cells (D). Data shown are the mean ± SEM and P values were calculated by two-way ANOVA.
BD Induced Changes in Gene Expression Related to Innate Immunity and Metabolism
Microarray analysis was performed to compare gene expression patterns from livers recovered from BD (n = 5) and NBD control NHP (n = 5). Pairwise statistical analysis was performed using an uncorrected t-test with a fold change cut-off of 1.5 (2811 probes up, 3175 probes down) or a t-test with a Benjamini and Hochberg correction for multiple testing (738 probes up, 862 probes down). A subset of the differentially expressed genes (n = 11) was validated using quantitative RT-PCR (qRT-PCR) (Suppl. Table 2). A majority of the 20 most highly up-regulated genes are linked to inflammatory pathways; including acute phase proteins (SOCS1, PTX3, ALDH1A3, PLA2G2A), chemokines (CXCL14, CCL23), matrix metalloproteinases (MMP1, MMP10), danger signals (S100A8, S100A9), and other known inflammatory processes (STEAP4, CALCA, IL2RA) (Suppl. Table 1).
KEGG Pathways
Genes were sorted into lists based upon up- or down-regulation in livers from BD animals compared with NBD controls. These lists were used to search pathways that were significantly enriched using Genesifter software and the DAVID Bioinformatics Resource (P < 0.05, t-test with Benjamini and Hochberg correction) [22, 23] (Suppl. Table 3). Pathways significantly up-regulated in the liver included apoptosis (P = 5.10 × 10−3), and the TLR signaling pathways (P = 4.22 × 10−2). Among the genes significantly up-regulated in the apoptosis pathway were pro-inflammatory cytokines (IL1A, IL1B), adaptor molecules that transfer signals from cell surface receptors to caspases (FADD, IRAK1, IRAK2) and caspases (3, 7, 8, 10). Genes significantly up-regulated in the TLR pathway included TLR2, TLR4, and TLR8, and proteins downstream of the TLR (IRAK, FADD, CASP8), MAP kinases, and NFKB signaling genes. Multiple metabolic pathways were down-regulated in livers of brain BD animals including the PPAR signaling pathway (P = 2.30 × 10−2), fatty acid metabolism (P = 1.41 × 10−8) and multiple amino acid synthesis pathways (Suppl. Table 3).
Gene Ontology Biological Functions
Analysis of Gene Ontology (GO) biological functions showed trends similar to the KEGG pathways (Table 1). Among the most significantly enriched were RNA metabolism and processing and protein and RNA transport. These biological functions also included genes involved in inflammation (Table 2). Genes involved in intracellular signaling were also highly up-regulated in livers from BD donors including the NFκB pathway and numerous MAPK genes, including p38MAPK and JNK. Metabolic pathways dominated the GO biological functions enriched in genes down-regulated after BD in the liver, including amino acid, fatty acid, alcohol, polysaccharide, and steroid metabolic pathways (Table 1).
TABLE 1.
Summary of Gene Ontology Categories Significantly Enriched in List of Genes Up- or Down-Regulated in the Liver After Brain Death
| Up-regulated | Count* | Benjamini† |
|---|---|---|
| RNA processing | 93 | 1.93E-08 |
| Ribonucleoprotein complex biogenesis and assembly | 51 | 1.98E-08 |
| Ribosome biogenesis and assembly | 31 | 4.55E-07 |
| mRNA metabolic process | 59 | 1.09E-04 |
| rRNA metabolic process | 23 | 2.43E-04 |
| Gene expression | 356 | 4.69E-03 |
| Nuclear transport | 36 | 4.92E-05 |
| Intracellular transport | 103 | 3.10E-04 |
| Cellular localization | 120 | 1.22E-03 |
| Protein transport | 96 | 2.10E-03 |
| Protein targeting | 41 | 2.91E-03 |
| Response to stress | 137 | 2.59E-04 |
| Immune response | 85 | 3.18E-03 |
| Immune system process | 106 | 6.61E-03 |
| Inflammatory response | 46 | 6.80E-03 |
| Defense response | 73 | 7.71E-03 |
| Regulation of programmed cell death | 76 | 3.09E-03 |
| Regulation of apoptosis | 75 | 3.79E-03 |
| Cell death | 106 | 7.14E-03 |
| Apoptosis | 100 | 9.39E-03 |
| I- κB kinase/NF-κB cascade | 34 | 5.09E-05 |
| Intracellular signaling cascade | 171 | 7.12E-03 |
| Down-regulated | Count* | Benjamini† |
| Organic acid metabolic process | 110 | 5.55E-13 |
| Carboxylic acid metabolic process | 108 | 2.22E-12 |
| Monocarboxylic acid metabolic process | 56 | 1.83E-09 |
| Lipid metabolic process | 128 | 1.10E-10 |
| Fatty acid metabolic process | 42 | 8.37E-07 |
| Nitrogen compound metabolic process | 79 | 1.30E-04 |
| Nitrogen compound catabolic process | 22 | 7.28E-04 |
| Amino acid and derivative metabolic process | 63 | 3.37E-04 |
| Amino acid catabolic process | 20 | 8.01E-04 |
| Amino acid metabolic process | 51 | 1.83E-03 |
| Alcohol metabolic process | 55 | 3.86E-04 |
| Generation of precursor metabolites and energy | 83 | 8.23E-04 |
| Electron transport | 62 | 4.84E-03 |
Number of genes in pathway up or down-regulated.
