

Abstract
Point-of-care (PoC) testing followed by personalized efficient therapy of infectious diseases may result in a considerable reduction of associated health care costs. Lab-on-a-chip (LoC) systems represent a potentially high efficient class of PoC tools. Here, we present a LoC system for automated pathogen analysis of respiratory viruses from nasopharyngeal specimens. The device prepares total nucleic acids from extracted swab samples using magnetic silica beads. After reverse transcription the co-purified viral RNA is amplified in accordance with the QIAplex multiplex PCR technology. Hybridized to corresponding QIAGEN LiquiChip beads and labelled with streptavidin R-phycoerythrin, the amplified target sequences are finally detected using a QIAGEN LiquiChip200 workstation. All chemicals needed are either stored freeze-dried on the disposable chip or are provided in liquid form in a reagent cartridge for up to 24 runs. Magnetic stir bars for mixing as well as turning valves with metering structures are integrated into the injection-moulded disposable chip. The core of the controlling instrument is a rotating heating bar construction providing fixed temperatures for fast cycling. PCR times of about half an hour (for 30 cycles) could be achieved for 120 μl reactions, making this system the fastest currently available high-volume PCR chip. The functionality of the system was shown by comparing automatically processed nasopharyngeal samples to ones processed manually according to the QIAGEN “ResPlex™ II Panel v2.0” respiratory virus detection kit. A prototype of the present instrument revealed slightly weaker signal intensities with a similar sensitivity in comparison to the commercially available kit and automated nucleic acid preparation devices, even without protocol optimization.
Keywords: Lab-on-a-chip, Point-of-care diagnostics, Analysis of respiratory diseases, On chip nested PCR, Microfluidic chip, Nasopharyngeal swab sample
Introduction
Kary Mullis’ 1983 invention of the polymerase chain reaction (PCR) marked the starting point of modern molecular diagnostics. Today, the PCR-based detection of nucleic acids is routinely used to diagnose a plethora of infectious diseases, to genetically test for cancer, and to monitor ongoing treatments (Ieven 2007). A wide range of commercially available specialized kits enable easy-to-perform protocols, but regarding the entire workflow from sample to result, the complex manual handling led to calls for more convenient or automated solutions. As a result, sequential steps previously executed manually in separate devices have gradually been combined within a single device. By now, fully integrated, as well as miniaturized solutions are available in the form of “lab-on-a-chip” (LoC) systems, the most elaborate of which are capable of processing diagnostic tests from sample to result. A major driver in their development is the reduction in the costs per analysis and per single test result afforded by them. Critical factors for the marketability of such devices, however, also include a high significance and reliability of the results as well as sensitivities and specificities that satisfy stringent clinical requirements. At this stage, many microfluidics-based concepts and technologies fail to meet these demands (e.g. Blow 2007, Chen et al. 2007, Dobson et al. 2007, Zhang and Xing 2007, Weigl et al. 2008, Sauer-Budge et al. 2009, Huckle 2006, Becker 2009, Kaigala et al. 2010, Lounsbury et al. 2010, Fujimoto et al. 2010, and references herein).
The initial preparation of samples, particularly the complexity involved in reducing original milliliter volumes to a few microliter, remains one of the major roadblocks in this regard. Here, we present a lab-on-a-chip system that integrates all processing steps for detecting respiratory viruses from nasopharyngeal samples. The device features, among other things, a combined lysis and nucleic acid binding step, a novel magnetic separation principle, “on-chip” nested multiplex amplification, and internal processing and amplification controls. Current cost-per-analysis (COG) estimates are in the $10–$15 range. The present system can easily be adapted to different pathogens and may thus offer a route to opening new markets for in-vitro diagnostics.
Materials & methods
Lab-on-a-chip concept
As first starting point, the assay had to be defined in a way that a fluidplan can be deduced. Several fluids have to be moved, merged, metered, mixed and heated in a controlled manner. To prevent cross-contamination during all steps T-junctions and overlapping structures were excluded on the chip. To ensure precise pumping and positioning of the fluids, the gas volume in the system has to be minimized as its compressibility may cause uncontrollable fluid movements. Homogeneous mixing and fast heat distribution within the reaction chambers has to be guaranteed. Since magnetic stirring known from standard laboratory equipment is a very effective mixing method, magnetic stir bars are integrated into the chip at several positions. Inside the lysis chamber mixing is done using a Teflon-coated magnetic stir bar. Within the SPE chamber, the PCR chambers, and the hybridization chamber a piece of foil-magnet is used, respectively.
