

Abstract
Background
Fibrinogen-like protein 2 (FGL2), a new member of the fibrinogen-like family, has recently been identified as a novel immunosuppressive molecule.
Aim
The purpose of this work was to investigate intestinal and peripheral expression of FGL2 in patients with inflammatory bowel disease (IBD), mainly ulcerative colitis (UC) and Crohn’s disease (CD).
Methods
FGL2 expression in mucosal biopsies from three groups (UC group (n = 61), CD group (n = 54), and controls group (n = 35)) was detected by immunohistochemistry. Concentrations of FGL2 in plasma from 50 UC patients, 45 CD patients, and 30 controls were analyzed by enzyme-linked immunosorbent assay. Western blot of FGL2 protein and real-time fluorescent quantitative PCR of FGL2 mRNA expression by peripheral mononuclear cells was performed. Correlations of FGL2 expression with disease type, activity, and location, and with measured laboratory data, including C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR), were examined.
Results
Intestinal and peripheral FGL2 protein data showed that FGL2 expression was significantly up-regulated in both UC and CD patients compared with controls (P < 0.001). Expression of FGL2 was higher in UC and CD patients with active disease than in those with inactive disease (P < 0.001). Moreover, FGL2 mRNA expression was significantly higher in patients with active disease than in those with inactive disease (P < 0.050). Expression of FGL2 protein was correlated with disease activity indices, CRP levels, and ESR levels.
Conclusion
Expression of FGL2 was up-regulated in IBD patients with active disease. Measurement of FGL2 may be used as a helpful biomarker for understanding immunopathogenesis and for assessment of IBD.
Keywords: Inflammatory bowel disease, Fibrinogen-like protein 2, Immunomodulation, Ulcerative colitis, Crohn’s disease, Regulatory T cell
Introduction
Inflammatory bowel disease (IBD) is a chronic relapsing and remitting inflammatory condition of the gastrointestinal (GI) tract that mainly includes two clinical entities, ulcerative colitis (UC) and Crohn’s disease (CD). Both are frequently associated with systemic manifestations and with increased risk of colon cancer. In recent decades there has been a continuing trend of greater incidence and prevalence of IBD throughout the world, particularly in East Asia [1], so more attention must be paid to effective diagnosis and treatment of IBD.
Although the exact causes and mechanisms of IBD have not been completely elucidated, the prevailing hypothesis of IBD pathogenesis is that the disease occurs in genetically susceptible individuals as the result of a complex interaction among environmental factors, microbial factors, and the intestinal immune system [2–4]. Furthermore, accumulated evidence indicates that an inappropriate and persistent immune response is central to the development of both major types of IBD [5–8]. An excess of inflammatory stimuli and mediators, and an inadequately low function or number of regulatory components that down-regulate the mucosal immune response can lead to a chronic, progressive inflammatory condition, and eventually to intestinal tissue damage [9, 10].
Fibrinogen-like protein 2 (FGL2), also known as fibroleukin, was first cloned from cytotoxic T lymphocytes and was classified as a member of fibrinogen superfamily because of its homology (36 %) with fibrinogen β and γ chains [11, 12]. Originally described as an immune coagulant with the ability to generate thrombin directly, FGL2 has been implicated in the pathogenesis of several inflammatory disorders, including viral hepatitis and experimental arthritis [13, 14]. It has recently been demonstrated that FGL2 is an effector molecule on regulatory T cells (Treg cells) [15–18]. FGL2 may bind to Fc gamma receptors (FcγR), which are expressed on antigen-presenting cells (APC), and then inhibits maturation of dendritic cells (DC) and induces apoptosis of B cells, eventually resulting in reduced ability to induce alloreactive T cell proliferation [16, 19]. All these findings collectively indicate FGL2 has potent immunosuppressant properties.
Because there has been no suggestion FGL2 is involved in the immunopathogenesis of IBD, we felt it was necessary to investigate expression of FGL2 in patients with IBD and discuss its involvement.
Materials and Methods
Mucosal Biopsy Specimens
The study was performed at the Department of Gastroenterology and Hepatology of the First Affiliated Hospital of Wenzhou Medical University from December 2006 to December 2012. Mucosal biopsy specimens were obtained from macroscopically inflamed areas of patients with UC (n = 61) or CD (n = 54), and from normal controls (NC, n = 35), i.e. from the normal areas of healthy subjects or patients with colonic polyp (Table 1).
Table 1.
Clinical details of the subjects included in the study (mucosal biopsy specimens)
| Characteristics | Normal controls | Ulcerative colitis | Crohn’s disease |
|---|---|---|---|
| Number | 35 | 61 | 54 |
| Gender | |||
| Male | 19 | 31 | 33 |
| Female | 16 | 30 | 21 |
| Mean age (years) | 40.8 | 44.2 | 37.2 |
| Active | 44 | 38 | |
| Inactive | 17 | 16 | |
| Disease localization | |||
| Proctitis (UC)/ileum (CD) | 28 | 10 | |
| Left sided colitis (UC)/colon (CD) | 14 | 22 | |
| Total colitis (UC)/ileum + colon (CD) | 19 | 22 | |
Peripheral Blood
Matched peripheral blood was available from patients with UC (n = 50) or CD (n = 45) at Department of Gastroenterology and Hepatology of First Affiliated Hospital of Wenzhou Medical University who were enrolled from January 2012 to April 2013 (Table 2). These were compared with samples from 30 normal controls, who were recruited from healthy blood donors, visitors of hospital wards, and normal hospital personnel.
