

Abstract
Purpose of review:
This review provides an up-to-date understanding of how peroxisome proliferator activated receptor γ (PPARγ) exerts its cardioprotective effect in the vasculature through its activation of novel PPARγ target genes in endothelium and vascular smooth muscle.
Recent findings:
In vascular endothelial cells, PPARγ plays a protective role by increasing nitric oxide bioavailability and preventing oxidative stress. RBP7 is a PPARγ target gene enriched in vascular endothelial cells, which is likely to form a positive feedback loop with PPARγ. In vascular smooth muscle cells, PPARγ antagonizes the renin-angiotensin system, maintains vascular integrity, suppresses vasoconstriction, and promotes vasodilation through distinct pathways. RhoBTB1 is a novel PPARγ target in vascular smooth muscle cells that mediates the protective effect of PPARγ by serving as a substrate adaptor between the Cullin-3 RING ubiquitin ligase and phosphodiesterase 5, thus restraining its activity through ubiquitination and proteasomal degradation.
Summary:
In the vasculature, PPARγ exerts its cardioprotective effect through its transcriptional activity in endothelium and vascular smooth muscle. From the understanding of PPARγ ‘s transcription targets in those pathways, novel hypertension therapy target(s) will emerge.
Keywords: Peroxisome proliferator activated receptor γ, Rho-related BTB domain containing protein 1, phosphodiesterase 5, vascular endothelial cells, vascular smooth muscle cells
Introduction: PPARγ and Hypertension
Peroxisome proliferator activator receptor gamma (PPARγ) is a ligand activated transcription factor which forms a heterodimer with retinoid X receptor (RXR) at PPAR response elements (PPRE) in regulatory regions of PPARγ target genes. In the absence of ligand, the heterodimer remains inactive on chromatin by binding to transcriptional co-repressors which results in transcriptional repression of target genes. Upon ligand binding, a conformational change in PPARγ leads to the dissociation of the co-repressors and recruitment of co-activators to facilitate the transactivation of these genes [1, 2]. PPARγ first gained prominence for its’ profound effect on adipogenesis and lipid metabolism [3]. Thiazolidinediones (TZDs), a class of anti-diabetic drugs which increase insulin sensitivity, are high affinity ligands of PPARγ [4]. Interestingly, clinical trials such as the PROactive (PROspective pioglitAzone Clinical Trial In macroVascular Events) and STARR (STudy of Atherosclerosis with Ramipril and Rosiglitazone) trials showed that TZDs can decrease blood pressure, improve vascular function and decrease carotid intima-media thickness in patients with type 2 diabetes and impaired glucose tolerance, respectively [5, 6].
Mounting evidence suggest that PPARγ plays a protective effect in hypertension (HT) development. First, administration of TZDs to a variety of hypertensive mouse and rat models lowered blood pressure [7–11]. Second, interference with, or ablation of PPARγ in a global or tissue-specific manner predisposes animals to various forms of HT and vascular diseases [12–14]. Finally, four different PPARγ mutations have been identified in humans (R165T, L339X, P467L, and V290M) which cause hypertension [15, 16]. Two of them (P467L and V290M) act dominant negatively, that is, interfere with the function of normal (wild type) PPARγ. Many have asked the question of how PPARγ exerts its protective effect? Studies from numerous laboratories, including ours, have suggested that the blood pressure lowering effect of PPARγ is primarily due to its actions in the vasculature [17, 18]. Of course, we cannot discount the positive cardiovascular effects of improved glycemic control in response to PPARγ activation, or the role of the vasculature within tissues such as the kidney. The purpose of this review is to focus on the vascular actions of PPARγ and one of its’ newest target gene, RhoBTB1, in the regulation of vasomotor function and blood pressure.
1. PPARγ in Vascular Endothelial cells (VEC)
Early studies suggested the PPARγ may control the release of nitric oxide (NO) from endothelial cells suggesting the hypothesis that PPARγ plays a protective role by enhancing vasodilation [19]. The first report of endothelial specific PPARγ deficient mice (Tie2Cre-PPARγflox/flox) suggested there was no difference in blood pressure at baseline [20]. A later study also utilizing Tie2Cre-PPARγflox/flox mice then reported a small, yet significant increase in baseline blood pressure, accompanied by decreased nitric oxide release, increased oxidative stress and an impaired endothelial dependent vasodilation in aorta [21]. It was suggested that the difference in blood pressure reported in the two studies might due to differences in the genetic background of the strains or methods used to measure blood pressure.
There is now compelling evidence that endothelial PPARγ plays a protective role under conditions of cardiovascular or metabolic stress. For example, in the study cited above, Tie2Cre-PPARγflox/flox mice exhibited increased sensitivity to angiotensin-II (Ang-II) induced HT [21]. We used mice expressing one of the dominant negative PPARγ mutations (V290M), which caused hypertension in humans, specifically in the endothelium (termed E-V290M mice). Similar to the endothelial-specific PPARγ deficient mice, E-V290M mice exhibited a modest increase in baseline blood pressure and an augmented pressor response to Ang-II [22]. Overnight incubation of carotid arteries from E-V290M mice with Ang-II in vitro caused an impaired vasodilation response to ACh which was restored by inhibitors of superoxide, NADPH oxidase, both Rho kinase 1 and 2, Rho kinase 2 alone, NF-κB, or IL-6 neutralizing antibody [22, 23]. Similarly, Basilar arteries from E-V290M mice showed decreased vasodilation response to angiotensin 1–7, a vasodilator peptide derived from Ang-II which is reported to act through the mas receptor [23]. The same vessels exhibited impaired vasodilation ACh in response to low salt diet-induced activation of the renin-angiotensin system, a response reversed by Tempol [**24]. More recently, we showed that male E-V290M offspring born from female mice whose pregnancy was complicated by hypertension (a model of preeclampsia) exhibited impaired vasodilation to a dose of Ang-II which does not raise blood pressure [**25].
Endothelial dysfunction is also a prominent feature in obesity and type 2 diabetes. Tie2Cre- PPARγflox/flox mice are more sensitive to high fat diet (HFD)-induced HT [20]. Similarly, basilar artery from E-V290M mice showed impaired vasodilation to ACh after 12-week of HFD feeding, whereas mice expressing wildtype PPARγ selectively in endothelial cells exhibited showed no difference in blood pressure after HFD. The endothelial dysfunction in basilar arteries from E-V290M mice was reversed by Tempol, indicating an increased oxidative stress [22].
With robust evidence supporting a protective role of endothelial PPARγ we sought to assess the transcriptional targets of PPARγ that mediate its’ protective effect. Using transcriptomic analysis, we identified retinol binding protein 7 (RBP7, previously known as CRBPIII) as a PPARγ target gene which is highly enriched in VEC [26–28]. RBP7 belongs to a larger family of fatty acid (FA) binding proteins (FABP) [29]. Some FABPs have been reported to translocate to the nucleus to co-regulate nuclear receptor transcription factors [30], and thus this suggests the hypothesis that like other FABPs, RBP7 may transport a retinol or fatty acid derivative into the nucleus where it may co-regulate PPARγ activity. Interestingly, RBP7 knockout (RBP7 KO) mice recapitulate the same vascular impairment as observed in E-V290M mice [31]. Notably, carotid arteries from RBP7 KO mice exhibited impaired ACh vasodilation after two-week subpressor Ang-II infusion, a response which was reversed by adiponectin. Adiponectin is also a PPARγ target which is well recognized for its cardiovascular protective effect and is also expressed in endothelium [32–34]. Our data suggest that RBP7 is required for PPARγ-mediated transactivation of adiponectin expression in endothelium. Based on this we hypothesized that PPARγ-RBP7 might form a positive feedback loop where PPARγ promotes RBP7 transcription which then delivers ligands to PPARγ on chromatin (Figure 1).