As we are aiming for a cost-efficient system, the chip is structured on one side only and features a single generic turning valve design. These multiway turning valves permit a chip design without T-junctions and overlapping structures. In addition, these valves not only allow for different channel connections but also for the integration of metering structures. One metering structure is needed to separate an aliquot of the first RT-PCR for processing in the second amplification step and a second metering structure separates an aliquot of the second PCR for the following hybridization step. To avoid moving tiny reagent volumes, we decided to store all enzymes and PCR reagents in freeze-dried form on the chip while all buffers needed in larger volumes are stored in liquid form in a reagent cartridge allowing 24 analyses. The buffers are moved into the chip using syringe pumps since those allow pulsation-free pumping. Contamination is prevented by collecting liquid waste in special waste chambers on the chip.
Although detection capabilities should ideally also be integrated into this system, for simplicity we use a LiquiChip 200 workstation which is an established tool for the detection of fluorescent-labelled targets. To this end, doubly labelled target DNA sequences are finally moved into a reservoir on chip and transferred to a LiquiChip 200 workstation with a conventional tube for detection.
Chip design
The developed microfluidic disposable processing cartridge (Fig. 1 left) consists of four individually moulded parts: 1. a basic chip with an “SBS” standard size (comparable to a 96-well plate), 2. a lysis chamber, 3. a cap for the lysis chamber, and 4. four turning valves. The basic chip is made from polycarbonate (PC). The slip-on lysis chamber and the corresponding cap are made from polypropylene (PP). The lysis chamber has a total volume of 3 ml enabling the procession of a sample volume up to 1 ml, a standard quantity in clinical molecular diagnostics.
Fig. 1.

Microfluidic disposable processing cartridge (left), 2-componend turning valve with interconnection channels and 5 μl metering loop (middle) and reagent cartridge (right)
Channel dimensions on the chip are 0.6 mm × 0.6 mm (WH). The reaction chambers for solid phase extraction (SPE), hybridization, and labelling are 0.6 mm high with a volume of 100, 50 and 30 μl, respectively, whereas the PCR reservoirs are 1 mm high with a volume of 120 μl.
Liquid movement is controlled by four turning valves each placed in a housing on the basic chip. The valve body is made from PC combined with a soft thermoplastic elastomer (TPE) for the structured sealing plate (Fig. 1 middle) manufactured by two-component injection-moulding. Short channels within the TPE form fluid channel connections. The pressure for tight connection is applied by the instrument.
The automated sample processing inside the chip operates in the following way.
The swab extract is put into the lysis chamber on the chip, and the lysis chamber is closed.
The chip is inserted into the device and “Start” button is pushed. All following steps are done automatically by the system.
Chaotropic lysis buffer is added into the lysis chamber and the chamber is heated up to 56°C.
Dried Proteinase K, the magnetic silica bead pellet and also the internal Processing and Amplification Control bead (iPAC) within the lysis chamber are resuspended by magnetic stirring. During incubation the sample material gets proteolytically digested.
After completing the lysis procedure and adjusting the nucleic acid binding conditions, the solution is moved through the SPE chamber to the waste, whereby RNA/DNA-bound magnetic particles are retained by the magnetic field within the SPE chamber.
RNA/DNA-bound magnetic particles get washed with washing buffer and rinsed by water.
Elution of RNA/DNA and transport to first PCR chamber.
Reverse transcription and outer primer-dependent amplification (dried reagents are rehydrated by the RNA/DNA solution).
Transport of PCR solution to waste, thereby metering an aliquot, flushing metered aliquot with water into the second PCR chamber.
Inner primer-dependent amplification (dried reagents are rehydrated by the PCR solution).
Transport of PCR solution to waste, thereby metering an aliquot.
Transfer of PCR aliquot with hybridization buffer into the hybridization chamber, resuspension of dried QIAplex beads.
Hybridization at 52°C to colour-coded QIAGEN LiquiChip beads.
Transfer of bead suspension into labelling chamber, rehydration of dried streptavidin-R-phycoerythrin and hybridization of double-stranded DNA to streptavidin-R-phycoerythrin.
Transport of dual-labelled complexes into storage well.
Transfer of the dual-labelled complexes to the detection system.
