

Abstract
Abstract
New series of Schiff’s bases, hydrazones, thiosemicarbazones, thiazoles, and thiocarbohydrazones of 5-fluoroisatin were synthesized by the reaction of 5-fluoroisatin with primary amines, hydrazine hydrate, and thiocarbohydrazides. Thiosemicarbazones were prepared by reacting hydrazone derivatives with isothiocyanates. Upon treatment of thiosemicarbazone derivatives with chloroacetone, the thiazole derivatives were obtained. Some of the prepared compounds exhibited antiviral activity.
Graphical abstract
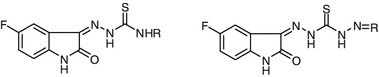
Keywords: Schiff bases, Carbonyl compounds, Thiosemicarbazones, Thiazoles
Introduction
The antiviral activities of isatin derivatives have been widely reported, especially their action against pox virus [1], vaccinia [2], rhino virus [3], Moloney leukemia virus [4], and SARS virus [5]. Previous works have also reported the inhibitory activity of isatin-thiosemicarbazones and isatin derivatives on HIV replication [6–8]. In addition, methisazone (an isatin-thiosemicarbazone) plays an important role as a prophylactic agent against several viral diseases [9]. There has been increasing interest in recent years concerning the introduction of fluorine into organic compounds [10, 11]. Incorporation of one or several fluorine atoms into an organic molecule can improve the pharmacokinetic and pharmacodynamic properties such as absorption, tissue distribution, secretion, the route and rate of biotransformation, toxicology, bioavailability, metabolic stability, and lipophilicity [12, 13]. Many of the compounds containing a fluorine atom have a significant therapeutic impact. Fluorinated isatin derivatives have become a focus in the development of new biologically active compounds [14].
In view of these observations and as a continuation of our effort in developing broad-spectrum chemotherapeutics [15–18], we report the preparation of some newer fluorinated iminoisatin derivatives in the hope of obtaining better antiviral drugs without side effects.
Results and discussion
Chemistry
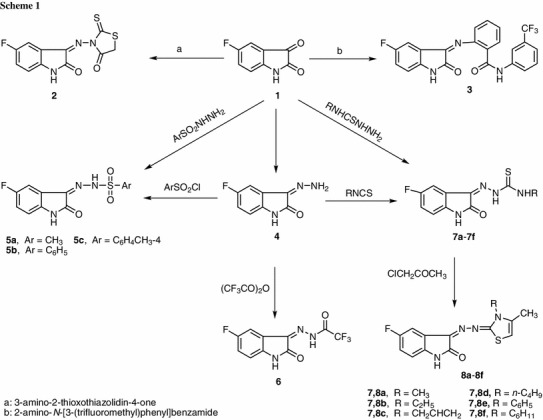
In the present investigation, the reactivity of 5-fluoroisatin (1) towards some different types of nitrogen nucleophile was studied with the objective of obtaining biologically active compounds. Thus, reaction of 5-fluoroisatin with 3-amino-2-thioxothiazolidin-4-one afforded the corresponding Schiff’s base 2. Reaction of 1 with a fluorinated aromatic amine, 2-amino-N-[3-(trifluoromethyl)phenyl]benzamide, afforded the corresponding Schiff’s base 3. The spectral data of the isolated products were in complete agreement with their structures. The aromatic signals were observed in all cases as multiplets for the 5-fluorisatin protons owing to F–H and H–H couplings. The structures of 2 and 3 were assigned the basis of analytical and spectral data. The IR spectrum of 2 displayed absorption bands at ν = 3,238 (NH), 1,700, 1,668 (C=O, amidic C=O) cm−1. Its 1H NMR spectrum exhibited a sharp singlet signal at δ = 5.80 ppm assigned to CH2 protons, three signals at 6.87, 8.60, and 7.30 ppm owing to indole protons, and a D2O-exchangeable singlet signal at 11.40 ppm due to NH proton.
Hydrazone derivative 4 was prepared according to a literature procedure by treatment of 5-fluoroisatin (1) with hydrazine hydrate in ethanol [19].
It was of interest to synthesize some new sulfonamide derivatives. Thus, sulfonamide derivatives 5a–5c were prepared by heating the hydrazone 4 with sulfonyl chloride derivatives (namely methanesulfonyl chloride, benzenesulfonyl chloride, or 4-methylbenzenesulfonyl chloride) in dioxane containing a catalytic amount of triethylamine. Sulfonamide derivative 5c was prepared previously by the reaction of 5-fluoroisatin with tolylsulfonylhydrazine and reported in a patent [20]. The IR spectrum of 5a revealed absorption bands at 3,178 and 3,106 cm−1 due to two NH groups, and a band near 1,660 cm−1 that was attributed to the carbonyl group. Its 1H NMR spectrum revealed singlet signals at 2.83 ppm due to CH3 protons beside two singlet signals in the region of 10.68 and 11.80 ppm due to two NH groups. The mass spectroscopic measurements of 7c showed m/z = 333 (M+, 5.8) which indicates structure 5.
Interaction of the hydrazone derivative 4 with trifluoroacetic anhydride afforded the corresponding hydrazone derivative 6. The spectral data of the isolated product was in complete agreement with its structure. The IR spectrum of 6 revealed absorption bands at 3,255 and 3,107 cm−1 (NH) and 1,696 and 1,661 cm−1 (C=O).