Figure 1. Protective Role of PPARγ in the Vasculature.
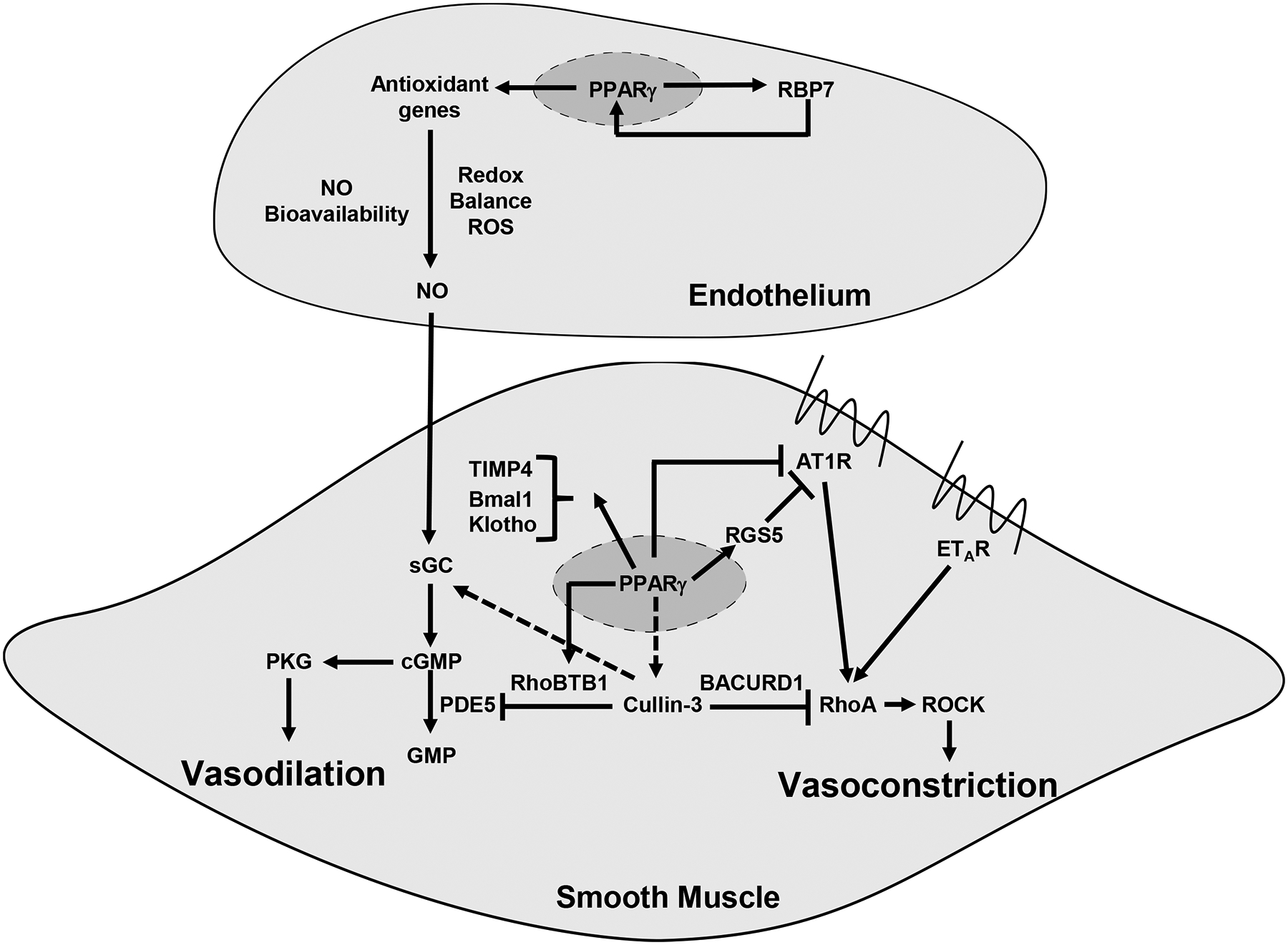
In vascular endothelial cells, PPARγ increases the bioavailability of NO and reduces ROS derived from the endothelium. RBP7, a PPARγ target gene, likely forms a positive feedback loop with PPARγ. We have hypothesized this may occur through a mechanisms by which RBP7 delivers PPARγ ligands to the PPARγ transcriptional complex in the nucleus. This interaction between PPARγ and RBP7 promotes transcription of antioxidant genes which maintains appropriate redox balance in favor of an anti-oxidant environment which promotes NO bioavailibility. In vascular smooth muscle cells, PPARγ sensitizes the vascular muscle to the effects of endothelium-derived NO. Here the effects of PPARγ are multifaceted. PPARγ promotes expression of RGS5 which controls the activity of AT1 receptor. PPARγ also prevents remodeling, prevents calcification, and maintains the cellular circadian rhythm, thorugh its targets TIMP4, Klotho and Bmal, respectively. PPARγ also engages the Cullin-3 E3 ubiquitin ligase to influence both vasodilation and vasoconstriction. Loss of PPARγ decreases Cullin-3 activity which promotes vasoconstriction through increased RhoA and ROCK activity. The mechanism is a loss of RhoA ubiquitination through the Cullin-3 complex. PPARγ promotes vasodilation by increasing expression of RhoBTB1 which acts as an adaptor to deliver PDE5 to the Cullin-3 complex for ubiquitination and proteasomal degradation. This tightly controls PDE5 activity and consequenctly the levels of cGMP in the cell.
2. PPARγ in Vascular Smooth Muscle Cells (VSMC)
Like its role in the endothelium, a review of the literature suggests that VSMC PPARγ is also protective by promoting vasodilation, restraining vasoconstriction, and maintaining structural integrity of the vasculature. VSMC specific PPARγ knockout mice were reported to exhibit impaired circadian rhythm of blood pressure in one study [35] and surprisingly hypotension in one another [36]. Inducible VSMC-specific deletion of PPARγ exaggerates vascular injury induced by either Ang-II [37] or endothelin-1 [38]. PPARγ also suppresses Ang-II signaling in VSMC by preventing Ang-II AT1 receptor transcription [16, 39, 40] and signal transduction [9, 41]. VSMC-specific deficient PPARγ mice also exhibited increased sensitivity to Ang-II induced vasoconstriction, hypertrophic remodeling, oxidative stress and inflammation [37].