Table 2.
Clinical details of the subjects included in the study (peripheral blood)
| Characteristics | Normal controls | Ulcerative colitis | Crohn’s disease |
|---|---|---|---|
| Number | 30 | 50 | 45 |
| Gender | |||
| Male | 17 | 28 | 26 |
| Female | 13 | 22 | 19 |
| Mean age (years) | 40.2 | 44.6 | 40.0 |
| Active | 35 | 30 | |
| Inactive | 15 | 15 | |
| Disease localization | |||
| Proctitis (UC)/ileum (CD) | 21 | 11 | |
| Left sided colitis (UC)/colon (CD) | 15 | 13 | |
| Total colitis (UC)/ileum + colon (CD) | 14 | 21 | |
Inclusion and Assessment of Patients
Diagnosis of UC and CD was based on standard criteria [20]. UC and CD patients with active disease enrolled in our study had not received any immunomodulatory medications for their disease. Most UC patients with inactive disease were undergoing maintenance treatment with low-dose 5-aminosalicylic acid; others had stopped all drugs. CD patients with inactive disease were undergoing maintenance treatment with low-dose azathioprine, or low-dose azathioprine and methylprednisolone. Disease activity in UC patients was evaluated by use of the Truelove–Witts criteria [21], For statistical purposes, the classification was quantitatively modified [22]. Active disease was defined as score >3 and inactive disease as score ≤3. For patients with CD, the severity of the disease was classified in accordance with the CDAI score [23]. Active disease was defined as CDAI score ≥150 and inactive disease as CDAI score <150. Disease activity was evaluated at the time of sample collection.
Laboratory Studies
Standard laboratory data, including red and white blood cell count, hemoglobin, hematocrit, platelet count, erythrocyte sedimentation rate (Alifax Test1; Italy) and C-reactive protein (Beckman Coulter detection kit; Japan) were routinely measured for all patients with UC and CD.
Preparation of Sections and Immunohistochemistry Staining
Mucosal biopsy specimens were fixed with 4 % paraform, processed into paraffin, and sectioned for immunohistochemical staining of FGL2. The sections were initially deparaffinated in xylene and rehydrated through ethanol to water. Nonspecific binding was blocked by sequential incubation of the sections in citrate buffer for 4 min at 100 °C and 16 min at 20 °C, then in 3 % hydrogen peroxidase solution for 15 min followed by 5 % normal goat serum in PBS at 37 °C for 30 min. Thereafter sections were incubated with mouse anti-human FGL2 monoclonal antibody (Abnova, Taiwan) at a dilution of 1:500 in PBS at 4 °C for 16 h. After washing with PBS, sections were incubated with immunoperoxidase-conjugated rabbit IgG fraction to mouse IgG Fc (Zhongshan, Beijing, China) at 37 °C for 30 min, followed by three washes in PBS. Finally, the sections were incubated with 3,3′-diaminobenzidine chromagen and counterstained with hematoxylin. A negative control was used in the experiment. For evaluation of FGL2 expression, ten random fields across each section were selected for semi-quantitative analysis of mean absorbance at a magnification of 200×.
Plasma Preparation and Enzyme-Linked Immunosorbent Assay
All blood samples were drawn between 6 and 9 am, after fasting. After a resting period of 20 min, non-traumatic venipuncture was performed in a standardized manner by trained operators. Blood (2.7 mL) was drawn into test tubes with 0.3 mL citrate. Within 2 h of sample collection, the samples were centrifuged for 15 min (3,000×g) at 4 °C and stored at −80 °C in plastic tubes. Before serial analysis, the plasma was thawed immediately in a water bath at 37 °C for 5 min. In accordance with the manufacturer’s instructions, plasma concentrations of FGL2 were measured by use of a commercially available enzyme-linked immunosorbent assay (ELISA; BioLegend, USA).
Protein Preparation and Western Blot Analysis
Peripheral blood was obtained and peripheral blood mononuclear cells (PBMC) were isolated by use of Ficoll density gradients (Solarbio, Shanghai, China) for Western blot analysis. Lysate protein (40 μg) extracted from PBMC was loaded on to 10 % SDS–polyacrylamide gels. After separation, the proteins were transferred to a nitrocellulose (NC) membrane. The membrane was blocked and probed with a monoclonal antibody against FGL2 (Abnova, Taiwan) at a dilution of 1:500 in 5 % milk in TBST. After washing with TBST, the blot was incubated with secondary antibodies conjugated to horseradish peroxidase (Biosharp, Hefei, China). Immunoreactive bands were detected with the enhanced chemiluminescence (ECL) reagent (Pierce Biotechnology, Shanghai, China). Protein levels, normalized against GAPDH, were determined by densitometric analysis using Quantity One Version 4.