In this study, the reactivity of hydrazone 4 towards isothiocyanate derivatives was investigated. Thus, when hydrazone 4 was left to react with different isothiocyanate derivatives in ethanol, the corresponding thiosemicarbazone derivatives 7a–7f were obtained. Thiosemicarbazone derivatives 7a–7f were prepared previously by treatment of 5-fluoroisatin with different N-substituted thiosemicarbazide derivatives [21]. We have extended our synthetic program to utilize structures 7a–7f as the key starting materials for alkylation with chloroacetone in ethanol containing fused sodium acetate to furnish the corresponding thiazole derivatives 8a–8f. The IR spectrum of compound 8a revealed absorption bands at 3,212 and 1,675 cm−1 characteristic for NH and C=O groups, respectively. Its 1H NMR spectrum revealed signals at 1.76 and 2.76 ppm due to two methyl groups and at 5.85 ppm due to the CH proton. Moreover the mass spectrum of compound 8c showed the molecular ion peak at 332 (M+, 84.2 %) (Scheme 1).
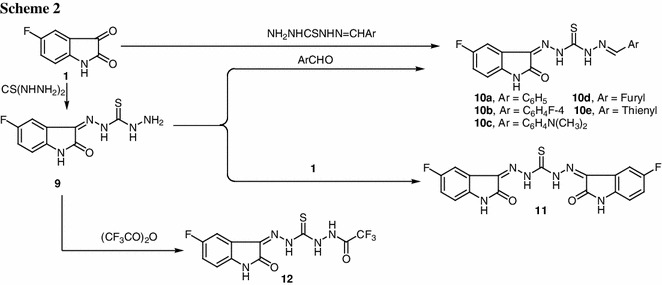
In continuation with our work in this area, we decided to prepare and evaluate the biological activity of some new isatin thiocarbohydrazone derivatives. When the 5-fluoroisatin was reacted with thiocarbohydrazide, 5-fluoroisatin thiocarbohydrazone (9) was obtained. The IR spectrum showed absorption bands at 3,270, 3,160, and 3,106 cm−1, corresponding to NH2 and NH functions. The 1H NMR spectrum revealed D2O-exchangeable signals at 2.51, 4.92, 10.40, and 12.54 ppm assignable to NH2 and NH protons. Thiocarbohydrazone 9 was used as a starting material in the synthesis of different types of isatin thiocarbohydrazone derivatives. Thus, when compound 9 was condensed with some selected aldehydes, the corresponding thiocarbohydrazones 10a–10e were obtained in good yield. The structure of 10 was established on the basis of its spectral data beside its independent synthesis via the reaction of N-thiocarbohydrazones with 5-fluoroisatin which afforded products identical in all respects with those obtained previously.
When 9 was condensed with 5-fluoroisatin, the corresponding bis-isatin thiocarbohydrazone 11 was obtained in quantitative yield. Finally, trifluoroacyl derivative 12 was prepared by the reaction of thiocarbohydrazone derivative 9 with trifluoroacetic anhydride. The structure of 11 was based of analytical and spectral data. Thus the mass spectrum of this product showed the molecular ion peak at m/z = 400 (M+, 12.6), with a base peak at 136. The IR spectrum revealed absorption bands at 3,256, 3,212, and 3,187 cm−1 (NH) and 1,667 cm−1 (C=O). The 1H NMR spectrum revealed signals at 7.10–7.64 ppm due to the aromatic protons. The IR spectrum of compound 12 showed a broad absorption band around 3,340–3,210 cm−1 due to NH groups besides two carbonyl absorption bands at 1,697 and 1,670 cm−1. Its 1H NMR spectrum revealed four D2O-exchangeable signals at 10.87, 11.45, 12.12, and 14.34 ppm due to four NH protons (Scheme 2).
Biological activity
Cytotoxicity and antiviral activities
The selected synthesized isatin derivatives 7a–7e, 10a–10e, and 11 were evaluated for their inhibitory effect on the replication of vesicular stomatitis virus (VSV) as well as cytotoxicity evaluation in Vero clone CCL-81 cell lines. Evaluation of the antiviral activity of isatin derivatives and interferon as standard was determined according to Shinji’s method [22] and the results are summarized in Table 1.
Table 1.
Cytotoxicity and antiviral activity for the selected compounds
| Compound | R | IC 50/μmol cm−3 | Antiviral activity | ||
|---|---|---|---|---|---|
| Virus titer log10 | Virus titer difference log | Effect | |||
| Control | – | – | 6.28 | – | – |
| 7a | –CH3 | 12.8 | 6.43 | 0.15 | Increased |
| 7b | –CH2CH3 | 25.3 | 6.33 | 0.05 | Increased |
| 7c | –CH2CH=CH2 | 24.8 | 5.40 | 0.88 | Decreased |
| 7d | –(CH2)3CH3 | 24.9 | 5.49 | 0.76 | Decreased |
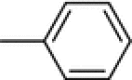
| 7e |
|
5.7 | 6.40 | 0.20 | Increased |
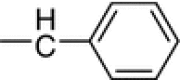
| 10a |
|
23.6 | 6.49 | 0.21 | Increased |
| 10b |
|
1.0 | 5.38 | 0.90 | Decreased |
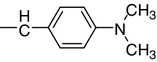
| 10c |
|
3.0 | 5.57 | 0.68 | Decreased |
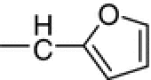
| 10d |
|
1.5 | 6.59 | 0.31 | Increased |
| 10e |
|
12.2 | 6.00 | 0.28 | Decreased |
| 11 |
|
12.6 | 6.11 | 0.17 | Decreased |
| Interferon | 4.40 | 1.88 | Decreased | ||
Certain aspects of the structure–activity relationships for these compounds can be more clearly highlighted using the general structures provided in Table 1. From the tabulated data, it was found that six compounds showed activities against the tested virus. In the Vero clone CCL-81 cell lines, compound 7c, which has an allyl moiety, was found to be the most active among compounds 7 against replication of VSV with a log virus titer difference of 0.88; when the allyl moiety of this compound was replaced by an n-butyl moiety, as in the case of compound 7d, the log virus titer difference decreased to 0.76. No positive activities were noticed in the other tested samples among the series of tested structure. Compound 10b, which has a 4-fluorophenyl moiety, showed the highest activity among compounds 10 against replication of VSV with a log virus titer difference of 0.90; when this moiety was replaced by a 4-dimethylphenyl moiety, as in the case of compound 10c, the activity decreased and showed a log virus titer difference of 0.68. Compounds 10e and 11 exhibited weak activity, whereas the other tested compounds showed no positive antiviral activity. In pharmacological terms, the presence of the 4-fluorophenyl moiety on the skeleton of structure 10 may be directly responsible for its antiviral activity, whereas the presence of the allyl moiety in compound 7c makes it a promising antiviral agent.