Chip fabrication
The fabrication is a multi-step process. The dried reagent beads for reverse transcription PCR (RT-PCR) 1, PCR 2, LiquiChip bead hybridization and streptavidin R-phycoerythrin labelling were each placed together with small magnetic stirring bars into the corresponding cavities on the chip. The stirring bars are punched out from a 0.5 mm thick magnetic polymer foil (Permaflex 928, Rheinmagner Horst Baermann GmbH) with a size of 7 mm × 1 mm × 0.5 mm (amplification chambers) and 3.5 mm × 0.5 mm × 0.5 mm (hybridization and labelling chambers). The chip is thermally bonded with a 175 μm PC foil. At the bottom of the lysis chamber, a polypropylene frit (~150 μm pore size) is placed to retain particles from the specimen. A 10 mm × 6 mm Teflon-coated mini stirring magnet, a dried silica magnetic bead pellet (containing QIAGEN’s MagAttract Suspension G beads and Proteinase K; pellet formulation undisclosed), and an iPAC (containing intact bacteriophage-fr particles; formulation undisclosed) are provided in the lysis chamber. The lysis chamber is closed with the “plugged on” cap sealed with an O-ring and directly attached onto the chip.
To ensure venting of the chip during fluid movement venting holes are inserted in the lysis and waste reservoirs. To avoid any contamination of the device by the sample the venting holes are covered with Teflon foils welded on the chip.
Reagent cartridge
While the processing cartridge is for one-time use and therefore discarded after each run, the reagent cartridge (Fig. 1 right) provides all liquids needed (e.g., lysis buffer, washing buffer, pure water, hybridization buffer) for 24 analyses. This multipart cartridge consists of a bottle holder (lower part) with twelve 30 ml high density polyethylene (HDPE) bottles and an upper part providing fluidic channels, three valves, two syringes (3 ml), and syringe adaptors. The upper plate is made out of two PC slides. The structures (0.6 mm × 0.8 mm, WH) are firstly milled, and afterwards both plates are laser-welded. The plastic capillaries for liquid aspiration from the bottle are glued onto the upper plate.
Operating device
To run the assay steps using the processing cartridge and the reagent cartridge, an automated operating device (Fig. 2) was developed. The main mechano-electronic features inside the controlling device are heating elements, syringe pumps, valve-driving motors, magnetic stirrers, and a unique “PCR engine”. The electronic components are controlled by a main board (National Instrument Corp.), which itself is controlled by a computer running a complex LabView (National Instrument Corp.) software protocol.
Fig. 2.
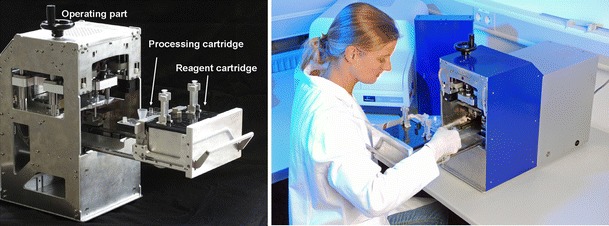
Operating device consisting of operating part and drawer for insertion of reagent cartridge and processing cartridge
The loaded drawer, containing the processing cartridge with up to 1 ml sample in the lysis chamber and the reagent cartridge, is pushed into the system, whereupon chip, reagent cartridge and instrument get connected. The processing cartridge gets connected to the operating device via the couplings of the turning valve driving motors. Additional connections to the reagent cartridge by Luer adaptors allow providing the liquid buffers. The reagent cartridge contains two syringe pumps which drive the fluids within the cartridges. By pushing the loaded drawer into the instrument the syringes get connected to linear drives within the instrument. The minimal pumping volume is 120 nl, which can be dispensed precisely into the chip. All fluids are guided to the addressed reservoirs via the on-chip integrated turning valves.
The lysis chamber is heated during lysis by a single side heating bracket (resistive heater) which comes in contact with the lysis chamber during insertion of the processing cartridge. The magnetic stir bar in the lysis chamber as well as the magnetic foil stirrer within the SPE chamber are actuated by motors with a permanent magnet (diametric polarity) mounted on the tip of a motor axis (Fig 3).
Fig. 3.

Magnetic Stirring within the SPE chamber: The SPE chamber contains magnetic beads and a foil magnet bar. (a) Without agitation the magnetic beads are attracted to the central foil magnet. (b) By magnetic stirring beads immediately homogenize within 2 sec. The black body underneath the SPE chamber is a screw-on version of the turning valve shown in Fig. 1
The core of the turning valves has on the opposite side of the TPE plate three asymmetrically arranged cavities which serve as receptacles for the three corresponding spring-loaded pins of a servo-motor. The zero position of the stepper- motor is detected by a photo-electric relay. After initializing the zero position, the motor precisely turns the valve according to the given number of moving steps.
