

Abstract
Essential for viral replication and highly conserved among poxviridae, the vaccinia virus I7L ubiquitin-like proteinase (ULP) is an attractive target for development of smallpox antiviral drugs. At the same time, the I7L proteinase exemplifies several interesting challenges from the rational drug design perspective. In the absence of a published I7L X-ray structure, we have built a detailed 3D model of the I7L ligand binding site (S2–S2′ pocket) based on exceptionally high structural conservation of this site in proteases of the ULP family. The accuracy and limitations of this model were assessed through comparative analysis of available X-ray structures of ULPs, as well as energy based conformational modeling. The 3D model of the I7L ligand binding site was used to perform covalent docking and VLS of a comprehensive library of about 230,000 available ketone and aldehyde compounds. Out of 456 predicted ligands, 97 inhibitors of I7L proteinase activity were confirmed in biochemical assays (∼20% overall hit rate). These experimental results both validate our I7L ligand binding model and provide initial leads for rational optimization of poxvirus I7L proteinase inhibitors. Thus, fragments predicted to bind in the prime portion of the active site can be combined with fragments on non-prime side to yield compounds with improved activity and specificity.
Keywords: Smallpox, Viral protease, Homology modeling, Covalent docking, Binding pocket, Virtual ligand screening, VLS, Ketone inhibitors
Introduction
Proteases comprise a prominent class of targets for rational drug design [1]. Human proteases have been implicated in a wide range of diseases, including cancer, cardiovascular and inflammatory disorders. Rationally designed protease inhibitors (PIs) have also proved utility in antiviral therapies against HIV-1 [2] and hepatitis C [3]. A number of other PIs, targeting such important disease agents such as Dengue, West Nile, Herpesviruses, rhinoviruses, and SARS coronavirus are in various stages of drug development [4].
One of the recently identified viral targets, I7L core proteinase, plays a critical role in replication of vaccinia and other orthopox viruses, such as variola (biowarfare agent smallpox) and monkeypox (emerging zoonotic) [5–7]. Biochemical studies established that I7L is a cysteine proteinase that cleaves the major structural and membrane proteins of the virus at a conserved AG/X motif, where X in the P1′ position has a preference for a small residue like Ala, Ser or Thr [7–9].
Highly conserved in orthopox viruses and within the Poxviridae family, I7L shares 99% amino acid identity between vaccinia, variola and monkeypox orthologues. Outside of the Poxviridae, I7L has only a distant homology to ubiquitin-like proteases (ULPs) of viral [10, 11] and eukaryotic [12–14] origin. Despite a sequence identity as low as ∼20%, all of these viral and eukaryotic proteinases share similar substrate specificity (GG/X or AG/X), overall 3D folding patterns, and identical catalytic core motifs (His-Gln-Cys). Like in other ubiquitin-like proteases, mutations in these conserved active site residues in vaccinia I7L completely abolish it’s catalytic activity [9].
Functional importance and conservation among poxviridae establishes I7L as an attractive target for the design of antivirals [7–9, 15, 16]. At the same time, the I7L proteinase represents several technical challenges for rational drug design.
First, the simplistic peptide cleavage specificity of I7L (AG/X) provides little basis for building selective peptidomimetic inhibitors—a traditional way of developing initial structure activity relationship (SAR) for protease targets [17]. Lack of peptidomimetic SAR and relatively low throughput of available biochemical assays makes large scale virtual ligand screening (VLS) of small molecule libraries the primary option for generating initial hits and SAR for I7L.
Second, there is no X-ray structure of the I7L proteinase available, and the low level of sequence identity (∼20%) with the structural template Ulp1 (PDB 1euv) [18] in most cases would only allow overall prediction of a protein fold. Fortunately, full conservation of several key residues that enclose the core binding site in ULPs [14, 18, 19], including the I7L proteinase, suggests the possibility for accurate modeling of the 3D structure of this target site comprising the S2, S1, S1′ and S2′ sub-pockets. Detailed analysis of the hypothetical model in this study validates its accuracy from several different perspectives, and also demonstrates its applicability to receptor based screening for I7L inhibitors.
The third challenge is related to the adequate modeling of covalent nature of binding inhibitors to protease active sites. The majority of PIs in the clinic or in clinical development employ some type of nucleophilic warhead to bind covalently to the protease catalytic residue [4]; the most successful warheads among cysteine protease inhibitors are moderately reactive ketones [20, 21]. Several studies show that an accurate modeling of PI covalent binding needs to account for the structural changes in the ligand upon reaction with the catalytic residue [22]. This requirement is especially relevant for the I7L active site, enclosed in a tight channel. Here we demonstrate an automated implementation of such covalent docking and VLS procedure in the framework of the ICM molecular modeling package (Molsoft LLC, http://www.molsoft.com).
In this study we used a detailed structural model of the I7L substrate binding site (S2–S2′) to perform covalent docking and VLS of a comprehensive library of about 230,000 available ketone compounds. Our biochemical assays confirmed 97 inhibitors of I7L proteinase activity out of 456 predicted candidates (∼20% hit rate). These experimental results both validate our 3D ligand binding model and provide initial leads for further rational optimization of poxvirus I7L proteinase inhibitors.