Conclusion
A new series of 5-fluoroisatin derivatives was synthesized. The antiviral screening of some selected compounds showed good inhibitory activities as antiviral agents. Among the tested compounds, N 1-(5-fluoro-2-oxoindolin-3-ylidene)-N 4-(p-fluoro- (or p-dimethylamino)benzylidene)thiocarbohydrazone and N-allyl (or n-butyl)-2-(5-fluoro-2-oxoindolin-3-ylidene) hydrazine carbothioamide were the most active members.
Experimental
All melting points were determined on an electrothermal Gallenkamp apparatus. The IR spectra were measured on a Mattson 5000 FTIR spectrophotometer as potassium bromide discs. The 1H NMR and 13C NMR spectra were recorded as solutions in DMSO-d 6 on a Bruker WP spectrometer (300 MHz for 1H NMR and 75 MHz for 13C NMR) with the chemical shifts downfield from TMS as an internal standard. The mass spectra were recorded on Finnegan MAT 212 instrument (the ionizing voltage was 70 eV) at the Faculty of Science, Cairo University. Elemental analyses were carried out by the microanalytical unit of the Faculty of Science, Cairo University.
3-(5-Fluoro-2-oxoindolin-3-ylideneamino)-2-thioxothiazolidin-4-one (2, C11H6FN3O2S2)
A mixture of 5-fluoroisatin (1, 0.01 mol) and 3-amino-2-thioxothiazolidin-4-one (0.01 mol) in 30 cm3 ethanol was heated under reflux for 1 h and left to cool. The solid product was filtered and recrystallized from ethanol to give 2 as brown crystals. Yield 85 %; m.p.: >300 °C; IR: = 3,238 (NH), 1,700, 1,668 (C=O) cm−1; 1H NMR: δ = 5.80 (s, 2H, CH2), 6.94 (m, 1H, indole C7-H), 7.21 (m, 1H, indole C6-H), 7.44 (m, 1H, indole C4-H), 11.40 (s, 1H, NH) ppm; 13C NMR: δ = 33.7, 108.1, 112.5, 117.7, 121.8, 131.4, 138.9, 158.6, 163.1, 174.5, 177.3 ppm.
2-(5-Fluoro-2-oxoindolin-3-ylideneamino)-N-[3-(trifluoromethyl)phenyl]benzamide (3, C22H13F4N3O2)
A mixture of 5-fluoroisatin (1, 0.01 mol) and 2-amino-N-[3-(trifluoromethyl)phenyl]benzamide (0.01 mol) in 30 cm3 ethanol was heated under reflux for 1 h. The solution was concentrated, then left to cool. The solid product was filtered and recrystallized from toluene to give 3 as pale yellow solid. Yield 86 %; m.p.: 176–177 °C; IR: = 3,235, 3,192 (2NH), 1,665 (C=O) cm−1; 1H NMR: δ = 7.13–7.76 (m, 11H, Ar–H), 9.12, 11.32 (2s, 2H, 2NH) ppm.
N′-(5-Fluoro-2-oxoindolin-3-ylidene)arylsulfonohydrazides5a–5c
A mixture of hydrazone derivative 4 (0.01 mol), the appropriate sulfonyl chloride (namely methanesulfonyl chloride, benzenesulfonyl chloride, or 4-methylbenzenesulfonyl chloride) (0.01 mol), and 0.5 cm3 triethylamine in 30 cm3 dioxane was heated under reflux for 2 h. The reaction mixture was poured into crushed ice. The resulting precipitate was filtrated, dried, and crystallized from ethanol to afford 5a–5c.
N′-(5-Fluoro-2-oxoindolin-3-ylidene)methanesulfonohydrazide (5a, C9H8FN3O3S)
Brown crystals; yield 76 %; m.p.: 248–250 °C; IR: = 3,178, 3,106 (2NH), 1,660 (C=O) cm−1; 1H NMR: δ = 2.83 (s, 3H, CH3), 6.84, 6.95, 7.14 (3 m, 3H of indolyl), 10.68, 11.80 (2s, 2H, 2NH) ppm; 13C NMR: δ = 40.4 (CH3), 109.8 (C4-indolyl), 112.9 (C7-indolyl), 114.4 (C6-indolyl), 123.5 (C4a-indolyl), 136.6 (C7a-indolyl), 156.5 (C5-indolyl), 158.9 (C=N-indolyl), 162.9 (C=O-indolyl) ppm.