The core of the controlling instrument is a PCR carrousel (Fig. 4), which enables a fast cycling of a high-volume (120 μl) PCR. Four resistive heating elements, each contained in one bracket, are symmetrically fixed on turnable discs. One disc is located above and one below the disposable chip. The temperatures of the corresponding heating brackets can be pairwisely set. Three pairs of these can be actively heated, while one corresponding pair of heating brackets serves as a passive cooling element having room temperature. An additional stepper motor moves two eccentric discs to travel the heating brackets towards and backwards the disposable processing chip. These eccentric discs are placed above the upper and below the lower heating elements. By bringing the heating elements directly in contact with the chip, the heat transfer is facilitated. After incubation for a given time (seconds), the heating elements are locked off, and the carrousel moves the turnable discs with the heating brackets to the next position. The next cycling step is accomplished by moving the corresponding pair of brackets towards the chip again. In order to increase the rate of cooling from e.g. 95°C to 52°C, the software programme moves for heat dissipation the non-heated pair of brackets towards the chip first, before the heating brackets preset to 52°C are attached to the cartridge to reach the final target temperature. Foil-magnet pieces within the PCR-chambers are moved by electromagnetic coils located at the lower part of the corresponding pair of heating brackets, supplying an alternating current. This enables a fast homogeneous heat transfer within the reaction mixture. The electromagnets run at AC mode with 50 Hz frequency. Using electromagnetic coils a tumbling action of the central magnets is generated, whereas the magnetic mini-stirrer induces a forced convection (Kim et al. 2009).
Fig. 4.

PCR carrousel with 4 heating clamps set on constant temperature. Left: Carrousel with chip inserted. Right: Detailed view on one heating clamp
Lysis and nucleic acid preparation
Flocked swabs from Copan Inc were used for sampling of nasopharyngeal specimen in accordance with manufacturers sample collection protocol. Within that, the swab is placed in a UTM transport medium tube filled with 1 ml of Copan’s Universal Transfer Medium (UTM) and glass beads for sample release and homogenization. The swab gets broken at its specified breakpoint, the tube is capped, and vortexed for at least 30 sec at the highest speed in order to release and homogenize the sample material from the swab. The performance of the developed LoC system was compared with a manually processed similar workflow in accordance with the ResPlex™ II Panel. As target either an in-vitro transcribed RNA fragment or a commercially available standard derived from the Zeptometrix “NATrol Respiratory Validation Panel” were spiked into UTM medium containing cell material derived from an eluted nasopharyngeal swab. One half of this artificial sample was processed with the LoC device, the other half was processed according to reference protocols.
For automated on-chip sample processing, 500 μl (1 volume) of the UTM sample extract was filled into the lysis chamber. The processing cartridge is inserted into the operating device, and the automated analysis is started. Within the lysis chamber, the sample gets mixed with 0.5 volume of a high-molar chaotropic salt containing lysis buffer (Buffer ML; QIAGEN). Beads of dried magnetic silica particles with Proteinase K and the iPAC are dissolved by the liquids, supported via mixing with the stirring bar at 1000 rpm. The sample is lysed by the buffer and by Proteinase K digestion at 56°C for 10 min. Another 0.5 volume of Buffer ML is pumped into the lysis chamber and incubated for further 5 min with stirring. Then, 1 volume of pure isopropanol is dispensed into the lysis mixture to adjust proper nucleic acid (NA) binding conditions and mixed for 1 min. Bead suspension is pumped with 0.6 ml/min through the SPE chamber into the waste. The magnetic beads are retained in the SPE chamber by the magnetic stirring bar and the permanent magnet underneath. During this transfer step, the magnetic stirring in the lysis chamber is turned on to prevent bead attraction to the central stirring magnet. After bead transfer into the SPE chamber, beads are serially flushed with 0.8 ml wash-buffer AW1 and 0.8 ml AW2 (both QIAGEN). Wash efficiency is increased by homogenizing the beads via magnetic stirring. All used wash buffers are pumped into the waste. In order to remove residual ethanol, beads are rinsed with 1 ml water without mixing. Elution of the bound NA is performed by mixing the beads with the remaining 100 μl water of the water rinse for 40 sec. The beads are magnetically separated by turning off the stirring motor. Finally, the entire volume of eluted NA is pumped into PCR chamber 1 for rehydration of the dried amplification reagents.