Methods
Protein modeling
Homology modeling of the I7L proteinase domain was based on the ULP1 protease structural template (PDB code 1euv) [18], and performed using ICM sequence-structure alignment and BuildModel algorithm [23–25]. ICM sequence-structure alignment is based on ZEGA sequence alignment [26] (Needleman and Wunsch algorithm with zero gap end penalties). To account for the template structural features, positional weights are adjusted for surface accessibility and secondary structure. The structural model of I7L was subsequently built by placing the aligned residues of the I7L polypeptide chain with ideal covalent geometry onto the corresponding residues of the structural template. Conformations of non-identical side chains and loops shorter that 4 amino acids were predicted using energy-based optimization of the model [24, 25]. Longer loops of the I7L proteinase located further than 20 Å from the active site are not expected to affect conformation of the active site, and were not predicted.
Active site refinement
Side chains of 15 residues forming the S3–S2′ portion of the substrate binding site in I7L were optimized through additional runs of energy minimization with Ala-Ala-Gly-Ser peptide substrate. The substrate conformation in the binding site was inferred from the Ulp1 complex with substrate. Heavy atoms of I7L conserved residues were tied to corresponding atoms in the Ulp1 structure with soft harmonic tethers. All torsion angles of the substrate and torsion angles in the selected fifteen side chains of the I7L model were optimized with respect to both conformational energy of the complex and energy of tethers. Three independent runs of refinement procedure were performed, each run including 100,000 Monte Carlo Minimization steps. The ICM Protein Health tool was used to check the quality of the refinement.
ICM docking and virtual screening
Virtual Screening was performed using the ICM fast docking procedure [27, 28]. The ICM employs a Monte-Carlo Minimization algorithm to find the optimal conformation of a flexible ligand in the receptor binding side represented by a pre-calculated potential grid. The ligand is described in internal coordinates with flexible torsion variables [29]. Best conformations of the ligand are assigned an ICM binding Score, which takes into account the following free energy terms: van der Waals, hydrogen bonding, Poisson electrostatic, desolvation and entropy. The ICM VLS screening procedure takes less than 1 min per compound per 3 GHz Intel Xeon processor. Comparative benchmarks [30–32] show robust performance and excellent accuracy of the ICM docking method in most applications.
Covalent docking procedure
The standard ICM VLS procedure was modified to reflect covalent binding of inhibitors to the active site Cys328, as illustrated in Fig. 1.
Fig. 1.
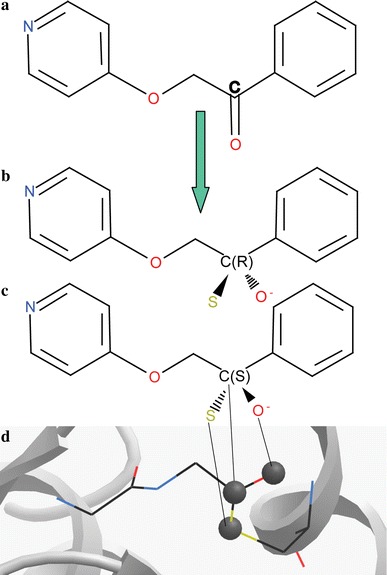
Covalent docking setup in ICM: (a) each ketone compound is converted, (b, c) yielding two possible thio-acyl stereoisomers. (d) Three thio-acyl atoms of the ligand intermediate are tethered to the corresponding three atoms of the template, derived form the X-ray structure of Ulp1-substrate complex (1euv)
In this algorithm, the SP2 ketone carbon in each compound was converted to a SP3 carbon, yielding two different thio-acyl stereoisomers. The Cys328 side chain was removed from the I7L receptor model to avoid apparent steric clash. Instead, a separate three atom “covalent bonding template” was created corresponding to the thio-acyl moiety of the substrate-Ulp1 X-ray structure (1euv) [18]. The corresponding three atoms in the ligand were tethered to this covalent bonding template. Some flexibility in the covalent bonding was allowed by using a harmonic tether penalty term. The tether penalty was calculated only at the docking stage, but not included in binding score calculations. Both modified stereoisomers were docked separately into the active site and the best scoring one was used for further analysis. Note that our scoring function intentionally does not take into account the chemical energy of ligand–thiol covalent bond formation (see Discussion).
Biochemical assay
A quantitative I7L in-vitro cleavage assay has been developed in this work, which employs measurement of fluorescence quenching time dependencies for detection of proteolytic cleavage. The assay uses extracts of vaccinia virus (VV) infected BSC40 cells as the source of enzyme, as described in our previous work [15]. For the negative controls we used extracts from cells transfected with pI7LH241A plasmid carrying inactivated I7L mutant, as well as a cell only extract control. The peptide substrate contains the defined AGT cleavage site of P4a with an ortho-aminobenzoyl (Abz) fluorescent group conjugated to the amino terminus and a diaminopropionamide 2,4-dinitrophenyl (Dap-Dnp) quench group conjugated to the carboxy terminus (Anaspec). Reactions were performed at room temperature in a final volume of 100 μL containing 10 μM (10 μL) of peptide substrate (Abz-T-N-A-G-T-C-T-V-Dap(dnp)-K-K-NH2 from Anaspec), 40 μL of enzyme extract, and 50 μL of buffer (20 mM HEPES (pH 7.4), 2 mM CaCl2, 2.5 mM EDTA).
