

Abstract
Swine performance in the face of disease challenge is becoming progressively more important. To improve the pig’s robustness and resilience against pathogens through selection, a better understanding of the genetic and epigenetic factors in the immune response is required. This review highlights results from the most recent transcriptome research, and the meta-analyses performed, in the context of pig immunity. A technological overview is given including wholegenome microarrays, immune-specific arrays, small-scale high-throughput expression methods, high-density tiling arrays, and next generation sequencing (NGS). Although whole genome microarray techniques will remain complementary to NGS for some time in domestic species, research will transition to sequencing-based methods due to cost-effectiveness and the extra information that such methods provide. Furthermore, upcoming high-throughput epigenomic studies, which will add greatly to our knowledge concerning the impact of epigenetic modifications on pig immune response, are listed in this review. With emphasis on the insights obtained from transcriptomic analyses for porcine immunity, we also discuss the experimental design in pig immunity research and the value of the newly published porcine genome assembly in using the pig as a model for human immune response. We conclude by discussing the importance of establishing community standards to maximize the possibility of integrative computational analyses, such as was clearly beneficial for the human ENCODE project.
Electronic supplementary material
The online version of this article (doi:10.1007/s00335-014-9549-4) contains supplementary material, which is available to authorized users.
Keywords: Digital Gene Expression, Tiling Array, System Biology Approach, Swine Leukocyte Antigen, Porcine Genome
Introduction
Anticipating the need to efficiently feed an estimated 9 billion people, pig farms have significantly increased in size, which also unfortunately increases the risk of disease incidence. A better understanding of the immune system in swine is essential, because susceptibility to infectious diseases has great influence on pig performance (Mellencamp et al. 2008; Boddicker et al. 2012). Selection for improvement in economical traits such as feed conversion and prolificacy is already routine practice, and improvement of immune capacity through selection is gradually catching up (Edfors-Lilja et al. 1998; Uddin et al. 2011; Lu et al. 2012). In addition, pigs are an important biomedical model for humans as they share great similarity in anatomy, physiology, genetics, and genomics, including many genes in the immune system (Dawson 2011; Dawson et al. 2013), which is very helpful when modeling human immune responses and diseases (Lunney 2007; Fairbairn et al. 2011; Kapetanovic et al. 2012; Meurens et al. 2012). Moreover, pigs have recently been genetically modified to serve as an improved model for human disease (Suzuki et al. 2012). For more genetically modified pig models for human diseases, we refer the reader to the reviews of Ross and Prather (2011) and Walters et al. (2012).
At the genomics and transcriptomics level, the pig is a very appealing model since one can extrapolate more easily to human due to the high homology in gene sequence and chromosomal structure (Groenen et al. 2012). The immune system components of the pig genome (immunome) have become the best-annotated genes in the current genome (Dawson et al. 2013). This improved pig genome annotation will greatly aid creation of biomedical models. When the cause of a medical condition in humans is found, the corresponding mutation(s) can be genetically engineered in pigs to mimic the disease, and (novel) drug therapies can be tested (Walters et al. 2012). Differences in genotype associated with a different immune response phenotype in pig can easily be examined with the use of the porcine 60 K high-density SNP chip (Boddicker et al. 2012; Lu et al. 2012; Wang et al. 2012a). The last decade witnessed a steep increase in the amount of transcriptomic pig immune response data using whole porcine genome microarrays. Meanwhile, custom-made arrays enriched in annotated genes of the pig immune system were used to address specific research questions (Gao et al. 2010). In addition, novel techniques are surfacing in studies focusing on the porcine immune response transcriptome. High-density oligonucleotide tiling arrays and Next Generation Sequencing (NGS) have been further broadening the knowledge of the pig’s immunome (Mockler et al. 2005; Morozova and Marra 2008). These techniques are able to identify and quantify known and unknown transcripts and can be used to extend recent efforts made on the pig genome annotation (Gao et al. 2012). The understanding of the function of diverse miRNAs will profit immensely from NGS, and papers on the porcine miRNAome are being published (Li et al. 2010a; Sharbati et al. 2010), as surely soon will follow on epigenetic changes such as DNA methylations or histone modifications. Several research groups have graciously provided information to our review on their recently published and unpublished studies, including whole genome microarrays to RNA-seq, miRNA-seq, RIP-ChIP, and some of those methylation and histone modification studies (Table 1).
Table 1.
Recently published and unpublished transcriptomic and epigenomic studies in pig immunity, contributed by community members
| Laboratory institution | Contact info | Type of analysis tool | Tissues/cell types surveyed | Research focus | Publication status |
|---|---|---|---|---|---|
| USDA ARS NADC | M. Bandrick (meggan.bandrick@ars.usda.gov) and T. Stanton (thad.stanton@ars.usda.gov) | RNA-seq | Globin depleted blood, lymph node, duodenum, jejunum, ileum, colon | To study gene (especially as related to immune function) expression patterns between tissues | In preparation |
| Parco Tecnologico Padano | S. Botti (sara.botti@tecnoparco.org) and B. Badaoui (bouabid.badaoui@tecnoparco.org) | RNA-seq | Porcine alveolar macrophages (PAMs) | To study the transcriptome profile of PAM infected in vitro with low virulent (Lylestad) and high virulent (Lena) PRRSV strains. We quantified the expression levels of genes, isoforms, alternative transcript starting sites and we highlighted a complex transcriptional and post-transcriptional genes regulation during PAM infection | Badaoui et al. (2014) |
| Humboldt-Universität Berlin | G. A. Brockmann (gudrun.brockmann@agrar.hu-berlin.de) | RNA-seq and miRNA-seq | Mesenteric lymph nodes (Jejunum, Ileum), Peyer’s Patches (Jejunum, Ileum) | To study effects of probiotics and zinc on immune cells | In preparation |
| University of Nebraska | D. C. Ciobanu (dciobanu@unl.edu) | Affymetrix GeneChip Porcine Array | Whole blood | To study differences in gene expression between pigs that display differences in growth and viremia following experimental infection with PCV2b | In preparation |
| University of Cordoba | J. J. Garrido (ge1gapaj@uco.es) | Affymetrix GeneChip Porcine Array | Neutrophils | To study the transcriptomic response of porcine neutrophils to Salmonella typhimurium and Salmonella choleraesuis | In preparation |
| INRA—GABI laboratory, France | E. Giuffra (elisabetta.giuffra@jouy.inra.fr) | RNA-seq, Small RNA-seq, RIP-Chip | Trigeminal ganglia | To study the role of miRNAs in Pig:pseudorabies virus interaction during latency | In preparation |