N′-(5-Fluoro-2-oxoindolin-3-ylidene)benzenesulfonohydrazide (5b, C14H10FN3O3S)
Brown crystals; yield 76 %; m.p.: 287–288 °C; IR: = 3,178, 3,106 (2NH), 1,660 (C=O) cm−1; 1H NMR: δ = 7.40–7.96 (m, 8H, Ar–H), 9.56, 11.43 (2s, 2H, 2NH) ppm; 13C NMR: δ = 110.3, 112.3, 116.4, 118.6, 125.4 (2C), 127.5, 129.8 (2C), 136.7, 139.2, 154.4, 158.0, 161.0 ppm.
4-Methyl-N′-(5-fluoro-2-oxoindolin-3-ylidene)benzenesulfonohydrazide (5c, C15H12FN3O3S)
Brown crystals; yield 75 %; m.p.: 198–199 °C; IR: = 3,178, 3,106 (2NH), 1,660 (C=O) cm−1; 1H NMR: δ = 2.36 (s, 3H, CH3), 7.40–7.98 (m, 7H, Ar–H), 10.96, 11.80 (2s, 2H, 2NH) ppm; 13C NMR: δ = 21.9 (CH3), 110.3, 112.3, 116.4, 118.6, 125.4 (2C), 127.5, 129.8 (2C), 136.7, 139.2, 154.4 (12 Ar–C), 158.0 (C=N of indolyl), 161.0 (C=O of indolyl) ppm; MS: m/z (%) = 333 (M+, 5.8), 299 (6.2), 271 (15.3), 257 (5.2), 179 (42.4), 151 (52.5), 136 (55.80), 108 (100).
2,2,2-Trifluoro-N′-(5-fluoro-2-oxoindolin-3-ylidene)acetohydrazide (6, C10H5F4N3O2)
A mixture of the hydrazone 4 (0.01 mol) and 5 cm3 trifluoroacetic anhydride was stirred for 1 h at room temperature. The solution was concentrated under vacuum. The resultant precipitate was collected by filtration and crystallized from ethanol to give compound 6 as orange crystals. Yield 71 %; m.p.: >300 °C; IR: = 3,255, 3,107 (2NH), 1,696, 1,661 (C=O) cm−1; 1H NMR: δ = 6.82 (m, 1H of indolyl), 7.36 (m, 1H of indolyl), 7.66 (m, 1H of indolyl), 11.46, 14.05 (2s, 2H, 2NH) ppm; 13C NMR: δ = 111.0 (C4-indolyl), 113.4 (C7-indolyl), 114.7 (C6-indolyl), 123.5 (C4a-indolyl), 136.5 (C7a-indolyl), 157.2 (C5-indolyl), 158.9 (C=N-indolyl), 163.2 (C=O-indolyl) ppm.
Synthesis of thiosemicarbazone derivatives7a–7f
A mixture of hydrazone 4 (0.01 mol) and the requisite isothiocyanate (namely methyl-, ethyl-, allyl-, n-butyl-, phenyl-, cyclohexylisothiocyanate) (0.01 mol) in 20 cm3 ethanol was heated under reflux for 1 h and left to cool. The resultant precipitate was collected and crystallized to give 7a–7f.
2-(5-Fluoro-2-oxoindolin-3-ylidene)-N-methylhydrazinecarbothioamide (7a)
Yellow crystals; yield 87 %; m.p.: 271–273 °C (Ref. [21] 271–274 °C).
N-Ethyl-2-(5-fluoro-2-oxoindolin-3-ylidene)hydrazinecarbothioamide (7b)
Greenish yellow; yield 92 %; m.p.: 243–245 °C (Ref. [21] 245–246 °C).
N-Allyl-2-(5-fluoro-2-oxoindolin-3-ylidene)hydrazinecarbothioamide (7c)
Yellow crystals; yield 74 %; m.p.: 219–220 °C (Ref. [21] 219–220 °C).
N-Butyl-2-(5-fluoro-2-oxoindolin-3-ylidene)hydrazinecarbothioamide (7d)
Greenish yellow crystals; yield 81 %; m.p.: 211–212 °C (Ref. [21] 211–212 °C).
2-(5-Fluoro-2-oxoindolin-3-ylidene)-N-phenylhydrazinecarbothioamide (7e)
Yellow crystals; yield 79 %; m.p.: 254–255 °C (Ref. [21] 218 °C).
N-Cyclohexyl-2-(5-fluoro-2-oxoindolin-3-ylidene)hydrazinecarbothioamide (7f)
Yellow crystals; yield 89 %; m.p.: 249–250 °C (Ref. [21] 249–250 °C).
Synthesis of thiazole derivatives8a–8f
A mixture of the thiosemicarbazide derivatives 7a–7f (0.01 mol), chloroacetone (0.01 mol), and fused sodium acetate (0.02 mol) in 40 cm3 ethanol was heated under reflux for 5 h and left to cool. The obtained product was collected by filtration and recrystallized from the appropriate solvent to give 8a–8f.
3-[(3,4-Dimethylthiazol-2(3H)-ylidene)hydrazono]-5-fluoroindolin-2-one (8a, C13H11FN4OS)
Orange crystals from ethanol; yield 63 %; m.p.: 297–298 °C; IR: = 3,212 (NH), 2,986–2,876 (CH-aliph), 1,675 (C=O) cm−1; 1H NMR: δ = 1.76, 2.76 (2s, 6H, 2CH3), 5.85 (s, 1H, thiazole-H), 6.92 (m, 1H, indole), 7.32 (m, 1H, indole-H), 7.63 (m, 1H, indole-H), 10.41 (s, H, NH) ppm; 13C NMR: δ = 21.2, 31.3 (2 CH3), 95.3, 148.3, 162.2 (3C-thiazole), 117.4, 118.8, 122.5, 131.3, 135.3, 154.7 (6C-indole), 164.4 (C=N indole), 165.1 (indole C=O) ppm.