Other studies suggest that PPARγ maintains vascular structural integrity by suppressing oxidative stress [38, 42], inflammation [38, 43], calcification [44] and remodeling [45, 46]. Intriguingly, VSMC PPARγ was reported to maintain perivascular adipose tissue (PVAT), which is known for its anticontractile effect [47]. There is an absence of PVAT in VSMC specific PPARγ knockout mice [48]. Interestingly, mice expressing constitutively active PPARγ selectively in VSMC exhibited decreased contractile activity but enhanced lipid deposition in the vascular media and extravascular tissues suggesting the level of PPARγ in VSMC needs to be tightly regulated to mediate all of its protective effects [49].
Like the mice selectively expressing dominant negative PPARγ selectively in the endothelium, we generated mice expressing the same hypertension-causing mutation PPARγ selectively in VSMC. These mice, termed S-P467L, developed HT, arterial stiffness and vascular dysfunction [50, **51]. When compared with endothelium where PPARγ acted to promote an antioxidant environment through its interaction with RBP7 and adiponectin, our data suggests the protective effects of PPARγ in VSMC are mediated by fundamentally different mechanisms (Figure 1).
Aorta from S-P467L mice exhibited increased vasoconstriction to endothelin-1 (ET-1), PGF2α and 5-HT. There was no change in vasoconstriction in mice selectively expressing wild type PPARγ [50]. Rho kinase activity was increased in aorta from these mice. Consistent with this, the ROCK inhibitor Y-27632 blunted the augmented vasoconstriction response to ET-1. Increased ROCK activity occurred concomitantly with increased RhoA protein, but not RhoA mRNA [52]. This suggested a possible defect in RhoA turnover. Cullin-3 RING ligase (CRL3) is a E3 ubiquitin ligase which promotes RhoA ubiquitination and degradation using BACURD as the substrate adaptor [53, 54]. Interestingly, protein levels of both activated and total Cullin-3 were decreased in aorta from S-P467L mice while Cullin-3 mRNA was not significantly changed [52]. Thus, our studies suggested a novel mechanistic link between PPARγ and the CRL3 complex.
Mesenteric arteries from S-P467L mice exhibited increased myogenic tone, which was accompanied by a decreased expression of regulator of G protein signaling 5 (RGS5), another PPARγ gene. RGS5 negatively regulates Ang-II AT1 receptor (AT1R) activation [55]. RGS5 knockdown increases the myogenic tone in mesenteric arteries and increases Ang-II induced PKC phosphorylation [56]. Thus, PPARγ mechanistically regulates vasoconstriction and myogenic tone through its effects on both Cullin-3 and RGS5, respectively.
The other distinctly novel mechanism of vascular protection was uncovered by studies of the NO/cGMP pathway in S-P467L mice. Aorta from these mice exhibited impaired vasodilation to ACh, SNP and cGMP analogue 8-Br-cGMP [50]. Similar impairment was also observed in cerebral arteries from S-P467L mice. The impaired SNP and cGMP response were surprising as it is atypical in hypertension. Mechanistic studies lead to the identification of another novel PPARγ target gene RhoBTB1.
3. RhoBTB1
3.1. RhoBTB1 Structure
RhoBTB1, along with the homologous family members RhoBTB2 and RhoBTB3, were first identified by Rivero et al in 2001 as a new subfamily of Rho GTPases [57]. RhoBTB1 is composed of a GTPase domain, a Proline-rich domain, two BTB (bric a brac, tramtrack and broad complex) domains and a C-terminal domain (Figure 2A). Canonical Rho GTPases, such as RhoA and CDC42, are in active conformation upon GTP binding and become inactive when the GTPase domain hydrolyze GTP into GDP. However, the GTPase domain of RhoBTB1 contains several sequence changes from the consensus Rho GTPase sequence. It was initially reported that RhoBTB2 does not bind to GTP, and since RhoBTB1 is 80% similar to RhoBTB2, it was assumed that RhoBTB1 also does not function as a Rho GTPase [58]. However, it was later reported that RhoBTB2 can bind to GTP [59]. It is unclear if RhoBTB1 exhibits GTPase activity in VSMC. Distal to the GTPase domain is a Proline-rich domain which features a PxxP motif (x indicates any amino acid). These motifs frequently interact with the Src homology 3 domain (SH3 domain) containing proteins involved in signal transduction and cytoskeletal organization [60]. Further investigations are needed to understand the role of this domain in RhoBTB1.
Figure 2. The structure and molecular function of RhoBTB1.
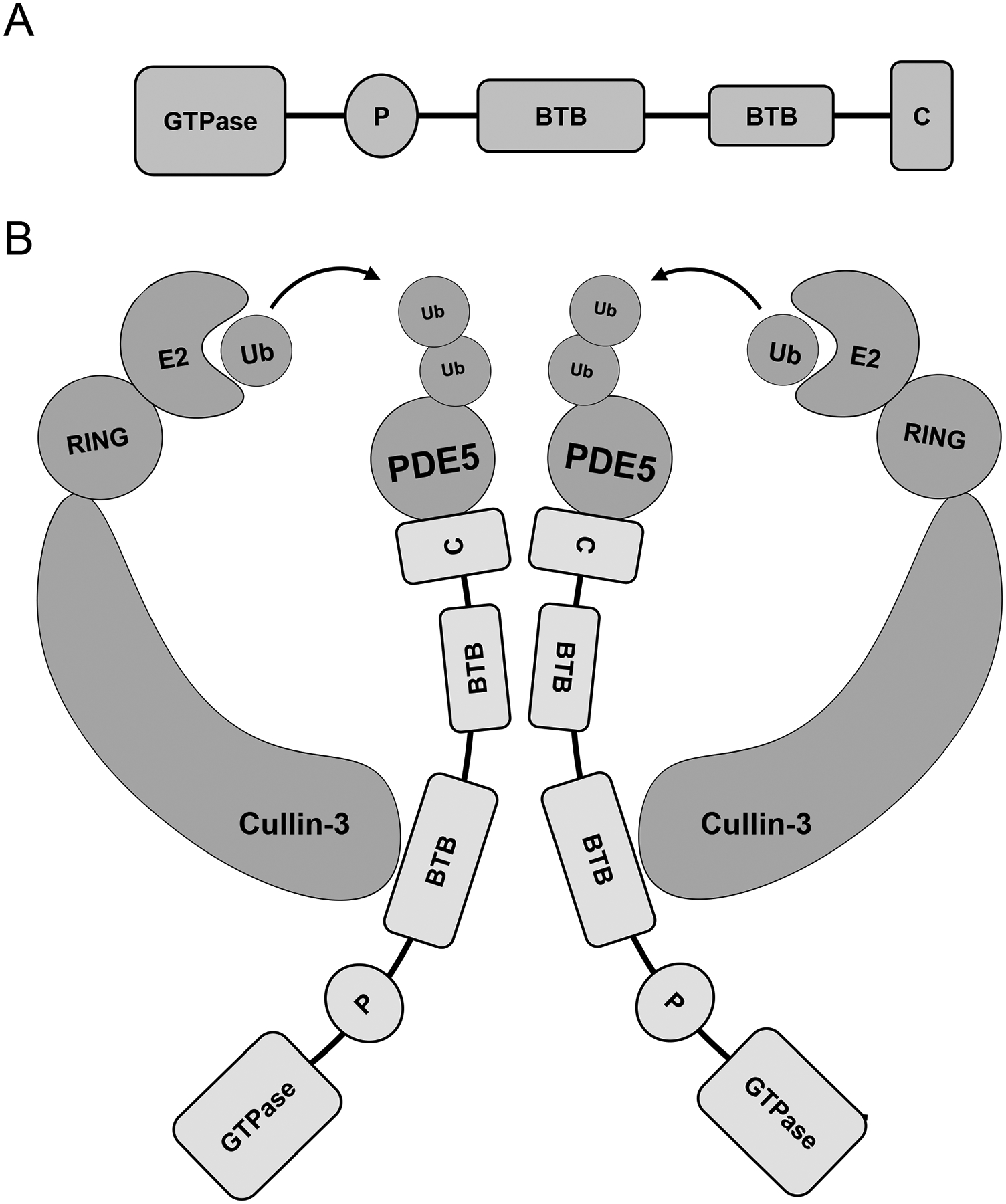
A) RhoBTB1 is composed of a GTPase domain, a Proline-rich domain, two BTB domains and a C-terminal. B) RhoBTB1 serves as a substrate adaptor for Cullin-3-RING ubiquitin ligase and promotes PDE5 ubiquitination. The BTB domains are involved in the RhoBTB1:Cullin-3 interaction as well as the formation of the dimeric complex required to mediate its activity. PDE5 presumably interacts with the C terminal of RhoBTB1 but this has not been tested experimentally. The formation of the productive complex results in transfer of ubiquitin from the E2 enzyme to PDE5 which targets PDE5 for proteasonal degradation.