RNA Preparation and Real-Time Fluorescent Quantitative PCR
Total RNA was isolated from PBMC by use of TRIzol reagent (Invitrogen, Shanghai, China) in accordance with the manufacturer’s procedure. The concentration and purity of RNA were determined by measurement of absorbance at 260 and 280 nm. Subsequently, the cDNAs were synthesized (TaiGen Biotechnology, China). The nucleotide sequences of the primers for PCR amplification of the 169 bp fragment of FGL2 were: sense primer, 5′-ACTGTGACATGGAGACCATG-3′, and antisense primer, 5′-TCCTTACTCTTGGTCAGAAG-3′. The amplified 145 bp fragment of GAPDH was used as an internal control to ensure equal loading with forward primer, 5′-TCCCATCACCATCTTCCAGG-3′ and reverse primer, 5′-GATGACCCTTTTGGCTCCC-3′ (Life Technologies, Shanghai, China). In the PCR reaction the cDNA was denatured at 95 °C for 3 min, and amplified over 40 cycles of 95 °C (15 s), 64 °C (1 min). The real-time PCR reactions were performed in SYBR Green Real-time PCR Master Mix Plus (Toyobo, Japan) by use of an ABI 7500 Sequence-Detection System (Applied Biosystems, Carlsbad, CA, USA). The specificity of the PCR reaction was verified by dissociation-curve analysis. FGL2 mRNA relative quantification was calculated by use of the method.
Ethical Considerations
The protocol of this study was approved by the clinical research ethics committee of the First Affiliated Hospital of Wenzhou Medical University. All the subjects enrolled in our study had been informed and had given written consent.
Statistical Analysis
All results were expressed as mean ± SD. Statistical analysis was conducted with SPSS 16.0 software. Continuous measurements among the three diagnostic groups were compared by one-way ANOVA. Post-hoc multiple comparisons were performed by use of Dunn’s test. The same tests were used for comparisons of disease activity or disease location among the different groups. The association between FGL2 expression and disease activity indices or other laboratory data, including CRP levels and ESR levels, was examined by non-parametric correlation (Spearman’s r). A level of P < 0.050 was considered to be statistically significant.
Results
FGL2 Expression Was Higher in Mucosal Biopsy Tissue from Patients with IBD
Compared with normal controls, FGL2 expression was significantly up-regulated in inflamed mucosal biopsy tissues from patients with UC and CD (both UC vs NC and CD vs NC, P < 0.001) (Figs. 1a–l, 2a). Expression of FGL2 was significantly higher in UC and CD patients with active disease than in those with inactive disease (P < 0.001) (Figs. 1e–p, 2b, c). FGL2 expression in patients with inactive disease was, however, still higher than that in controls (both inactive UC vs NC, inactive CD vs NC, P < 0.001) (Figs. 1e–p, 2b, c). Disease location in UC and CD patients was no different. There was an association between FGL2 expression and clinical indices of activity—Truelove–Witts for UC and CDAI for CD (r = 0.528, P < 0.001; r = 0.331, P = 0.015, respectively). Moreover, our results suggested that enhanced FGL2 expression was correlated with ESR levels in both UC and CD patients (r = 0.491, P < 0.001; r = 0.357, P = 0.008, respectively) but not with levels of CRP.
Fig. 1.
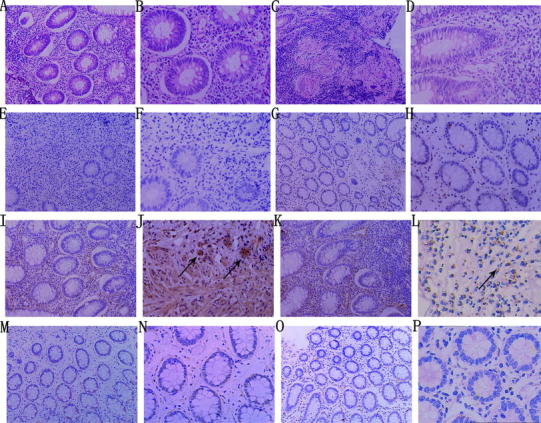
Representative photographs of hematoxylin–eosin (HE) staining and immunohistochemical staining of FGL2 in mucosal biopsy tissues. a–d HE staining of tissues from patients with UC and CD; a large number of monocytes and neutrophils infiltrating the intestinal tissues were observed (a UC ×200, b UC ×400, c CD ×200, d CD ×400). e, f Negative controls showed no positive expression of FGL2 (e ×200, f ×400). g, h Normal controls showed little or no FGL2 expression (g ×200, h ×400). i–l FGL2 was strongly expressed in inflammatory infiltrating cells and endothelial cells (black arrows) of tissues from patients with active UC or CD (i active UC ×200, j active UC ×400, k active CD ×200, l active CD ×400). m–p Expression of FGL2 was less in patients with inactive UC or CD (m inactive UC ×200, n inactive UC ×400, o inactive CD ×200, p inactive CD ×400)
Fig. 2.
