

Abstract
ORF 3a of human severe acute respiratory syndrome corona virus (SARS-CoV) has been identified as a 274 amino acid membrane protein. When expressed in Xenopus oocytes the protein forms channels. Based on bioinformatics approaches the topology has been identified to include three transmembrane domains (TMDs). Since structural models from experiments are still lacking, computational methods can be challenged to generate such models. In this study, a ‘sequential approach’ for the assembly is proposed in which the individual TMDs are assembled one by one. This protocol is compared with a concerted protocol in which all TMDs are assembled simultaneously. The role of the loops between the TMDs during assembly of the monomers into a bundle is investigated. Molecular dynamics simulations for 20 ns are performed as a short equilibration to assess the bundle stability in a lipid environment. The results suggest that bundles are likely with the second TMD facing the putative pore. All the putative bundles show water molecules trapped within the lumen of the pore with only occasional events of complete crossing.
Electronic supplementary material
The online version of this article (doi:10.1007/s00894-011-1092-6) contains supplementary material, which is available to authorized users.
Keywords: Docking, Ion channel, Membrane protein, Molecular dynamics, Protein assembly, 3a of SARS-CoV
Introduction
SARS-CoV has a single positive strand RNA genome carrying 14 open reading frames (ORFs), encoding viral structural proteins (such as spike, envelope, membrane, and nucleocapsid proteins), replicases, and accessory proteins [1]. ORF 3a of SARS-CoV is identified as a 274 amino acid (a.a.) structural protein, which is located between S and E proteins [2]. ORF 3a protein harbors three transmembrane domains (TMDs) at its N-terminal side and a longer intracellular C-terminal region of about 148 amino acids. The central region of 3a protein consists of cysteine-rich domain (a.a. 127–133), Yxxϕ domain (a.a. 160–163) and diacidic domain (a.a. 171–173) [1–3]. 3a protein is suggested to form a homotetramer via monomer disulfide bridges (Cys-133 [4]) forming a dimer and the noncovalent assembly of two of the dimers forming the functional tetramer [5]. Structural information about the protein or its biological role in the cellular life cycle of the virus is still in the dark.
Viral channel forming proteins, have also been found for other viruses [6–8], such as M2 from influenza A [9–12], Vpu from HIV-1 [13, 14], 8a from SARS-CoV [5, 15], protein p7 from HCV [16, 17], 2B from Polio virus [18, 19] 3a and E proteins from SARS-CoV [4, 20], just to mention some of them, are also known to homo-oligomerize. The number of TMDs increases going from M2, Vpu and 8a, with a single TMD per monomer, to two TMDs for p7 and 2B and finally to three TMDs for 3a. Albeit the emergence of more and more structural information derived from experiments (for a review see [7, 21]) modeling the assembly of the proteins is still a challenge.
In general, a two-staged mechanism for helix-bundle membrane protein folding is suggested [22]: (i) the fold of the membrane domain into its secondary structure, a helix, (ii) the assembly of the inserted helices in the membrane. This model is expanded to include a third stage in which co-factor insertion, folding of the extramembrane parts and quaternary assembly is included [23]. This model description also holds on the energy landscape for membrane protein folding [24]. Along the line of this mechanism some computational assembly protocols are designed by using rigid body movements to explore energy landscapes [25–27]. These methods allow improving the quality of sampling conformational space via the step width of structure placements. Another approach reported in the literature includes extended replica exchange molecular dynamics simulations to assembly homooligomers [28]. Another almost unbiased approach is achieved if helices can freely diffuse within the lipid bilayer. This approach has been demonstrated for the assembly of TMDs into dimers using coarse-grained MD simulations techniques [29]. Still the full story of membrane protein folding remains to be elucidated [30].
Previous work proposed an assembly methodology to search the conformational space of all possible assemblies for the preferable structure, taking symmetry considerations into account [31]. The methodology has been tested on M2 from influenza A showing agreement with experimentally derived structure. The structure of protein 3a from SARS-CoV was first time predicted based on this methodology. Although it’s good agreement with the experiments, the mechanism of simultaneous assembly is still hard to imagine. One idea proposed herein is that there are two assembly methods, concerted and sequential [32], for comparison to search the most preferable bundle models of 3a from SARS-CoV. Loops between the transmembrane domains (TMDs) are also predicted for comparison. MD simulations of possible bundle models are performed for confirmation.
Computational methods
Ideal helices, of the TMDs of 3a [31], TMD139-59 (AS40 LPFGWLVIGV50 AFLAVFQSA), TMD279-99 (FI80 CNLLLLFVTI90 YSHLLLVAA), and TMD3105-125 (FLYLYA110 LIYFLQCINA120 CRIIM), were generated with backbone dihedrals of ϕ = −65° and φ = −39° using the program MOE (Molecular Operation Environment, www.chemcomp.com) and its integrated protein builder.
Equilibration of the TMDs
Each of the ideal helix was embedded into a fully hydrated POPC lipid bilayer (16:0−18:1 diester PC, 1-palmitory-2-oleoyl-sn-glycero-3-phospho-chloine) for 10 ns MD simulation to derive a relaxation of the conformation. POPC topology parameters were taken from [33]. And the bilayer has undergone a 70 ns MD simulation to be equilibrates as much as possible [34]. After insertion a stepwise energy minimization and equilibration protocol was adapted [34]. The system was heated gradually to 310 K in 500 ps, and then five stages of equilibration were performed where all the heavy atoms of the bundle were restrained in their initial positions by applying a harmonic force in x, y and z directions (1000, 500, 250, 100, and 10 kJ/mol/nm). These runs were to adjust the lipid to the inserted bundle.
Prior to assembly
A principal component analysis (PCA) over the backbone atoms of all frames of the last 3 ns of each of the TMDs has been done. A structure was calculated averaging over the first few eigenvectors. PCA was accomplished using the program g_covar from the GROMACS-4.0.5 package. Rotational and translational motions were removed by fitting the peptide structure of each time frame to the starting structure.
Assembly
The equilibrated TMDs derived from MD simulations are used to generate tetrameric assemblies via various routes (Fig. 1).
Fig. 1.
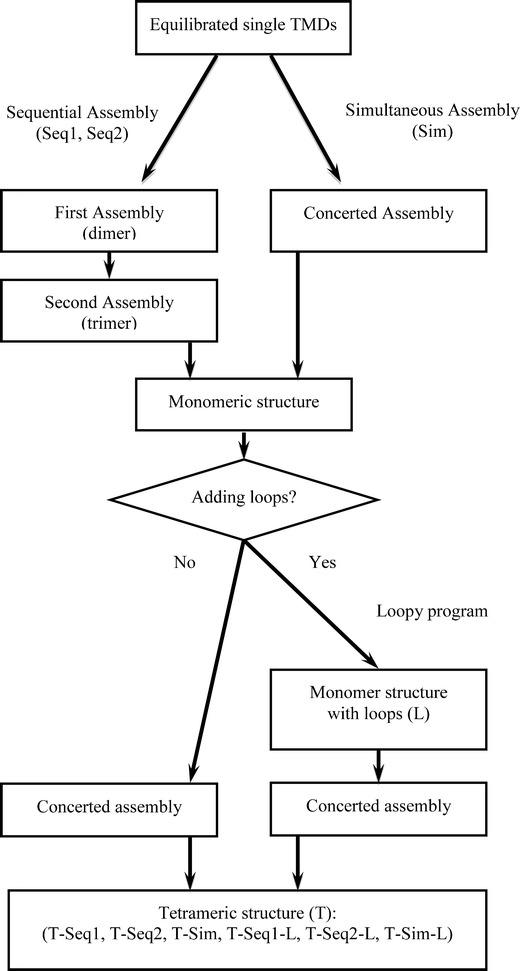
Flowchart of the steps involved forming the tetrameric assemblies. Single TMDs are either assembled in a sequential (Seq1, Seq2) or concerted (Sim) manner to form monomeric structures. The monomers are assembled into tetramers (T) either with loops (L) or without prior to assembly
Monomer assembly
-
Sequential assembly
The helical backbone structure from PCA analysis is aligned along the z-axis. Two methods were used to assemble the monomer (Fig. 1): sequential 1 (Seq1, assembly from C-terminus to N-terminus) and sequential 2 (Seq2, assembly from N-terminus to C-terminus). For Seq1, TMD3 and TMD2 were assembled first becoming a new TMD unit (TMD3 + TMD2). This unit is consequently assembled with TMD1 to form a monomeric subunit ((TMD3 + TMD2) + TMD1). Herein, one of the TMDs was fixed and the second TMD was rotated around the other TMD (rotational angle 2) and around its own helical axis (rotational angle 1), then tilted and translated to the other TMD. The same rotation protocol was adopted for the generated unit of two TMDs being kept fixed whilst the third TMD was rotated around the unit. Similarly, for Seq2, TM1 is first assembled with TM2 to form a new TMD unit followed by the assembly with TMD3 to form a monomer ((TMD1 + TMD2) + TMD3).