The presence of two BTB (bric a brac, tramtrack and broad complex) domains are a unique feature of the RhoBTB proteins that separates them from other Rho GTPases [57]. BTB domains are known to mediate protein-protein interactions and are the signature of adaptor proteins which target protein substrates for ubiquitination through Cullin-3 E3 RING ubiquitin ligase (CRL3) [61]. The presence of the BTB domain was perhaps the most informative piece of information in hypothesizing a function for RhoBTB1. Indeed, RhoBTB1 interacts with the N-terminal of Cullin-3 through its first BTB domain [62]. As a result, it was hypothesized that all three RhoBTB proteins serve as substrate adaptors for proteins destined for ubiquitination by CRL3 complex (discussed below).
The function of the C-terminal domain of RhoBTB1 remains poorly understood. It was suggested that the C-terminal domain resembles a motif called a 3-box, which is similar to the F-box in the Skp1-Cullin-1-F-box (SCF) E3 ubiquitin ligase complex [63]. In the SCF model, Skp1 binds to the N-terminal of Cullin-1, and the F-box serves as a bridge between protein substrates and Skp1. Interestingly, the BTB domain shares structural homology with Skp1 [64]. Thus, RhoBTB1 in CRL3 might serve the functions of both Skp1 and F-box in SCF, using the first BTB domain to bind Cullin-3 and the C-terminal to interact with the substrate (Figure 2B).
3.2. RhoBTB1 Expression and Regulation
The expression patterns of RhoBTB1 in human and mouse are similar: RhoBTB1 is highly expressed in reproduction system (placenta, testis, uterus) and muscular tissues (skeletal muscle, heart, stomach, kidney) [62]. Later our lab also showed that RhoBTB1 is abundant in the vasculature [52].
We previously determined that RhoBTB1 is a PPARγ target gene regulated at the transcriptional level [52]. The evidence for this is four-fold. First, RhoBTB1 mRNA and protein was decreased in the aorta from S-P467L mice. Second, expression of RhoBTB1 protein was increased in aorta from transgenic mice overexpressing wild-type PPARγ in smooth muscle. Third, PPARγ and retinoid X receptor (RXR) binding sites were detected at the RhoBTB1 locus by genome-wide chromatin immunoprecipitation. Fourth, PPARγ and RXR bind to the RhoBTB1 locus using chromatin immunoprecipitation in differentiated 3T3–L1 cells which endogenous express PPARγ.
MicroRNAs (miRNAs) are a class of small endogenous non-coding RNAs. By binding to the 3’ untranslated region (UTR) of a target mRNA, miRNAs can induce either mRNA turnover or translation repression. Studies show that RhoBTB1 can be regulated by miR-31 in human colon cancer HT29 cells, cutaneous squamous cell carcinoma A-431 cells, and HEK293 cells [65–67]. Similarly, miR-31a-5p was reported to target RhoBTB1 in neonatal rat ventricular cardiomyocyte to regulate proliferation [67].
3.3. RhoBTB1 May be a Target of CRL3-mediated Turnover
There is data suggesting that RhoBTB1 might itself be a target of Cullin-3-mediated ubiquitination and degradation [53]. This is based on the observation that inhibition of Cullin-3 either by inhibiting nedd-8-ylation or dominant negative Cullin-3 mutation caused an increase in RhoBTB1 protein, whereas Cullin-3 overexpression caused a decreased RhoBTB1 protein. Similarly, Keap1, another Cullin-3 substrate adaptor for NRF2, along with three other BTB-Kelch proteins (GAN1, ENC1, and Sarcosin) were reported to be ubiquitinated by CRL3 in an in vitro ubiquitination assay [68]. Thus, in addition to mechanisms which regulate RhoBTB1 at the transcriptional or posttranscriptional level, it may also be regulated at the posttranslational level.
3.4. RhoBTB1 in Hypertension
We identified RhoBTB1 as a target gene of the nuclear receptor transcription factor PPARγ by transcriptome analysis of aorta of S-P467L mice [50, 52]. RhoBTB1 expression was repressed by dominant negative PPARγ. Recall S-P467L mice exhibit hypertension, vascular dysfunction and arterial stiffness. The hypothesis that RhoBTB1 may be an important regulator of BP was strengthened by human genetic studies reporting that RhoBTB1 was identified among several loci for BP in a GWAS of nearly 35,000 individuals, and more recently, as an interacting locus for HT in GWAS of 1 million subjects [69, 70].
It is notable that RhoBTB1 was only one of over two-hundred genes whose expression was altered by PPARγ mutation. Thus, we devised a strategy to assess the functional significance of RhoBTB1. To accomplish this and test the hypothesis that the hypertension, vascular dysfunction and arterial stiffness observed in S-P467L mice was a result of RhoBTB1-deficiency, we generated transgenic mice expressing VSMC-specific, tamoxifen inducible RhoBTB1 [51]. The goal was to inducibly restore RhoBTB1 level in a PPARγ-independent manner. We reported that restoration of RhoBTB1 in VSMC by this genetic complementation schema reversed HT and vascular dysfunction within 2 weeks of its restored expression. The arterial stiffness began to regress during the first week and was completely reversed after 3 weeks [51].