The assay reactions were carried out in 96-well Costar black-sided black-bottom plates. A background reading was taken prior to addition of inhibitor, various concentrations of compound were added, and the reaction was measured over a 4-h time course. Fluorescence was measured using a Wallac Victor2 V Multilabel HTC counter (Perkin Elmer) with an excitation wavelength of 340 nm and plates were read at 420 nm. The slope of the enzyme plus peptide reactions was calculated as 100% and the 50% inhibition for each compound was calculated in reference to that slope.
Results
Structural model and analysis of I7L ligand binding site
A 3D model of the vaccinia virus I7L protein, presented in Fig. 2, was built with the yeast Ulp1 cysteine proteinase X-ray structural template, as described in the Methods. The Ulp1 proteinase is the closest homologue of I7L with a high resolution X-ray structure (PDB code 1euv). Though the sequence homology between I7L and Ulp1 is weak (∼20% identical amino acids), there are several factors that make it possible to generate an all-atom model of the I7L “core” binding pocket corresponding to S2–S2′ sub-sites.
Fig. 2.
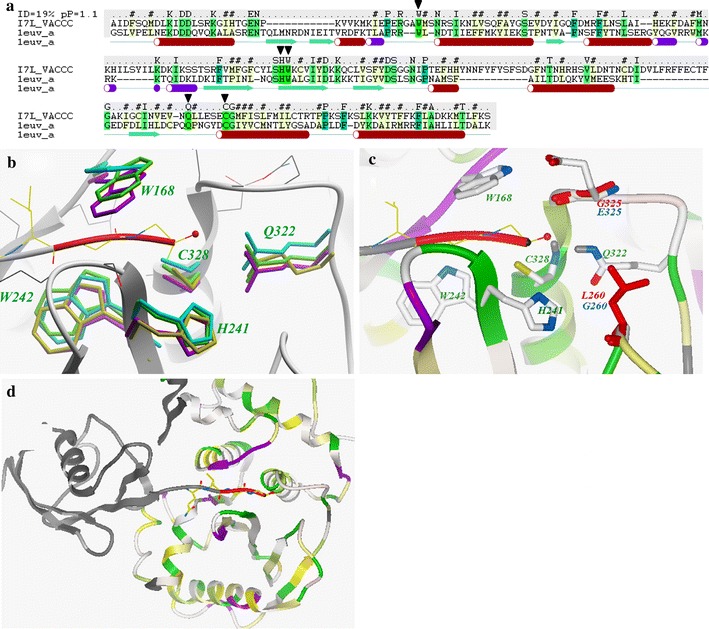
Structural model of I7L and its active site, based on the Ulp1 cysteine protease X-ray structure (PDB code 1euv). (a) Sequence-structure alignment between I7L and Ulp1, with highly conserved ULP residues “W-HW-Q-C” marked by ▼. (b) Superimposition of “W-HW-Q-C” motif side chains in four different ULPs structures (green: 1euv, magenta: 1xt9, cyan: 1th0, yellow: 1avp). Backbone contact residues of Ulp1 shown in thin wire representation with grey carbons. (c) Close up of I7L model active site. Five conserved residues are shown as sticks colored by atom type with green labels. Two residues of I7L active site (G260 and E325), not conserved in Ulp1 are shown as sticks with blue labels, the corresponding Ulp1 residues shown as red sticks. (d) Predicted overall structure of the I7L proteinase; ribbon is colored according residue identity with Ulp1 (green is fully conserved, yellow—similar residue). The Ulp1 substrate is shown as grey ribbon with C-terminal Gly-Gly residues highlighted in red
Conservation of key contact residues in Ulp1 and I7L active sites
Analysis of the Ulp1-substrate X-ray structure reveals that there are only five side chains of the proteinase in direct contact with the P2, P1 and P1′ positions of the substrate (Fig. 2a, b). All five residues in this “core” binding site motif (W-HW-Q-C) are identical between I7L and Ulp1. In the nomenclature of I7L, this key motif includes C328 and H241 as the catalytic side chains; Q322 side chain, which together with the backbone amino group of C328 forms a subtilisin-like oxyanion hole; as well as side chains W242 and W168 that form a narrow channel for the substrate. Other P2–P1′ core substrate contacts occur with backbone atoms of the Ulp1 binding pocket.
Structural conservation of key residues in ULP family
While the above sequence-structure alignment suggests identical P2–P1′ core substrate contacts in Ulp1 and I7L, it does not guarantee conservation of spatial positioning of W-HW-Q-C motif residues. More evidence towards such conservation comes from structural comparison of X-ray structures of three of the closest ULPs structural homologues: Sentrin Specific Proteases SSP8 and SSP2, as well as Adenovirus Core Protease AdV-CP (Fig. 2b and Table 1). While the sequence identity level between these ULPs is as low as between Ulp1 and I7L, we found remarkable preservation of 3D coordinates in W-HW-Q-C motif residues with RMSD ∼0.3 Å. In addition, the backbone conformation of all 10 residues in ULPs making direct (backbone or side chain) contacts with the P2–P1′ substrate site is highly preserved too.
Table 1.