3-[(3-Ethyl-4-methylthiazol-2(3H)-ylidene)hydrazono]-5-fluoroindolin-2-one (8b, C14H13FN4OS)
Brown crystals from ethanol; yield 65 %; m.p.: 276–277 °C; IR: = 3,218 (NH), 2,978–2,865 (CH-aliph), 1,668 (C=O) cm−1; 1H NMR: δ = 1.23 (t, 3H, ethyl-CH3), 1.78 (s, 3H, CH3), 3.68 (q, 2H,ethyl-CH2), 5.87 (s, 1H, thiazole), 6.89 (m, 1H, indole-H), 7.20 (m, 1H, indole-H), 7.68 (m, 1H, indole-H), 10.98 (s,1H, NH) ppm; 13C NMR: δ = 14.2 (ethyl-CH3), 22.3 (C-CH3), 40.1 (ethyl-CH2), 93.1, 157.3, 161.2 (3C-thiazole), 116.3, 119.2, 121.8, 125.4, 131.4, 152.6 (6C-indolyl), 159.9 (C=N indole), 163.5 (C=O indole) ppm.
3-[(3-Allyl-4-methylthiazol-2(3H)-ylidene)hydrazono]-5-fluoroindolin-2-one (8c, C15H13FN4OS)
Brown crystals from ethanol; yield 61 %; m.p.: 278–279 °C; IR: = 3,232 (NH), 2,967–2,864 (CH-aliph), 1,676 (C=O) cm−1; 1H NMR: δ = 1.76 (s, 3H, CH3), 4.28 (t, 2H, allyl C1-H), 5.14 (d, 1H, allyl-H), 5.45 (d, 1H, allyl C3-H), 5.67–5.85 (m, 1H, allyl C2-H), 6.21(s, 1H, thiazol-H), 6.93 (m, 1H, indole-H), 7.19 (m, 1H, indole-H), 7.51 (m, 1H, indole-H), 11.20, (s, 1H, NH) ppm; 13C NMR: δ = 21.2 (C-CH3), 78.7, 120.2, 120.2 (3C-propenyl), 93.4, 157.7, 161.5 (3C-thiazole), 116.3, 119.4, 121.8, 126.3, 131.4, 155.6 (6C-indolyl), 159.9 (C=N indole), 163.5 (C=O indole) ppm.
3-[(3-Butyl-4-methylthiazol-2(3H)-ylidene)hydrazono]-5-fluoroindolin-2-one (8d, C16H17FN4OS)
Red crystals from methanol; yield 66 %; m.p.: 248–250 °C: IR: = 3,232 (NH), 2,967–2,864 (CH-aliph), 1,676 (C=O) cm−1; 1H NMR: δ = 1.02 (t, 3H, butyl-CH3), 1.35–1.37 (m, 2H, butyl C3-H), 1.67 (m, 2H, butyl C2-H), 1.75 (s, 3H, CH3), 3.69 (q, 2H, butyl C1-H), 6.12 (s, 1H, thiazole-H), 6.67 (m, 1H, indole-H), 7.16 (m, 1H, indole-H), 7.64 (m, 1H, indole-H), 10.54 (s, 1H, NH) ppm; 13C NMR: δ = 14.2 (butyl CH3), 20.3 (butyl C3), 31.3 (butyl C2), 46.6 (butyl-C1), 23.2 (C-CH3), 98.5, 157.3, 164.3 (3C-thiazole), 112.4, 117.6, 121.8, 125.4, 131.3, 155.7 (6C-indole), 162.4 (C=N-indole), 163.4 (indole C=O) ppm; MS: m/z (%) = 332 (M+, 84.2), 276 (12.3), 262 (6.8), 247 (36.5), 177 (29.3), 162 (28.4), 134 (35.2), 94 (29.4), 55(100).
5-Fluoro-3-[(4-methyl-3-phenylthiazol-2(3H)-ylidene)hydrazono]indolin-2-one (8e, C18H13FN4OS)
Red crystals from methanol; yield 62 %; m.p.: 265–267 °C; IR: = 3,236 (NH), 2,964–2,862 (CH-aliph), 1,667 (C=O) cm−1; 1H NMR: δ = 1.75 (s, 3H, CH3), 5.64 (s, 1H, CH-thiazole), 6.93–7.65 (m, 8H, Ar–H), 11.27 (s, 1H, NH) ppm; 13C NMR: δ = 22.3, 108.1, 112.5, 117.3 (2C), 118.4, 121.8, 125.7, 126.3 (2C), 129.4, 131.5, 138.9, 157.3, 158.7, 161.2, 165.2, 166.1 ppm.