-
Simultaneous assembly
In the simultaneous assembly three TMDs are assembled in a concerted fashion to form a monomeric subunit [31]. This assembly method is hither forth referred to as Sim. According to the symmetry all single TMD backbones were rotated around their own helical axis in the same sense with respect to the central pore axis, and were also tilted simultaneously. The construction of a trimer followed basic geometry with inter-helical separation angles of 120°. Besides, to cover all weak and tight packing inter-helical distances in the range from 8.5 to 12.0 Å were sampled for each monomer assembly method. The distance data are referred to as the distance between the center of mass of each TMD.
-
Adding loops
Monomeric models ‘with loops’ obtained their ‘loops’ using the program Loopy [35, 36]. Two loops (loop1: residues 60–78; loop2: residues 100–104) were added on the monomeric subunit accordingly. The lowest energy structures are named Seq1-L, Seq2-L, Sim-L.
Tetramer assembly
The monomeric subunits were assembled into a tetrameric bundle using the Sim protocol. According to the protocol used for the monomeric subunit the tetrameric bundles are referred to as T-Seq1, T-Seq2 and T-Sim (with added loops T-Seq1-L, T-Seq2-L and T-Sim-L). The interhelical separation angle was set to 90° and the interhelical distances sampled in the range of 18 to 24 Å to cover all possible packing modes.
To further sample conformational space, several degrees of freedom were varied systematically, such as interhelical distance by 0.25 Å, rotational angle by 5°, and tilt angle (hither forth called tilt) by 2°. After each positioning, side chain atoms were reconstructed, followed by an energy minimization of 5 steps of steepest descend and 10 steps of conjugated gradient. Potential energy of each conformation/position was evaluated based on the Amber 94 force field in an implicit lipid environment characterized by a dielectric constant of ε = 2. With this protocol hundreds of thousands of different conformations were generated.
MD simulations
The selected tetrameric bundle was then embedded into a POPC lipid bilayer system by removing overlapping lipids and waters molecules. After energy minimization, 4 or 16 Cl- ions were added to compensate for the positive net charge of each monomer. Finally, the whole system without adding loops to the bundle consisted of the bundle (2624 atoms), 462 POPC- and 14616 SPC-water molecules including 4 Cl- (70500 atoms in total). The system with added loops consisted of the 3640 bundle atoms, 462 POPC-, and 14604 SPC-water molecules, including 16 Cl- (71492 atoms in total). The MD simulation protocol was as followed, after energy minimization (see above), 20 ns production runs were carried out without any constraint on the bundle.
GROMACS-4.0.5 with the Gromos96 (ffG45a3) force field was used for the simulations. The simulations were conducted in the NPT ensemble employing the velocity-rescaling thermostat at constant temperature 310 K, and 1 bar. The temperature of the protein, lipid and the solvent were separately coupled with a coupling time of 0.1 ps. Semi-isotropic pressure coupling was applied with a coupling time of 0.1 ps and a compressibility of 4.5 x 10−5 bar−1 for the xy-plane as well as for the z-direction. Long range electrostatics calculated using the particle-mesh Ewald (PME) summation algorithm with grid dimensions of 0.12 nm. Lennard-Jones and short-range Coulomb interactions were cut off at 1.4 and 0.8 nm, respectively.
The simulations were run on a DELL Precision T5400 workstation, and a cluster consisting of 32 cores (Xeon 2.26 GHz). Plots and pictures were generated using xmgrace, VMD and MOE.
Results
Equilibration
All three TMDs show stable root mean square deviation (RMSD) values over the entire duration of the simulation. Within the last 4 ns of the 10 ns simulation values between 0.1 and 0.2 nm are calculated identifying that the short run deliver reasonably equilibrated structures (Fig. 2a). The root mean square fluctuation (RMSF) of the individual residues of the TMDs shown in Fig. 2b is indicative for low dynamic of the amino acids with higher fluctuation at either end of the TMDs. The residues of the core region of the TMD, albeit at very low level, exhibit slightly higher dynamics than the residues in the head group region (appr. residues 10 – 25 and 55 – 70) giving the graph a w-like shape. A sequence of residues from Asn-82 to Leu-85 and around Leu-94 to Leu-96 of TMD2 shows a localized area of larger RMSF values.
Fig. 2.
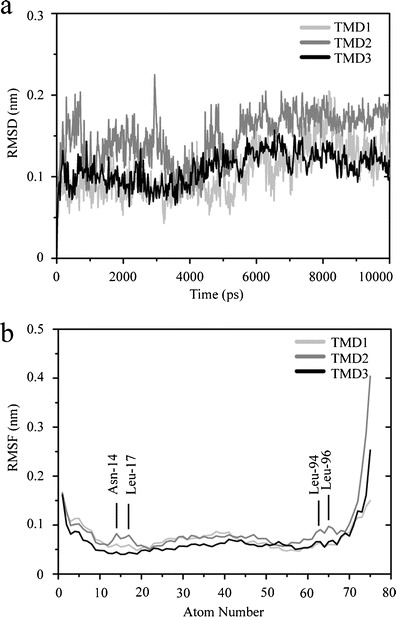
Root mean square deviation (RMSD) of the Cα backbones of the single TMDs, TMD1, TMD2 and TMD3, referring to the respective starting structure (a). Root mean square fluctuation (RMSF) of the atoms of the amino acids (b). The TMDs are overlaid so that the atom numbers match for each TMD. Values for TMD1 are shown in light gray, those for TMD2 in gray and the values for TMD3 in black
Assembly
Generation of the monomer
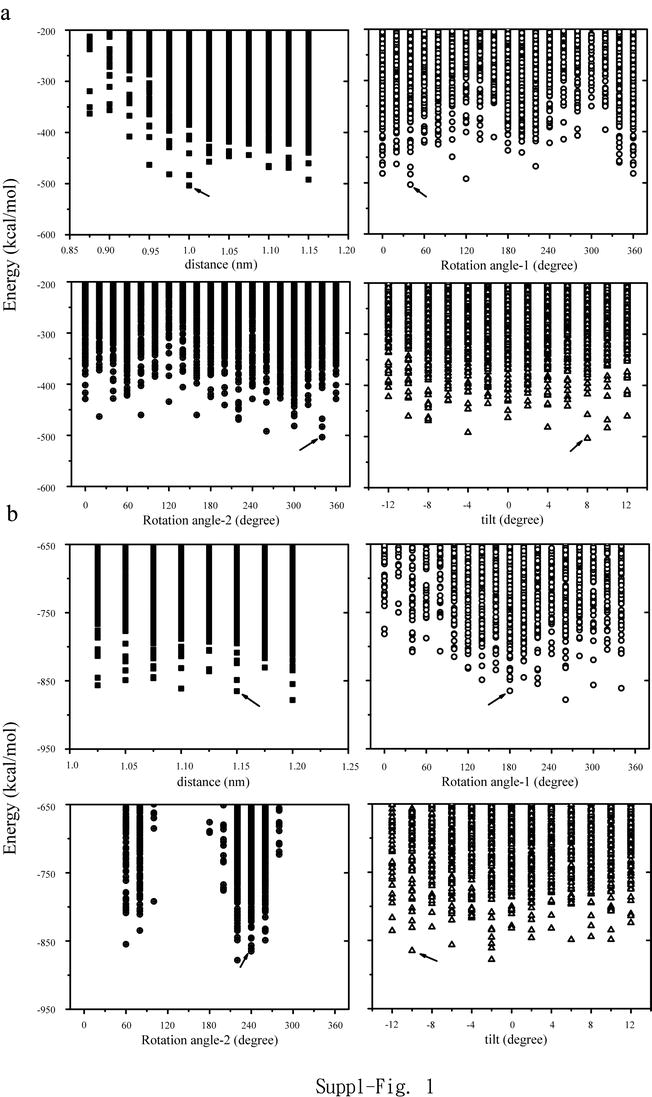
Analysis of the energetic of the monomer assemblies (Suppl. Fig. 1) reveals mostly some close clustering of lowest values independent of the sequence used. Using the first part of Seq1, assembling TMD2 and TMD3, reveals a dimer with lowest energy of −503.6 kcal mol−1 (Table 1 and Suppl. Fig. 1a). Assembling the third TMD, TMD3, results in two low energy structures calculated with values of −878.1 kcal mol−1 and −865.12 kcal mol−1 and interhelical distances of 1.2 and 1.15 nm, respectively (Suppl. Fig. 1b). Both structures are separated by their individual rotational angles but adopt the same tilt direction of −2° and −10°, respectively.