| University of Alberta | L. L. Guan (lelou.guan@ualberta.ca) | RNA-seq and miRNA-seq | Whole blood | To study the role of miRNAs in in response to Salmonella Typhimurium | (Bao et al. 2014; Kommadath et al. 2014) |
| The Roslin Institute, University of Edinburgh | D. Hume (david.hume@roslin.ed.ac.uk) | RNA-seq and CAGE | Macrophages from lungs and bone marrow | To study gene expression differences in response to lipopolysaccharide (LPS) | In preparation |
| University of Copenhagen | H.N. Kadarmideen (hajak@sund.ku.dk) | RNA-seq | Porcine subcutaneous fat from obese and lean pigs | To study transcriptomic profiles to elucidate differential and co-regulation and to integrate with 60 k SNP data to detect eQTLs, biological pathways and biomarkers for obesity and metabolic phenotypes in a porcine model | In preparation & WCGALP 2014 Conference paper |
| USDA ARS NADC | L. Miller (laura.miller@ars.usda.gov) | Digital Gene expression Tag profiling (DGETP) | Porcine Trancheobronchial lymph nodes (TBLNs) | Comparative transcript expression snalysis in tracheobronchial lymph nodes of PRRSV-, PCV-2, PRV and SIV-Infected pigs | Miller et al. (2014) |
| Kansas State University | Y. Sang (ysang@vet.k-state.edu) and F. Blecha (blecha@vet.k-state.edu) | RNA-seq | Polarized macrophages | To profile signature genes and gene response pathways for antiviral regulation in monocytic cells | Sang et al. (2014) |
| Iowa State University | C. K. Tuggle (cktuggle@iastate.edu) | RNA-seq | Globin depleted blood | To study gene expression differences between residual feed intake extremes in response to endotoxin | In preparation |
| University of Bonn | M. J. Uddin (judd@itw.uni-bonn.de) | RNA-seq | Porcine dendritic cells | To study the gene expression of DCs in response to PRRSV challenge in vitro | In preparation |
| University of Bonn | M. J. Uddin (judd@itw.uni-bonn.de) | Affymetrix GeneChip Porcine Array | Porcine PBMCs | To study the transcriptomic profile of porcine PBMCs in response to PRRSV vaccine in vivo | Qu et al. (2014) |
| University of Bonn | M. J. Uddin (judd@itw.uni-bonn.de) | Epigenetic study: DNA methylation, HDAC activity, Histone acetylation and miRNAs study | Porcine monocyte-derived dendritic cells (MoDCs) and spleenic dendritic cells (SDCs) | To study the epigenetic modulation of TLR4 in response to lipopolysaccharide (LPS) | In preparation |
| University of Bonn | M. J. Uddin (judd@itw.uni-bonn.de) | Epigenetic study: DNA methylation, HDAC activity, Histone acetylation and miRNAs study | Porcine alveolar macrophages (PAMs) | To study the epigenetic modulation of CD14 in response to lipopolysaccharide (LPS) | In preparation |
| Leibniz-Institute for Farm Animal Biology | K. Wimmers (wimmers@fbn-dummerstorf.de) | Affymetrix GeneChip Porcine Array | Peripheral blood mononuclear cells (PBMCs) | To study transcriptomic response of PBMC of pigs with divergent humoral immune responses and coping behavior | In preparation |
The aim of this review is to summarize the use and results of recent transcriptomic research in pig immunity, identify current needs in the field, and anticipate future areas of progress. An overview will be given of studies using whole genome microarrays and immune-specific arrays, small-scale high-throughput expression methods, high-density tiling array, and NGS (Table 2; Fig. 1). Experimental design concerns and studies using the newest sequencing techniques and epigenetic tools together with a systems biology approach to interpret the data will also be discussed. For this review, we searched the Entrez Pubmed database, using keyword search terms such as pig transcriptomics, pig immunity, microarray, immune-specific array, high-throughput qPCR, RNA-seq and miRNA-seq, and systems biology. In addition, references from the articles obtained by this method were checked for additional relevant material. The ArrayExpress database from EBI was consulted to create Supplementary Table 1, using Sus scrofa as organism and filtered on ‘Array assay’ as technology.
Table 2.
Transcriptomic and epigenomic tools, their advantages and disadvantages, and interesting examples with regard to immune response research in swine
| Specific tool | Advantage | Disadvantage | Example | Example analysis |
|---|---|---|---|---|
| Whole genome array | ||||
| Qiagen-NRSP8 13297 probes | Whole genome screening | Restricted to probes available on chip and limited to the chip’s annotation | Zhao et al. (2005) | Array was validated in porcine liver, muscle, uterine endothelium and brain stem |
| Pigoligo 20400 probes | Steibel et al. (2009) | Array was validated in porcine liver, muscle, small intestine and lung | ||
| Affymetrix 23937 probes | Huang et al. (2011) | Pathways mediated by IFNγ, TNF, and NFκB are upregulated due to S. typhimurium infection | ||
| Agilent 43803 probes | Bao et al. (2012) | 18 genes were differentially expressed after infection with enterotoxigenic E. coli (ETEC) with F18 | ||
| Snowball 47485 probes | Freeman et al. (2012) | Array was validated in 62 porcine tissue/cell types and expression correlation analysis revealed specific clustering | ||
| Immune-specific array | ||||
| Macroarray 63 probes | Ideal for specific interest focus on immunity genes | Restricted to probes available on chip and limited to the chip’s annotation. The chips are of use primarily to the specific research question asked | Ledger et al. (2004) | An upregulation of several immune genes is observed after PMA/ionomycin stimulation of PBMCs |
| Peyer’s Patches array 2400 probes | Specialized tool for specific tissue | Dvorak et al. (2006) | A differential expression has been found in Peyer Patches after stimulation with Salmonella, LPS + Cholera toxin and PMA | |
| Jejunum array 3468 probes | Specialized tool for specific tissue | Niewold et al. (2005) | ETEC upregulates 220 genes in 4-week-old and only 80 genes in 12-week-old pigs present on the array | |
| Immune array 2880 probes | Specialized tool for pathogen infection | Zhang et al. (2006) | Macrophages stimulated with an African Swine Fever Virus change the expression of 125 genes on this array | |
| Immune array 373 probes | Specialized tool for pathogen infection | Skovgaard et al. (2010) | Inoculation with Actinobacillus pleuropneumonia reveals a differential expression of 53 genes in the liver | |
| Qiagen + SLA/PrV | Whole genome screening, but with focus on immunity genes in the SLA region and simultaneous data on two species (pig and pseudorabies virus) | Flori et al. (2008) | After 2 hpi a viral gene encoding a TAP inhibitor is upregulated, after 4 until 12 hpi porcine genes involved in SLA presentation are downregulated, showing a mechanism used by the virus to escape the porcine immune system | |
| SLA-RI/NRSP8-13 K | Whole genome screening, but with focus on immunity genes, in and outside the SLA region | Gao et al. (2010) and Solinhac et al. (2011) | PBMCs stimulated with LPS an PMA/ionmycin were examined. While stimulation with LPS triggers a general inflammation response, PMA/ionomycin stimulation favors a T cell activation over a B-cell response | |
| Tiling array | ||||
| Affymetrix tiling array | No prior assumptions (previous annotation not necessary) and thus possible to refine annotation | Use of a very large number of probes, no whole genome re-sequencing possible with this technique | Gao et al. (2012) | With the use of 386620 probes, 97 genes in the SLA region were found to be differentially expressed between control PBMCs and PBMCs stimulated with PMA/ionomycin |