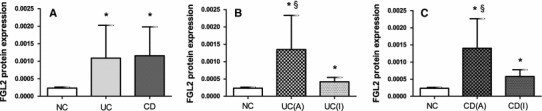
FGL2 expression in mucosal biopsy tissues. a Column diagrams represent expression of FGL2 in the normal controls group (NC group, n = 35), ulcerative colitis group (UC group, n = 61), and Crohn’s disease group (CD group, n = 54). b Expression of FGL2 in the NC group (n = 35), the active UC group (n = 44), and the inactive UC group (n = 17). c Expression of FGL2 in the NC group (n = 35), the active CD group (n = 38), and the inactive CD group (n = 16). The clinical details of the subjects are listed in Table 1. Data are presented as mean ± SD. UC (A) active UC, UC (I) inactive UC, CD (A) active CD, CD (I) inactive CD. *Significance compared with NC (P < 0.001); §significance compared with patients with inactive disease (P < 0.001)
Level of FGL2 Was Elevated in Plasma from Patients with IBD
Mean plasma FGL2 levels were 9.373 ± 4.482 ng/mL (95 % CI 7.700–11.047) for normal controls, 27.068 ± 13.542 ng/mL (95 % CI 23.220–30.916) for UC patients, and 29.974 ± 17.074 ng/mL (95 % CI 24.844–35.104) for CD patients. The differences between the groups were statistically significant (P < 0.001). Multiple comparisons tests showed that FGL2 levels were significantly higher in both UC and CD patients than in controls (P < 0.001), but there was no difference between UC and CD patients (Fig. 3a). Mean plasma FGL2 levels were 32.132 ± 12.828 ng/mL (95 % CI 27.726–36.539) for active UC patients, 15.251 ± 5.3344 ng/mL (95 % CI 12.2974–18.205) for inactive UC patients, 36.172 ± 17.592 ng/mL (95 % CI 29.603–42.741) for active CD patients, and 15.579 ± 5.314 ng/mL (95 % CI 14.636–20.522) for inactive CD patients. FGL2 levels were significantly higher for patients with active UC or CD than for those with inactive disease (P < 0.001), and FGL2 levels were significantly higher for patients with inactive disease than for controls (both inactive UC vs NC, inactive CD vs NC, P < 0.001) (Fig. 3b, c). Disease location in UC and CD patients was no different. There was an association between FGL2 levels and clinical indices of activity—Truelove–Witts for UC and CDAI for CD (r = 0.791, P < 0.001; r = 0.561, P < 0.001, respectively). Furthermore, the enhanced FGL2 levels were correlated with CRP levels for both UC and CD patients (r = 0.284, P = 0.046; r = 0.352, P = 0.018, respectively), and with levels of ESR (r = 0.440, P < 0.001; r = 0.400, P < 0.001, respectively).
Fig. 3.
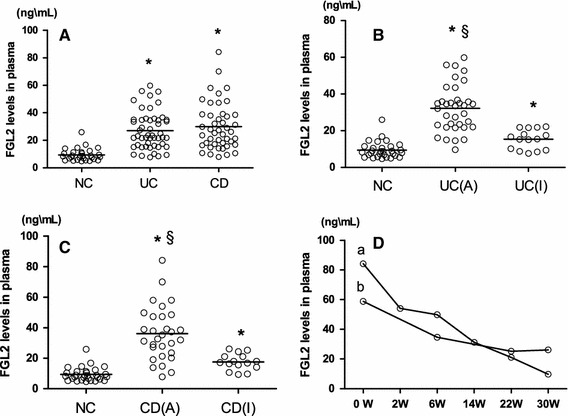
FGL2 levels in plasma. Each individual patient or control is shown as a circle, and bold lines are the mean values. a Levels of FGL2 in the NC group (n = 30), UC group (n = 50), and CD group (n = 45). b Levels of FGL2 in the NC group (n = 30), active UC group (n = 35), and inactive UC group (n = 15). c FGL2 levels in the NC group (n = 30), active CD group (n = 30), and inactive CD group (n = 15). The clinical details of the subjects are listed in Table 2. d Two CD patients with active disease, in both of whom the ileum and colon were involved, were both undergoing anti-TNF-α and azathioprine therapy. The line graphs labeled a and b represent the FGL2 levels of a 20-year-old female patient and a 24-year-old male patient, respectively, during the period of treatment. Each time is shown as a circle. UC (A) active UC, UC (I) inactive UC, CD (A) active CD, CD (I) inactive CD. *Significance compared with NC (P < 0.001); §significance compared with patients with inactive disease (P < 0.001)
In addition, two CD patients with active disease, both of whom underwent anti-tumor necrosis factor α (TNF-α) and azathioprine therapy, were followed-up regularly in our study. The levels of FGL2 in one patient were 84.200, 54.000, 49.750, 31.200, 21.050, and 9.700 ng/mL during the period of treatment on the 0th, the 2nd, the 6th, the 14th, the 22nd and the 30th week respectively. The FGL2 levels of another patient were 58.800 ng/mL before the treatment, 34.60 ng/mL on the 6th week, 25.200 ng/mL on the 22nd week, 26.100 ng/mL on the 30th week of treatment. The decreased FGL2 levels were in parallel with the clinical cut-down CDAI scores (Fig. 3d).