Our series of experiments showed that restoration of RhoBTB1 improved the vasodilator response to both acetylcholine, an endothelial-dependent vasodilator, and sodium nitroprusside, an endothelium-independent vasodilator, both of which activate the cGMP pathway in VSMC. A further series of pharmacological studies suggested that RhoBTB1 regulates the level of cGMP in VSMC. Specifically, we first showed that production of cGMP was blunted in isolated aorta from S-P467L mice which was normalized upon RhoBTB1 restoration. This initially suggested a defect in cGMP production, but the levels of soluble guanylate cyclase subunits, the enzyme responsible for production of cGMP, was normal. Thus, we turned our attention to cGMP stability and turnover. PDE5 regulates the NO/cGMP pathway by enzymatically degrading cGMP into GMP. Indeed, PDE activity was increased in aorta from S-P467L mice, which was normalized by RhoBTB1 restoration. Consistent with this, vasodilation of aorta from S-P467L to cGMP analogs that are sensitive to PDE5 was impaired whereas the response to a cGMP analog which is resistant to PDE5-mediated degradation was normal. Solidifying the important of PDE5 in S-P467 mice, the PDE5-specific inhibitor Tadalafil lowered blood pressure and mimicked the protective effect of RhoBTB1 restoration in S-P467L mice. These data suggested that RhoBTB1 exerts its protective role in VSMC by restraining the activity of phosphodiesterase 5 (PDE5).
How does RhoBTB1 restrain PDE5 activity? The clue comes from the hypothesis that BTB-domain containing proteins function as substrate adaptors of Cullin-3 RING E3 ubiquitin ligase. RhoBTB1 is no exception as it was reported to be co-immunoprecipitated with Cullin-3 in transfected COS7 and HEK293 cells [53, 71]. We found that RhoBTB1 was reciprocally immunoprecipitated with PDE5, and endogenous Cullin-3 was also present in the co-precipitate [51]. Moreover, RhoBTB1 promoted PDE5 ubiquitination in the presence of Cullin-3, which was blunted upon the treatment with an inhibitor (MLN4924) of neddylation, a requirement for Cullin-3 activation. Consistent with this, blocking Cullin activity with MLN4924 increased PDE5 activity in aorta from normal C57BL/6J mice. This suggests that RhoBTB1 mediates PDE5 ubiquitination in a Cullin-3-dependent manner and represents the first direct evidence of a role for RhoBTB1 in ubiquitination. We also demonstrated that RhoBTB1 was insufficient to inhibit PDE5 activity independently of Cullin-3 in vitro further supporting the hypothesis that RhoBTB1 is a substrate adaptor for Cullin-3 RING ubiquitin ligase (Figure 2B). A model illustrating the PPARγ-RhoBTB1-Cullin-3-PDE5 pathway is shown in Figure 1.
Notably, whereas the aorta from S-P467L mice exhibited both enhanced vasoconstriction and impaired vasodilation, RhoBTB1 restoration only normalized the vasodilation response. Enhanced contractile activity was preserved in S-P467L mice even when RhoBTB1 expression was restored. We previously demonstrated that S-P467L mice exhibit increased RhoA and Rho kinase activity. Interestingly, RhoA is also a substrate of Cullin-3; but its substrate adaptor is not RhoBTB1 but another BTB-domain containing adaptor called BACURD1 [54]. Thus, RhoA protein and RhoA activity remains elevated even in the presence of RhoBTB1. This data is conceptually interesting as it implies that the hypertension and arterial stiffness in S-P467L mice is associated with the vasodilator pathway as they were both corrected despite preservation of enhanced vasoconstrictor responses.
The protective role of RhoBTB1 is also applicable to other forms of HT. Ang-II infusion decreases RhoBTB1 mRNA in aorta [51]. Transgenic mice expressing RhoBTB1 selectively in VSMC partially prevented the pressor effects of Ang-II. VSMC RhoBTB1 also prevented impaired vasodilator function normally caused by Ang-II. But, like S-P467L mice, RhoBTB1 did not prevent increased contraction of the aorta to some contractile agonists in Ang-II treated mice.
3.5. RhoBTB1 in Other Diseases
RhoBTB1 first gained attention as a potential tumor suppressor gene [72]. The level of expression of RhoBTB1 was found to be decreased in human head and neck cancer, colon cancer, breast, kidney and stomach tumor samples [71, 72]. RhoBTB1 knockdown in human colon cancer HT29 cell line increased proliferation and colony formation [65]. Similarly, RhoBTB1 knockdown in the epidermoid carcinoma A-431 cell line exhibited increased proliferation and invasion in a trans-well assay [66].
RhoBTB1 knockdown in HeLa cells resulted in fragmentation in the Golgi apparatus and a significant decrease of methyltransferase like 7B (METTL7B), a Golgi-associated methyltransferase, suggesting RhoBTB1 might be required to maintain integrity of the Golgi apparatus [73]. A comparison of three human breast cancer cell lines (MDA-MB-231, T47D, MCF7) to the normal human mammary epithelial cell line MCF10A found that T47D not only exhibited marked Golgi fragmentation but also the lowest level of RhoBTB1 and METTL7B. Lentiviral restoration of RhoBTB1 increased RhoBTB1 (but not to normal) levels and concomitantly improved Golgi integrity and reduced invasion into 3D matrix gel. It remains unclear if the effects of RhoBTB1-deficiency in cancer and Golgi fragmentation are the result of its role as a Cullin-3 adaptor or another undiscovered role.
Other studies reported an association between RhoBTB1 mRNA and/or protein in human diseases. For example, RhoBTB1 mRNA was decreased by osteoprotegerin in osteoclasts in a model of bone resorption [74], and in cells from peripheral blood of Turkish population with metabolic syndrome [75]. RhoBTB1 protein was decreased in esophagus and tongue from rats with zinc-deficiency [76]. On the contrary, RhoBTB1 mRNA was upregulated in the cumulus cells from in vitro matured oocytes that successfully developed to blastocyst stage compared to those failed [77], and in peripheral blood mononuclear cells of patients with locoregional breast cancer who develop chemotherapy-induced fatigue [78]. As above, it remains unclear if these effects are being mediated by its role as a Cullin-3 adaptor protein. Given the range of phenotypes associated with RhoBTB1-deficiency or over-expression, it may be predicted that RhoBTB1 regulates a fundamental cellular process that is common to all cells, or alternatively has many substrates and that each condition is due to changes in the way RhoBTB1 interacts with and regulates the level of those substrates.
Conclusion
In the vasculature, the endothelium communicates with VSMC through the release of many substances including vasoconstrictors (such as ET-1), vasodilators (such as NO), inflammatory mediators (such as cytokines), and in response to stress, reactive oxygen species (ROS). PPARγ plays a protective role not only by increasing the bioavailability of NO and reducing ROS derived from the endothelium (through the PPARγ-RBP7-adiponectin pathway), but also by sensitizing the vascular muscle to the effects of NO (through RhoBTB1) and minimizing the effects of vasoconstrictors (by regulating RhoA and Rho kinase activity). Identifying these and other downstream targets of PPARγ may help identify novel pathways that can lead to the discovery of novel anti-hypertension targets that would preserve the most beneficial effects of PPARγ activation while circumventing the adverse effects of TZD-mediated PPARγ activation. Moreover, the potent anti-hypertensive effect of RhoBTB1 suggest the importance of dilatory pathway in alleviating end organ damage such as arterial stiffness. Future studies are needed to understand RhoBTB1 function in other organs and other diseases.
Key points:
PPARγ plays a protective role in hypertension.