Amplification and detection of purified nucleic acids
The multiplex amplification for the detection of respiratory RNA viruses is done on the basis of the one-tube RT-PCR QIAplex method (Han et al. 2006, patent: WO002005038039A2). QIAplex-mediated detection involves two pairs of gene-specific nested primers at extremely low concentration. These primers are used for reverse transcription followed by the initial target specific enrichment. Efficient amplification is mediated with Super Primers, which are universal primers for all targets and bind to the complementary sequence of the 5’-tags of the inner nested primer pair. For detection purposes using a LiquiChip 200 workstation, the reverse Super Primer is labelled with 5’-biotin. All primer and detection oligo sequences were adopted from the QIAGEN ResPlex II Panel v2.0 (Brunstein and Thomas 2006). This panel is able to detect the following viruses: Respiratory syncytial viruses A or B (RSVA, RSVB); Influenza A virus (INFA); Influenza B virus (INFB); Parainfluenza viruses (PIV1, PIV2, PIV3, PIV4); Human metapneumoviruses A and B (hMPV); Coxsackieviruses/Echovirus (CVEV); Rhinovirus (RHV); Adenoviruses A or B (ADVB, ADVE); Coronaviruses (NL63, HKU1, 229E, OC43); Bocavirus (BOCV). In addition; an internal control (IC) and a positive control (IDS) can be amplified using this panel. The IDS is represented by a human X-chromosomal target serving for the detection of human DNA derived from the swab sample material.
The QIAplex nested-amplification principle was split into two separate amplification reactions. The reaction mixtures for RT-PCR 1 and PCR 2 are provided as freeze-dried pellets also containing primers and enzymes. These pellets are based on the QuantiTect Virus Kit (QIAGEN) in PCR chamber 1 and the QuantiFast Probe PCR Kit (QIAGEN) in PCR chamber 2, supplemented with cryo-protectives and bulking agents (formulation and freeze-drying protocols undisclosed). After reconstitution of the freeze-dried pellet with the entire eluate from the NA preparation in PCR chamber 1, the reverse transcription and the outer primer-dependent amplification take place. The cycling conditions are 10 min 52°C, 5 min 95°C, 20 × {10 sec 95°C, 40 sec 52°C, 10 sec 72°C} and 1 min 72°C. For the second PCR reaction, a volume of 1.6 μl of RT-PCR 1 is metered by a small interconnecting channel within the turning valve. This aliquot is flushed with water into the second PCR chamber to rehydrate the dried reagents contained therein. The cycling conditions for PCR 2 are 5 min 95°C, 5 × {10 sec 95°C, 30 sec 70°C}, 30 × {10 sec 95°C, 5 sec 52°C, 5 sec 70°C} and 1 min 70°C. During all incubation steps the magnetic stirring bars in PCR chamber 1 and PCR chamber 2 are moved.
For subsequent detection, PCR 2 is pumped through a 5 μl metering loop. This aliquot is transferred with 50 μl hybridization buffer into the hybridization chamber whereby the dried QIAplex beads (formulation undisclosed) are resuspended utilizing a magnetic stirrer. Here, amplicons are hybridized for 10 min at 52°C to colour-coded QIAGEN LiquiChip beads derived from the ResPlex™ II Panel kit. The bead suspension is then transferred into the labelling chamber containing dried streptavidin-R-phycoerythrin (formulation undisclosed). After 5 min incubation at 52°C, the dual-labelled complex is transferred into the storage well and kept at 52°C. By a tubing connection the sample is transferred into a QIAGEN LiquiChip 200 workstation for quantification. Here, the amount of phycoerythrin (secondary fluorescence) corresponding to the respective bead code (primary fluorescence) is used to determine the presence of each amplified target sequence. The detection of the hybridized and labelled target specific fragments is performed according to the LiquiChip® Applications Handbook and the ResPlex™ II Handbook (both QIAGEN).
Results and discussion
Amplification
To increase the sensitivity of the analysis, a high-volume nested PCR (120 μl) was implemented. While such volume exceeds clearly those typical for microfluidic systems, a reduction could be easily achieved with standard microfabrication techniques as the case may demand. However, the low volumes advantageous for microfluidic systems are rarely found in applications other than pure analytical testing and thus that we feel the present apparatus reflects real-world requirements. Also, the limited number of target molecules in low volumes often is a reason for poor sensitivity. Our choice of a 120 μl volume is further supported by the fact that for example the Roche LightCycler® SeptiFast Test, among others, uses a very similar volume of 100 μl in their PCR. Realistically, even the best sample preparation techniques cannot remove all PCR inhibitors present in a sample. However, the larger the PCR volume processed the less relevant are these inhibitors.