FGL2 Protein Was Up-Regulated in PBMC from IBD Patients with Active Disease
Western blot analysis revealed that FGL2 protein expression, normalized against GAPDH, in PBMC was 0.738 ± 0.386 (95 % CI 0.593–0.882) for patients with UC, and 0.907 ± 0.3686 (95 % CI 0.767–1.0469) for patients with CD, compared with 0.662 ± 0.311 (95 % CI 0.546–0.778) for controls. It was markedly elevated in patients with CD in comparison with normal controls (P = 0.023), and there was no statistically significant difference between patients with UC and controls (Fig. 5a). FGL2 protein expression was 0.926 ± 0.395 (95 % CI 0.716–1.136) and 1.107 ± 0.289 (95 % CI 0.958–1.255), respectively, in UC and CD patients with active disease, and 0.738 ± 0.386 (95 % CI 0.382–0.662) and 0.624 ± 0.272 (95 % CI 0.451–0.797), respectively, in UC and CD patients with inactive disease. Expression of FGL2 protein was higher in UC and CD patients with active disease than in those with inactive disease (P < 0.001). However, expression of FGL2 was no higher in patients with inactive disease than in controls (Figs. 4, 5b, c).
Fig. 5.
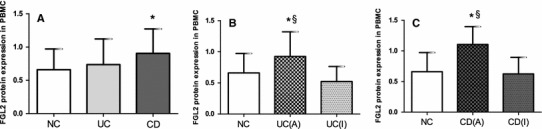
FGL2 protein expression in peripheral blood mononuclear cells (PBMC). a Column diagrams represent expression of FGL2 in the NC group, UC group, and CD group. The NC group (n = 30) contained 18 males and 12 females; the mean age was 40.5 years. The UC group (n = 30) contained 17 males and 13 females; the mean age was 44.3 years. The CD group (n = 29) contained 17 males and 12 females; the mean age was 35.4 years. b Expression of FGL2 in the NC group, active UC group, and inactive UC group. The active UC group (n = 16) contained 9 males and 7 females; the mean age was 48.3 years. The inactive UC group (n = 14) contained 8 males and 6 females; the mean age was 40.3 years. c Expression of FGL2 in the NC group, active CD group, and inactive CD group. The active CD group (n = 17) included 10 males and 7 females; the mean age was 38.9 years. The inactive CD group (n = 12) contained 7 males and 5 females; the mean age was 32.0 years. Data are presented as mean ± SD. UC (A) active UC, UC (I) inactive UC, CD (A) active CD, CD (I) inactive CD. *Significance compared with NC (P < 0.001); §significance compared with patients with inactive disease (P < 0.001)
Fig. 4.

Representative photographs of Western blot analysis of FGL2 protein expression. The molecular mass of FGL2 is 64 kDa, and that of GAPDH is 37 kDa. NC normal controls, UC (A) active UC, UC (I) inactive UC, CD (A) active CD, CD (I) inactive CD
FGL2 mRNA Was Up-Regulated in PBMC from IBD Patients with Active Disease
Relative expression of FGL2 mRNA in PBMC, normalized against 1 for controls, was 1.286 ± 0.926 (95 % CI 0.919–1.652) for patients with UC and 1.299 ± 0.818 (95 % CI 0.981–1.616) for patients with CD. Differences between the three groups were not statistically significant (Fig. 6a). Relative expression of FGL2 mRNA was 1.686 ± 0.994 (95 % CI 1.156–2.215) and 1.698 ± 0.738 (95 % CI 1.331–2.065), respectively, for UC and CD patients with active disease, and 0.704 ± 0.350 (95 % CI 0.469–0.939) and 0.580 ± 0.294 (95 % CI 0.370–0.791), respectively, for UC and CD patients with inactive disease. FGL2 mRNA expression was significantly higher for UC and CD patients with active disease than for those with inactive disease (P < 0.050) (Fig. 6b, c). However, FGL2 mRNA expression was lower for patients with inactive disease than for controls (both inactive UC vs NC, inactive CD vs NC, P < 0.050) (Fig. 6b, c).
Fig. 6.