PPARγ suppresses oxidative stress in vascular endothelial cells while promoting vasodilation, and suppresses vasoconstriction and maintains vascular integrity in vascular smooth muscle cells.
RBP7 is a novel transcription target of PPARγ in vascular endothelial cells, which forms a positive feedback with PPARγ.
RhoBTB1 is a novel transcription target of PPARγ in vascular smooth muscle cells, which promotes vasodilatory NO/cGMP pathway by restraining PDE5 activity.
RhoBTB1 serves as a substrate adaptor for Cullin-3-RING ligase and supports PDE5 ubiquitination.
Financial Support and sponsorship
This work was supported through research grants from the National Institutes of Health (HL084207, HL144807) and American Heart Association (15SFRN23480000).
Footnotes
Conflicts of interest
None
References
- 1.Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19(5):557–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lefterova MI, Haakonsson AK, Lazar MA, Mandrup S. PPARgamma and the global map of adipogenesis and beyond. Trends Endocrinol Metab. 2014;25(6):293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10(4):355–61. [DOI] [PubMed] [Google Scholar]
- 4.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem. 1995;270(22):12953–6. [DOI] [PubMed] [Google Scholar]
- 5.Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366(9493):1279–89. [DOI] [PubMed] [Google Scholar]
- 6.Lonn EM, Gerstein HC, Sheridan P, Smith S, Diaz R, Mohan V, et al. Effect of ramipril and of rosiglitazone on carotid intima-media thickness in people with impaired glucose tolerance or impaired fasting glucose: STARR (STudy of Atherosclerosis with Ramipril and Rosiglitazone). J Am Coll Cardiol. 2009;53(22):2028–35. [DOI] [PubMed] [Google Scholar]
- 7.Walker AB, Chattington PD, Buckingham RE, Williams G. The thiazolidinedione rosiglitazone (BRL-49653) lowers blood pressure and protects against impairment of endothelial function in Zucker fatty rats. Diabetes. 1999;48(7):1448–53. [DOI] [PubMed] [Google Scholar]
- 8.Iglarz M, Touyz RM, Amiri F, Lavoie MF, Diep QN, Schiffrin EL. Effect of peroxisome proliferator-activated receptor-alpha and -gamma activators on vascular remodeling in endothelin-dependent hypertension. Arterioscler Thromb Vasc Biol. 2003;23(1):45–51. [DOI] [PubMed] [Google Scholar]
- 9.Benkirane K, Viel EC, Amiri F, Schiffrin EL. Peroxisome proliferator-activated receptor gamma regulates angiotensin II-stimulated phosphatidylinositol 3-kinase and mitogen-activated protein kinase in blood vessels in vivo. Hypertension. 2006;47(1):102–8. [DOI] [PubMed] [Google Scholar]
- 10.Diep QN, El Mabrouk M, Cohn JS, Endemann D, Amiri F, Virdis A, et al. Structure, endothelial function, cell growth, and inflammation in blood vessels of angiotensin II-infused rats: role of peroxisome proliferator-activated receptor-gamma. Circulation. 2002;105(19):2296–302. [DOI] [PubMed] [Google Scholar]
- 11.Ryan MJ, Didion SP, Mathur S, Faraci FM, Sigmund CD. PPAR(gamma) agonist rosiglitazone improves vascular function and lowers blood pressure in hypertensive transgenic mice. Hypertension. 2004;43(3):661–6. [DOI] [PubMed] [Google Scholar]
- 12.Beyer AM, Baumbach GL, Halabi CM, Modrick ML, Lynch CM, Gerhold TD, et al. Interference with PPARgamma signaling causes cerebral vascular dysfunction, hypertrophy, and remodeling. Hypertension. 2008;51(4):867–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Modrick ML, Kinzenbaw DA, Chu Y, Sigmund CD, Faraci FM. Peroxisome proliferator-activated receptor-gamma protects against vascular aging. Am J Physiol Regul Integr Comp Physiol. 2012;302(10):R1184–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsai YS, Kim HJ, Takahashi N, Kim HS, Hagaman JR, Kim JK, et al. Hypertension and abnormal fat distribution but not insulin resistance in mice with P465L PPARgamma. J Clin Invest. 2004;114(2):240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402(6764):880–3. [DOI] [PubMed] [Google Scholar]
- 16.Auclair M, Vigouroux C, Boccara F, Capel E, Vigeral C, Guerci B, et al. Peroxisome proliferator-activated receptor-gamma mutations responsible for lipodystrophy with severe hypertension activate the cellular renin-angiotensin system. Arterioscler Thromb Vasc Biol. 2013;33(4):829–38. [DOI] [PubMed] [Google Scholar]
- 17.Ketsawatsomkron P, Pelham CJ, Groh S, Keen HL, Faraci FM, Sigmund CD. Does peroxisome proliferator-activated receptor-gamma (PPAR gamma) protect from hypertension directly through effects in the vasculature? J Biol Chem. 2010;285(13):9311–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sigmund CD. Endothelial and vascular muscle PPARgamma in arterial pressure regulation: lessons from genetic interference and deficiency. Hypertension. 2010;55(2):437–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calnek DS, Mazzella L, Roser S, Roman J, Hart CM. Peroxisome proliferator-activated receptor gamma ligands increase release of nitric oxide from endothelial cells. Arterioscler Thromb Vasc Biol. 2003;23(1):52–7. [DOI] [PubMed] [Google Scholar]
- 20.Nicol CJ, Adachi M, Akiyama TE, Gonzalez FJ. PPARgamma in endothelial cells influences high fat diet-induced hypertension. Am J Hypertens. 2005;18(4 Pt 1):549–56. [DOI] [PubMed] [Google Scholar]
- 21.Kleinhenz JM, Kleinhenz DJ, You S, Ritzenthaler JD, Hansen JM, Archer DR, et al. Disruption of endothelial peroxisome proliferator-activated receptor-gamma reduces vascular nitric oxide production. Am J Physiol Heart Circ Physiol. 2009;297(5):H1647–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beyer AM, de Lange WJ, Halabi CM, Modrick ML, Keen HL, Faraci FM, et al. Endothelium-specific interference with peroxisome proliferator activated receptor gamma causes cerebral vascular dysfunction in response to a high-fat diet. Circ Res. 2008;103(6):654–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Silva TM, Hu C, Kinzenbaw DA, Modrick ML, Sigmund CD, Faraci FM. Genetic Interference With Endothelial PPAR-gamma (Peroxisome Proliferator-Activated Receptor-gamma) Augments Effects of Angiotensin II While Impairing Responses to Angiotensin 1–7. Hypertension. 2017;70(3):559–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **24.Nair AR, Agbor LN, Mukohda M, Liu X, Hu C, Wu J, et al. Interference With Endothelial PPAR (Peroxisome Proliferator-Activated Receptor)-gamma Causes Accelerated Cerebral Vascular Dysfunction in Response to Endogenous Renin-Angiotensin System Activation. Hypertension. 2018;72(5):1227–35. Mice with endothelial-specifc expression of dominant negative PPARγ mutation showed augmented cerebral vascular dysfunction induced by low salt diet.