Sequence and structural similarities between Ulp1 proteinase and its closest ULP homologues
| Ulp1 (1euv) compared to | Identical residues (Similar residues) | RMSD of WHWQC residues (all heavy atoms), Å | Backbone RMSD for 10 backbone contacts, Å |
|---|---|---|---|
| SSP8 (1xt9) | 19% (35%) | 0.28 | 0.23 |
| SSP2 (1th0) | 31% (55%) | 0.47 | 0.29 |
| AdenoV Core Protease (1avp)a | 13% (34%) | 0.7b | 0.84 |
aLower resolution structure (2.60 Å)
bResidue corresponding to W168 is not conserved in AdV-CP
Such a high level of local structural similarity, normally observed only in very close protein homologues points to functional significance of this conservation. Therefore, we expect that the structure of the corresponding S2–S1′ sub-site in I7L is preserved with comparable accuracy.
Identical core substrate specificity between I7L and ULPs
Substrate specificity for I7L has been defined as AG↓X [8, 9], and for most ULPs it is GG↓X [14, 18], where P1′ position X has a preference for small residues (Ala, Ser or Thr). Interestingly, although the natural Ulp1 substrates have Gly in the P2 position, our analysis of the X-ray model shows that this binding site can easily accommodate Ala in the P2 pocket, creating favorable van der Waals contacts. Thus, the substrate specificity of I7L and Ulp1 is practically identical in the P2–P1′ positions, which further validates the model of the I7L binding site.
Potential importance of S2′ sub-site for I7L selectivity
Conformation of the adjacent S2′ sub-site of I7L can also be predicted using structural modeling. In Ulp1 and other ULPs this site is highly exposed to solvent and is controlled by only three residues—conserved H241and Q322 and a variable residue in position 260 (in I7L nomenclature). While in eukaryotic ULPs, this variable position 260 is occupied by a large residue (Leu in Ulp1, Met in SSP2, His in SSP8), in I7L it is replaced by Gly (shown in Fig. 2c with blue labels for I7L and red labels for Ulp1). The absence of an amino acid side chain in this position creates an additional small cleft in our model, which can be exploited to design I7L selectivity vs. other ULPs. Conformation of the only other variable side chain in close proximity of the active site, Glu325 (in Ulp1 it is Gly), was inferred from other ULPs structure (Ser in 1xt9) and additional conformational modeling.
Refinement of the I7L binding pocket model
The side chain refinement procedure was performed for the model of I7L binding pocket as described in the Methods. Comparative modeling above suggests no significant deviations for the I7L “core” binding site side chains from their conformations in the Ulp1 model. The refinement was performed to verify that conformations of these five residues are compatible with the adjacent residues of the I7L model and binding of the peptide substrate. Our results show very good convergence of the refinement procedure, so that the models from three independent runs had RMSD not exceeding 0.6 Å when the heavy atoms of the optimized 15 residues were compared. The ICM Protein Health tool shows no significant clashes in these 15 residues of the model. The five “core” residues in the model had conformations within 0.2 Å RMSD from the corresponding Ulp1 residues. The non-conserved Glu325 side chain was predicted to form an H-bond with the W168 side chain and not to have substrate contacts.
The described active site of I7L represents a channel-like binding pocket with a volume of about 400 Å3, suitable for rational drug design. While this core binding pocket is fully conserved between I7L and some ULP proteinases, the non-conserved residues on both sides of the S2–S1′ core can provide a basis for design of I7L selective inhibitors. We must note here that while the above structural analysis strongly supports our Ulp1-based model of I7L ligand-binding pocket, the model should be considered hypothetical—until its utility in the design of I7L inhibitors is demonstrated.
Covalent docking and virtual ligand screening
Candidate I7L inhibitors were predicted using a docking-based VLS procedure implemented in the ICM molecular modeling package (Molsoft, LLC). The standard ICM docking algorithm was modified to reflect chemical changes in ketone ligands upon covalent binding to the active site Cys328, as described in the Methods.
Selection of available ketone and aldehyde compounds
The ketone compounds were selected from a non-redundant library of ∼3.5 M compounds available from 40 major vendors, as well as from our in-house diverse library of ∼208 K compounds. All ketones found in these libraries (total of ∼230 K compounds) were divided into four groups based on their chemical reactivity with the I7L active site thiol: Aliphatic Aldehydes (AL(al), Aromatic Aldehydes (AL(ar), Aliphatic Ketones (KE(al) and Aromatic Ketones (KE(ar)). As expected, aliphatic aldehydes represent the fewest compounds in our drug-like database as they are considered to be the most reactive, while the least reactive aromatic ketones represent 84% of all available ketone compounds. Note that conjugation to aromatic rings usually renders ketones and aldehydes more chemically stable, though their reactivity can be also modulated by electron-withdrawing substitutions in the ring.
Docking and VLS results
More than 230 K available ketones and aldehydes were converted into corresponding thio-acyl intermediates and the resulting stereoisomers were docked into the I7L proteinase binding site model. Binding scores of the docked compounds were predicted and VLS “hits” were identified with an ICM Score better than −32 kJ/mol. The docking procedure was repeated 3 times for each compound and the results of these 3 runs compared. We found good reproducibility of docking poses and overall in silico results with at least 83% of “hits” identical between independent runs.