3-[(3-Cyclohexyl-4-methylthiazol-2(3H)-ylidene)hydrazono]-5-fluoroindolin-2-one (8f, C18H19FN4OS)
Orange crystals from methanol; yield 64 %; m.p.: 287–289 °C; IR: = 3,230 (NH), 2,960–2,868 (CH-aliph), 1,675 (C=O), 1,611, 1,602 (2 C=N) cm−1; 1H NMR: δ = 1.15, 1.28–1.35, 1.42–1.51, 1.64, 1.77, 1.92 (m, 10H, cycl), 1.78 (s, 3H, CH3), 4.18–4.21(m, 1H, cycl-C1-H), 5.89 (s, 1H, thiazole-H), 6.90 (m, 1H, indole-H), 7.32 (m,1H, indole-H), 7.76 (m, 1H, indole-H), 11.25 (s, 1H, indole NH) ppm; 13C NMR: δ = 25.3 (2C), 25.5, 31.9 (2C), 54.2 (6C-cycl), 117.7, 118.4, 121.8, 125.7, 131.5, 138.9, 158.7 (6C indole), 165.2 (C=N), 166.1 (indole C=O) ppm.
N1-(5-Fluoro-2-oxoindolin-3-ylidene)thiocarbohydrazone (9, C9H8FN5OS)
A mixture of isatin 1 (0.01 mol) and thiocarbohydrazide (0.01 mol) in 20 cm3 ethanol was heated under reflux for 0.5 h. The solid product so formed was filtered and recrystallized to give 9 as brown crystals. Yield 71 %; m.p.: >300 °C; IR: = 3,270, 3,160, 3,106 (NH, NH2), 1,665 (C=O) cm−1; 1H NMR: δ = 2.51 (s, 1H, NH), 4.92 (bs, 2H, NH2),7.34, 7.42, 7.96 (3 m, 3H of indolyl), 10.40, 12.54 (2s, 2H, 2NH) ppm; 13C NMR: δ = 116.3, 118.3, 124.8, 127.5, 132.5, 157.7 (6C of indolyl), 161.0 (C=N of indolyl), 162.4 (C=O of indolyl), 179.4 (C=S) ppm.
Synthesis of thiocarbohydrazones10a–10e
Method A: a solution of isatinthiocarbohydrazone 9 (0.01 mol) in 30 cm3 dioxane was treated with the requisite aldehyde (namely benzaldehyde, 4-fluorobenzaldehyde, 4-(dimethylamino)benzaldehyde, furan-2-carbaldehyde, or thiophene-2-carbaldehyde) (0.01 mol). The reaction mixture was heated under reflux for 1 h and left to cool. The solid product so formed was filtered and recrystallized to give 10a–10e.
Method B: a solution of isatin 1 (0.01 mol) and the requisite thiocarbohydrazone derivative (0.01 mol) in ethanol was heated under reflux for 1 h. The solid product was filtered and recrystallized (m.p. and mixed m.p.).
N4-Benzylidene-N1-(5-fluoro-2-oxoindolin-3-ylidene)thiocarbohydrazone (10a, C16H12FN5OS)
Orange crystals; yield 74 %; m.p.: 287–288 °C; IR: = 3,223, 3,160, 3,106 (NH), 1,665 (C=O) cm−1; 1H NMR: δ = 6.96 (m, 1H, indole-H), 7.22 (m, 1H, indole-H), 7.35 (m, 1H, indole-H), 7.48 (m, 3H, Ar–H), 7.48 (m, 2H, Ar–H), 8.34 (s, 1H, CH=N), 11.36, 12.44, 14.63 (3s, 3H, 3NH) ppm; 13C NMR: δ = 116.3, 118.3, 124.8, 125.4, 126.6, 127.5, 128.5, 129.5, 130.4, 131.4, 132.5, 157.7 (6C of indolyl + 6C-Ph), 159.6 (CH=N), 161.0 (C=N of indolyl), 162.8 (C=O of indolyl), 179.6 (C=S) ppm.
N1-(5-Fluoro-2-oxoindolin-3-ylidene)-N4-(4-fluorobenzylidene)thiocarbohydrazone (10b, C16H11F2N5OS)
Orange crystals; yield 75 %; m.p.: 265–266 °C; IR: = 33,412–3,110 (3NH), 1,697 (C=O) cm−1; 1H NMR: δ = 7.22–7.86 (m, 7H, Ar–H), 8.54 (s, 1H, CH=N), 11.28, 12.76, 14.62 (3s, 3H, 3NH) ppm; 13C NMR: δ = 116.3, 118.3, 124.8, 125.4, 126.6, 127.5, 128.5, 129.5, 131.4, 132.5, 156.4, 157.7 (6C of indolyl + 6C-Ph), 159.6 (CH=N), 161.4 (C=N of indolyl), 162.8 (C=O of indolyl), 179.6 (C=S) ppm.
N4-(4-Dimethylaminobenzylidene)-N1-(5-fluoro-2-oxoindolin-3-ylidene)thiocarbohydrazone (10c, C18H17FN6OS)
Reddish brown crystals; yield 70 %; m.p.: 277–278 °C; IR: = 3,243, 3,198, 3,161 (NH), 1,660 (C=O) cm−1; 1H NMR: δ = 2.88 (s, 6H, 2CH3), 7.18–7.76 (m, 7H, Ar–H), 8.11 (s, 1H, CH=N), 11.38, 12.60, 14.44 (3s, 3H, 3NH) ppm; 13C NMR: δ = 45.4, 45.4 (2CH3), 116.6, 118.4, 124.7, 125.6, 126.9, 127.8, 128.4, 129.5, 131.8, 132.2, 145.8, 157.9 (6C of indolyl + 6C-Ph), 159.6 (CH=N), 161.4 (C=N of indolyl), 162.8 (C=O of indolyl), 179.2 (C=S) ppm.