- **25.Nair AR, Silva SD Jr., Agbor LN, Wu J, Nakagawa P, Mukohda M, et al. Endothelial PPARgamma (Peroxisome Proliferator-Activated Receptor-gamma) Protects From Angiotensin II-Induced Endothelial Dysfunction in Adult Offspring Born From Pregnancies Complicated by Hypertension. Hypertension. 2019;74(1):173–83. Male mice with endothelial specifc expression of dominant negative PPARγ mutation, when born from a female with pre-eclampsia, showed enhanced endothelial dysfunction mediated byAng-II.
- 26.Keen HL, Halabi CM, Beyer AM, de Lange WJ, Liu X, Maeda N, et al. Bioinformatic analysis of gene sets regulated by ligand-activated and dominant-negative peroxisome proliferator-activated receptor gamma in mouse aorta. Arterioscler Thromb Vasc Biol. 2010;30(3):518–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caprioli A, Zhu H, Sato TN. CRBP-III:lacZ expression pattern reveals a novel heterogeneity of vascular endothelial cells. Genesis. 2004;40(3):139–45. [DOI] [PubMed] [Google Scholar]
- 28.Zizola CF, Schwartz GJ, Vogel S. Cellular retinol-binding protein type III is a PPARgamma target gene and plays a role in lipid metabolism. Am J Physiol Endocrinol Metab. 2008;295(6):E1358–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vogel S, Mendelsohn CL, Mertz JR, Piantedosi R, Waldburger C, Gottesman ME, et al. Characterization of a new member of the fatty acid-binding protein family that binds all-trans-retinol. J Biol Chem. 2001;276(2):1353–60. [DOI] [PubMed] [Google Scholar]
- 30.Hughes ML, Liu B, Halls ML, Wagstaff KM, Patil R, Velkov T, et al. Fatty Acid-binding Proteins 1 and 2 Differentially Modulate the Activation of Peroxisome Proliferator-activated Receptor alpha in a Ligand-selective Manner. J Biol Chem. 2015;290(22):13895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu C, Keen HL, Lu KT, Liu X, Wu J, Davis DR, et al. Retinol-binding protein 7 is an endothelium-specific PPARgamma cofactor mediating an antioxidant response through adiponectin. JCI Insight. 2017;2(6):e91738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Komura N, Maeda N, Mori T, Kihara S, Nakatsuji H, Hirata A, et al. Adiponectin protein exists in aortic endothelial cells. PLoS One. 2013;8(8):e71271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao Y, Tao L, Yuan Y, Jiao X, Lau WB, Wang Y, et al. Endothelial dysfunction in adiponectin deficiency and its mechanisms involved. J Mol Cell Cardiol. 2009;46(3):413–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong WT, Tian XY, Xu A, Yu J, Lau CW, Hoo RL, et al. Adiponectin is required for PPARgamma-mediated improvement of endothelial function in diabetic mice. Cell Metab. 2011;14(1):104–15. [DOI] [PubMed] [Google Scholar]
- 35.Wang N, Yang G, Jia Z, Zhang H, Aoyagi T, Soodvilai S, et al. Vascular PPARgamma controls circadian variation in blood pressure and heart rate through Bmal1. Cell Metab. 2008;8(6):482–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang L, Villacorta L, Zhang J, Garcia-Barrio MT, Yang K, Hamblin M, et al. Vascular smooth muscle cell-selective peroxisome proliferator-activated receptor-gamma deletion leads to hypotension. Circulation. 2009;119(16):2161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marchesi C, Rehman A, Rautureau Y, Kasal DA, Briet M, Leibowitz A, et al. Protective role of vascular smooth muscle cell PPARgamma in angiotensin II-induced vascular disease. Cardiovasc Res. 2013;97(3):562–70. [DOI] [PubMed] [Google Scholar]
- 38.Idris-Khodja N, Ouerd S, Trindade M, Gornitsky J, Rehman A, Barhoumi T, et al. Vascular smooth muscle cell peroxisome proliferator-activated receptor gamma protects against endothelin-1-induced oxidative stress and inflammation. J Hypertens. 2017;35(7):1390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takeda K, Ichiki T, Tokunou T, Funakoshi Y, Iino N, Hirano K, et al. Peroxisome proliferator-activated receptor gamma activators downregulate angiotensin II type 1 receptor in vascular smooth muscle cells. Circulation. 2000;102(15):1834–9. [DOI] [PubMed] [Google Scholar]
- 40.Sugawara A, Takeuchi K, Uruno A, Ikeda Y, Arima S, Kudo M, et al. Transcriptional suppression of type 1 angiotensin II receptor gene expression by peroxisome proliferator-activated receptor-gamma in vascular smooth muscle cells. Endocrinology. 2001;142(7):3125–34. [DOI] [PubMed] [Google Scholar]
- 41.Carrillo-Sepulveda MA, Keen HL, Davis DR, Grobe JL, Sigmund CD. Role of vascular smooth muscle PPARgamma in regulating AT1 receptor signaling and angiotensin II-dependent hypertension. PLoS One. 2014;9(8):e103786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bijli KM, Kleinhenz JM, Murphy TC, Kang BY, Adesina SE, Sutliff RL, et al. Peroxisome proliferator-activated receptor gamma depletion stimulates Nox4 expression and human pulmonary artery smooth muscle cell proliferation. Free Radic Biol Med. 2015;80:111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mukohda M, Lu KT, Guo DF, Wu J, Keen HL, Liu X, et al. Hypertension-Causing Mutation in Peroxisome Proliferator-Activated Receptor gamma Impairs Nuclear Export of Nuclear Factor-kappaB p65 in Vascular Smooth Muscle. Hypertension. 2017;70(1):174–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng L, Zhang L, Yang J, Hao L. Activation of peroxisome proliferator-activated receptor gamma inhibits vascular calcification by upregulating Klotho. Exp Ther Med. 2017;13(2):467–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ketsawatsomkron P, Keen HL, Davis DR, Lu KT, Stump M, De Silva TM, et al. Protective Role for Tissue Inhibitor of Metalloproteinase-4, a Novel Peroxisome Proliferator-Activated Receptor-gamma Target Gene, in Smooth Muscle in Deoxycorticosterone Acetate-Salt Hypertension. Hypertension. 2016;67(1):214–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wakino S, Kintscher U, Kim S, Yin F, Hsueh WA, Law RE. Peroxisome proliferator-activated receptor gamma ligands inhibit retinoblastoma phosphorylation and G1--> S transition in vascular smooth muscle cells. J Biol Chem. 2000;275(29):22435–41. [DOI] [PubMed] [Google Scholar]
- 47.Huang Cao ZF, Stoffel E, Cohen P. Role of Perivascular Adipose Tissue in Vascular Physiology and Pathology. Hypertension. 2017;69(5):770–7. [DOI] [PubMed] [Google Scholar]
- 48.Chang L, Villacorta L, Li R, Hamblin M, Xu W, Dou C, et al. Loss of perivascular adipose tissue on peroxisome proliferator-activated receptor-gamma deletion in smooth muscle cells impairs intravascular thermoregulation and enhances atherosclerosis. Circulation. 2012;126(9):1067–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kleinhenz JM, Murphy TC, Pokutta-Paskaleva AP, Gleason RL, Lyle AN, Taylor WR, et al. Smooth Muscle-Targeted Overexpression of Peroxisome Proliferator Activated Receptor-gamma Disrupts Vascular Wall Structure and Function. PLoS One. 2015;10(10):e0139756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Halabi CM, Beyer AM, de Lange WJ, Keen HL, Baumbach GL, Faraci FM, et al. Interference with PPAR gamma function in smooth muscle causes vascular dysfunction and hypertension. Cell Metab. 2008;7(3):215–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **51.Mukohda M, Fang S, Wu J, Agbor LN, Nair AR, Ibeawuchi SC, et al. RhoBTB1 protects against hypertension and arterial stiffness by restraining phosphodiesterase 5 activity. J Clin Invest. 2019;129(6):2318–32. RhoBTB1 plays a protective role in vascular smooth muscle cells. RhoBTB1 controls PDE5 activity by enabling PDE5 ubiquitination.