Table 1.
Lowest and second lowest energy structures of the dimer and finally the monomeric structures. Data represent the interhelical distance, rotational angle of each of the individual TMDs and the averaged (overall TMDs) tilt angle, as well as the energy calculated with MOE
| Method | Distance (nm) | Angle 1 (°) | Angle 2 (°) | Angle 3 (°) | Tilt (°) | Energy (kcal/mol) |
|---|---|---|---|---|---|---|
| Seq1 | 1.0 | 40 | 340 | 8 | −503.6 | |
| (TMD3 + TMD2) | ||||||
| Seq1 | 1.20 | 260 | 220 | −2 | −878.1 | |
| ((TMD3 + TMD2) + TMD1) | 1.15 | 180 | 240 | −10 | −865.1 | |
| Seq2 | 0.95 | 340 | 300 | 0 | −349.6 | |
| (TMD1 + TMD2) | ||||||
| Seq2 | 1.175 | 0 | 40 | 2 | −863.5 | |
| ((TMD1 + TMD2) + TMD3) | ||||||
| Sim | 1.075 | 60 | 180 | 340 | −4 | −730.5 |
| (TMD1 + TMD2 + TMD3) | 1.075 | 40 | 100 | 80 | 4 | −654.8 |
Seq2 reveals a dimer of TMD1 and TMD2 of −349.6 kcal mol−1. Adding TMD3 results in a monomer of −863.5 kcal mol−1, with an interhelical distance of 1.175 nm.
Finally for Sim the lowest energy structure (−730.5 kcal mol−1) is clearly distinct from the second lowest (−654.8 kcal mol−1) by 75.7 kcal mol−1 (data not shown). For these two structures the rotation difference of TMD2 is of up to 40° and with an opposite tilt direction (−4°) than the second lowest (+4°). Similar to the monomer Seq1 the lowest energy structure has a hydrophobic pore and therefore the second lowest monomer is considered further. The interhelical distance of the second lowest structure is 1.075 nm.
The monomeric structure from Seq1 (Fig. 3a) exhibits a hydrophilic stripe, which is due to residues of TM3 (Tyr-109, Tyr-113, Gln-116, and Asn-119) spanning the entire TM stretch. Residues like His-93, Tyr-89 and Asn-82 of TMD2 join toward the same direction. The monomeric structure assembled from Seq2 indicates two hydrophilic stripes, one from Ser-92, His-93, Thr-89, and Asn-82 (all TMD2), and the other from Tyr-109, Tyr-113, Gln-116, and Arg-122 (all TMD3) (shown in Fig. 3b). Similar to Seq1, Sim reveals a single line of hydrophilic residues due to hydrophilic residues of TMD3 (Fig. 3c).
Fig. 3.
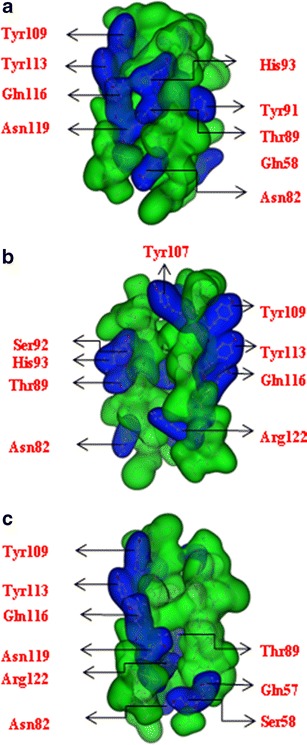
Monomers according to the assembly protocol: Seq1 (a), Seq2 (b) and Sim (c). Hydrophilic residues are highlighted in blue, hydrophobic residues in green. All models are drawn in a ‘Gaussian Contact’ illustration (MOE)
Tetramer assembly without loops
Assembling T-Seq1 a structure with a minimum energy (−4710.72 kcal mol−1, Table 2) is obtained for inter-monomer distance from the centers of mass of the monomers of 2.375 nm (Suppl. Fig. 2, I). The structure adopts a rotational angle of 320° and a tilt of 9°. TM2 is the pore lining with only one hydrophilic residue Tyr 91 (highlighted in Fig. 4a) facing the pore.
Table 2.
Lowest energy structures of the tetramer generated from monomers listed in Table 1. Data represent inter monomer distances, rotational and tilt angles, as well as the interaction energy calculated with MOE. The TMD of each of the monomer facing the pore is listed for each of the tetramers
| Method | Distance (nm) | Angle (°) | Tilt (°) | Energy (kcal/mol) | Pore lining |
|---|---|---|---|---|---|
| T-Seq1 | 2.375 | 320 | 9 | −4710.7 | TMD2 |
| T-Seq2 | 2..5 | 180 | 36 | −4724.8 | TMD2 |
| T-Sim | 2.10 | 190 | 24 | −4294.2 | TMD3 |
| T-Seq1-L | 2.025 | 310 | 24 | −5596.9 | TMD2 |
| T-Seq2-L | 2.20 | 150 | −36 | −6136.7 | TMD2 |
| T-Sim-L | 2.20 | 305 | 21 | −5543.0 | TMD2 |
Fig. 4.
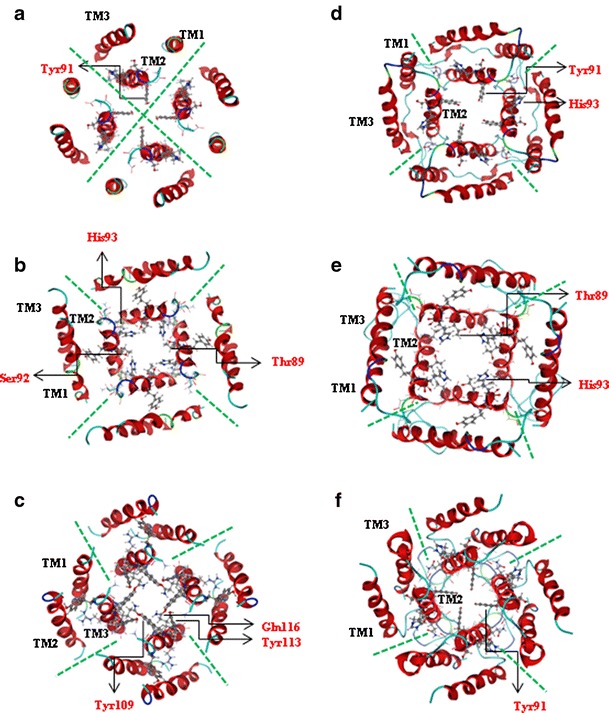
Tetramers according to the assembly protocols without loops: T-Seq1 (a), T-Seq2 (b), T-Sim (c); tetramers assembled with loops added after monomer assembly: T-Seq1-L (d), T-Seq2-L (e), T-Sim-L (f)
Alignment of T-Seq2 shows a Lennard-Jones type pattern for the low energy values with the lowest value −4724.84 kcal mol−1 of 2.15 nm interhelical distance (Suppl. Fig. 2, II). Although in T-Seq1 TMD2 is facing the pore, similar to T-Seq2, the rotational angle is different in T-Seq2, leading to more hydrophilic residues inside the pore lumen (Asn-82, Thr-89, Ser-92, and His-93, Fig. 4b).