| Agilent tiling array | ||||
| Nimblegen tiling array | ||||
| Digital gene expression | ||||
| Illumina Genome Analyzer platform | Whole genome sequencing (High-throughput SAGE) with tag at all sequences with a CATG recognition site, no prior assumptions | Restricted to detection of transcripts containing CATG recognition sites and the Tag should be long enough to be unique, otherwise results are ambiguous. Isoforms cannot be detected | Xiao et al. (2010a, b) | Lung expression profile was examined in control or PRRSv-infected pigs (both Highly Pathogenic (HP) and US strain infected) at 4dpi. An average of 6.1 million tags per library was obtained. 4520 significantly differentially expressed genes were found in the HP and 5430 in the US PRRSv-infected animals. Important down-regulated genes are SPI IFN and IFNα and CD163 was upregulated |
| mRNA-seq | ||||
| The SOLiD™system, Applied Biosystems | Whole genome sequencing, no prior assumptions and splice variants can be detected | With polyA tail library, possible loss of non polyadenylated mRNA sequences and depletion of highly abundant genes might be necessary | Miller et al. (2012) | Trachea-bronchial lymph nodes expression profile was examined in control or PRRSv-infected pigs (both Highly Pathogenic (HP) and US strain infected) at 13dpi. The RNA-Seq showed 5.6 million reads for the control, 4.3 million reads for the HP and 3.5 million for the US PRRSv-infected animals. 632 differentially expressed genes were found to be significant for the HP and 633 for the US PRRSv-infected animals. Among the top ten, upregulated genes are RETN, S100A8, S100A9, and S100A12 |
| Illumina HiSeq | Without polyA tail library, depletion of rRNA is necessary | |||
| PacBio | ||||
| Roche 454 life sciences | ||||
| miRNA-seq | ||||
| Same tools as used for mRNA-seq | Sequencing of all miRNAs, no prior assumptions and splice variants can be detected | Examines only miRNAs. Their influence on mRNA still has to be tested | Chen et al. (2011a) and Endale Ahanda et al. (2012) | Inverse expression values were found between miRNAs measured by miRNA-seq and their target mRNAs (measured by mRNA-seq) |
Whole genome microarray studies, the start of global high-throughput transcriptomics
The range of genes assayed with whole genome microarray depends on the array used (Table 2; Fig. 1). The first porcine whole genome array available, the Qiagen-NRSP8 array, was a result of collaboration between Qiagen-Operon and the USDA-NRSP-8 Swine Genome community (Zhao et al. 2005). Until then, human arrays were used to tackle porcine transcriptomic issues, or swine arrays were developed for a specific research question, as described in the next section. The Pigoligoarray was a second-generation porcine 70-mer oligonucleotide array (Steibel et al. 2009). Currently, the most well known are commercially available microarrays such as the Porcine Gene Expression Microarray of Agilent and the GeneChip Porcine Genome Array of Affymetrix (Tuggle et al. 2010; Huang et al. 2011; Zhou et al. 2011; Bao et al. 2012). Freeman and colleagues recently introduced the Affymetrix Snowball microarray, which is more comprehensive than the first Affymetrix chip in coverage both for transcript structure and for the transcriptome as a whole, containing more than double as many probes (Freeman et al. 2012). Details and examples of these whole genome arrays are shown in Table 2. An overview of pig immunity experiments submitted to the ArrayExpress database is given in Supplementary Table 1.
Since global porcine microarray studies have been extensively described earlier (Tuggle et al. 2007, 2010), we will limit this section by highlighting the importance of using microarrays in a systems biology approach; we do provide a comprehensive table on all porcine immune-related microarray experiments submitted to ArrayExpress (Supplementary Table 1). Several microarray studies have been conducted to study host response to porcine reproductive and respiratory syndrome (PRRS) using commercial or more specific pig annotated microarrays (Bates et al. 2008; Genini et al. 2008a; Ait-Ali et al. 2011; Wysocki et al. 2012). Badaoui et al. (2013) recently illustrated how the information of multiple PRRS studies could be used simultaneously to gain insight on host response to PRRSv challenges. They collected all publicly available microarray data covering multiple porcine immunology studies and including many different breeds, tissues, pathogens, and array platforms. The data of 779 general immune response arrays were assembled, and separate meta-analyses for differential expression were performed using these 779 arrays as well as a subset of 279 arrays specifically from PRRS experiments (Badaoui et al. 2013). To find PRRS-specific expression responses, they eliminated differentially expressed genes common to these two meta-analyses. Other meta-analysis studies, examining the immune response to Salmonella typhimurium, combine microarray information with data such as serum cytokine measurements or microbiota differences. The results of these meta-analyses performed on PRRS or S. typhimurium will be discussed in the section “Overall value of transcriptomics in important infectious swine diseases.”
In addition, whole genome microarrays were used to study pig response to Haemophilus parasuis infection by Zhao et al. (2013). In one of their earlier studies, a genome-wide Affymetrix array experiment revealed 931 differentially expressed genes between 3 non-infected and 3 H. parasuis-infected pigs in spleen at 7dpi (Chen et al. 2009). Among them were RETN, S100A8, S100A9, and S100A12, all important innate immunity genes marking inflammation. Chen et al. (2011b) reexamined the raw data of this first experiment and used a more robust GeneChip RMA (GCRMA) normalization (Wu and Irizarry 2004), and an improved annotation using ANEXdb (Couture et al. 2009). A network analysis revealed that the pS100A8/pS100A9-CASP3-SLC1A2 pathway played an important role in H. parasuis infection, and CEBPB may act as a transcription factor of the two S100 family members (Chen et al. 2011b). Later, Zhao and co-workers describe a systems biology approach starting from the same Affymetrix data. With the use of the KEGG and reactome databases, 1,999 transcripts from the Affymetrix chip were flagged as immunogenes. With this reduced dataset, exploratory analyses such as a principal component analysis (PCA) and a geneset enrichment analysis (GSEA) were conducted. With this PCA/GSEA method, they came to a core set of 16 differentially expressed genes, indicative of an H. parasuis infection (Zhao et al. 2013). To construct the immunologically important topology involved in an H. parasuis infection, the C3NET algorithm was used (Altay and Emmert-Streib 2010). Several networks were created, and the largest network had the complement gene C1R as hub; although C1R was not differentially expressed on the microarray, it was predicted to play a crucial role in the immune defense. With this immunome-focused method, more subtle differences could be pulled out, and much new information was obtained as compared to the earlier whole genome array analysis (Zhao et al. 2013).