- 52.Pelham CJ, Ketsawatsomkron P, Groh S, Grobe JL, de Lange WJ, Ibeawuchi SR, et al. Cullin-3 regulates vascular smooth muscle function and arterial blood pressure via PPARgamma and RhoA/Rho-kinase. Cell Metab. 2012;16(4):462–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ibeawuchi SR, Agbor LN, Quelle FW, Sigmund CD. Hypertension-causing Mutations in Cullin3 Protein Impair RhoA Protein Ubiquitination and Augment the Association with Substrate Adaptors. J Biol Chem. 2015;290(31):19208–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen Y, Yang Z, Meng M, Zhao Y, Dong N, Yan H, et al. Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol Cell. 2009;35(6):841–55. [DOI] [PubMed] [Google Scholar]
- 55.Holobotovskyy V, Manzur M, Tare M, Burchell J, Bolitho E, Viola H, et al. Regulator of G-protein signaling 5 controls blood pressure homeostasis and vessel wall remodeling. Circ Res. 2013;112(5):781–91. [DOI] [PubMed] [Google Scholar]
- 56.Ketsawatsomkron P, Lorca RA, Keen HL, Weatherford ET, Liu X, Pelham CJ, et al. PPARgamma regulates resistance vessel tone through a mechanism involving RGS5-mediated control of protein kinase C and BKCa channel activity. Circ Res. 2012;111(11):1446–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rivero F, Dislich H, Glockner G, Noegel AA. The Dictyostelium discoideum family of Rho-related proteins. Nucleic Acids Res. 2001;29(5):1068–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang FK, Sato N, Kobayashi-Simorowski N, Yoshihara T, Meth JL, Hamaguchi M. DBC2 is essential for transporting vesicular stomatitis virus glycoprotein. J Mol Biol. 2006;364(3):302–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Manjarrez JR, Sun L, Prince T, Matts RL. Hsp90-dependent assembly of the DBC2/RhoBTB2-Cullin3 E3-ligase complex. PLoS One. 2014;9(3):e90054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kay BK, Williamson MP, Sudol M. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14(2):231–41. [PubMed] [Google Scholar]
- 61.Pintard L, Willems A, Peter M. Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. EMBO J. 2004;23(8):1681–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramos S, Khademi F, Somesh BP, Rivero F. Genomic organization and expression profile of the small GTPases of the RhoBTB family in human and mouse. Gene. 2002;298(2):147–57. [DOI] [PubMed] [Google Scholar]
- 63.Ji W, Rivero F. Atypical Rho GTPases of the RhoBTB Subfamily: Roles in Vesicle Trafficking and Tumorigenesis. Cells. 2016;5(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stogios PJ, Prive GG. The BACK domain in BTB-kelch proteins. Trends Biochem Sci. 2004;29(12):634–7. [DOI] [PubMed] [Google Scholar]
- 65.Xu RS, Wu XD, Zhang SQ, Li CF, Yang L, Li DD, et al. The tumor suppressor gene RhoBTB1 is a novel target of miR-31 in human colon cancer. Int J Oncol. 2013;42(2):676–82. [DOI] [PubMed] [Google Scholar]
- 66.Lin N, Zhou Y, Lian X, Tu Y. MicroRNA-31 functions as an oncogenic microRNA in cutaneous squamous cell carcinoma cells by targeting RhoTBT1. Oncol Lett. 2017;13(3):1078–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xiao J, Liu H, Cretoiu D, Toader DO, Suciu N, Shi J, et al. miR-31a-5p promotes postnatal cardiomyocyte proliferation by targeting RhoBTB1. Exp Mol Med. 2017;49(10):e386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang DD, Lo SC, Sun Z, Habib GM, Lieberman MW, Hannink M. Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J Biol Chem. 2005;280(34):30091–9. [DOI] [PubMed] [Google Scholar]
- 69.Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41(6):666–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50(10):1412–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Berthold J, Schenkova K, Ramos S, Miura Y, Furukawa M, Aspenstrom P, et al. Characterization of RhoBTB-dependent Cul3 ubiquitin ligase complexes--evidence for an autoregulatory mechanism. Exp Cell Res. 2008;314(19):3453–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beder LB, Gunduz M, Ouchida M, Gunduz E, Sakai A, Fukushima K, et al. Identification of a candidate tumor suppressor gene RHOBTB1 located at a novel allelic loss region 10q21 in head and neck cancer. J Cancer Res Clin Oncol. 2006;132(1):19–27. [DOI] [PubMed] [Google Scholar]
- 73.McKinnon CM, Mellor H. The tumor suppressor RhoBTB1 controls Golgi integrity and breast cancer cell invasion through METTL7B. BMC Cancer. 2017;17(1):145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Song R, Gu J, Liu X, Zhu J, Wang Q, Gao Q, et al. Inhibition of osteoclast bone resorption activity through osteoprotegerin-induced damage of the sealing zone. Int J Mol Med. 2014;34(3):856–62. [DOI] [PubMed] [Google Scholar]
- 75.Tabur S, Oztuzcu S, Oguz E, Korkmaz H, Eroglu S, Ozkaya M, et al. Association of Rho/Rho-kinase gene polymorphisms and expressions with obesity-related metabolic syndrome. Eur Rev Med Pharmacol Sci. 2015;19(9):1680–8. [PubMed] [Google Scholar]
- 76.Alder H, Taccioli C, Chen H, Jiang Y, Smalley KJ, Fadda P, et al. Dysregulation of miR-31 and miR-21 induced by zinc deficiency promotes esophageal cancer. Carcinogenesis. 2012;33(9):1736–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu Y, Zhou T, Shao L, Zhang B, Liu K, Gao C, et al. Gene expression profiles in mouse cumulus cells derived from in vitro matured oocytes with and without blastocyst formation. Gene Expr Patterns. 2017;25–26:46–58. [DOI] [PubMed] [Google Scholar]
- 78.de Alcantara BBR, Cruz FM, Fonseca FLA, da Costa Aguiar Alves B, Perez MM, Varela P, et al. Chemotherapy-induced fatigue is associated with changes in gene expression in the peripheral blood mononuclear cell fraction of patients with locoregional breast cancer. Support Care Cancer. 2019;27(7):2479–86. [DOI] [PubMed] [Google Scholar]