EASE score, a modified Fisher exact P value with a Benjamini and Hochberg multiple testing correction.
TABLE 2.
List of Genes Changed in the Inflammatory Gene Ontology
| Ratio | Gene name | Gene ID | P value | Adj. P value |
|---|---|---|---|---|
| 32.13 | Calcitonin-related polypeptide α | CALCA | 2.09E-03 | 3.85E-02 |
| 14.47 | S100 calcium binding protein A8 | S100A8 | 3.31E-03 | 4.42E-02 |
| 9.23 | Prokineticin 2 | PROK2 | 1.61E-03 | 3.66E-02 |
| 5.88 | Chemokine (C-X-C motif) ligand 11 | CXCL11 | 1.71E-02 | 7.88E-02 |
| 5.58 | S100 calcium binding protein A12 | S100A12 | 7.52E-04 | 2.85E-02 |
| 5.47 | Oncostatin M receptor | OSMR | 3.12E-03 | 4.34E-02 |
| 5.34 | NLR family, pyrin domain containing 12 | NLRP12 | 9.08E-03 | 6.11E-02 |
| 4.75 | Chemokine (C-X-C motif) ligand 2 | CXCL2 | 3.17E-02 | 1.01E-01 |
| 4.07 | Interleukin 18 receptor accessory protein | IL18RAP | 1.23E-02 | 6.95E-02 |
| 3.73 | Indoleamine 2,3-dioxygenase 1 | IDO1 | 1.04E-03 | 3.16E-02 |
| 3.59 | Tumor necrosis factor, α-induced protein 6 | TNFAIP6 | 4.70E-02 | 1.22E-01 |
| 3.59 | Interleukin 1, α | IL1A | 1.97E-02 | 8.30E-02 |
| 2.81 | Arachidonate 5-lipoxygenase-activating protein | ALOX5AP | 3.28E-03 | 4.41E-02 |
| 2.79 | N-myc (and STAT) interactor | NMI | 9.67E-03 | 6.22E-02 |
| 2.73 | Prostaglandin-endoperoxide synthase 2 | PTGS2 | 1.85E-02 | 8.06E-02 |
| 2.71 | Formyl peptide receptor 2 | FPR2 | 9.05E-03 | 6.06E-02 |
| 2.58 | Interleukin-1 receptor-associated kinase 2 | IRAK2 | 4.01E-03 | 4.63E-02 |
| 2.56 | UDP-Gal:betaGlcNAc beta 1,4- galactosyltransferase, polypeptide 1 | B4GALT1 | 7.59E-03 | 5.95E-02 |
| 2.53 | Receptor-interacting serine-threonine kinase 2 | RIPK2 | 5.21E-04 | 2.80E-02 |
| 2.50 | Histone deacetylase 4 | HDAC4 | 3.65E-04 | 2.32E-02 |
| 2.25 | Serpin peptidase inhibitor, clade A (α-1 antiproteinase, antitrypsin), member 1 | SERPINA1 | 1.04E-02 | 6.44E-02 |
| 2.23 | B-cell CLL/lymphoma 6 | BCL6 | 1.84E-03 | 4.26E-02 |
| 2.17 | CD46 molecule, complement regulatory protein | CD46 | 1.08E-03 | 3.22E-02 |
| 2.03 | Mannan-binding lectin serine peptidase 1 | MASP1 | 4.26E-03 | 4.70E-02 |
| 1.99 | Phospholipase A2, group IVC | PLA2G4C | 1.25E-02 | 6.96E-02 |
| 1.91 | Nuclear factor of κ light polypeptide gene enhancer in B-cells 1 | NFKB1 | 5.45E-03 | 5.06E-02 |
BD Induces Key Indicators of Pro-inflammatory Cytokine Activation and Innate Immunity
To further establish a link between brain death and pro-inflammatory cytokine activation of inflammation, we assessed liver protein extracts for the expression of SOCS3 and S100A9. Western blot analysis demonstrated a significant increase in both SOCS3 and S100A9 protein in BD versus NBD donor NHP liver (Fig. 4A).