The immune-specific array, a focused microarray
Besides using commercially available or custom-made transcriptome-wide microarrays, or examining only immunogenes on those arrays, arrays focusing only on the genes involved in the porcine immune response are also used (Table 2). In the past, several of these arrays were constructed to specifically focus on key immune genes (Ledger et al. 2004), to explore gene expression in a specific immune tissue (Dvorak et al. 2006; Machado et al. 2005; Niewold et al. 2005), or to examine host interactions with a specific pathogen (Zhang et al. 2006; Skovgaard et al. 2010). More information on these studies is given in Table 2. Next to these small-scale arrays, other immune-specific arrays were made using an existing whole genome array and adding probes examining immune-related genes. In a study by Flori and co-workers, the Qiagen-NRSP8 array was extended by adding probes for Pseudorabies Virus RNAs as well as probes for transcripts from the porcine Major Histocompatibility Complex, also called the Swine Leukocyte Antigen (SLA) complex, referred to as the Qiagen + SLA/PrV in Fig. 1. This array was the first to simultaneously examine viral transcripts and porcine immune transcripts (Flori et al. 2008). Gao and colleagues used the NRSP8-13K chip enriched with SLA genes and immunity genes outside the SLA complex and called it the SLA-RI/NRSP8-13K chip (Gao et al. 2010). In the future, when using microarrays to examine gene expression patterns, whole genome arrays or the extended versions of them are clearly preferred. When the objective of the study is to examine only a relative small number of genes, other techniques such as Fluidigm Digital Array or Nanostring (described in more detail below) can be performed. These low cost, user-friendly, and rapid approaches make the small-scale in-house printed arrays redundant.
Fig. 1.
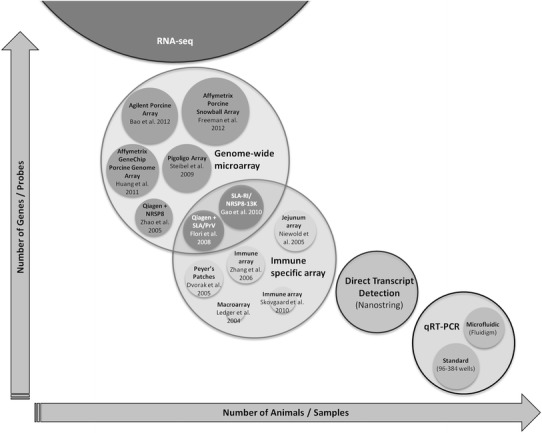
Different transcriptomic tools used in porcine immune response studies, graphed with respect to the size of the dataset and the depth of the analysis possible. Nanostring has not yet been used in porcine immune response studies
Upcoming sub-genomic-scale, high-throughput methods: Fluidigm and NanoString
Even though qRT-PCR is often used as the routine method to confirm microarray and RNA-seq findings, it has been used in many pig immune studies as a primary means to measure expression levels of a dozen or more immune genes (Borca et al. 2008; Lastra et al. 2009; Islam et al. 2012a, b; Uddin et al. 2012; Martins et al. 2013a, b; Uddin et al. 2013), together with suitable housekeeping genes for normalization (Cinar et al. 2012, 2013). Although it is a very accurate and sensitive method, standard qRT-PCR has the major drawback that only a few genes in a few samples can be examined at once as compared to high-throughput methods (Fig. 1). Further, significant amounts of money and time are spent controlling sample and assay variation by using appropriate—and preferably multiple—housekeeping genes and validating PCR efficiencies or correcting for them by using standard curves. The use of high-throughput qRT-PCR methods, such as the Fluidigm Digital Array, can help overcome these issues.
The Fluidigm Digital Array integrated fluidic circuit (Spurgeon et al. 2008) is an example of a nanofluidic chip through which up to 9,216 qRT-PCR reactions can be performed at once, which represents 96 genes tested on 96 samples (Ramakrishnan et al. 2013) (Fig. 1). Beyond sheer speed of analysis, an important advantage of nanotechnology is that a much smaller amount of RNA per sample is necessary to perform 96 assays. In livestock, the first Fluidigm experiments have been published only recently, as a stand-alone expression study or as a validation of a microarray/RNA-seq experiment (Robic et al. 2011; Skovgaard et al. 2013; Sorg et al. 2013; Pilcher et al. 2014). In a H1N2 influenza virus study, Skovgaard and co-workers used Fluidigm to examine the expression of several pattern recognition receptors, IFN and IFN-induced genes, cytokines, and acute response protein genes after 24 hpi, 72 hpi, and 14 dpi. They expanded their study by including data on differentially expressed miRNAs and mRNAs in the lungs and saw several miRNAs potentially targeting the differentially expressed mRNAs (Skovgaard et al. 2013).
Another high-throughput method uses the NanoString nCounter system by which color-coded probes are counted to digitally measure gene expression (Fig. 1). This non-amplification method can be seen as a small-scale, hybridization-based platform. Per gene of interest, two sequence-specific probes are needed, a capture probe that has an affinity tag such as biotin to capture the gene on a surface, and a reporter probe with a multiple-fluorescent tag code that acts as a unique detector (Geiss et al. 2008). Its strength lies in the non-enzymatic approach, with no reverse transcriptase and no PCR amplification. This approach essentially eliminates primer–primer interactions, and up to 800 transcripts can be measured at a time. The technique is not described in livestock species thus far. However, with the possibility to use degraded RNA samples such as RNA from formalin-fixed paraffin-embedded tissue samples, this method could become a very useful expression measuring tool.
High-density oligonucleotide tiling array for gene expression and genome annotation
High-density oligonucleotide tiling arrays have diverse genomic applications, such as transcriptome mapping and quantification. In general, tiling arrays are similar to microarrays as they both utilize oligonucleotide probes. However, through utilization of large numbers of probes, tiling arrays can span large genomic sequences without being restricted to annotated sequences (Gao et al. 2012). Thus, tiling arrays may contain probes covering the entire genome in an unbiased manner (Liu 2007), and such partially overlapping probes can be used to find expressed genes, previously annotated or not. Gao and co-workers used a Nimblegen tiling array (386,620 probes) for the analysis of the transcription map of the SLA complex (Gao et al. 2012). Ninety-seven genes were found to be differentially expressed between un-stimulated Peripheral Blood Mononuclear Cells (PBMCs) and PBMCs stimulated with Phorbol Myristate Acetate (PMA)/ionomycin. These results are in nearly complete agreement with their previous experiment using the SLA-RI/NRSP8-13K chip. Furthermore, with the tiling array, they were able to confirm and refine the previous annotation of the SLA genes (Gao et al. 2012).
Next generation sequencing
Transcriptome sequencing
RNA-seq is a recently developed method enabling the examination of the complete set of transcripts in a cell using a sequencing by synthesis approach (Wang et al. 2009). With this technique, mRNA is converted into a library of cDNA fragments, and at one or both ends of these fragments, adaptors are ligated. Each fragment is read in a highly parallel manner, and typical read lengths are between 30 and 700 bp, and go up to an average of 4,000 bp and more with the PacBio RS sequencer (Ferrarini et al. 2013). The resulting reads are aligned to a reference genome of the organism examined, and a genome-wide transcription map can be made describing the structure and enumerating the expression level of each gene. De novo assembly of a transcriptome is also possible if the reference genome is not available or poorly annotated (Martin and Wang 2011); integrated approaches are also possible.