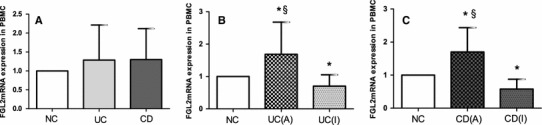
FGL2 mRNA expression in peripheral blood mononuclear cells (PBMC). a Column diagrams represent expression of FGL2 in the NC group, the UC group, and the CD group. The NC group (n = 24) contained 14 males and 10 females; the mean age was 40.3 years. The UC group (n = 27) contained 14 males and 13 females; the mean age was 46.1 years. The CD group (n = 28) contained 16 males and 12 females; the mean age was 36.9 years. b Expression of FGL2 mRNA in the NC group, the active UC group, and the inactive UC group. The active UC group (n = 16) contained 9 males and 7 females; the mean age was 50.8 years. The inactive UC group (n = 11) contained 5 males and 6 females; the mean age was 41.4 years. c Expression of FGL2 mRNA in the NC group, the active CD group, and the inactive CD group. The active CD group (n = 18) contained 10 males and 8 females; the mean age was 42.5 years. The inactive CD group (n = 10) contained 6 males and 4 females; the mean age was 31.3 years. Data are presented as mean ± SD. UC (A) active UC, UC (I), inactive UC, CD (A) active CD, CD (I) inactive CD. *Significance compared with NC (P < 0.050); §significance compared with patients with inactive disease (P < 0.050)
Discussion
The intestinal lumen is home to more than 500 strains of bacteria, so the GI mucosal immune system must strike a delicate homeostatic balance between maintaining tolerance toward the commensal microflora and remaining poised to mount an aggressive immune response against invading pathogens. Under normal conditions, the epithelial barrier continually samples antigen from the lumen and presents it to lymphocytes in the Peyer’s patches to promote tolerance. When it encounters invasion of pathogens, APCs in the mucosa, including macrophages, DC, and intestinal epithelial cells, phagocytose the invading pathogens and present their components to naive CD4+ T cells. The T cells then undergo activation and differentiation to an effector phenotype (i.e., Th1, Th2, or Th17) or a regulatory phenotype (i.e., Th3 or Treg) [9]. While effector T-helper cells mount specific immune responses by expression of unique cytokines to combat the invading pathogens, Treg cells suppress inflammation and restore homeostasis to the mucosal tissues by expression of regulatory cytokines, for example interleukin-10 (IL-10) and transforming growth factor (TGF-β).
However, there is an inherent defect in the mucosal immune system of IBD patients [6]. Although the exact nature of this defect has not been identified, mounting evidence indicates that IBD occurs in the presence of dubious antigens, most likely normal resident luminal bacterial and/or food-derived, which initiate a dysregulated immune response within the intestinal mucosa and the eventual development of chronic intestinal inflammation [8]. Furthermore, there has been sustained interest in the importance of Treg cells in IBD. It seems that fewer circulating Treg cells are present in patients with active IBD, but insufficiently increased in the intestinal mucosa [24]. Subsequent research suggested the increased apoptosis of Treg cells in IBD can be reversed by anti-TNF-α therapy, and this would be a critical factor in the recurrence of disease [25, 26]. Nevertheless, the clearly core mechanism of Treg-mediated suppression is still controversial. Although IL-10, TGF-β and some other molecules have been reported to account for the regulatory activity of Treg cells, antibodies to these molecules in some cases had no effect or only minimally inhibited Treg cells activity in vitro [27–29]. Recent findings have revealed the possible importance of the contribution of FGL2 to the suppressive activity of Treg cells [18, 30, 31].
FGL2, consists of 432 aa and contains a C-terminal fibrinogen-related domain (FRED), which is a highly conserved region and is characteristic of proteins within the fibrinogen superfamily [11]. These functionally diverse proteins, including fibrinogen, tenascin, angiopoietin, and ficolin, have been shown to have immunoregulatory activity [32, 33]. Similarly, much research has recently revealed that FGL2, secreted by T cells, also has immunomodulatory activity [17]. FGL2 mRNA increased in CD4+CD25+Foxp3+ Treg cells, along with mRNA for several known Treg suppression effector molecules [15, 34]. Furthermore, Shalev et al. [18] reported that the suppressive activity of FGL2−/− Treg cells was significantly impaired in FGL2−/− mice, and antibody to FGL2 completely inhibited the activity of FGL2+/+ Treg cells in vitro. Consistent with the contribution of FGL2 to the activity of Treg cells, targeted deletion of the FGL2 gene led to an increase in immune reactivity of DC, T cells, and B cells, and the development of autoimmune glomerulonephritis in aged FGL2−/− mice. The regulatory activity of FGL2 also contributes to inhibition of allograft rejection and the pathogenesis of experimental and human viral infections, including HIV, severe acute respiratory syndrome (SARS), hepatitis B virus, and hepatitis C virus [18, 30, 31, 35]. All this evidence strongly supports the hypothesis that FGL2 is an effector molecule of Treg cells. The mechanism whereby FGL2 exerts its immunomodulatory activity has been demonstrated by Liu et al. [19]. FGL2 may bind to FcγRIIB, which is expressed on APCs, and then inhibit DC maturation and induce B cells apoptosis, eventually resulting in reduced ability to induce alloreactive T cell proliferation.