FIG. 4.
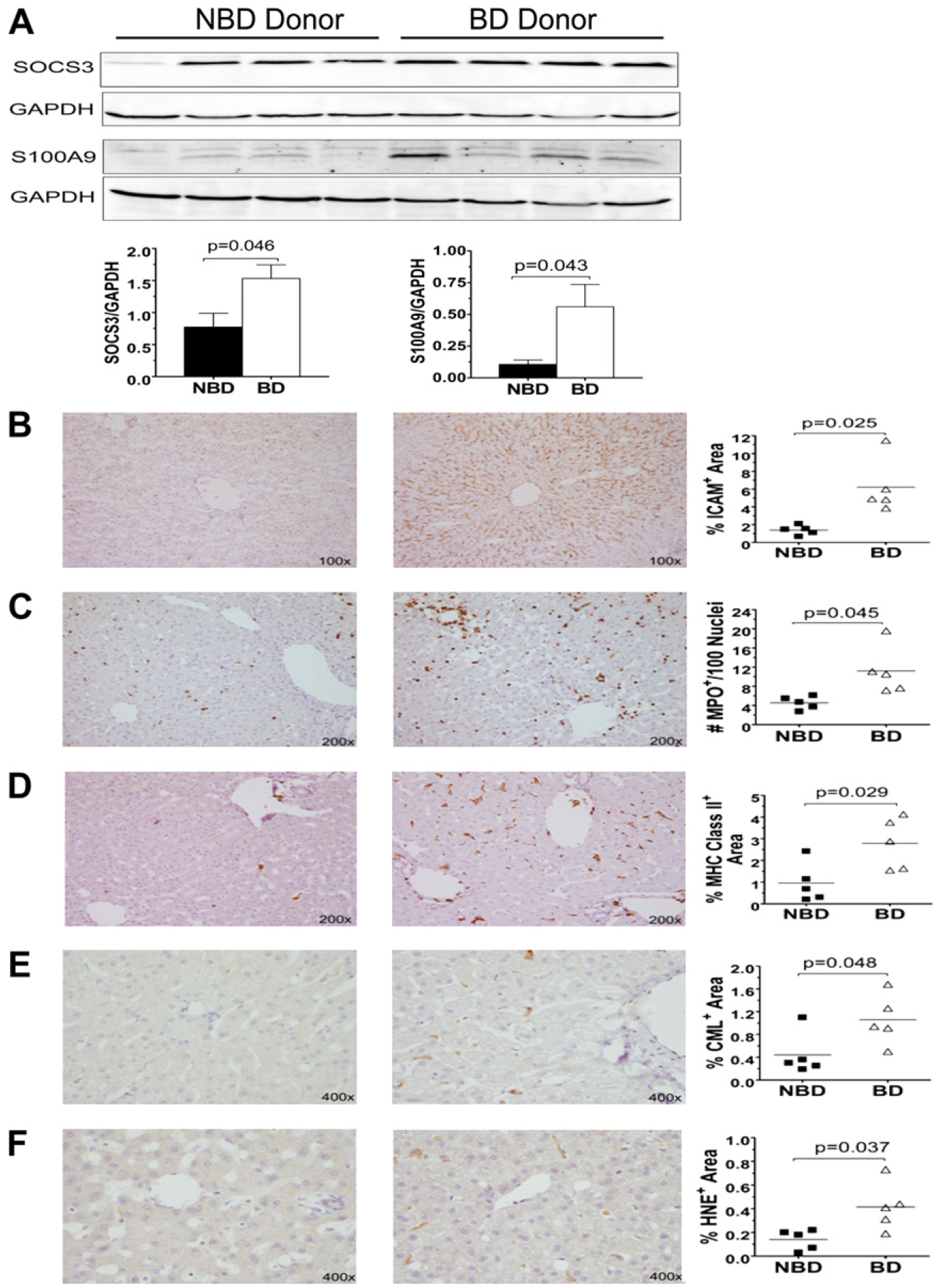
Brain death induces increases in level of proteins involved in cells adhesion, inflammatory processes, and accumulation of oxidative stress products in the liver. Liver protein from NBD donor “Control” and “Brain Dead” NHP were analyzed by Western blotting for the expression of SOCS3 and S100A9 proteins (A). Immunohistochemical assessment of liver biopsies taken from NBD donor controls and brain dead NHP stained for ICAM-1 (B), MPO (C), MHC Class II DR (D), CML (E), and HNE (F). Immunoblot quantification of the identified bands was performed by densitometry and normalized to GAPDH protein levels with p values determined by Student’s t-test. Quantification of staining was performed by image analysis (see the Methods section) and data shown are the individual values and means (line). P values were calculated by Student’s t-test.