RNA-seq has already proven to be a cost-effective way to investigate the sequence of all mRNA transcripts in a specific tissue and/or for a specific physiological condition. Not only is the sequence obtained together with the quantitative level of transcripts, but also splice variants can be detected, which is a major advantage in contrast with techniques such as microarrays. Moreover, with RNA-seq, there is very low, if any, background noise, and no transcript specific primers or probes are required, so no prior knowledge is needed (Wang et al. 2009). This latter attribute is important when working with livestock species for which genomic sequence assemblies are incomplete (Bauersachs and Wolf 2012). As such, Esteve-Codina and colleagues reported hundreds of un-annotated protein coding genes in the porcine genome while studying the gonad transcriptome (Esteve-Codina et al. 2011). However, projects costs are influenced by the research question asked. If the goal is to examine general expression pattern differences in well-annotated, moderately expressed genes, sequencing does not have to be very deep, and samples can be multiplexed in one lane. For novel transcripts or genes with a low expression level, higher coverage is needed, and costs will increase (The ENCODE Consortium Project 2011a). One may also consider depleting highly abundant genes of little interest, such as alpha and beta globin in a blood sample, prior to sequencing in order to improve detection sensitivity (Choi et al. submitted).
In 2010, Xiao and collaborators performed a 3’ tag digital gene expression (DGE) analysis of the porcine lung transcriptome on pigs infected with the PRRS virus (Xiao et al. 2010a, b). This DGE technique can be seen as a predecessor of RNA-seq. Whereas RNA-seq will sequence all transcripts containing poly-A+ tails, DGE detects those transcripts containing CATG recognition sites because the transcripts are ‘tagged’ through digestion of cDNAs at a NlaIII restriction site (Hong et al. 2011). With DGE, only a portion of the transcript is analyzed instead of the nearly full transcripts as seen with RNA-seq (Wang et al. 2009). Both techniques offer similar estimates of gene expression levels but RNA-seq has the advantage to be able to provide information about transcript structure and consequently can detect splice variants (Hong et al. 2011). Xiao and colleagues sequenced lung RNA libraries of control non-infected pigs (n = 3), PRRS-affected pigs necropsied at 4dpi (n = 3,) and PRRS-affected pigs necropsied at 7dpi (n = 3) after infection with the classical North American PRRSv type (NPRRSv) (Xiao et al. 2010a) or the highly virulent PRRSv (HPRRSv), typically found in Asia (Xiao et al. 2010b) and found 4,520 (HPRRSv) and 5,430 (NPRRSv) differentially expressed genes. For both types of PRRSv, a higher expression of anti-apoptotic genes and a lower expression of pro-apoptotic genes could be seen as a viral strategy for replication and spread (Xiao et al. 2010a, b). As such, suppressed expression of short type I interferon (SPI IFN) and IFNα, both important innate anti-viral genes, was detected. Additionally, there was an upregulation of CD163 noted, which could indicate an increase in internalization of PRRSv since a positive correlation is described between the expression of this PRRSv receptor gene and PRRSv infectivity (Patton et al. 2009). Patton and colleagues described that treatment with modulators such as lipopolysaccharide (LPS) or IL10, affecting cells to express CD163, as a consequence also affected the susceptibility of the host for PRRSv, thereby increasing or decreasing viral infectivity (Patton et al. 2009).
Miller and colleagues examined gene expression profile data obtained by SAGE tag analysis of trachea-bronchial lymph nodes of sham- or PRRSv-infected pigs at 13dpi. Infection occurred with both highly pathogenic HPRRSv (n = 8) as well as North American type NPRRSv (n = 7) (Miller et al. 2012). The HPRRSv strain showed a more altered gene expression profile with higher fold change differences relative to the controls than the NPRRSv, indicating the increased pathogenicity of the HPRRSv. Among the top 10 genes that were upregulated were three serum amyloid A2 acute-phase isoforms, resistin (RETN), and three S100 calcium binding proteins, S100A8, S100A9, and S100A12 (Miller et al. 2012). Jiang et al. (2013) used the GO, KEGG, and REACTOME databases to analyze the data on NPRRSv infection further and identified six biological system categories affected by PRRSv, including cellular processes, genetic information processing, environment information processing, metabolism, organismal systems, and human diseases (Jiang et al. 2013). In a large-scale PRRSv infection study aimed at finding genes controlling variation in immune response outcomes, Boddicker and colleagues identified a region on SSC4, containing SNP marker WUR10000125, was strongly associated with both weight gain and PRRS viral load for 21 days post-infection (Boddicker et al. 2012, 2013). Eisley and co-workers performed an RNA-seq analysis of all genes in this WUR10000125 region through comparing blood RNA from 8 pairs of littermates with one of two different WUR10000125 genotypes. They identified a strong candidate gene differentially expressed between favorable and unfavorable genotypes (Eisley et al. 2014).
Several other immune-related RNA-seq studies are forthcoming (Table 1). Their objective is to examine viral or bacterial immune responses in pig macrophages, dendritic cells, lymph nodes, globin depleted whole blood, and tissue samples from the gastrointestinal tract. Undoubtedly, the knowledge of pig immune responses at the transcriptome level will greatly benefit from these recently published and to-be-published studies.
miRNA-seq
Next to exploring the “traditional” transcriptome, miRNAome analyses are also progressing in the pig (McDaneld 2009; Liu et al. 2010). MicroRNAs (miRNAs) are small non-coding RNAs that can regulate gene expression through degrading or interfering with the respective mRNA sequence, often at the 3’ un-translated region (3’UTR) of the gene. During the last few years, high-throughput sequencing has lead to a steep rise in discovering novel porcine miRNAs (Xie et al. 2011). Moreover, NGS can facilitate distinguishing between miRNAs and other small RNA fragments, and thus, NGS is the most promising technique for exploring the miRNAome. Criteria to distinguish true miRNA from other RNA fragments are listed in Kozomara and Griffiths-Jones (2011). miRNA sequences and predicted targets for all important livestock animals can be found in miRBase, a comprehensive miRNA information database (Griffiths-Jones et al. 2006). With regard to immunological responses in pigs, recent studies have reported that porcine miRNA can intricately engage itself in host-virus interaction networks (He et al. 2009; Loveday et al. 2012; Guo et al. 2013). Conversely, miRNAs expressed by the viral pathogen can promote a favorable host cell environment for enhanced viral replication by targeting porcine mRNAs (Skalsky and Cullen 2010). An example of this involving response to Pseudorabies virus was published by Anselmo and co-workers. Using NGS, they analyzed both viral and host miRNA expressions in infected dendritic cells, and identified 5 viral miRNAs, 156 known porcine and 27 new porcine miRNAs (Anselmo et al. 2011). Another group used NGS techniques to analyze miRNA expression profiles in Pseudorabies virus infected porcine epithelial cell lines (Wu et al. 2012). Eleven miRNAs were detected in the viral genome, and 209 known and 39 novel miRNAs assigned to the porcine genome were also found. Wu and colleagues mainly focused on the viral miRNAs, which were associated with regulation of viral gene transcription but also proposed to control gene expression in the host of genes annotated for immune processes, viral replication, cell death, as well as other processes. Podolska and colleagues compared the miRNAome in necrotic and visually unaffected pieces of lungs from piglets infected with A. pleuropneumoniae and found 169 conserved and 11 candidate novel miRNAs. Twenty-nine were significantly up- or down-regulated between necrotic and unaffected tissue (Podolska et al. 2012). Timoneda and colleagues noted differences in miRNA expression between Aujeszky’s disease or suid herpesvirus type 1 (SuHV-1) virus-infected and mock-infected animals, as well as differences when looking at a virulent strain of SuHV-1 compared to an attenuated one (Timoneda et al. 2014).