N1-(5-Fluoro-2-oxoindolin-3-ylidene)-N4-(2-furylmethylene)thiocarbohydrazone (10d, C14H10FN5O2S)
Brown crystals; yield 75 %; m.p.: 235–236 °C; IR: = 3,240, 3,212, 3,173 (NH), 1,668 (C=O) cm−1; 1H NMR: δ = 6.87–7.56 (m, 6H, Ar–H), 8.11 (s, 1H, CH=N), 11.22, 12.61, 14.41 (3s, 3H, 3NH) ppm; 13C NMR: δ = 111.3, 113.3, 116.6, 118.4, 125.6, 131.8, 144.2, 144.3, 145.8, 157.9 (6C of indolyl + 4C-furyl), 159.7 (CH=N), 161.6 (C=N of indolyl), 163.1 (C=O of indolyl), 179.2 (C=S) ppm.
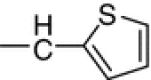
N1-(5-Fluoro-2-oxoindolin-3-ylidene)-N4-(2-thienylmethylene)thiocarbohydrazone (10e, C14H10FN5O2S2)
Brown crystals; yield 74 %; m.p.: 282–284 °C; IR: = 3,248, 3,223, 3,190 (NH), 1,665 (C=O) cm−1; 1H NMR: δ = 7.18–7.98 (m, 6H, Ar–H), 8.54 (s, 1H, CH=N), 11.67, 12.43, 14.00 (3s, 3H, 3NH) ppm; 13C NMR: δ = 116.6, 118.4, 123.3, 125.6, 126.1, 126.4, 127.6, 127.8, 134.2, 157.9 (10C of indolyl), 159.7 (CH=N), 161.6 (C=N of indolyl), 163.4 (C=O of indolyl), 179.4 (C=S) ppm.
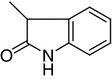
N1,N4-Bis(5-fluoro-2-oxoindolin-3-ylidene)thiocarbohydrazone (11, C17H10F2N6O2S)
A mixture the thiocarbohydrazone 9 (0.01 mol) and 5-fluoroisatin (0.01 mol) in 20 cm3 dioxane was heated under reflux for 2 h. The solid product so formed was filtered and recrystallized from dioxane to give 11 as red crystals. Yield 68 %; m.p.: >300 °C; IR: = 3,256, 3,212, 3,187 (NH), 1,667 (C=O) cm−1; 1H NMR: δ = 7.10–7.64 (m, 6H, indole-H), 10.76, 11.54, 12.23, 14.11 (4s, 4H, 4NH) ppm; 13C NMR: δ = 116.6, 116.7, 118.4, 118.7, 122.4, 123.3, 125.4, 126.1, 134.1, 134.2, 156.5, 157.9 (12C of indolyl), 161.6, 161.9 (2C=N of indolyl), 163.1, 163.4 (2C=O of indolyl), 178.1 (C=S) ppm; MS: m/z (%) = 400 (M+, 12.6), 373 (32.26), 221 (36.4), 179 (86.4), 164 (46.8), 136 (99.28), 108 (100), 94 (48.24), 81 (52.35).
2,2,2-Trifluoro-N′-(2-(5-fluoro-2-oxoindolin-3-ylidene)hydrazinecarbonothioyl)acetohydrazide (12, C11H7F4N5O2S)
A solution of the thiocarbohydrazone 9 (0.01 mol) in 10 cm3 trifluoroacetic anhydride was stirred at room temperature for 1 h. The solution was concentrated under vacuum. The solid product so formed was filtered off, washed with ethanol, dried well, and recrystallized to give 12 as red crystals. Yield 79 %; m.p.: >300 °C; IR: = 3,340–3,210 (br-NH), 1,697, 1,670 (C=O) cm−1; 1H NMR: δ = 7.18–7.56 (3 m, 3H, indole-H), 10.87, 11.45, 12.12, 14.34 (4s, 4H, 4NH) ppm; 13C NMR: δ = 116.6, 118.4, 123.3, 125.6, 127.8, 134.2, 157.9 (6C of indolyl + CF3), 161.6 (C=N of indolyl), 163.4 (C=O of indolyl), 169.7 (CO-CF3), 179.4 (C=S) ppm.
Antiviral and cytotoxicity assays
Vero clone CCL-81 cells were grown in 199 E-Hepes buffer growth medium supplemented with 10 % inactivated fetal calf serum, 5 mM HEPES buffer, and antibiotics (100 U of penicillin/cm3, 100 g of streptomycin/cm3) at 37 °C and incubated in 5 % CO2 atmosphere.
Cytotoxicity assay
Organic compounds (50 μmol) were dissolved in 2 cm3 phosphate buffer saline, pH 7.2 ± 0.2. Test materials were filtered through 0.2-μm membranes using Millipore disposable syringe filters. The cytotoxicity assay of the tested organic compounds was performed according to the previous reports [23], whereby sterile filtrated organic test materials were twofold serially diluted in MEM and added to precultured Vero cells. A negative cell control was included. Plates were incubated at 37 °C for 24 h. Cell culture-treated plates were microscopically examined using an inverted microscope for detection of cellular changes. The test compounds treatment medium was discarded. Plates were washed using PBS. Cell viability was evaluated using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). The cytotoxicity of each compound was expressed as the 50 % cytotoxic concentration (CC50), which is the concentration required to reduce cell viability to 50 % of the control.