Activation of Endothelium and Kupffer Macrophages and Neutrophil Infiltration in the Liver
Liver biopsies were stained for adhesion molecules (ICAM-1), neutrophils (myeloperoxidase, MPO), macrophages (CD68), MHC Class II DR, and products of oxidative stress (carboxy methyl lysine, CML and 4-hydroxynonenal, HNE). BD NHP liver showed significantly elevated expression of ICAM-1 localized primarily to sinusoidal endothelium surrounding the central veins (Fig. 4B). Significantly greater numbers of MPO+ neutrophils were detected within the central veins, sinusoids, and parenchyma of BD NHP liver (Fig. 4C). Blood-derived and resident Kupffer macrophage numbers, indicated by CD68+ staining did not differ significantly between the two groups (data not shown). However, expression of MHC Class II DR was up-regulated in BD NHP liver compared with controls (Fig. 4D). The expression of MHC Class II DR was observed on multiple cell types (macrophages, endothelium, and lymphocytes) within the livers of BD NHP. Staining for oxidative stress markers, advanced glycation end product CML and the lipid peroxide HNE, was significantly higher in BD NHP liver (Fig. 4E and F).
DISCUSSION
Brain death resulting from acute neurologic injury is characterized by disturbances in physiologic, biochemical, and immunologic functions that lead to impairment of peripheral organs and graft dysfunction in the post-transplant period. Sustained hypoperfusion induces tissue necrosis prompting the release of damage associated molecular pattern signal molecules (DAMPs) from activated inflammatory leukocytes and/or damaged cells [24]. DAMPs have been implicated in the regulation of inflammation after hepatic ischemia/reperfusion injury through activation of the TLR family and the receptors for advanced glycation end products (RAGE) [25, 26]. This activation leads to activation of MAPK and NF-Kβ signaling, which regulate cytokine and chemokine production as well as endothelial cell activation and leukocyte mobilization during inflammation, resulting in further damage to already ischemic tissue.
We developed a non-human primate BD model with the objective of closely mimicking the human organ donor environment and characterizing early molecular and cellular changes that are hypothesized to predispose the liver to decreased post-transplant function and survival. The liver is central to the relationship between BD, systemic inflammation, and organ damage. The data described here were integrated into a model, discussed in detail below, which links brain death with immune system pathways in the peripheral blood and liver, which are consistent with activation of innate immunity (Fig. 5).
FIG. 5.
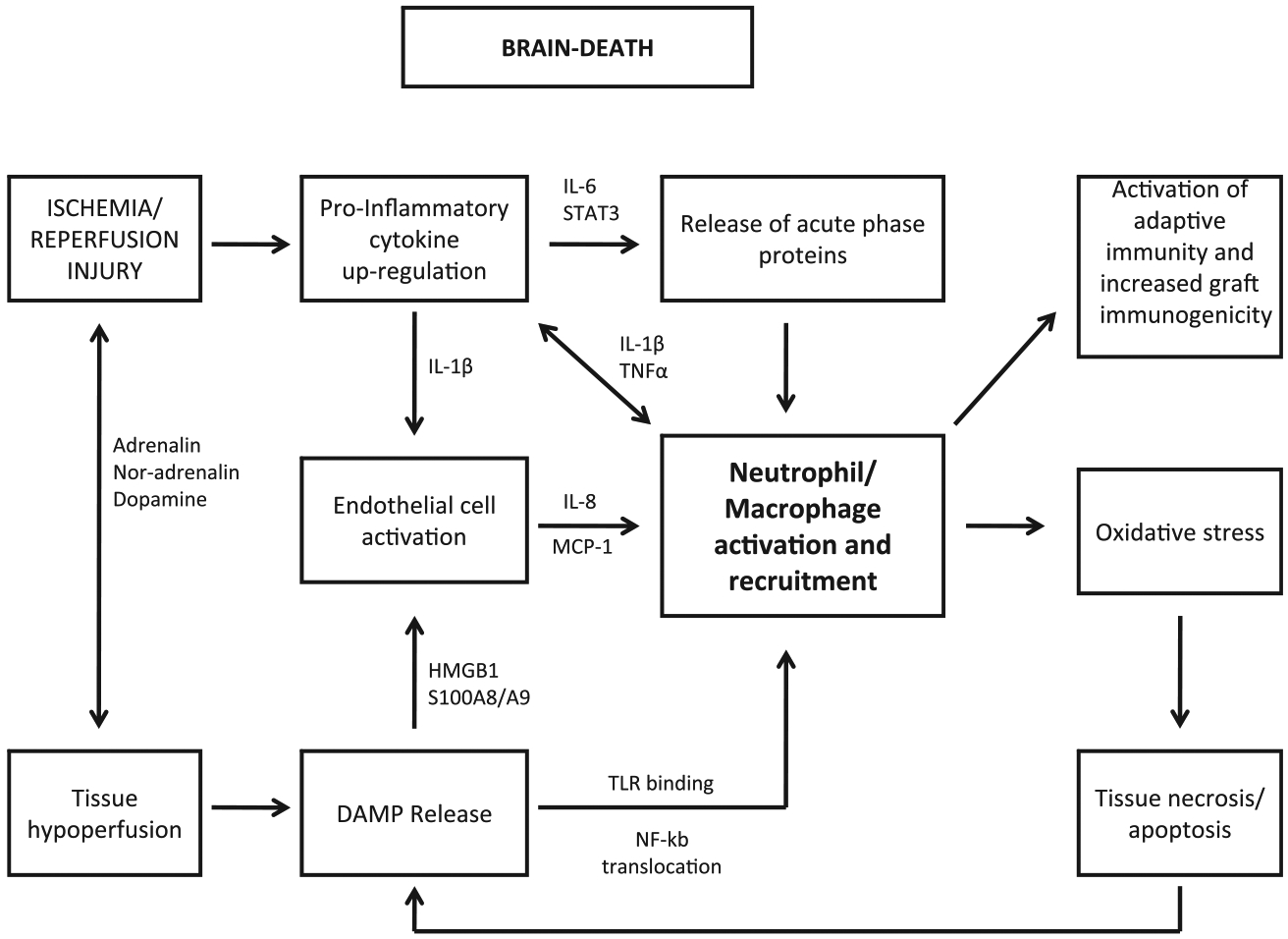