mRNA-seq and miRNA-seq: a powerful combination
Toward the elucidation of miRNA-RNA interactions, Endale Ahanda and colleagues analyzed the 3’UTR variants of all genes of the SLA region by analyzing RNA-seq data. In this way, SNPs in miRNA target sequences, potentially impacting gene expression, could be revealed. To investigate the co-expression between mRNA and miRNA, mRNA-seq and miRNA-seq data from an earlier study looking at liver, longissimus dorsi, and abdominal fat were used (Chen et al. 2011a; Endale Ahanda et al. 2012). Negative correlation between expression levels of miRNAs and their predicted target genes could be found, which suggested that the prediction algorithms used were reliable (Endale Ahanda et al. 2012).
Since miRNAs can play an important role in host–pathogen interactions (Scaria et al. 2006), Gao and colleagues looked by means of miRNA-seq at host miRNAs that could target PRRSv transcripts. Deep sequencing was performed on PAMs inoculated with a mock dose or with PRRSv. The resulting data were mapped against all known miRNAs listed in the current miRBase. miRNA target prediction revealed that one miRNA family, the miR-181 members, seemed to suppress PRRSv replication in vivo at the early stage. One miR-181c target is the 3’UTR of CD163 mRNA, which encodes an important PRRSv receptor. miR-181c is able to downregulate CD163 expression and thus interferes with viral attachment and penetration (Gao et al. 2013). Similar results were seen for other miR-181 members.
There is also good evidence that expression profile differences with regard to an S. typhimurium infection may be partially controlled by miRNAs. Huang and co-workers found that miR-155 was decreased in persistently shedding (PS) pigs in comparison with low shedding (LS) pigs (Huang et al. 2011) after an S. typhimurium infection. miR-155 targets transcription factors CEBPB and SPI1, which in turn control the expression of important immunogenes (Adamik et al. 2013; Wei et al. 2014). Bao and co-workers specifically investigated potential miRNA-mRNA regulatory interactions occurring during challenge with S. typhimurium. mRNA-seq and miRNA-seq data were collected on whole blood samples of LS and PS animals (Bao et al. unpublished). They found 37 and 24 miRNAs up- and down-regulated at 2 dpi when looking at LS and PS pigs together. Several of them were thought to be involved in innate and adaptive immune responses. They also discovered 3 miRNAs to be higher expressed in LS than in PS pigs at day 2, which could be interesting candidates for biomarkers for selection toward low shedders. Bao and colleagues subsequently used the sequence-based miRNA target prediction software miRanda to propose miRNAs–mRNA regulatory relationships associated with the S. typhimurium infection (Bao et al. unpublished). Ye and colleagues searched for factors controlling susceptibility for enterotoxigenic E. coli with fimbria 18 (ETEC-F18) in intestinal tissue in the Sutai pig and found 58 differentially expressed miRNAs, and after examining regulatory networks with differentially expressed mRNAs that are target of one of those miRNAs, 12 of them were shown as hubs for an enriched list of differentially expressed immune-related genes (Ye et al. 2012). Several other porcine miRNAome studies are to be published soon focusing on the impact of miRNAs after bacterial or viral infections (Table 1).
Experimental design
In addition to different expression measurement techniques to examine pig immune responses, various experimental designs to study immunity have been used in these studies. First, there are in vitro versus in vivo studies. Whereas in vitro studies are performed outside the living organism, and thus can be more controlled, in vivo studies usually better reflect the underlying biology. Choices can be made to compare challenged and non-challenged or vaccinated versus non-vaccinated pigs, and whenever possible preferably littermates are chosen for such comparisons. To reduce genetic background variation even more, treated and untreated tissue within animal can be compared, as seen with the small intestinal segment perfusion (SISP) technique (Hulst et al. 2013). One can also choose to challenge all animals with a specific pathogen, and not using separate un-infected animals as controls, but contrast high and low responders, as was done in the Salmonella experiments (Uthe et al. 2009; Huang et al. 2011; Knetter et al. 2014). Less controlled, but perhaps more realistic, are studies performed after an on-farm outbreak, comparing healthy and diseased pigs or low and high responders (Serao et al. 2014).
Another key variable concerns the type of samples to be collected. Most pig immune studies conducted to identify host response to common porcine pathogens or to immune response stimulators such as LPS or PMA/ionomycin described in this review provide gene expression data from a single tissue or isolated cell type, and this at a limited number of times post-infection/stimulation. To study specific components of immune response, it is clear that dissection and analysis of primary or secondary immune tissue are required. In human and mouse studies, significant effort has been taken a step further—the analysis of the transcriptome of highly specific cell types. Such samples are isolated on the basis of cell surface marker expression. The parameters for cell selection and isolation are often complex, utilizing a multifactorial list of cell surface markers to identify a highly refined cell population (Novershtern et al. 2011; Shay and Kang 2013). Reports on, and options for, specific cell subsets are limited in swine (Genini et al. 2008a; Kapetanovic et al. 2012) and are mostly due to the relative lack of immune-targeted reagents critical for such detailed cell phenotyping.
Examining the whole blood transcriptome has several advantages, including ease of collection and repeated sampling of the same individual during response to a stimulus, which is especially useful in controlling for baseline variation in the study of immune responses. Blood RNA profiling is advantageous in screening for biomarkers as well; it can be used to study variation in immune response and develop gene signatures predictive of inflammatory and/or disease status. An example is given by the PRRS host genome consortium (PHGC) project, where blood of 200 infected animals per trial is collected at day 0 and 8 different times post-infection (Rowland et al. 2012). However, since whole blood comprises a number of cell types, gene expression differences should be handled with great caution. With the aid of complete blood counts, the transcriptional response data can be deconvoluted to help identify the unique regulatory control of specific cellular responses to pathogens (Shen-Orr et al. 2010).