Alignment of T-Sim derives a lowest energy structure of −4294.2 kcal mol−1 with an inter monomer distance of 2.1 nm (Fig. 4c). A second low energy model (see Suppl. Fig. 2, III) does not expose any hydrophilic residues into the pore. In T-Sim TMD3 is pore lining, with several hydrophilic residues facing the pore (Tyr-109, Tyr-113, Gln-116, and Asn-119).
Tetramer assembly with added loops
In another approach the monomeric units are assembled in the presence of the loops between TMD1 and TMD2 as well as between TMD2 and TMD3. Assembling four copies of the Seq1-L monomer delivers a low energy structure with distances of around 2.025 nm (−5596.99 kcal mol−1, T-Seq1-L) (Table 2). In T-Seq1-L bundle TMD2 is pore lining with Tyr-91 inside the pore lumen and His-93 facing outside the pore (Fig. 4d).
Assembling Seq2-L into a tetramer shows a low energy model with an monomer distance of 2.2 nm (−6136.76 kcal mol−1), and a tilt angle of −36° T-Seq2-L is shown in Fig. 4e with two hydrophilic residues, Thr-89 and His-93, of TMD2 face the pore.
Screening the energy landscape of Sim-L, the model with the lowest energy (−5543 kcal mol−1, T-Sim-L) has an inter monomer distance of 2.2 nm (data not shown). The tilt of its monomers adopts 21°. The lowest energy bundle, T-Sim-L, exposes hydrophilic residue Tyr 91 of TMD2 to the pore (Fig. 4f). Although the T-Sim-L is similar to T-Seq1-L with one with Tyr 91 inside the pore and His 93 outside the pore, the pore of T-Sim-L has more hydrophilic residues pointing into the pore than T-Seq1-L.
Comparing the energy values amongst the monomers reveals that Seq1 and Seq2 generate monomers with minimum energies around −860 kcal mol−1 to −880 kcal mol−1 whilst Sim generates monomers with higher values of around −650 kcal mol−1 and −730 kcal mol−1 (Suppl. Fig. 1). The bundle models reflect this trend independent of the presence of the loops (Suppl. Fig. 2). Whilst energies for bundles similar to T-Seq1 and T-Seq2 both are calculated to be around −4700 kcal mol−1, the respective values for bundles similar to T-Seq1-L and T-Seq2-L show lower values for the bundles similar to T-Seq2-L: around −6100 kcal mol−1 (T-Seq2-L) versus −5600 kcal mol−1 (T-Seq1-L). The energy values for the bundles similar to T-Seq1-L are indistinguishable from those for bundles according to T-Sim-L. As a result, T-Seq2-L is the bundle with the low interaction energy.
MD simulations
All six tetrameric assembled structures of 3a from SARS CoV (Fig. 4) are run for 20 ns of a MD simulation embedded into a bilayer of POPC to equilibrate the structures further. The RMSD plot for Cα atoms of the tetrameric bundles without loops is shown in Fig. 5a. The data reveals a progressive rising for all structures and consequent stable fluctuation after the first 5 ns (Fig. 5a, I). All RMSD values remain in a range of 0.1 – 0.3 nm. In order to know how each TMD affects the stabilization of the structure, the RMSD of each TMD for the three bundles are shown individually. For T-Seq1 the RMSD of all TMDs are within the same range of 0.2 – 0.3 nm. (Fig. 5a, II). The RMSD values for TMD1 and TMD3 of T-Seq2 are higher (∼ 0.24 nm for TMD1, and ∼0.26 nm for TMD3) than for TMD2 (∼ 0.15) (Fig. 5a, III). The same situation can also be found in T-Sim with TMD3 pore lining (RMSD ∼ 0.19 nm) and TMD1 (∼ 0.23 nm) and TMD2 (∼ 0.25 nm) at the outside of the bundle (Fig. 5a, IV). Super positioning the final structure (green, Fig. 6a-c) with the initial structures (red, Fig. 6a-c) indicates the result of the RMSD calculations in as much as the bundles do not deviate from each other very much, but show a pattern that the non-pore lining TMDs experience larger deviation from the initial structure than the pore lining residues.
Fig. 5.
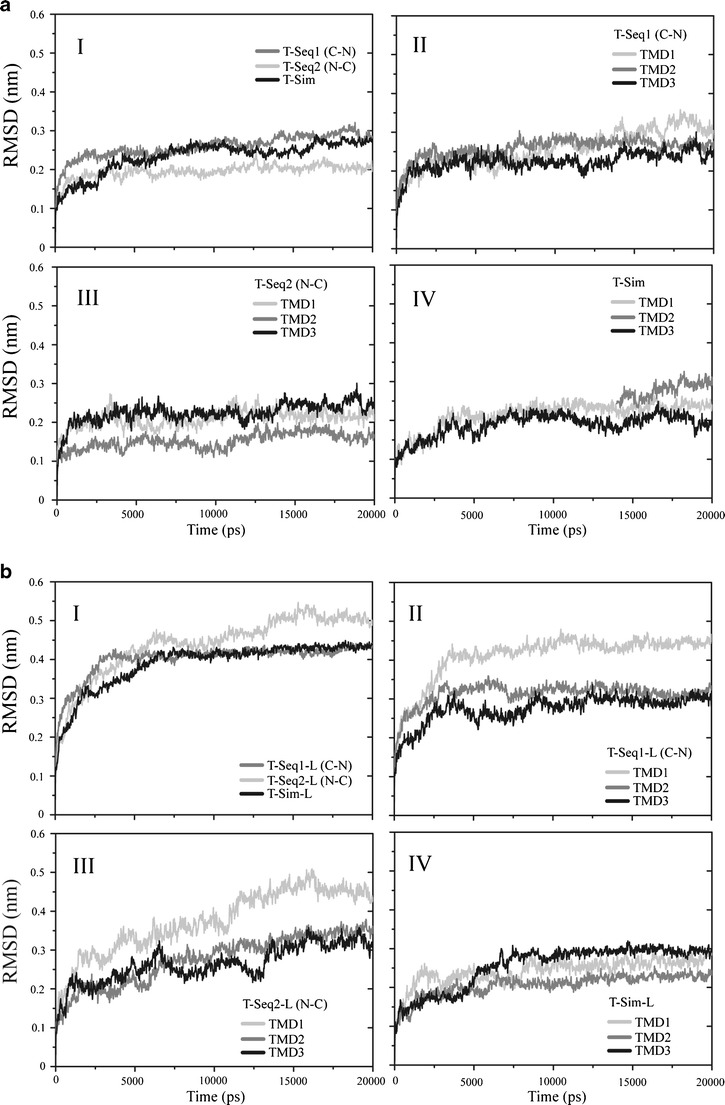
Root mean square deviation (RMSD) of the of Cα backbones of the bundle structures referring to the starting structure. T-Seq1 (gray), T-Seq2 (light gray) and T-Sim are shown (aI). The respective RMSD values for the individual TMDs of each simulation (TMD1 in light gray, TMD2 in gray, TMD3 in black) are shown separately (aII-IV). RMSD values of the bundles including the loops are shown for T-Seq1-L, TSeq2-L and T-Sim-L (b). Color coding and arrangement of the panels like in (a)
Fig. 6.