As illustrated in Fig. 3, the high-scoring compounds have their thio-acyl moiety accurately fitting the template anchor. For predicted hits the average RMSD between the thio-acyl template and the corresponding O–C–S atoms of these compounds was estimated at ∼0.23 Å, with a maximum deviation at 1.1 Å. The deprotonated acyl oxygen of these ligands was also found to be properly positioned in the oxyanion hole. Beyond the thio-acyl intermediate “core”, a variety of specificity fragments, extending into both the prime and non-prime sites was identified. Figure 3 gives three examples of predicted inhibitors with different coverage of the I7L binding pocket. The first candidate ligand occupies mainly the S2–S1 (non-prime) sub-sites, with only a methyl group extending into the S1′ site. The second ligand is located primarily in the S1′–S2′ sub-site, while the third stretches across both the prime and non-prime sub-sites.
Fig. 3.
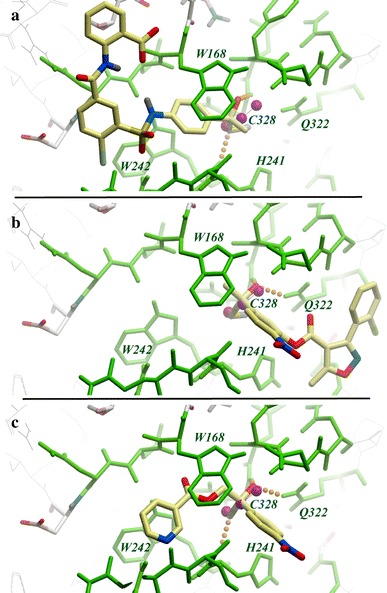
Examples of predicted binding modes for the three most active I7L inhibitors. Conserved residues of I7L binding site are shown as green sticks. Positions of the thio-acyl template atoms are shown as magenta balls. Inhibitors are shown as sticks with yellow carbons. (a) ST-0194802 in non-prime pockets (S3, S2, S1) of the active site, (b) ST-0194680 in prime pockets (S1′, S2′), and (c) ST-0194797 in both prime and non-prime pockets (S2–S1–S1′)
Overall, more than 900 cmpds were identified with ICM Scores better than −32 kJ/mol (a default threshold for ICM VLS). After filtering out compounds with bad chemical properties and removing redundant compounds, we selected a set of about 500 compounds. A total of 456 compounds were received for biochemical testing.
Inhibition of I7L protease in biochemical assay
Predicted compounds were tested in a biochemical assay for their ability to inhibit the proteolytic activity of I7L. The assay is based on a fluorescence-quench pair of probes that are conjugated to the amino and carboxy-terminal ends of an I7L cleavage peptide, respectively. Cleavage of the peptide and removal of the quench group is monitored through an increase in the fluorescent signal over the baseline.
All 456 candidate compounds were initially screened through the biochemical assay at a single concentration of 200 μM. The overall results of the primary assays are presented in Table 2, which gives an account of compounds in each of the four chemical groups, the number of high scoring compounds and experimental hit rates. A total of 97 inhibitors of I7L were identified, yielding a hit rate of more than 20%. While the hit rate for aromatic ketones is lower than the average (17%), this group of compounds still represents the majority of the hits.
Table 2.
Summary of docking and primary biochemical assay results for compounds with different warheads
| Warhead type | Total in librariesa | Docking Score < −32 kJ/molb | Compounds tested | I7L inhibition >50% at 200 μM | Hit rate (%) |
|---|---|---|---|---|---|
| AL(al) | 2,600 | 5 | 2 | 1 | ∼50 |
| AL(ar) | 11,400 | 44 | 29 | 14 | 50 |
| KE(al) | 17,000 | 75 | 71 | 22 | 31 |
| KE(ar) | 196,000 | 782 | 354c | 60 | 17 |
aIn-house library and Molsoft database of available drug-like compounds from 40 chemical vendors (∼3.5 million non-redundant compounds)
bDefault threshold in ICM VLS for likely inhibitors is −32 kJ/mol
cOnly compounds with VLS Score < −34 kJ/mol were ordered and tested
In contrast, we found that out of 20 randomly selected compounds with suboptimal docking Scores (ICM Score between −20 and −30 kJ/mol) none has shown significant activity in the primary assay.
Those 26 compounds that were found to inhibit I7L proteolytic activity by more than 70% at 200 μM concentrations were then re-tested to determine their IC50 value. The results of these secondary assays are summarized in Fig. 4 and Table 3, chemical structures of the compounds are illustrated in Fig. 5. Most compounds were found to have IC50 values below 200 μM, six of them with IC50 values <50 μM. Again, five of the best six inhibitors have aromatic ketone warheads, which have the most favorable activity/specificity profile, as discussed below.
Fig. 4.
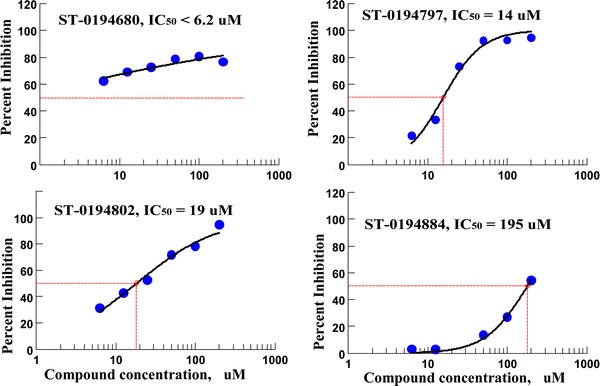
Inhibition curves for the three most active inhibitors of I7L proteolytic activity (compounds ST-680, ST-797, ST-802) and one of the “marginal” inhibitors (ST-884)
Table 3.