Overall value of transcriptomics in important infectious swine diseases
At the end of the day, the ultimate goal is to see how the results of all these individual transcriptomic studies fit into an improved understanding of porcine immune response. Recently, such a meta-analysis was performed by combining results of several microarray-based pig immune studies to find PRRS-specific responses (Badaoui et al. 2013). This meta-analysis successfully summarized the general pathway(s) believed to be induced by PRRSv. Several interferon regulatory factors (IRFs) were highlighted in this analysis, and interferons clearly play an important role in viral infections. In agreement with the digital gene expression experiment (Xiao et al. 2010a), in several microarray and qPCR experiments, a dampened expression type I IFN response can be seen, which indicates an inadequate stimulation of the innate anti-viral immune response (Genini et al. 2008a; Xiao et al. 2010a; Ait-Ali et al. 2011; Garcia-Nicolas et al. 2014). Genini and colleagues observed a strong elevation of IFNβ at 9hpi, but only a slightly elevated expression of IFNα (Genini et al. 2008a). Ait-Ali and colleagues noted a similar low IFNα expression, but an, albeit late, accumulation of IFNβ expression. They stated that PRRSv could delay type I interferon transcriptional response in an attempt to counteract the host’s early immune response (Ait-Ali et al. 2011). Van Reeth and colleagues measured the IFNα levels in bronchoalveolar fluids during PRRSv infection and saw that its presence was low, a thousand-fold lower than with an infection with swine influenza virus or porcine respiratory coronavirus (Van Reeth et al. 1999). However, different PRRSv isolates were shown to invoke different (and sometimes significantly higher) IFNα expression levels, but no detectable IFNα protein levels were found by ELISA (Lee et al. 2004). Zhang and colleagues found that PRRSv does not fail to induce IFNα or IFNβ mRNA expression in monocyte-derived dendritic cells, but the protein seems to be blocked post-transcriptionally (Zhang et al. 2012). Although all described data point out to a weakened IFN response, greatly responsible for a persistent viral infection, the data by these last two studies demonstrate the incomplete information achieved from looking solely at transcriptomic data.
Another overall PRRS finding is the induction of pro-inflammatory chemokines and cytokines. The differential expression of a cell surface receptor involved in cytokine regulation, TREM1, was found through the meta-analyses by Badaoui et al. (2013) and was also present in the list of top ten upregulated transcripts in the RNA-seq experiment of Miller et al. (2012). In the meta-analysis study, TREM1 changed, among others, the expression of chemokines such as CCL2 and CCL3, interleukins IL6, IL18, and IL1β, and toll-like receptors TLR2 and TLR4. Xiao et al. (2010a) showed an upregulation in the inflammatory response toll-like receptor genes TLR2, TLR4, cytokines (among which IL1β), and chemokines. The acute-phase protein SAA2 and inflammasome genes RETN, s100A8, s100A9, and s100A12 were upregulated in the RNA-seq experiment by Miller and colleagues.
For S. typhimurium, as mentioned earlier, transcriptomic studies in blood have examined differences between LS and PS pigs. In the in vivo study by Knetter and colleagues, when looking at cytokine presence in serum at 2 dpi, the pro-inflammatory cytokines IL1β, TNFα, and IFNγ levels were higher in PS pigs compared to LS and control pigs, and the anti-inflammatory IL10 was upregulated in both LS and PS pigs, while only CXCL8 was elevated in the LS animals (Knetter et al. 2014). Also, Uthe and colleagues saw a correlation between IFNγ levels and shedding status. It seems that the PS animals have a much more extreme inflammatory response, as if the PS animals respond less quickly and thus extend their inflammatory response (Uthe et al. 2009). Additionally, looking at gene expression differences at 2 dpi compared to 0 dpi, PS animals showed a more extensive transcriptomic response, both in number of differentially expressed genes, as well as in level of expression compared to the LS animals. The most overrepresented regulation networks in PS animals at 2dpi involve the STAT1, IFNβ1, and IFNγ networks, showing a complex pro-inflammatory profile (Knetter et al. 2014). The genes CASP1, TNFα, and IL10 were also found upregulated in these networks, and hence, a nice correlation between serum cytokines and gene expression could be noted. Other important regulators CEBPB, SPI1, and TLR4 in the PS upregulated expression pathways as well as the TNFα and IFNγ pathways were earlier reported by Huang and colleagues in similar challenge studies (Huang et al. 2011). Not surprisingly, the microbiota that differ between LS and PS on 2dpi point to microbiota that play a role in gastrointestinal inflammation (Bearson et al. 2013). There are, however, regional differences in the inflammatory response to S. typhimurium expression pattern in the gut on 2dpi (Collado-Romero et al. 2010). While cytokine genes such as TNFα, IL6, IL1β, and IFNγ are upregulated in the jejunum and colon, they are not induced in the ileum. Collado-Romero and co-workers proposed that the ileum mucosa reacts slowly against the pathogen. Martins and colleagues and Wang and colleagues both examined transcriptomic differences due to S. typhimurium in mesenteric lymph nodes. Martins and colleagues describe an elevation of IFNγ, IL1β, CXCL2, CXCL8, CASP1, SLC11A, DEFB2, TLR8, and NOD2 at 2dpi. NFκB was significantly down-regulated at 2 and 6dpi (Martins et al. 2013a). Wang and colleagues also note an early repression of the NFκB pathway from 24 to 48 hpi in mesenteric lymph nodes (Wang et al. 2007). When looking at gene expression at only 2, 4, or 8hpi in jejunal scrapings, elevated gene expression was observed of inflammatory genes such as IL8, IL1β, PAP, and S100A9 (Hulst et al. 2013). They also noted an upregulation of NFKBIA, an NFκB inhibitor at this early time point.
A study using in vitro stimulation with endotoxin of blood of animals prior to infection was also able to find cytokine differences between LS and PS pigs, showing an attenuated response in LS animals, in contrast to a clear pro-inflammatory response in PS pigs (Knetter 2013). Interestingly, while gene expression on day 0 showed a similar magnitude of response in LS and PS pigs, response differences to LPS in blood at 2 dpi between LS and PS pigs were dramatic. Only 14 probesets were differentially expressed in LS animals after endotoxin stimulation, while 959 probesets in PS animals changed significantly, showing an apparent tolerization mechanism in LS animals. Since differences in gene expression patterns between LS and PS animals on day 0 were not significant enough to create predictor sets of genes in this and earlier studies, Kommadath and co-workers used the more sensitive weighted gene co-expression network analysis (WGCNA) technique after RNA-seq profiling of 8 LS and 8 PS animals. WGCNA creates modules of co-expressed genes that are significantly correlated with shedding levels. They include interesting genes such as cytokine genes, genes involved in TLR, NFκB, and NOD-like receptor pathways and genes linked to bacterial infections (Kommadath et al. 2014).
Future directions: a clear need for standard bioinformatic annotation!
For over a decade, microarrays have provided an enormous amount of information concerning different immune-related questions in pig transcriptomics. They have become increasingly inexpensive tools to search for gene expression differences between distinct immune phenotypes. Recently, RNA-seq, and other sequence-based methods such as miRNA-seq, BS-seq, ChIP-seq, and MeDIP-seq, experiments have become more cost-effective. The advantage of gaining information about all expressed and modified genes, different regulation of splice variants, as well as information about sequence-specific and histone code regulation, is a big plus for RNA-seq over microarrays. However, in the near future, the biggest challenge lies in comparing all existing data from many different kinds of platforms, so as to integrate such orthogonal data and better understand the physiology behind the disease phenotype and to find regulatory networks or biomarkers for disease resistance. Such meta-analyses use a set of statistical techniques to combine results from different studies (Badaoui et al. 2013), requiring only that the platform elements can be matched. It is not necessary that the exact same questions were addressed; e.g., an experiment looking at high and low responders to an infection can be compared to a study with infected versus control animals. Or an acute response study can be matched to a chronic response study. Adding to this, the possibility to integrate new (and broader) information obtained from upcoming next generation sequencing studies would really improve our transcriptomic and epigenomic insights into pig immunity.