Model of systemic and localized inflammation affecting the liver in a non-human primate model of brain death. The initial process of BD results in the Cushing response including dysfunction of the hypothalamic-pituitary-adrenal axis and central autonomous dysregulation. The resulting hypoperfusion leads to endothelial cell activation, chemokine secretion and release of DAMPs from necrotic cells and activated innate immune cells. DAMPs bind to and activate TLR and result in NFKB activation followed by production and release of pro-inflammatory cytokines (PIC). PIC release results in further activation of endothelial cells and up-regulation of adhesion molecules including VCAM, ICAM, and P-selectin. PIC also induce systemic release of IL-6 leading to STAT3 activation and production of acute phase proteins by hepatocytes. These events culminate in the recruitment and migration of PMN and monocytes from bone marrow into peripheral circulation and then into the ischemic tissue. Once in the liver, innate immune cells prime and amplify the immune response, produce reactive oxygen species, and induce secondary tissue damage that ultimately leads to decreased organ function after recovery and an increased immunogenic potential.
We and others have documented the severe hyperdynamic response to brain stem herniation, which involves massive catecholamine release and autonomic dysregulation [27–31]. During ischemia/reperfusion excess ROS are produced leading to oxidative stress and tissue damage. Evidence of ROS production in livers from BD NHP was indicated by IHC detection of increased lipid peroxidation (HNE) and oxidation of carbohydrates (CML). We propose that oxidative stress may be one of the BD initial inflammatory signals resulting from endothelial and Kupffer macrophage activation, a finding documented by several other publications [32]. Further supporting this conclusion was the observation that the majority of CML and HNE staining co-localized with macrophages lining the sinusoids.
In our model, the S100A8 and S100A9 genes were both highly up-regulated in the liver after BD. Furthermore, S100A9 protein was present in significantly greater levels in the livers from BD than NBD control NHP as evidenced by immunoblotting. S100 proteins are novel DAMPs found in phagocytes and the observation of increased S100A8/A9 may partially reflect the increased number of neutrophils observed in hepatic tissue after 6 h of BD [33]. However, due to the large difference observed in both mRNA and protein levels, increased neutrophil infiltration is unlikely to be the only mechanism for protein up-regulation in our model. Furthermore, microarray analysis showed that genes in the TLR signaling pathway were significantly up-regulated in the liver following brain death induction. These findings are consistent with well-known mechanisms of innate immune activation in the context of tissue injury and in clinical and experimental models of BD have been linked to graft dysfunction and accelerated rejection in the post transplant period [12, 34, 35].