Given that IBD shares immunologic features with autoimmune diseases to some extent, it is logical to hypothesize that the immunomodulatory activity of FGL2 may also be involved in the immunopathogenesis of IBD. However, our study demonstrated that intestinal and peripheral expression of FGL2 was significantly higher in UC and CD patients with active disease, and decreased in inactive disease. Moreover, expression of FGL2 was positive correlated with disease activity indices, CRP levels, and ESR levels. Up-regulation of FGL2 in both UC and CD patients with active disease may be an insufficient compensation secreted by Treg cells which fails to counter chronically activated effector T cells, and which leads to inappropriate immune responses in IBD. Reduced FGL2 expression in patients with inactive disease may be because successful treatment, including immune modifiers, anti-TNF-α therapy, and other anti-inflammatory treatment, made up for the deficiency of Treg cells, or reversed the deficiency to exert sufficient suppression against other subsets of T lymphocytes through multiple other suppression molecules. This assumption is in agreement with the results of Maul et al. [24], which reported a decrease of peripheral Treg cells and an insufficient increase in intestinal lesions, and also consistent with therapeutic strategies in which immune modifiers [1] or anti-TNF-α therapy [25] are used.
In conclusion, the results of our study of FGL2 expression in IBD have important theoretical implications for our understanding the immunopathogenesis of IBD, and practical implications for its therapy. FGL2 may be a helpful biomarker of the pathogenesis of IBD and for assessment of the disease. To confirm this, we have now initiated further investigation of the involvement of FGL2 among patients with IBD.
Acknowledgments
The authors are grateful to Chengcheng Chen, Silu Wang, and Fajing Yang, Department of Surgery Laboratory, the First Affiliated Hospital of Wenzhou Medical University, for assistance with technical support and consulting. This work was supported by the Science and Technology Project of Wenzhou City, China (Y20130037).
Conflict of interest
None.
References
- 1.Bernstein CN, Fried M, Krabshuis JH, et al. World Gastroenterology Organization practice guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis. 2010;16:112–124. doi: 10.1002/ibd.21048. [DOI] [PubMed] [Google Scholar]
- 2.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagalingam NA, Lynch SV. Role of the microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2012;18:968–984. doi: 10.1002/ibd.21866. [DOI] [PubMed] [Google Scholar]
- 4.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 5.Rb S. Pathagenesis and immune mechanisms of chronic inflammatory bowel diseases. Am J Gastroenterol. 1997;92:5S–11S. [PubMed] [Google Scholar]
- 6.Matricon J. Immunopathogenesis of inflammatory bowel disease. Med Sci. 2010;26:405–410. doi: 10.1051/medsci/2010264405. [DOI] [PubMed] [Google Scholar]
- 7.Scarpa M, Stylianou E. Epigenetics: concepts and relevance to IBD pathogenesis. Inflamm Bowel Dis. 2012;18:1982–1996. doi: 10.1002/ibd.22934. [DOI] [PubMed] [Google Scholar]
- 8.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arseneau KO, Tamagawa H, Pizarro TT, Cominelli F. Innate and adaptive immune responses related to IBD pathogenesis. Curr Gastroenterol Rep. 2007;9:508–512. doi: 10.1007/s11894-007-0067-3. [DOI] [PubMed] [Google Scholar]
- 10.O’Garra A, Vieira P. Regulatory T cells and mechanisms of immune system control. Nat Med. 2004;10:801–805. doi: 10.1038/nm0804-801. [DOI] [PubMed] [Google Scholar]
- 11.Koyama THL, Haser WG, Tonegawa S, Saito H. Structure of a cytotoxic T-lymphocyte-specific gene shows a strong homology to fibrinogen beta and gamma chains. Proc Natl Acad Sci USA. 1987;84:1609–1613. doi: 10.1073/pnas.84.6.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruegg C, Pytela R. Sequence of a human transcript expressed in T-lymphocytes and encoding a fibrinogen-like protein. Gene. 1995;160:257–262. doi: 10.1016/0378-1119(95)00240-7. [DOI] [PubMed] [Google Scholar]
- 13.Levy GA, Liu M, Ding J, et al. Molecular and functional analysis of the human prothrombinase gene (HFGL2) and its role in viral hepatitis. Am J Pathol. 2000;156:1217–1225. doi: 10.1016/S0002-9440(10)64992-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melnyk MC, Shalev I, Zhang J, et al. The prothrombinase activity of FGL2 contributes to the pathogenesis of experimental arthritis. Scand J Rheumatol. 2011;40:269–278. doi: 10.3109/03009742.2010.536163. [DOI] [PubMed] [Google Scholar]
- 15.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 16.Marazzi S, Blum S, Hartmann R, et al. Characterization of human fibroleukin, a fibrinogen-like protein secreted by T lymphocytes. J Immunol. 1998;161:138–147. [PubMed] [Google Scholar]