Experimental IC50 values for the secondary assay
| Compound ID | Catalytic warhead | Binding pocket | Docking Score | IC50 (μM) |
|---|---|---|---|---|
| ST-0194680 | AL(ar) | S1′–S2′ | −33 | 6.2 ± 1.5 |
| ST-0194797 | KE(ar) | S2–S1–S1′ | −34 | 14 ± 2.3 |
| ST-0194802 | KE(ar) | S3–S2–S1 | −37 | 19 ± 3 |
| ST-0194780 | KE(ar) | S2–S1–S1′ | −43 | 39 ± 12 |
| ST-0194766 | KE(ar) | S2–S1–S1′ | −39 | 48 ± 10 |
| ST-0194951 | KE(ar) | S2–S1–S1′ | −35 | 50 ± 12 |
| ST-0194671 | AL(ar) | S1′–S2′ | −31 | 63 ± 18 |
| ST-0194763 | KE(ar) | S2–S1–S1′ | −35 | 70 ± 10 |
| ST-0194456 | KE(al) | S2–S1–S1′ | −32 | 73 ± 14 |
| ST-0195032 | KE(ar) | S2–S1–S1′ | −34 | 82 ± 6 |
| ST-0194672 | AL(al) | S3–S2–S1 | −31 | 82 ± 14 |
| ST-0194782 | KE(ar) | S2–S1–S1′ | −40 | 98 ± 25 |
| ST-0194466 | KE(al) | S2–S1 | −41 | 100 ± 30 |
| ST-0194691 | KE(al) | S1–S1′–S2′ | −35 | 102 ± 13 |
| ST-0194692 | KE(al) | S1–S1′–S2′ | −34 | 113 ± 30 |
| ST-0194497 | KE(al) | S3–S2–S1 | −32 | 122 ± 30 |
| ST-0194479 | KE(al) | S3–S2–S1 | −40 | 125 ± 35 |
| ST-0194682 | AL(ar) | S3–S2–S1 | −30 | 158 ± 40 |
| ST-0194681 | AL(ar) | S1′–S2′–S3′ | −31 | 160 ± 31 |
| ST-0195086 | KE(ar) | S3–S2–S1 | −34 | 198 ± 50 |
| ST-0194884 | KE(ar) | S2–S1–S1′ | −35 | 195 ± 43 |
| ST-0194457 | KE(ar) | S2–S1–S1′ | −37 | 250 ± 40 |
| ST-0194459 | KE(al) | S2–S1 | −37 | 260 ± 40 |
| ST-0194689 | KE(al) | S2–S1–S1′ | −35 | 300 ± 80 |
| ST-0194486 | KE(al) | S3–S2–S1 | −30 | 500 ± 200 |
| ST-0194800 | KE(ar) | S2–S1–S1′ | −33 | 650 ± 150 |
Predicted ICM docking score, position in the I7L binding pocket and catalytic warhead are also shown. Chemical structures of compounds are presented in Fig. 5
Fig. 5.
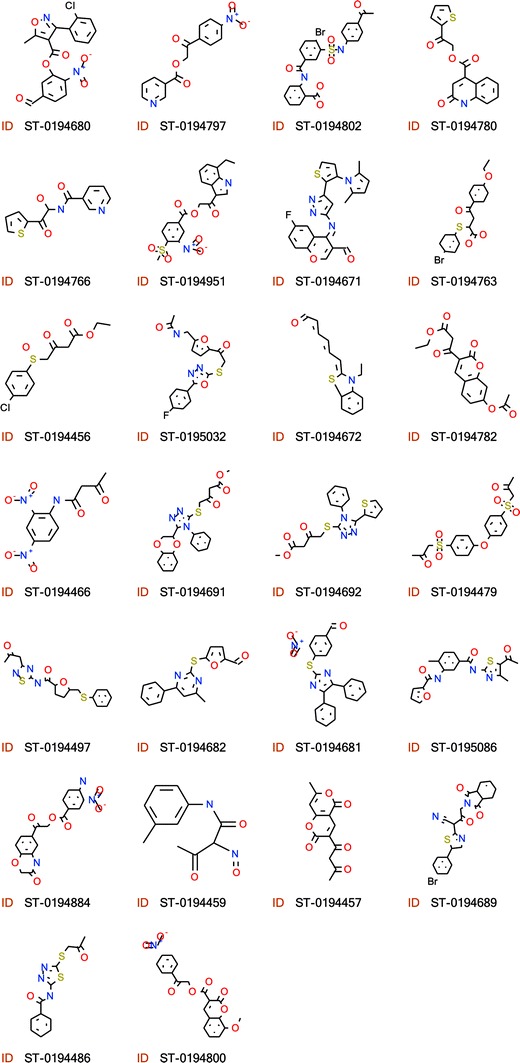
Chemical structures of new I7L inhibitors and their ID numbers as shown in Table 3
Discussion
Choice of ketones as chemical warheads
Covalent binding of ligands to protease nucleophilic active groups, especially thiols, remains the dominant strategy in drug discovery. Note that 7 out of 8 cysteine protease inhibitors in clinical and preclinical development employ covalent binding to the thiol; of those, 5 candidates are based on ketone warheads [33]. In the case of I7L and other ULPs strong covalent binding of ligand is especially important, because it has to compete with strong substrate binding, reinforced by large protein–protein interface (see Fig. 2d). In the Ulp1-substrate complex, this substrate recognition interface decoupled from the active site, amounts to ∼1,900 Å2.