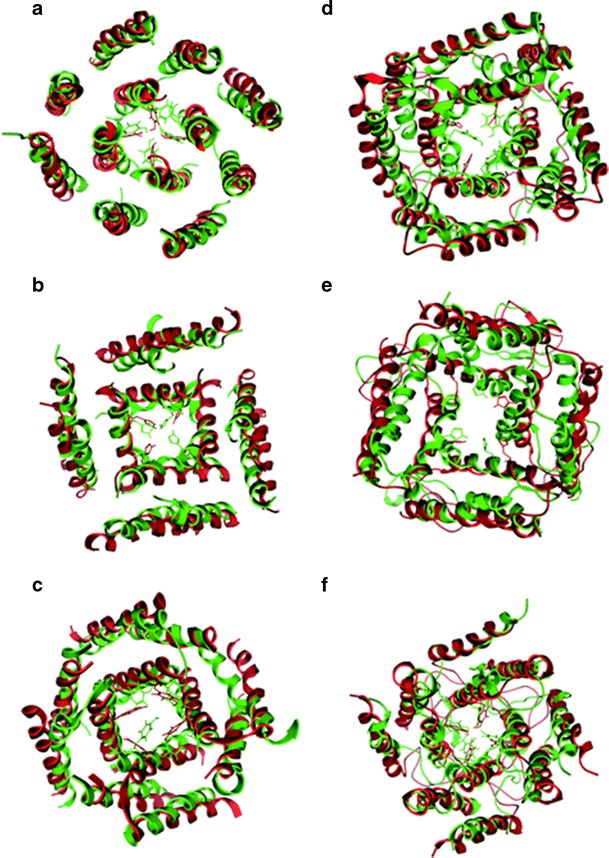
Models of T-Seq1 (a), T-Seq2 (b) and T-Sim (c), T-Seq1-L (d), T-Seq2-L (e) and T-Sim-L (f) are shown in their starting conformation (green) and after 20 ns of MD simulation (red)
RMSD values for the bundles with loops indicate deviations in the range of 0.35 – 0.5 nm (Fig. 5b, I). T-Seq1; The large deviation is due to TMD1 in T-Seq1-L (∼ 0.43 nm) and T-Seq2-L (∼ 0.45 nm) shown in Fig. 5b, II and III. TMDs 2 and 3 in both bundles almost not deviate from each other. The RMSD values for TMDs of T-Sim-L are in a close range (0.24 ∼ 0.30 nm) (Fig. 5b, IV). There is a tendency for increased values in the order TMD2 < TMD1 < TMD3. Indicating TMD2, which is pore lining to exhibit the lowest deviation. The superposition of the initial and final bundle for the structures with loops reflect the RMSD data that at least one of the TMD outside the pore has a large deviation, most likely TMD1. Less deviation is observed for the second outer TMD and the pore lining TMD.
Pore-radius analysis
The pore radii of the first 25 structures (covering five hundred pico second simulation in steps of 20 ps, Fig. 7, light lines) are compared to the radii derived toward the end of the simulation, taking the last 25 structures in steps of 20 ps for all the bundles (Fig. 7, thick lines). For T-Seq1 bundle, inside the membrane there are three local minima in the initial structure (Fig. 7a, thin line), caused by rings of Phe-87 (at −1.5 nm), Tyr-91 (at −0.5 nm), and Leu-94 (at 0.6 nm). The minimum pore radius is at Tyr-91, about 0.02 nm. Toward the end of the simulation only the region around Leu-94 is closed causing a minimum pore radius of 0.05 nm. For T-Seq2 (Fig. 7b), minima are caused by His-93 at position 0.3 nm and Leu-96 (at 1.2 nm). The minimum pore radius is at His-93, with about 0.04 nm. After 20 ns the pore radius is calculated to be around 0.02 nm around both, His-93 and Leu-96. T-Sim minima cover the stretch along Gln-116 (position −0.7 nm), Tyr-113 (position −0.1 nm), and Tyr-109 (position 1.0 nm) in the initial configuration (Fig. 7c). The minimum pore radius is at Gln-116, with about 0.026 nm. At the end of the simulation the entire stretch around Tyr-113 to Gln-116 retains a narrow pore passage with even Phe-105 at position 1.75 nm closing in at the mouth of the pore inducing almost a closure of the pore (minimum radius 0.03 nm).
Fig. 7.
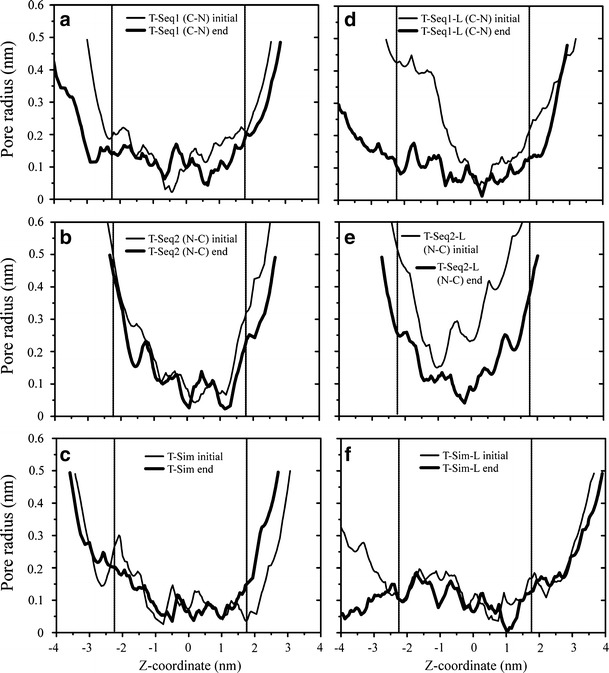
Pore radii calculated using the software HOLE [56]. The values of the first 25 structures, covering 500 ps simulation in steps of 20 ps, are averaged and depicted in light lines. A similar average has been calculated covering the last 500 ps of the simulations (thick lines). Models of T-Seq1 (a), T-Seq2 (b) and T-Sim (c), T-Seq1-L (d), T-Seq2-L (e) and T-Sim-L (f) are shown
The starting structure of T-Seq1-L bundle indicates a very narrow passage around Tyr-91 at position 0.45 nm with a minimum radius of 0.04 nm (Fig. 7d, thin line). After 20 ns the whole pore collapses and the minimum pore radius around Tyr-91 is at 0.013 nm (Fig. 7d, thick line). In T-Seq2-L a smallest pore radius is found around Leu-85 (position −1.1 nm) with about 0.15 nm (Fig. 7e). Two more space confinements are around Thr-89 (position 0.0 nm) with a radius of 0.2 nm and His-93 (position 0.85 nm) adopting a radius of 0.4 nm. During the simulation pore confines around Thr-89 at around −0.2 nm with a radius of 0.04 nm. For T-Sim-L the minimum pore radius of initial average structure is located at Tyr-91 at position 0.8 nm with a radius of 0.04 nm (Fig. 7f). At the end of the simulation the tyrosines have closed the pore. Constriction is at 1.0 nm due to the flexibility of the aromatic side chains.
Water molecules trajectories analysis
Water molecules do show three different kind of behaviors, (i) they get trapped in the pore found for T-Seq1 (data not shown), T-Sim-L, and T-Seq2-L (ii) they enter the pore on either side and escape on the same side found especially for T-Seq2 and T-Sim, and T-Seq1-L (iii) water molecules traverse the pore completely as found only for T-Seq2-L (5 water molecules in total). Adding the loop to the bundles results in pores with the likely hood of enabling a water passage across the bundle.
Discussion
Biological considerations
Experiments with 3a have identified the protein as a tetrameric unit enabling ion flux across the plasma membrane of infected Xenopus oocytes [4] which can also be inhibited by emodin [37]. Based on the experimental evidence the idea is to suggest a potential channel assembly based on experience in assembling smaller channel forming proteins [15, 31, 38–40]. Similar to other channel forming proteins such as Vpu from HIV-1 [41], also 3a is reported to interact with host factors [42]. Therefore these proteins are also called accessory proteins. The term implies that the presence of the protein helps the virus, but the virus is not dependent on it. Based on electrophysiological measurements the formation of channels cannot be ruled out at this stage and has to be considered also for drug development.