A well-known consequence of IL-6 induction is the increased expression of acute phase genes. In this study, we identified multiple acute phase genes that were up-regulated in the brain dead liver and detected increased levels of IL-6 in peripheral blood and ICAM-1 within liver sinusoids. SOCS genes are up-regulated during inflammation in response to activation of the JAK-STAT pathway by interferon signaling (SOCS1) or IL6 signaling (SOCS3). PTX3 is produced locally at sites of inflammation and interacts with multiple ligands, including complement components, extracellular matrix proteins, growth factors, apoptotic cells, and pathogens [36]. Also, PLA2G2A is secreted from liver cells in response to inflammatory stimuli including IL-6 and is involved in removal of anionic extracellular cell debris [37]. It has been widely described that ischemia reperfusion and hemorrhage induce an inflammatory response to tissue injury that is characterized by the rapid mobilization of immature innate immune cells from the bone marrow and spleen into peripheral circulation [38, 39]. In our model, peripheral blood neutrophil numbers increased, while myeloid dendritic cells (mDC) decreased. Moreover, the population of neutrophils, monocytes, and mDC present at 6 h post-brain death indicated a predominance of immature phenotypes, based upon chemokine receptor expression, which is considered by some to be a direct indicator of leukocyte activation [40]. Our data suggest that mature leukocytes were recruited out of the peripheral circulation while new immature leukocytes were probably released from the bone marrow and spleen and were likely being chemoattracted to the inflammatory site. Further evidence of activation and recruitment of inflammatory leukocytes was obtained by observation of significantly increased numbers of MPO+ cells [primarily polymorphonuclear (PMN) granulocytes] in the livers of BD animals. This supports the hypothesis of immune activation and leukocyte migration triggered by BD, and is consistent with findings from multiple studies in which BD elicits activation of endothelium, innate immunity and recruitment of PMN into tissues [12, 41–43]. This finding is important as BD induced leukocytic infiltration has been associated with increased immunogenicity of the donor graft and a higher incidence of acute rejection [7].
Our data also revealed that energy and amino acid metabolism genes were down-regulated in livers from brain dead NHP. Colombo et al. used a cDNA microarray technique to compare liver tissue from human brain dead donors to those from normal tissue obtained during resection of benign focal lesions. In their analysis, they also observed decreased expression of genes related to lipid, amine, and nucleic acid metabolism [44]. Inflammatory pathways have previously been shown to modify expression of metabolic genes, including those involved in fatty acid oxidation. Maitra et al. [45] showed that the decrease in fatty acid oxidation occurs through a TLR4 dependent pathway that is mediated through IRAK1. As a result, IRAK1 reduces levels of PPARα In the liver from BD donors the PPAR signaling pathway, specifically PPARα was significantly down-regulated compared with NBD control. PPAR is an important regulator of cellular energetics, including β-oxidation of fatty acids and inflammation including the production of cytokines such as IL-6 [46, 47]. This is consistent with our microarray data, which shows that the TLR pathway, including the genes for TLR4 and IRAK1, were up-regulated. The connection between BD, inflammation, metabolic gene down-regulation, and post-transplant organ function is relatively unexplored and may be important in addressing delayed graft function or primary non-function in liver transplantation.
In summary, BD produces multiple changes in the liver that may negatively impact post transplant function. In this communication, we have extensively characterized the innate immune response to traumatic brain injury and brain death using a clinically translatable large animal model. We have shown that a cascade of events occurring early during BD resulted in increased inflammation in the liver, including release of pro-inflammatory DAMPs, cytokines, endothelial cell activation, TLR pathway up-regulation, and recruitment of inflammatory leukocytes to the site of injury. It has been shown previously that organ inflammation, leukocyte infiltration, and apoptosis can significantly impact pre-transplant organ viability and determine organ quality and function in the post-transplant period. In this study, we have reproduced events that have been characterized in clinical practice, and our results support the utility of this pre-clinical model for evaluating targeted interventional donor management strategies with the objective of improving pre-transplant graft quality and, ultimately, liver transplant outcomes.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Wayne Davis, Kim Maurer, Chelsea Rentmeester, and Stacey Jenkins-Bryant for expert technical assistance. They thank Juan Contreras for guidance in the development of the NHP brain death model. This research was funded by internal funding from the Department of Surgery, Division of Transplantation, University of Wisconsin-Madison.
Footnotes
SUPPLEMENTARY DATA
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.jss.2011.10.042.
REFERENCES
- 1.Department of Health and Human Services, H. R. a. S. A., Healthcare Systems Bureau, Division of Transplantation, Rockville, MD, United Network for Organ Sharing, R., VA, and University Renal Research and Education Association, A. A., MI. 2009. Annual Report of the U.S. Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients: Transplant Data 1999-2008.