While a number of new promising warheads have been introduced recently [21], ketones are still considered optimal due to their moderate warhead reactivity, proven ADME/Tox profiles, and good chemical tractability Also, ketones are among the few warheads suitable for targeting both primed and non-primed sub-sites simultaneously, which is critical for optimization of binding specificity for I7L inhibitors.
Covalent docking procedure
While some docking studies do not explicitly account for the covalent nature of ligand-receptor binding, we found this aspect critical for accurate modeling of I7L-ligand binding.
A standard molecular docking procedure does not reflect structural changes in the ligand, where a flat SP2 ketone carbon is converted into the tetrahedral (SP3) carbon. Our initial docking results indicate that the ketone moiety does not fit into the narrow I7L active site channel and can not properly position itself for nucleophilic attack by Cys328 without inducing significant conformational changes in the binding pocket. To simulate the proper transition state for ketone binding to I7L, a modified “covalent docking” procedure, similar to the method described in [22] was implemented in this work. Our procedure converts the ketone into the thio-acyl intermediate and links it up to the active site thiol of the I7L proteinase with flexible tethers.
On the other hand, the docking score in our procedure does not account for the change in covalent energy upon the ketone–thiol reaction. Though this portion of ΔG can contribute significantly in strong ligand binding to the I7L protease, it is hard to properly account for this aspect of binding in the model. Moreover, higher reactivity also contributes to non-specific interaction of the ligands, compromising bioavailability and safety profiles of the drug [22]. Therefore, we believe that the chemical bonding contribution should be considered separately from conformational contributions when screening for protease inhibitors.
Differences in the reactivity of the four types of ketone warheads are reflected in the “hit rate” of predicted candidates as presented in Table 2. One can notice that for the same predicted conformational binding threshold (Score < −32 kJ/mol), more reactive aldehydes have about 50% hit rate, while the rate drops to 30% for aliphatic ketones and 17% for aromatic ketones. The latter hit rate is still high enough, so that the least reactive (and presumably most specific) aromatic ketone compounds dominate the list of I7L inhibitors.
Further strategies for rational design of I7L small molecule inhibitors
The high observed hit rate for the predicted I7L inhibitors (more than 21% overall) successfully validates the suggested structural model of I7L and shows its applicability to rational drug design. Note that most identified here inhibitors do not reach the specificity-defining S2′ sub-site; therefore we do not expect them to be selective to I7L over other ULP proteinases. To take full advantage of the core binding pocket and to achieve such selectivity, we suggest synthesis of compounds that combine prime and non-prime moieties of the first generation hits, shown in Table 3. Since binding of prime and non-prime parts of ketones to I7L is virtually independent in most cases due to the fixed position of the ketone–thiol bond, we can expect additive effects on binding energy. Though such fragment based design strategy requires expensive de-novo synthesis, it is likely to bring about 2nd generation I7L inhibitors with significantly improved affinity and specificity.
Conclusions
Perfect structural conservation of S2–S1′ binding site in the ULP family allows generation of a detailed model of the vaccinia virus I7L proteinase binding site, suitable for docking and VLS. A modified ICM covalent docking procedure was implemented to reflect the covalent nature of ketone inhibitor binding to the I7L active site thiol. About 450 candidate inhibitors of I7L were selected in this work from 230,000 available ketones and aldehydes.
Biochemical assays confirmed I7L inhibition for 97 of these candidates (∼21% success rate), some with activity better than 20 μM. These results provide validation for both the I7L model and the covalent docking procedure, as well as extensive QSAR for hit optimization. Further improvement in activity and specificity can be achieved for ligands combining various “primed” and non-primed moieties, identified in this work.
Acknowledgements
This work was supported by U.S. Army Medical Research and Material Command contract (DAMC 17-03-C-0040).