Considerations about the assembly protocol
A specific protocol is used to generate the tertiary structure of the TMD of a membrane protein [31]. It takes the secondary structural elements of the TMD which are helices in this study and screens the interactions of these helices in 2D. Upon each positioning in 2D the potential orientation of the side chains at each position is taken from rotational library integrated in the program MOE. Each position is allowed to relax via energy minimization prior to energy calculations. Screening in 3D with a rigid body approach as done by other programs (e.g., [43]) has been omitted due to biological reasons as vertical movement of TMDs within a lipid bilayer is very much limited. It is anticipated that, e.g., adjustment to lipid dynamics is rather achieved by changes in tilt angles which is taken care of in the present assembly approach. With the assembly protocol at hand it is possible to evaluate different kind of routes of assembly.
Assembly of membrane proteins and especially the TMDs can go two ways, either they are done ‘ab initio’, or they are done taking biological considerations into account. The first approach has been demonstrated to deliver results on other viral channel proteins which are in agreement with experiments [31]. Another approach is to assume biological pathways such as the TMDs once released from the translocon assembly step-by-step, in a sequential way. After another short period of time they find the other monomers to assemble finally in the functional form. In the protocol described at the stage of assembling the tetramer, the concerted protocol is used. A sequential assembly at this stage, however, does not need to be ruled out. Assembly at this level may follow another biological pathway: The monomer can be in equilibrium between “free” and “raft” or “protein attached” states. Raft association has been proposed for M2 [44] and Vpu [45]. Thus also a raft attached state could be the seed for assembly of more monomers. In addition, the same scenario could be followed attaching to a host factor first or even to generate the covalent link between two of these monomers. All of these routes would be necessary to be taken into account. In the lack of any information about these scenarios the concerted assembly at this stage seems to be reasonable. It is assumed that the approach samples all low energy structures which inevitably impose constraints also on the “biological” pathways.
During the sequential assembly two routes are assumed, from the C to N termini and the opposite direction. The assembly route from C to N termini reflects the idea that TMD1 escapes the translocon first, ‘diffuses’ away and allows the other two TMDs to be assembled first. This idea may be synonymous for a “loose” packing of the helices. The opposite route takes its rationale from the consideration that TMD1 may be retained near or at the translocon despite the longer loop between the TMD1 and the consecutive TMD2. Consequently TMD2 is manufactured and assembled with TMD1, followed by the assembly of TMD3. This route could be seen as a “constraint” packing.
Bundle and pore structure
All bundles in common are a pore lining TMD2 except for the bundle without loops built from the monomer using the simultaneous assembly protocol (T-Sim). TMD2 as the pore lining domain creates a Tyr-only (using Seq1 and Sim with loops) and a His/Tyr (Seq2) motif within the pore, whilst TMD3 creates a Tyr/Gln motif (using Sim without loops). A histidine within a pore has been found for M2 from influenza A [46, 47] and is proposed for p7 from HCV [39]. Tyrosines lining the pore may rather be unusual. Tyrosines may catch a cation via cation/π interaction [48] and impose an ion trap along the pathway. Together with histidine this energy we think may be overcome making the bundle derived from Seq2 protocol the most likely one. Similar to M2 in respect to the number of monomers it seems to be likely that 3a may even be conducting protons rather than ions or should at least be pH dependent in its mechanism of function similar to the same proposal for p7 [39]. The Tyr/Gln motif may adopt the same mechanism as assumed for the bundle with the Tyr/His motif. At this stage it cannot be discriminated which motif would be the most effective one in respect to ion or proton conductance.
In a configuration of TMD2 the pore lining domain and TMD3 at the outside allows conformational freedom to enable covalent Cys-Cys linkage within the extramembrane part (Cys-133 in [4]) of two monomers without constraining the packing of the overall bundle in respect to the pore lining configuration.
Previously we have assembled in simultaneous mode, and got TMD3 pore lining. With a ‘biological route’ we suggest TMD2 pore lining.
Bundle dynamics
The results from the short equilibration dynamics of the bundles without loops deliver the picture that the inner helices of the bundles remain constrained relative to the TMDs at the outside of the bundle facing the lipid environment (T-Seq2, T-Sim). In all simulations of the bundles generated with the loops TMD1 shows the largest deviation from the starting structure whilst the values for TMD3 go almost in concord with those for TMD2 (T-Seq1-L, T-Seq2-L). This suggests that the assembly protocol delivers a structurally stable pore motif whilst the outer TMDs still need an extended equilibration. With the outer TMDs adjusting during the MD simulation, the pore lining TMDs are unaffected by the dynamics of the outer TMDs for the bundles without loop. It further implies that the short loop between TMD2 and TMD3 restrains the dynamics of TMD3. The findings suggest that the outer TMDs could be susceptible and allowing for some dynamics without affecting the inner helices.
In respect to the dynamics of the TMDs, analysis of the temperature (B) factor a series of crystal structures of known channel and pore proteins reveals a pattern in which helical TMDs surrounded by other helical TMDs show lower temperature (B) factors [49–53]. In the case of the mechanosensitive channel [54], the closed state model of pentameric ligand gated ion channel (LGIC) [49] and the glutamate receptor [51] a similar gradient of the temperature (B) factor for the TMDs across the membrane exists. For the mechanosensitive channel lower factors are found in the center of the TMDs and higher factors to both sides whilst for pLGIC and the glutamate receptor the temperature factor decreases within the TMDs toward the extramembrane domain of the channel. These data suggest that central TMDs adopt some rigidity whilst outer TMDs allow for some dynamics.
Water molecules in the pore
During the short equilibration the pore radius in all models fall below the radius of a sodium ion (e.g., 0.1 nm [55]) implying sever constraints onto the putative passage of ions. Only the bundle generated according to Seq2 with loops (T-Seq2-L) allows some water molecules to traverse. The water molecules remain on the level of the ring of His-93 for several ns before they leave the place in the other direction. All bundles have in common that not only hydrophilic stretches but also the rings of tyrosine impose special constrains on the passage through the pore. The findings for T-Seq2-L with water molecules crossing the pore and tyrosines restricting the pore it is likely that T-Seq2-L is the bundle of choice in this study. It may further underpin the suggestion of 3a to be proton conducting or at least sensitive to and triggered by the pH of the environment.
The lack of a continuous water column, which exists over the entire simulations in any of the bundles, imposes the question what are the necessities to generate and maintain such a column. At this stage it is speculated that ions are necessary to “stabilize” the pore and similar to the finding for the K+-channel are essential for ion conductance.
At this stage any conductance of substrates has to be ruled out making the protein rather more ion channel like than pore like.
Role of the loops
Throughout the protocol we do not find a major impact of the loops on the structural modeling. The only exception is that in T-Sim TMD3 is suggested to be pore lining. However in the light of missing dynamics of the loops during assembly T-Sim may be rather a conformational exception. This underpins the idea that structural features can be independently modeled from the rest of the protein. Any extramembrane parts can be added after assembly. Possibly proteins are built in either of the environments, hydrophilic or hydrophobic, and then assembled. This leaves the question of the dynamics of the linker region between these two segments open for debate.
It is evident that the bundles with loops added have lower energy than those without loops. This is an indication that the addition of the loops improves the stability of the bundle.
Conclusions
Modeling of a membrane protein from ab initio conditions delivers a reasonable model of 3a prior to experimental calculations. Model generation is based on a combination of pure energetic considerations and the implementation of biological manufacturing praxis. As expected the computational approach delivers not a single result but the plurality can be reduced by considering further calculations on the proposed structural models. At the current level of calculations it is suggested that 3a adopts a bundle structure with TMD2 facing the putative pore albeit a TMD3 pore lining cannot be completely ruled out. The configuration delivers a Tyr and/or His motif to line the pore. It is further concluded based on the low pore radii generated by the protocol that ions embedded within the pore may be necessary to stabilize the pore and enabling ion flux. With histidine as part of the pore motif, 3a may also be a proton channel or at least sensitive or triggered by the pH around it. The pore architecture as presented would rule out 3a to be a substrate conducting pore.