Antiviral activity
Antiviral activity of the synthetic compounds and interferon against VSV isolate was determined according to the method reported by Shinji [22], whereby nontoxic concentrations of test chemical compounds and rh-IFN (10 IU/0.1 cm3) as a positive control were used for the treatment of precultured Vero cells for 24 h at 0.1 cm3/well. One plate was maintained and left untreated for viral control titration. A virus infectivity titer was determined according to a reported method [24]; VSV was tenfold serially diluted in 199 E-Hepes buffer (10−1–10−8). Each dilution was dispensed as 100 mm3/well onto preculture Vero cells. Plates were inoculated using 10 CCID50 of VSV isolate tested. Plates were incubated at 37 °C. Seven days post infection the 50 % end point (CCID50) was determined. Antiviral activity was determined by subtracting the VSV mean titer in the treated and non-treated cell titers. The difference between both titers refers to the antiviral activity.
References
- 1.Katz E. Rev Clin Basic Pharm. 1987;6:119. doi: 10.1515/jbcpp.1987.6.2.119. [DOI] [PubMed] [Google Scholar]
- 2.Cooper JA, Moss B, Katz E. Virology. 1979;96:381. doi: 10.1016/0042-6822(79)90096-5. [DOI] [PubMed] [Google Scholar]
- 3.Webber SE, Tikhe J, Worland ST, Fuhrman SA, Hendrickson TF, Matthews DA, Love RA, Patick AK, Meador JW, Ferre RA, Brown EL, DeLisle DM, Ford CE, Binford SL. J Med Chem. 1996;39:5072. doi: 10.1021/jm960603e. [DOI] [PubMed] [Google Scholar]
- 4.Ronen D, Sherman L, Barnun S, Teitz Y. Antimicrob Agents Chemother. 1987;31:1798. doi: 10.1128/AAC.31.11.1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen LR, Wang YC, Lin YW, Chou SY, Chen SF, Liu LT, Wu YT, Kuo CJ, Chen TS, Juang SH. Bioorg Med Chem Lett. 2005;15:3058. doi: 10.1016/j.bmcl.2005.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teitz Y, Ronen D, Vansover A, Stematsky T, Riggs JL. Antivir Res. 1994;24:305. doi: 10.1016/0166-3542(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 7.Bal TR, Anand B, Yogeeswari P, Sriram D. Bioorg Med Chem Lett. 2005;15:4451. doi: 10.1016/j.bmcl.2005.07.046. [DOI] [PubMed] [Google Scholar]
- 8.Sriram D, Bal TR, Yogeeswari P. J Pharm Pharm Sci. 2005;8:565. [PubMed] [Google Scholar]
- 9.Richman DD. Antiviral Res. 2006;71:117. doi: 10.1016/j.antiviral.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Saeed A, Shaheen U, Hammed A, Kazmi F. J Fluorine Chem. 2010;13:333. doi: 10.1016/j.jfluchem.2009.11.005. [DOI] [Google Scholar]
- 11.Dolbier WR., Jr J Fluorine Chem. 2005;126:157. doi: 10.1016/j.jfluchem.2004.09.033. [DOI] [Google Scholar]
- 12.Smart BE. J Fluorine Chem. 2001;109:3. doi: 10.1016/S0022-1139(01)00375-X. [DOI] [Google Scholar]
- 13.Isanbor C, O’Hagan D. J Fluorine Chem. 2006;127:303. doi: 10.1016/j.jfluchem.2006.01.011. [DOI] [Google Scholar]
- 14.Podichetty AK, Faust A, Kopka K, Wagner S, Schober O, Schäfers M, Haufe G. Bioorg Med Chem. 2009;17:2680. doi: 10.1016/j.bmc.2009.02.048. [DOI] [PubMed] [Google Scholar]
- 15.Farag AA, Abd-Alrahman SN, Ahmed GF, Ammar RM, Ammar YA, Abbas SY. Arch Pharm Life Sci. 2012;345:703. doi: 10.1002/ardp.201200014. [DOI] [PubMed] [Google Scholar]
- 16.Farag AA, Khalifa EM, Sadik NA, Abbas SY, Al-Sehemi AG, Ammar YA. Med Chem Res. 2013;22:440. doi: 10.1007/s00044-012-0046-6. [DOI] [Google Scholar]
- 17.Abbas SY, El-Sharief MAMS, Basyouni WM, Fakhr IMI, El-Gammal EW. Eur J Med Chem. 2013;64:111. doi: 10.1016/j.ejmech.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 18.Aly MM, Mohamed YA, El-Bayouki KhAM, Basyouni WM, Abbas SY. Eur J Med Chem. 2010;45:3365. doi: 10.1016/j.ejmech.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 19.Lv K, Wang L, Zhou X, Fan S, Liu H, Zheng Z, Li S, Liu M. Bioorg Med Chem Lett. 2011;21:3062. doi: 10.1016/j.bmcl.2011.03.031. [DOI] [PubMed] [Google Scholar]
- 20.Chen L, Huang M, Feng L, He Y, Yun H. Novel cyclopropane indolinone derivatives. Patent WO2011/69298 A1, Jun 16, 2001. Chem Abstr. 2011;155:93890. [Google Scholar]
- 21.Karal N, Gursoy A, Kandemirli F, Shvets N, Kaynak FB, Suheyl K, Ozbey S, Kovalishyne V, Dimoglo A. Bioorg Med Chem. 2007;15:5888. doi: 10.1016/j.bmc.2007.05.063. [DOI] [PubMed] [Google Scholar]
- 22.Shinji H. Biochem J. 2005;392:191. doi: 10.1042/BJ20051069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vijayan P, Raghu C, Ashok G, Dhanaraj SA, Suresh B. Indian J Med Res. 2004;120:24. [PubMed] [Google Scholar]
- 24.Steven S, Richard LH, Stephen AY, Weidbrauk DL. Clinical virology manual. 4. Washington DC: ASM; 2009. [Google Scholar]