Abbreviations
- PI
Protease inhibitor
- ULP
Ubiquitin-like proteases
- SAR
Structure-activity relationship
- VLS
Virtual ligand screening
- ICM
Molecular modeling package from Molsoft, LLC
References
- 1.Klebe G. J Mol Med. 2000;78:269–281. doi: 10.1007/s001090000084. [DOI] [PubMed] [Google Scholar]
- 2.Gago F. IDrugs. 1999;2:309–320. [PubMed] [Google Scholar]
- 3.Thomson JA, Perni RB. Curr Opin Drug Discov Devel. 2006;9:606–617. [PubMed] [Google Scholar]
- 4.Hsu JT, Wang HC, Chen GW, Shih SR. Curr Pharm Des. 2006;12:1301–1314. doi: 10.2174/138161206776361110. [DOI] [PubMed] [Google Scholar]
- 5.Kane EM, Shuman S. J Virol. 1993;67:2689–2698. doi: 10.1128/jvi.67.5.2689-2698.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrd CM, Hruby DE. Virol J. 2005;2:4. doi: 10.1186/1743-422X-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ansarah-Sobrinho C, Moss B. J Virol. 2004;78:6335–6343. doi: 10.1128/JVI.78.12.6335-6343.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Byrd CM, Bolken TC, Hruby DE. J Virol. 2002;76:8973–8976. doi: 10.1128/JVI.76.17.8973-8976.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byrd CM, Bolken TC, Hruby DE. J Virol. 2003;77:11279–11283. doi: 10.1128/JVI.77.20.11279-11283.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding J, McGrath WJ, Sweet RM, Mangel WF. Embo J. 1996;15:1778–1783. [PMC free article] [PubMed] [Google Scholar]
- 11.Andres G, Alejo A, Simon-Mateo C, Salas ML. J Biol Chem. 2001;276:780–787. doi: 10.1074/jbc.M006844200. [DOI] [PubMed] [Google Scholar]
- 12.Li SJ, Hochstrasser M. Nature. 1999;398:246–251. doi: 10.1038/18457. [DOI] [PubMed] [Google Scholar]
- 13.Fujiwara T, Saito A, Suzuki M, Shinomiya H, Suzuki T, Takahashi E, Tanigami A, Ichiyama A, Chung CH, Nakamura Y, Tanaka K. Genomics. 1998;54:155–158. doi: 10.1006/geno.1998.5554. [DOI] [PubMed] [Google Scholar]
- 14.Reverter D, Lima CD. Structure. 2004;12:1519–1531. doi: 10.1016/j.str.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 15.Byrd CM, Hruby DE. Virol J. 2005;2:63. doi: 10.1186/1743-422X-2-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Byrd CM, Bolken TC, Mjalli AM, Arimilli MN, Andrews RC, Rothlein R, Andrea T, Rao M, Owens KL, Hruby DE. J Virol. 2004;78:12147–12156. doi: 10.1128/JVI.78.22.12147-12156.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borodovsky A, Ovaa H, Meester WJ, Venanzi ES, Bogyo MS, Hekking BG, Ploegh HL, Kessler BM, Overkleeft HS. Chembiochem. 2005;6:287–291. doi: 10.1002/cbic.200400236. [DOI] [PubMed] [Google Scholar]
- 18.Mossessova E, Lima CD. Mol Cell. 2000;5:865–876. doi: 10.1016/S1097-2765(00)80326-3. [DOI] [PubMed] [Google Scholar]
- 19.Reverter D, Wu K, Erdene TG, Pan ZQ, Wilkinson KD, Lima CD. J Mol Biol. 2005;345:141–151. doi: 10.1016/j.jmb.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 20.Leung-Toung R, Li W, Tam TF, Karimian K. Curr Med Chem. 2002;9:979–1002. doi: 10.2174/0929867024606704. [DOI] [PubMed] [Google Scholar]
- 21.Leung-Toung R, Zhao Y, Li W, Tam TF, Karimian K, Spino M. Curr Med Chem. 2006;13:547–581. doi: 10.2174/092986706776055733. [DOI] [PubMed] [Google Scholar]
- 22.Lindvall MK. Curr Pharm Des. 2002;8:1673–1681. doi: 10.2174/1381612023394142. [DOI] [PubMed] [Google Scholar]
- 23.Cardozo T, Totrov M, Abagyan R. Proteins. 1995;23:403–414. doi: 10.1002/prot.340230314. [DOI] [PubMed] [Google Scholar]
- 24.Cardozo T, Batalov S, Abagyan R. Comput Chem. 2000;24:13–31. doi: 10.1016/S0097-8485(99)00044-3. [DOI] [PubMed] [Google Scholar]
- 25.Abagyan R, Batalov S, Cardozo T, Totrov M, Webber J, Zhou Y. Proteins. 1997;Suppl 1:29–37. doi: 10.1002/(SICI)1097-0134(1997)1+<29::AID-PROT5>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 26.Abagyan RA, Batalov S. J Mol Biol. 1997;273:355–368. doi: 10.1006/jmbi.1997.1287. [DOI] [PubMed] [Google Scholar]
- 27.Totrov M, Abagyan R. Proteins. 1997;Suppl 1:215–220. doi: 10.1002/(SICI)1097-0134(1997)1+<215::AID-PROT29>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 28.Totrov M, Abagyan R. In: Drug-receptor thermodynamics: introduction and applications. Raffa RB, editor. New York: John Wiley & Sons, Ltd; 200. pp. 603–624. [Google Scholar]
- 29.Abagyan R, Totrov M, Kuznetsov D. J Comput Chem. 1994;15:488–506. doi: 10.1002/jcc.540150503. [DOI] [Google Scholar]
- 30.Bursulaya BD, Totrov M, Abagyan R, Brooks CL., 3rd J Comput Aided Mol Des. 2003;17:755–763. doi: 10.1023/B:JCAM.0000017496.76572.6f. [DOI] [PubMed] [Google Scholar]
- 31.Perola E, Walters WP, Charifson PS. Proteins. 2004;56:235–249. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- 32.Chen H, Lyne PD, Giordanetto F, Lovell T, Li J. J Chem Inf Model. 2006;46:401–415. doi: 10.1021/ci0503255. [DOI] [PubMed] [Google Scholar]
- 33.Abbenante G, Fairlie DP. Med Chem. 2005;1:71–104. doi: 10.2174/1573406053402569. [DOI] [PubMed] [Google Scholar]