Short equilibration runs using MD simulations are indicative for an excellent packing of the inner helices. The outer TMDs still need an extended equilibration to adjust for the bundle architecture.
With the more complex architecture 3a must be able to harbor a more precise activation mechanism. With this the role of the channel protein could be more specific and triggered by a more specific modulation mechanism underpinning is status as an ion/proton channel rather than a pore.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
Potential energies (MOE) of the assemblies to generate the monomer Seq1: TMD3 and TMD2 displayed in respect to distance rotational angle_1, rotational angle_2, and tilt (a). Potential energies of the assemblies of (TMD3+TMD2) with TMD1 are shown as for (a) to form Seq1 (b). The structure used for further studies is marked by the arrow (JPEG 117 kb)
{kind=link}
Potential energies of the assembled tetramers without loops. Values for generating all the sequences are shown in dependence of distance (left panels), rotational angles (middle) and tilt (right panels). The arrows indicate models T-Seq1 (I), T-Seq2 (II), T-Sim (III) (JPEG 137 kb)
Acknowledgments
WBF acknowledges National Yang-Ming University, the government of Taiwan and the National Science Council of Taiwan (NSC) for financial support. Thanks to D. Willbold (Jülich, D) for valuable discussions. We thank the National Center for High-Performance Computing of Taiwan (www.nchc.org.tw) for providing computer time.
References
- 1.Marra MA, Jones SJM, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YSN, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Gordon Robertson A, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M, Petric M, Skowronski DM, Upton C, Roper RL. The genome sequence of the SARS-Associated Coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 2.Zeng R, Yang R-F, Shi M-D, Jiang M-R, Xie Y-H, Ruan H-Q, Jiang X-S, Shi L, Zhou H, Zhang L, Wu XD, Lin Y, Ji YY, Xiong L, Jin Y, Dai EH, Wang XY, Si BY, Wang J, Wang HX, Wang CE, Gan YH, Li YC, Cao JT, Zuo JP, Shan SF, Xie E, Chen SH, Jiang ZQ, Zhang X, Wang Y, Pei G, Sun B, Wu JR. Characterization of the 3a protein of SARS-associated coronavirus in infected Vero E6 cells and SARS patients. J Mol Biol. 2004;341:271–279. doi: 10.1016/j.jmb.2004.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan YJ, Teng E, Shen S, Tan THP, Goh PY, Fielding BC, Ooi EE, Tan HC, Lim SG, Hong W. A novel severe acute respiratory syndrome coronavirus protein, U274, is transported to the cell surface and undergoes endocytosis. J Virol. 2004;78:6723–6734. doi: 10.1128/JVI.78.13.6723-6734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu W, Zheng BJ, Xu K, Schwarz W, Du L, Wong CKL, Chen J, Duan S, Deubel V, Sun B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc Natl Acad Sci USA. 2006;103:12540–12545. doi: 10.1073/pnas.0605402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen CY, Ping YH, Lee HC, Chen KH, Lee YM, Chan YJ, Lien TC, Jap TS, Lin CH, Kao LS, Chen YMA. Open reading frame 8a of the Human severe acute respiratory syndrome coronavirus not only promotes viral replication but also induces apoptosis. J Infect Dis. 2007;196:405–415. doi: 10.1086/519166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fischer WB, Sansom MSP. Viral ion channels: structure and function. Biochim Biophys Acta. 2002;1561:27–45. doi: 10.1016/S0304-4157(01)00009-0. [DOI] [PubMed] [Google Scholar]
- 7.Fischer WB, Hsu HJ. Viral channel forming proteins - modelling the target. Biochim Biophys Acta. 2011;1808:561–571. doi: 10.1016/j.bbamem.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer WB, Krüger J. Viral channel forming proteins. Int Rev Cell Mol Biol. 2009;275:35–63. doi: 10.1016/S1937-6448(09)75002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allen H, McCauley J, Waterfield M, Gethering M. Influenza virus RNA segment 7 has the coding capacity for two polypeptides. Virol. 1980;107:548–551. doi: 10.1016/0042-6822(80)90324-4. [DOI] [PubMed] [Google Scholar]
- 10.Holsinger LJ, Nichani D, Pinto LH, Lamb RA. Influenza A virus M2 ion channel protein: a structure-function analysis. J Virol. 1994;68:1551–1563. doi: 10.1128/jvi.68.3.1551-1563.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mould JA, Li HC, Dudlak CS, Lear JD, Pekosz A, Lamb RA, Pinto LH. Mechanism for proton conduction of the M2 ion channel of influenza A virus. J Biol Chem. 2000;275:8592–8599. doi: 10.1074/jbc.275.12.8592. [DOI] [PubMed] [Google Scholar]
- 12.Lin T, Schroeder C. Definitive assignment of proton selectivity and attoampere unitary current to the M2 ion channel protein of influenza A virus. J Virol. 2001;75:3647–3656. doi: 10.1128/JVI.75.8.3647-3656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schubert U, Ferrer-Montiel AV, Oblatt-Montal M, Henklein P, Strebel K, Montal M. Identification of an ion channel activity of the Vpu transmembrane domain and its involvement in the regulation of virus release from HIV-1-infected cells. FEBS Lett. 1996;398:12–18. doi: 10.1016/S0014-5793(96)01146-5. [DOI] [PubMed] [Google Scholar]
- 14.Ewart GD, Sutherland T, Gage PW, Cox GB. The Vpu protein of human immunodeficiency virus type 1 forms cation-selective ion channels. J Virol. 1996;70:7108–7115. doi: 10.1128/jvi.70.10.7108-7115.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen CC, Krüger J, Sramala I, Hsu HJ, Henklein P, Chen YMA, Fischer WB. ORF 8a of severe acute respiratory syndrome coronavirus forms an ion channel: experiments and molecular dynamics simulations. Biochim Biophys Acta. 2011;1808:572–579. doi: 10.1016/j.bbamem.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griffin SDC, Beales LP, Clarke DS, Worsfold O, Evans SD, Jäger J, Harris MPG, Rowlands DJ. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, amantadine. FEBS Lett. 2003;535:34–38. doi: 10.1016/S0014-5793(02)03851-6. [DOI] [PubMed] [Google Scholar]
- 17.Pavlovic D, Neville DCA, Argaud O, Blumberg B, Dwek RA, Fischer WB, Zitzmann N. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proc Natl Acad Sci USA. 2003;100:6104–6108. doi: 10.1073/pnas.1031527100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agirre A, Barco A, Carrasco L, Nieva JL. Viroporin-mediated membrane permeabilization. Pore formation by nanostructural poliovirus 2B protein. J Biol Chem. 2002;277:40434–40441. doi: 10.1074/jbc.M205393200. [DOI] [PubMed] [Google Scholar]
- 19.de Jong AS, Wessels E, Dijkman HBPM, Galama JMD, Melchers WJG, Willems PHGM, van Kuppeveld FJM. Determinants for membrane association and permeabilization of the coxsackievirus 2B protein and the identification of the Golgi complex as the target organelle. J Biol Chem. 2003;278:1012–1021. doi: 10.1074/jbc.M207745200. [DOI] [PubMed] [Google Scholar]
- 20.Pervushin K, Tan E, Parthasarathy K, Lin X, Jiang F-L, Yu D, Vararattanavech A, Soong TW, Liu D-X, Torres J. Structure and inhibition of the SARS Coronavirus envelope protein ion channel. PLoS Pathog. 2009;5:e1000511. doi: 10.1371/journal.ppat.1000511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cook GA, Zhang H, Park SH, Wang Y, Opella SJ. Comparative NMR studies demonstrate profound differences between two viroporins: p7 of HCV and Vpu of HIV-1. Biochim Biophys Acta. 2011;1808:554–560. doi: 10.1016/j.bbamem.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Popot JL, Engelman DM. Membrane protein folding and oligomerization: the two-stage model. Biochemistry. 1990;29:4031–4037. doi: 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- 23.Engelman DM, Chen Y, Chin CN, Curran AR, Dixon AM, Dupuy AD, Lee AS, Lehnert U, Matthews EE, Reshetnyak YK, Senes A, Popot JL. Membrane protein folding: beyond the two stage model. FEBS Lett. 2003;555:122–125. doi: 10.1016/S0014-5793(03)01106-2. [DOI] [PubMed] [Google Scholar]