As described above, to allow for fast cycling of the high-volume PCR reactions, fast switching of temperatures was enabled by a turnable disc placing the heating elements above and underneath the right chambers as the PCR schedule requires. The lower limit of that switching time was determined (aside from biological or chemical limitations) by physical or technical factors (e.g. inertia of the system due to mass and mechanics, data rate transfer) pertaining to the construction of the PCR device or of the consumable cartridges. Flat chamber geometry with a large area of contact to the heating elements was chosen for efficient heat transfer and avoidance of temperature gradients within the reaction mixture. A typical time-dependent PCR temperature profile comprising clamp, fluid, and target temperatures is shown in Fig. 5. The targeted temperatures and corresponding durations are: 24 sec at 95°C, 40 sec at 56°C, 30 sec at 72°C, where each interval includes heating/cooling and hold times. The heating and cooling times are: approx. 8 sec for cooling from 95°C to 52°C yielding a cooling rate of 5.4°C/s, approx. 17 sec for heating from 52°C to 72°C yielding a heating rate of 1.2°C/s, and approx. 12 sec for heating from 72°C to 95°C yielding a heating rate of 1.9°C/s. By sequentially moving heating clamps set to constant temperatures to the PCR chamber, PCR times of around 30 min (30 cycles) can be reached (Fig. 5).
Fig. 5.
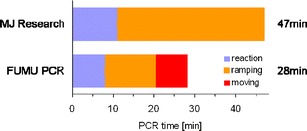
PCR cycling. Comparison of time consumption for 30 cycles using a conventional lab cycler system (MJ Research) and the heating carrousel (“Fumu PCR”) where times needed for the reaction itself, the temperature ramping and the movement of the clamps are indicated with different colours
For comparison of the overall time of a standard cycler (MJ Research) and the PCR carrousel, a PCR reaction was carried out using 40 pg/μl human gDNA as template and 30 cycles (Fig. 5). The MJ Research cycler took 47 min for a 25 μl reaction, while the PCR carrousel took 28 min for a 120 μl reaction, both amplifying a 66 bp fragment with comparable target yield after cycling (data not shown). By accelerating the mechanical moving steps the overall process time could be reduced even further.
One reason for the different heat transfer rates is that for the annealing step the forth heating clamp set to room temperature is used. A further reason for not identical heat transfer rates is a varying contact pressure of the different clamps to the processing cartridge. By having a sufficient thermal mass in the clamps it is made sure that they can transfer enough energy to the PCR chamber while not cooling down significantly. The thickness of the foil covering the PCR chambers is also important. On one hand, it should be as thin as possible so as to aid thermal energy transfer to the fluid, on the other hand, however, it should not be too thin to be deformed by the pressure of the heated clamps. The foil we used for bonding the cartridge is 175 μm thick in order to fulfil the discussed issues.
To further enhance heat transfer, the reactions are stirred during the incubation steps by small magnetic foil stirrers placed inside the chambers. So, simultaneous to the effect of an optimized homogenization of all components within the reaction, a uniform temperature distribution can be achieved. With this approach an overall PCR time of around 30 min for 30 cycles at a volume of 120 μl positions this system at the fast end of the spectrum of currently available PCR chips (Zhang and Ozdemir 2009).
Comparative performance experiments
The individual protocol steps performed in the LoC system (sample lysis, nucleic acid preparation, amplification, hybridization) were compared with standard methods (Fig. 6). In order to investigate the performance of the LoC chip regarding lysis and extraction, 4 million bacteriophage-fr particles per preparation were spiked in a swab sample in UTM. For the conventional approach the QIAGEN EZ1 Advanced system utilizing the EZ1 Virus Mini Kit for NA preparation was used according to the EZ1 virus Handbook. NA yields were compared via RT-PCR and PCR assays for bacteriophage fr and IDS targets using the QuantiTect Virus Kit (QIAGEN) for RNA and the QuantiFast Probe PCR Kit for DNA templates. The RNA yield was slightly lower (81%) and the DNA yield slightly higher (131%) for the LoC system compared to the EZ1 prepared RNA and DNA yields (Fig. 6 top left). These small differences might result from the assay specific performance based on the sample preparation method, but overall both preparation approaches revealed similar yields in RNA and DNA.
Fig. 6.

Efficiencies of module performance. Top left: swab sample extracts spiked with 4x106 PFU of bacteriophage-fr were lysed, RNA/DNA isolated and quantified via RT-qPCR and qPCR against bacteriophage-fr and human targets. Top right: 103 copies RSVB in vitro transcript were amplified in a nested multiplex RT-PCR, once conventionally in a reaction tube (pos. ctrl.) and once on-chip (on-chip RT-PCR), while the negative control (neg. ctrl.) was amplified in a reaction tube without in vitro transcripts. Bottom left: Influence of blocking reagents on the nested multiplex RT-PCR on-chip. Reactions were carried out on-chip without the use of blocking reagents (LoC-V1), use of blocking reagents in RT-PCR 1 and PCR 2 (LoC-V2) or use of blocking reagents only in RT-PCR 1 (LoC-V3). Positive control was done conventionally in reaction tubes without blocking reagents. Bottom right: DNA obtained by ResPlex II multiplex RT-PCR was hybridised and labelled for LiquiChip detection either conventionally in a reaction tube (Reference) or using dried reagents on–chip (LoC). (MFI = Median fluorescence intensities values measured by QIAGEN LiquiChip 200 workstation). (Per graph one representative experiment is shown.)