- 17.Chan CW, Kay LS, Khadaroo RG, et al. Soluble fibrinogen-like protein 2/fibroleukin exhibits immunosuppressive properties: suppressing T cell proliferation and inhibiting maturation of bone marrow-derived dendritic cells. J Immunol. 2003;170:4036–4044. doi: 10.4049/jimmunol.170.8.4036. [DOI] [PubMed] [Google Scholar]
- 18.Shalev I, Liu H, Koscik C, et al. Targeted deletion of fgl2 leads to impaired regulatory T cell activity and development of autoimmune glomerulonephritis. J Immunol. 2008;180:249–260. doi: 10.4049/jimmunol.180.1.249. [DOI] [PubMed] [Google Scholar]
- 19.Liu H, Shalev I, Manuel J, et al. The FGL2-FcgammaRIIB pathway: a novel mechanism leading to immunosuppression. Eur J Immunol. 2008;38:3114–3126. doi: 10.1002/eji.200838338. [DOI] [PubMed] [Google Scholar]
- 20.Lennard-Jones JE. Classification of inflammatory bowel disease. Scand J Gastroenterol. 1989;170:2–6. doi: 10.3109/00365528909091339. [DOI] [PubMed] [Google Scholar]
- 21.Truelove SC, Witts LJ. Cortisone in ulcerative colitis; final report on a therapeutic trial. Br Med J. 1955;2:1041–1048. doi: 10.1136/bmj.2.4947.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vrij AA, Rijken J, Van Wersch JW, Stockbrugger RW. Platelet factor 4 and beta-thromboglobulin in inflammatory bowel disease and giant cell arteritis. Eur J Clin Invest. 2000;30:188–194. doi: 10.1046/j.1365-2362.2000.00616.x. [DOI] [PubMed] [Google Scholar]
- 23.Best WR, Becktel JM, Singleton JW, Kern F., Jr Development of a Crohn’s disease activity index. National cooperative Crohn’s disease study. Gastroenterology. 1976;70:439–444. [PubMed] [Google Scholar]
- 24.Maul J, Loddenkemper C, Mundt P, et al. Peripheral and intestinal regulatory CD4+CD25 (high) T cells in inflammatory bowel disease. Gastroenterology. 2005;128:1868–1878. doi: 10.1053/j.gastro.2005.03.043. [DOI] [PubMed] [Google Scholar]
- 25.Veltkamp C, Anstaett M, Wahl K, et al. Apoptosis of regulatory T lymphocytes is increased in chronic inflammatory bowel disease and reversed by anti-TNFalpha treatment. Gut. 2011;60:1345–1353. doi: 10.1136/gut.2010.217117. [DOI] [PubMed] [Google Scholar]
- 26.Li Z, Arijs I, De Hertogh G, et al. Reciprocal changes of Foxp3 expression in blood and intestinal mucosa in IBD patients responding to infliximab. Inflamm Bowel Dis. 2010;16:1299–1310. doi: 10.1002/ibd.21229. [DOI] [PubMed] [Google Scholar]
- 27.Kullberg MC, Hay V, Cheever AW, et al. TGF-beta1 production by CD4+CD25+ regulatory T cells is not essential for suppression of intestinal inflammation. Eur J Immunol. 2005;35:2886–2895. doi: 10.1002/eji.200526106. [DOI] [PubMed] [Google Scholar]
- 28.Bachmann MF, Gallimore A, Jones E, Ecabert B, Acha-Orbea H, Kopf M. Normal pathogen-specific immune responses mounted by CTLA-4-deficient T cells: a paradigm reconsidered. Eur J Immunol. 2001;31:450–458. doi: 10.1002/1521-4141(200102)31:2<450::AID-IMMU450>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 29.Yang XF. Factors regulating apoptosis and homeostasis of CD4+CD25 (high) FOXP3+ regulatory T cells are new therapeutic targets. Front Biosci. 2008;13:1472–1499. doi: 10.2741/2775. [DOI] [PubMed] [Google Scholar]
- 30.Shalev I, Wong KM, Foerster K, et al. The novel CD4+CD25+ regulatory T cell effector molecule fibrinogen-like protein 2 contributes to the outcome of murine fulminant viral hepatitis. Hepatology. 2009;49:387–397. doi: 10.1002/hep.22684. [DOI] [PubMed] [Google Scholar]
- 31.Foerster K, Helmy A, Zhu Y, et al. The novel immunoregulatory molecule FGL2: a potential biomarker for severity of chronic hepatitis C virus infection. J Hepatol. 2010;53:608–615. doi: 10.1016/j.jhep.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 32.Chiquet-Ehrismann R, Hagios C, Matsumoto K. The tenascin gene family. Perspect Dev Neurobiol. 1994;2:3–7. [PubMed] [Google Scholar]
- 33.Procopio WN, Pelavin PI, Lee WM, Yeilding NM. Yeilding, angiopoietin-1 and -2 coiled coil domains mediate distinct homo-oligomerization patterns, but fibrinogen-like domains mediate ligand activity. J Biol Chem. 1999;274:30196–30201. doi: 10.1074/jbc.274.42.30196. [DOI] [PubMed] [Google Scholar]
- 34.Herman AE, Freeman GJ, Mathis D, Benoist C. CD4+CD25+ T regulatory cells dependent on ICOS promote regulation of effector cells in the prediabetic lesion. J Exp Med. 2004;199:1479–1489. doi: 10.1084/jem.20040179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mendicino M, Liu M, Ghanekar A, et al. Targeted deletion of Fgl-2/fibroleukin in the donor modulates immunologic response and acute vascular rejection in cardiac xenografts. Circulation. 2005;112:248–256. doi: 10.1161/CIRCULATIONAHA.105.534271. [DOI] [PubMed] [Google Scholar]