- 2.Hong JC, Yersiz H, Farmer DG, et al. Long-term outcomes for whole and segmental liver grafts in adult and pediatric liver transplant recipients: A 10-year comparative analysis of 2988 cases. J Am Coll Surg 2009;208:682 discusion 689. [DOI] [PubMed] [Google Scholar]
- 3.Van der Hoeven J, Lindell S, van Schilfgaarde R, et al. Donor brain death reduces survival after transplantation in rat livers preserved for 20 hours. Transplantation 2001;72:1632. [DOI] [PubMed] [Google Scholar]
- 4.Pratschke J, Wilhelm MJ, Kusaka M, et al. Accelerated rejection of renal allografts from brain-dead donors. Ann Surg 2000; 232:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barklin A Systemic inflammation in the brain-dead organ donor. Acta Anaesthesiol Scand 2009;53:425. [DOI] [PubMed] [Google Scholar]
- 6.Pratschke J, Wilhelm MJ, Kusaka M, et al. Brain death and its influence on donor organ quality and outcome after transplantation. Transplantation 1999;67:343. [DOI] [PubMed] [Google Scholar]
- 7.Jassem W, Koo DD, Cerundolo L, et al. Cadaveric versus living-donor livers: Differences in inflammatory markers after transplantation. Transplantation 2003;76:1599. [DOI] [PubMed] [Google Scholar]
- 8.Weiss S, Kotsch K, Francuski M, et al. Brain death activates donor organs and is associated with a worse I/R injury after liver transplantation. Am J Transplant 2007;7:1584. [DOI] [PubMed] [Google Scholar]
- 9.Kotsch K, Ulrich F, Reutzel-Selke A, et al. Methylprednisolone therapy in deceased donors reduces inflammation in the donor liver and improves outcome after liver transplantation: A prospective randomized controlled trial. Ann Surg 2008; 248:1042. [DOI] [PubMed] [Google Scholar]
- 10.Hoeger S, Reisenbuechler A, Gottmann U, et al. Donor dopamine treatment in brain dead rats is associated with an improvement in renal function early after transplantation and a reduction in renal inflammation. Transpl Int 2008;21:1072. [DOI] [PubMed] [Google Scholar]
- 11.Kusaka M, Pratschke J, Wilhelm MJ, et al. Activation of inflammatory mediators in rat renal isografts by donor brain death. Transplantation 2000;69:405. [DOI] [PubMed] [Google Scholar]
- 12.Takada M, Nadeau KC, Hancock WW, et al. Effects of explosive brain death on cytokine activation of peripheral organs in the rat. Transplantation 1998;65:1533. [DOI] [PubMed] [Google Scholar]
- 13.Contreras JL, Eckstein C, Smyth CA, et al. Brain death significantly reduces isolated pancreatic islet yields and functionality in vitro and in vivo after transplantation in rats. Diabetes 2003;52:2935. [DOI] [PubMed] [Google Scholar]
- 14.Bittner HB, Kendall SW, Campbell KA, et al. A valid experimental brain death organ donor model. J Heart Lung Transplant 1995;14:308. [PubMed] [Google Scholar]
- 15.Cooper DK, Novitzky D, Wicomb WN. The pathophysiologic effects of brain death on potential donor organs, with particular reference to the heart. Ann R Coll Surg Engl 1998;71:261. [PMC free article] [PubMed] [Google Scholar]
- 16.Mangino MJ, Kosieradzki M, Gilligan B, et al. The effects of donor brain death on renal function and arachidonic acid metabolism in a large animal model of hypothermic preservation injury. Transplantation 2003;75:1640. [DOI] [PubMed] [Google Scholar]
- 17.Barklin A, Larsson A, Vestergaard C, et al. Does brain death induce a pro-inflammatory response at the organ level in a porcine model? Acta Anaesthesiol Scand 2008;52:621. [DOI] [PubMed] [Google Scholar]
- 18.Skrabal CA, Thompson LO, Potapov EV, et al. Organ-specific regulation of pro-inflammatory molecules in heart, lung, and kidney following brain death. J Surg Res 2005;123:118. [DOI] [PubMed] [Google Scholar]
- 19.Compagnon P, Wang H, Lindell S, et al. Brain death does not affect hepatic allograft function and survival after orthotopic transplantation in a canine model. Transplantation 2002; 73:1218. [DOI] [PubMed] [Google Scholar]
- 20.van Der Hoeven J, Ter Horst G, Molema G, et al. Effects of brain death and hemodynamic status on function and immunologic activation of the potential donor liver in the rat. Ann Surg 2000; 232:804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doniger SW, Salomonis N, Dahlquist KD, et al. MAPPFinder: Using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol 2003; 4:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang da W, Sherman BT, Zheng X, et al. Extracting biological meaning from large gene lists with DAVID. Curr Protoc Bioinformatics Chapter 13: Unit 13 11, 2009. [DOI] [PubMed] [Google Scholar]