The efficiency of the nested RT-PCR reaction on the microchip was comparable to the manually performed standard procedures (Fig. 6 top right). As template for RT-PCR 1 1000 copies of RSVB in vitro transcripts were used. 1 μl of the first RT-PCR reaction was used as template for the second PCR reaction. 5 μl of the second PCR reaction were then processed and analyzed in the LiquiChip 200 workstation off-chip. Surprisingly, blocking reagents sustain the efficiency of the RT-PCR reaction but were unfavourable in the second PCR reaction (Fig. 6 bottom left). The efficiency of the nested RT-PCR was lower on-chip than in the conventional setup without the prior addition of blocking reagents (on-chip V1). Upon adding such blocking reagents in both PCR reactions (on-chip V2) it was further decreased. If blocking reagents are added to RT-PCR 1 only (on-chip V3), the resulting efficiency on-chip is comparable to that of conventional tubes.
For hybridisation and labelling experiments amplicons obtained by ResPlex II multiplex RT-PCR were used as sample. The on-chip labelling and hybridisation was carried out with freeze-dried colour-coded QIAGEN LiquiChip beads and streptavidin-R-phycoerythrin. Efficiency of this step was reduced using the dried reagents to about 60% of the results obtained with the conventional procedure and liquid reagents (Fig. 6 bottom right). This might be caused by activity losses of the beads and the labelling dye due to the freeze-drying protocol. To increase efficiency of this step it is necessary to investigate the drying procedure further on to identify appropriate conditions that preserve activities.
To investigate the performance of the complete sample-to-answer assay using the developed LoC system, a nasopharyngeal swab sample transferred into transport medium was spiked with 10 billion copies of in-vitro transcript of the target fragment of RSVB. We performed the complete processing protocol manually and with the LoC device using this medium as sample input material. All manual performed steps were done similar to the steps performed in the LoC device, applying freeze-dried reagents, too. For detection, the median fluorescence intensity values were measured with the LiquiChip 200 workstation. The via LoC system processed sample showed approx. 50% of the MFIs when compared to the manually processed sample. Optimizing the complex protocol further the different parameters will merge even better and better results will be achievable.
Additional benchmark tests were done to compare the performance of the prototype device with commercially available kits and nucleic acid preparation devices (preparation: BioRobot EZ1 Workstation with EZ1 Virus Mini Kit extraction protocol; amplification and detection: QIAGEN ResPlex II Panel v2.0). As a “positive” sample, we spiked 1 ml of Copan UTM medium that already contained the biomaterial from a nasopharyngeal swab with an ADV Standard from ZeptoMetrix Corp. The signal intensity of the via LoC system processed sample showed 81% of the signal intensity of the references. Since the experimental results vary strongly for the references already (see, e.g., MFIs in Figs. 6 and 7), results should be determined as signal ratios rather than absolute signal values.
Fig. 7.
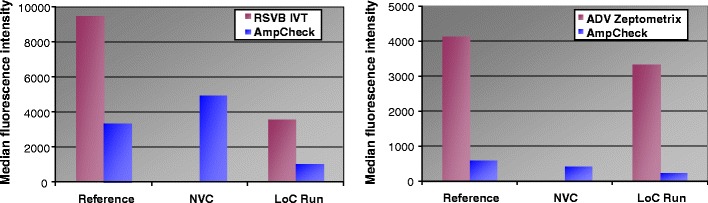
MFI measured by QIAGEN LiquiChip 200 workstation. Left: MFI of the RSVB in vitro transcripts and the internal controls (“Amp Check”). “Reference” samples were prepared manually. “LoC Run” denotes the fully integrated microsystem procedure. The non-virus-control (NVC) is the negative control (prepared on the operating device) without RSVB RNA added. Right: MFI of ADV Standard from ZeptoMetrix Corp. and the internal controls (“Amp Check”). “Reference” samples were prepared using the BioRobot EZ1 for automated sample preparation and the QIAGEN ResPlex II Panel v2.0 for amplification and detection. (In each figure, one representative experiment is shown.)