- 24.White SH, Wimley WC. Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 25.Sansom MSP, Kerr ID. Influenza virus M2 protein: a molecular modelling study on the ion channel. Protein Eng. 1993;6:65–74. doi: 10.1093/protein/6.1.65. [DOI] [PubMed] [Google Scholar]
- 26.Kukol A, Adams PD, Rice LM, Brunger AT, Arkin IT. Experimentally based orientational refinement of membrane protein models: a structure for the influenza A M2 H+ channel. J Mol Biol. 1999;286:951–962. doi: 10.1006/jmbi.1998.2512. [DOI] [PubMed] [Google Scholar]
- 27.Cordes F, Kukol A, Forrest LR, Arkin IT, Sansom MSP, Fischer WB. The structure of the HIV-1 Vpu ion channel: modelling and simulation studies. Biochim Biophys Acta. 2001;1512:291–298. doi: 10.1016/S0005-2736(01)00332-7. [DOI] [PubMed] [Google Scholar]
- 28.Bu L, Im W, Brooks CL., III Membrane assembly of simple helix homo-oligomers studied via molecular dynamics simulations. Biophys J. 2007;92:854–863. doi: 10.1529/biophysj.106.095216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Psachoulia E, Marshall DP, Sansom MSP. Molecular dynamics simulations of the dimerization of transmembrane α-helices. Acc Chem Res. 2010;43:388–396. doi: 10.1021/ar900211k. [DOI] [PubMed] [Google Scholar]
- 30.Bowie JU. Solving the membrane protein folding problem. Nature. 2005;438:581–589. doi: 10.1038/nature04395. [DOI] [PubMed] [Google Scholar]
- 31.Krüger J, Fischer WB. Assembly of viral membrane proteins. J Chem Theor Comput. 2009;5:2503–2513. doi: 10.1021/ct900185n. [DOI] [PubMed] [Google Scholar]
- 32.Wahba K, Schwab D, Bruinsma R. Statistical mechanics of integral membrane protein assembly. Biophys J. 2010;99:2217–2224. doi: 10.1016/j.bpj.2010.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chandrasekhar I, Kastenholz M, Lins RD, Oostenbrink C, Schuler LD, van Gunsteren WF. A consistent potential energy parameter set for lipids: dipalmitoylphosphatidylcholine as a benchmark of the GROMOS96 45A3 force field. Eur Biophys J. 2003;32:67–77. doi: 10.1007/s00249-002-0269-4. [DOI] [PubMed] [Google Scholar]
- 34.Krüger J, Fischer WB. Exploring the conformational space of Vpu from HIV-1: a versatile and adaptable protein. J Comput Chem. 2008;29:2416–2424. doi: 10.1002/jcc.20986. [DOI] [PubMed] [Google Scholar]
- 35.Xiang Z, Soto CS, Honig B. Evaluating conformational free energies: the colony energy and its application to the problem of loop prediction. Proc Natl Acad Sci USA. 2002;99:7432–7437. doi: 10.1073/pnas.102179699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soto CS, Fasnacht M, Zhu J, Forrest L, Honig B. Loop modeling: sampling, filtering, and scoring. Proteins. 2008;70:834–843. doi: 10.1002/prot.21612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwarz S, Wang K, Yu W et al. (2011) Emodin inhibits current through SARS-associated coronavirus 3a protein. Antiviral Res (ahead of print) [DOI] [PMC free article] [PubMed]
- 38.Cordes FS, Tustian AD, Sansom MS, Watts A, Fischer WB. Bundles consisting of extended transmembrane segments of Vpu from HIV-1: computer simulations and conductance measurements. Biochemistry. 2002;41:7359–7365. doi: 10.1021/bi025518p. [DOI] [PubMed] [Google Scholar]
- 39.Patargias G, Zitzmann N, Dwek R, Fischer WB. Protein-protein interactions: modeling the hepatitis C virus ion channel p7. J Med Chem. 2006;49:648–655. doi: 10.1021/jm050721e. [DOI] [PubMed] [Google Scholar]
- 40.Patargias G, Barke T, Watts A, Fischer WB. Model generation of viral channel forming 2B protein bundles from polio and coxsackie viruses. Mol Membr Biol. 2009;26:309–320. doi: 10.1080/09687680903164101. [DOI] [PubMed] [Google Scholar]
- 41.Skasko M, Tokarev A, Chen C-C et al. (2011) BST-2 is rapidly down-regulated from the cell surface by the HIV-1 protein Vpu: evidence for a post-ER mechanism of Vpu-action. Virology (ahead of print) [DOI] [PMC free article] [PubMed]
- 42.Narayanan K, Huang C, Makino S. SARS coronavirus accessory proteins. Virus Res. 2008;133:113–121. doi: 10.1016/j.virusres.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ausiello G, Cesareni G, Helmer-Citterich M. ESCHER: a new docking procedure applied to the reconstruction of protein tertiary structure. Proteins. 1997;28:556–567. doi: 10.1002/(SICI)1097-0134(199708)28:4<556::AID-PROT9>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 44.Schroeder C, Heider H, Moncke-Buchner E, Lin TI. The influenza virus ion channel and maturation cofactor M2 is a cholesterol-binding protein. Eur Biophys J. 2005;34:52–66. doi: 10.1007/s00249-004-0424-1. [DOI] [PubMed] [Google Scholar]
- 45.Ruiz A, Hill MS, Schmitt K et al. (2010) Membrane raft association of the Vpu protein of human immunodeficiency virus type 1 correlates with enhanced virus release. Virology (in press) [DOI] [PMC free article] [PubMed]
- 46.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cady SD, Schmidt-Rohr K, Wang J, Soto CS, DeGrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dougherty DA. Cation - π interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science. 1996;271:163–168. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- 49.Hilf RJC, Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- 50.Hilf RJC, Dutzler R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature. 2009;457:115–119. doi: 10.1038/nature07461. [DOI] [PubMed] [Google Scholar]
- 51.Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mueller M, Grauschopf U, Maier T, Glockshuber R, Ban N. The structrure of a cytolytic alpha-helical toxin pore reveals its assembly mechanism. Nature. 2009;459:726–730. doi: 10.1038/nature08026. [DOI] [PubMed] [Google Scholar]
- 53.Waight AB, Love J, Wang DN. Structure and mechanism of a pentameric formate channel. Nat Struct Biol. 2010;17:31–37. doi: 10.1038/nsmb.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steinbacher S, Bass R, Strop P, Rees DC. Structures of the prokaryotic mechanosensitive channels MscL and MscS. Curr Top Membr. 2007;58:1–24. doi: 10.1016/S1063-5823(06)58001-9. [DOI] [Google Scholar]
- 55.Stein WD. Channels, carriers and pumps. An introduction to membrane transport. San Diego, CA: Academic Press Inc; 1990. [Google Scholar]
- 56.Smart OS, Neduvelil JG, Wang X, Wallace BA, Sansom MSP. Hole: a program for the analysis of the pore dimensions of ion channel structural models. J Mol Graph. 1996;14:354–360. doi: 10.1016/S0263-7855(97)00009-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Potential energies (MOE) of the assemblies to generate the monomer Seq1: TMD3 and TMD2 displayed in respect to distance rotational angle_1, rotational angle_2, and tilt (a). Potential energies of the assemblies of (TMD3+TMD2) with TMD1 are shown as for (a) to form Seq1 (b). The structure used for further studies is marked by the arrow (JPEG 117 kb)
Potential energies of the assembled tetramers without loops. Values for generating all the sequences are shown in dependence of distance (left panels), rotational angles (middle) and tilt (right panels). The arrows indicate models T-Seq1 (I), T-Seq2 (II), T-Sim (III) (JPEG 137 kb)