- 23.Dennis G Jr, Sherman BT, Hosack DA, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003;4:P3. [PubMed] [Google Scholar]
- 24.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol 2008;8:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsung A, Klune JR, Zhang X, et al. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med 2007;204:2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pardo M, Budick-Harmelin N, Tirosh B, et al. Antioxidant defense in hepatic ischemia-reperfusion injury is regulated by damage-associated molecular pattern signal molecules. Free Radic Biol Med 2008;45:1073. [DOI] [PubMed] [Google Scholar]
- 27.Bittner HB, Kendall SW, Chen EP, et al. The effects of brain death on cardiopulmonary hemodynamics and pulmonary blood flow characteristics. Chest 1995;108:1358. [DOI] [PubMed] [Google Scholar]
- 28.Chen EP, Bittner HB, Kendall SW, et al. P. Hormonal and hemodynamic changes in a validated animal model of brain death. Crit Care Med 1996;24:1352. [DOI] [PubMed] [Google Scholar]
- 29.Herijgers P, Leunens V, Tjandra-Maga TB, et al. Changes in organ perfusion after brain death in the rat and its relation to circulating catecholamines. Transplantation 1996;62:330. [DOI] [PubMed] [Google Scholar]
- 30.Mertes P, el Abassi K, Jaboin Y, et al. Changes in hemodynamic and metabolic parameters following induced brain death in the pig. Transplantation 1994;58:414. [DOI] [PubMed] [Google Scholar]
- 31.Fugate JE, Rabinstein AA, Wijdicks EF. Blood pressure patterns after brain death. Neurology 2001;77:399. [DOI] [PubMed] [Google Scholar]
- 32.Jaeschke H Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. J Gastroenterol Hepatol 2001; 26(Suppl 1):173. [DOI] [PubMed] [Google Scholar]
- 33.Foell D, Wittkowski H, Vogl T, et al. S100 proteins expressed in phagocytes: A novel group of damage-associated molecular pattern molecules. J Leukoc Biol 2007;81:28. [DOI] [PubMed] [Google Scholar]
- 34.van Der Hoeven JA, Ter Horst GJ, Molema G, et al. Effects of brain death and hemodynamic status on function and immunologic activation of the potential donor liver in the rat. Ann Surg 2000;232:804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKeating EG, Andrews PJ, Signorini DF, et al. Transcranial cytokine gradients in patients requiring intensive care after acute brain injury. Br J Anaesth 1997;78:520. [DOI] [PubMed] [Google Scholar]
- 36.Mantovani A, Garlanda C, Doni A, et al. Pentraxins in innate immunity: From C-reactive protein to the long pentraxin PTX3. J Clin Immunol 2008;28:1. [DOI] [PubMed] [Google Scholar]
- 37.Crowl RM, Stoller TJ, Conroy RR, et al. Induction of phospholipase A2 gene expression in human hepatoma cells by mediators of the acute phase response. J Biol Chem 1991;266:2647. [PubMed] [Google Scholar]
- 38.Swirski FK, Nahrendorf M, Etzrodt M, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009;325:612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Yuan Y, Li Y, et al. Interacting neuroendocrine and innate and acquired immune pathways regulate neutrophil mobilization from bone marrow following hemorrhagic shock. J Immunol 2009;182:572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murdoch C, Finn A. Chemokine receptors and their role in inflammation and infectious diseases. Blood 2000;95:3032. [PubMed] [Google Scholar]
- 41.Smith M Physiologic changes during brain stem death—lessons for management of the organ donor. J Heart Lung Transplant 2004;23:S217. [DOI] [PubMed] [Google Scholar]
- 42.van der Hoeven JA, Ploeg RJ, Postema F, et al. Induction of organ dysfunction and up-regulation of inflammatory markers in the liver and kidneys of hypotensive brain dead rats: A model to study marginal organ donors. Transplantation 1999;68:1884. [DOI] [PubMed] [Google Scholar]
- 43.Wilhelm MJ, Pratschke J, Beato F, et al. Activation of the heart by donor brain death accelerates acute rejection after transplantation. Circulation 2000;102:2426. [DOI] [PubMed] [Google Scholar]
- 44.Colombo G, Gatti S, Turcatti F, et al. Alteration in the transcriptional profile of livers from brain-dead organ donors. Transplantation 2006;82:69. [DOI] [PubMed] [Google Scholar]
- 45.Maitra U, Chang S, Singh N, et al. Molecular mechanism underlying the suppression of lipid oxidation during endotoxemia. Mol Immunol 2009;47:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 2008; 454:470. [DOI] [PubMed] [Google Scholar]
- 47.Ahmed W, Ziouzenkova O, Brown J, et al. PPARs and their metabolic modulation: New mechanisms for transcriptional regulation? J Intern Med 2007;262:184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.