Conclusion
A LoC system is presented which allows automated pathogen analysis of respiratory viruses from nasopharyngeal samples. Next to functionality another focus of this development laid on the manufacturability of the system. By permanent checking manufacturability and availability of the components used, this approach led to a LoC system suitable for serial production. The system can easily be adapted to different pathogens and may thus offer a route to open new markets for in-vitro diagnostics. One remaining major challenge of LoC systems in general, is to make them more robust yet keep them affordable. It appears that simplifying both the assay and the instrument will be the key to achieve this goal (Kim et al. 2009). As it turned out during this work, much of what is today considered the state of the art in microfluidic construction strategies fails to meet said demands. We therefore think that our valve design and simple stirring process represent a clear improvement over established construction principles in terms of ease of production, robustness, and reliability.
Also, our PCR carrousel strategy produces cycling rates for 120 μl solutions that clearly outperform those of commercial end-point thermal cyclers. To our knowledge, our system is currently the fastest high-volume PCR chip. There are other chip developments exhibiting noticeably shorter cycling times, but these reactions are either based on pre-amplified DNA or have reaction volumes of less then 20 μl (Zhang and Xing 2007, Zhang and Ozdemir 2009 and references herein).
First tests using the developed system revealed that the overall performance of the LoC system is comparable with standard lab procedures, whereas there are differences in the efficiency of the individual steps. The differences in the overall results are most probably due to the complex protocol where different parameters have to fit exactly to each other. The functionality was demonstrated by the direct comparison of samples processed manually vs. automatically using the “ResPlex Panel II v2.0” for the detection of respiratory viruses from nasopharyngeal samples. Experiments showed comparable results of the developed system in comparison to conventional lab procedures and therefore provide an excellent basis for further optimization. It is envisaged to reduce complexity of the chip by simplifying the assay and the electro-mechanical device and to integrate the detection to end up with an all-in-one LoC-system.
Acknowledgement
We acknowledge support from the German Federal Ministry for Education and Research (BMBF) under grant agreement FKZ 16SV3318.
References
- Becker H. Lab Chip. 2009;9:2119. doi: 10.1039/b911553f. [DOI] [PubMed] [Google Scholar]
- Blow N. Nat. Meth. 2007;4(8):665. doi: 10.1038/nmeth0807-665. [DOI] [Google Scholar]
- Brunstein J, Thomas E. Diagn. Mol. Pathol. 2006;15:169. doi: 10.1097/01.pdm.0000210430.35340.53. [DOI] [PubMed] [Google Scholar]
- Chen L, Manz A, Day PJR. Lab Chip. 2007;7:1413. doi: 10.1039/b708362a. [DOI] [PubMed] [Google Scholar]
- Dobson MG, Galvin P, Barton DE. Expert Rev. Mol. Diagn. 2007;7(4):359. doi: 10.1586/14737159.7.4.359. [DOI] [PubMed] [Google Scholar]
- Fujimoto T, Konagaya M, Enomoto M, Tsuboi K, Hashimoto K, Taniguchi K, Kodama T, Okabe N. Jpn J. Infect. Dis. 2010;63(1):31. [PubMed] [Google Scholar]
- Han J, Swan DC, Smith SJ, Lum SH, Sefers SE, Unger ER, Tang YW. J. Clin. Microbiol. 2006;44(11):4157. doi: 10.1128/JCM.01762-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckle D. Expert Rev. Med. Devices. 2006;3(4):421. doi: 10.1586/17434440.3.4.421. [DOI] [PubMed] [Google Scholar]
- Ieven M. J. Clin. Virol. 2007;40:259. doi: 10.1016/j.jcv.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaigala GV, Behnam M, Bidulock ACE, Bargen C, Johnstone RW, Elliott DG, Backhouse CJ. Analyst. 2010;135:1606. doi: 10.1039/b925111a. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Wang F, Burns MA, Kurabayashi K. Anal. Chem. 2009;81(11):4510. doi: 10.1021/ac900512x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- J.A. Lounsbury, N. Coult, P. Kinnon, D. Saul, and J.P. Landers, Proc. μTAS 2010, 773, ISBN: 978-0-9798064-3-8 (2010)
- Sauer-Budge F, Mirer P, Chatterjee A, Klapperich CM, Chargin D, Sharon A. Lab Chip. 2009;9:2803. doi: 10.1039/b904854e. [DOI] [PubMed] [Google Scholar]
- Weigl B, Domingo G, LaBarre P, Gerlach J. Lab Chip. 2008;8:1999. doi: 10.1039/b811314a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Ozdemir P. Anal. Chim. Acta. 2009;638(2):115. doi: 10.1016/j.aca.2009.02.038. [DOI] [PubMed] [Google Scholar]
- Zhang C, Xing D. NAR. 2007;35(13):4223. doi: 10.1093/nar/gkm389. [DOI] [PMC free article] [PubMed] [Google Scholar]