An example of combining microarray studies for deeper insight is given by Pérez-Montarelo and co-workers. A meta-analysis was performed on 20 independent gene expression studies, using data from 480 of the same Affymetrix array. By doing so, the expression of 12,320 genes could be checked in 27 tissues and they could identify tissue-specific genes and tissue-specific regulatory networks and transcription factors (Perez-Montarelo et al. 2012). The meta-analysis study by Badaoui and colleagues described above illustrates how a meta-analysis experiment could be achieved for a PRRS-specific research question even across different platforms. Disparate microarray elements that mapped to the same IPA cDNA assembly (Couture et al. 2009) were considered to be comparable. 30,504 such elements could be compared across all 779 datasets used, of which more than a third was investigating PRRS. To facilitate these kinds of meta-analyses, lessons can be learned from the human ENCODE project by conducting experiments in a similar way and processing and archiving data using standard procedures (The ENCODE Consortium Project 2011b; Birney 2012; Landt et al. 2012). In this way, data quality is assured, data utility can be extended, and it becomes easier to compare datasets, combine computational analyses, and consequently perform meta-analysis.
Besides cross-platform meta-analyses, also cross-species meta-analyses are very promising, and R packages are freely available to conduct them (Kuhn et al. 2008; Kristiansson et al. 2013). However, until now, the main goal of these cross-species expression comparisons is often to employ model organisms for human diagnostic or therapeutic research (Yu et al. 2012; Grigoryev et al. 2013). Kapetanovic and colleagues show how the clustering software BioLayout Express 3D (BE3D) can be used to visualize inter-species expression comparisons (Kapetanovic et al. 2013). When comparing differential gene expression in mouse, human, and pig macrophages after LPS stimulation, they noted that pig macrophages act more human-like than do mouse macrophages. In a first paper, this group used BE3D to identify and visualize sets of genes responding similarly over time to LPS. They found that a subset of these genes had similar patterns of induction with human macrophage response, but not with mouse, where much lower stimulation or even repression was observed (Kapetanovic et al. 2012). They extended this work in a later paper by using the larger Snowball array. Taking only the differential genes between mouse and human, and removing those that did not have pig orthologues on the Snowball array, BE3D showed a large upregulated cluster in human compared to mouse macrophages which contained IDO1 as the hub gene. This cluster was highly upregulated in pig macrophages as well. Other genes such as those clustering around NOS2A also behaved in a murine-specific manner, with an upregulated expression in mouse macrophages while no differential expression in human or pig (Kapetanovic et al. 2013).
In addition to cross-platform and cross-species comparisons, it is important to investigate simultaneously different biological levels influencing the pig’s immune status. In the past, disease research very often focused only on one part of the host–pathogen interaction (Smits and Schokker 2011). One examined a portion of the host response, and looked at its ability to fight off an infection and examining its degree of disease susceptibility. Or one focused solely on the pathogen, describing level of virulence among different pathogen variants. As well, very often, only a small part of the host immune response was measured, such as a specific cell type or tissue, or only one particular timeframe was targeted. Narrow time windows or even single stages can be quite limiting, as immune response varies dramatically over time, and thus, time is a particularly crucial variable in an expression study of immune responses. With a systems biology approach, many levels of knowledge are gathered on both host and pathogen in a challenge study (genomic, transcriptomic, epigenomic, and metabolomic) and at different time points (Smits and Schokker 2011; Tuggle et al. 2011). The ultimate goal is to combine that data to fully explain host-pathogen interactions and discover emergent properties of the system that are difficult to reveal with current approaches. To disseminate public data and improve transcriptomic and epigenomic data mining, a livestock expression and epigenetic database (EpiDB) is under development. EpiDB includes data from chickens, cattle, pigs, sheep, and horses and provides a useful repository source, as well as tools to process and visualize expression and epigenetic data (Koltes et al. 2014). Examples of systems biology approaches integrating transcriptomic and epigenomic data can already be found in many miRNA-seq experiments, where one miRNA can regulate a network of several mRNAs (Giles et al. 2013; Valdmanis et al. 2013; Szeto et al. 2014; Yang et al. 2014). A porcine mRNA-seq study often precedes a miRNA-seq experiment on the same sample set to test for correlations between mRNA and miRNA in order to predict influences of specific miRNAs on components of the whole transcriptome (Bao et al. unpublished; Endale Ahanda et al. 2012).
However, the accuracy and depth of understanding stand or fall with the quality of the pig draft genome assembly and its annotation. Recently, the Immunome Response Annotation Group (IRAG) was able to improve the characterization of the pig immunome by manual annotation of almost 3,500 transcripts mapping to over 1,400 genes (Dawson et al. 2013). This was accomplished using the latest swine genome assembly version 10.2. For the genes without porcine RNA sequence evidence, RNA sequences of other species (often of human) were used to annotate more than 1,100 transcripts using the alignment tools in Otterlace (Searle et al. 2004; Loveland et al. 2012). Furthermore, gene expression clustering after infection or stimulation for many independent challenge experiments provided evidence for the involvement of over 500 genes not previously annotated for function in immune response processes (Dawson et al. 2013). On-going improvements of the draft assembly and additional annotation of the immunome will greatly improve the value of pig disease transcriptomic studies as well as further support the pig as model for human immune response.
Since immune networks are very complex (Gardy et al. 2009), a deeper understanding of such complexity is needed for advancements in unraveling porcine disease response mechanisms and in developing the pig as a viable model for human immunity. It is encouraging that substantial new high-throughput data have been reported in this area, and that analysis of such data is moving toward a systems biology approach by integrating different methods and combining multiple datasets. With the even higher throughput whole genome techniques coming to the forefront and performed at a relatively low cost, these comprehensive experiments will become more commonplace in the near future.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Contributor Information
Martine Schroyen, Email: schroyen@iastate.edu.
Christopher K. Tuggle, Phone: 1-515-294-4252, Email: cktuggle@iastate.edu
References
- Adamik J, Wang KZ, Unlu S, Su AJ, Tannahill GM, Galson DL, O’Neill LA, Auron PE. Distinct mechanisms for induction and tolerance regulate the immediate early genes encoding interleukin 1beta and tumor necrosis factor alpha. PLoS One. 2013;8:e70622. doi: 10.1371/journal.pone.0070622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler M, Murani E, Brunner R, Ponsuksili S, Wimmers K. Transcriptomic response of porcine PBMCs to vaccination with tetanus toxoid as a model antigen. PLoS One. 2013;8:e58306. doi: 10.1371/journal.pone.0058306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler M, Murani E, Ponsuksili S, Wimmers K. PBMC transcription profiles of pigs with divergent humoral immune responses and lean growth performance. Int J Biol Sci. 2013;9:907–916. doi: 10.7150/ijbs.6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ait-Ali T, Wilson AD, Carre W, Westcott DG, Frossard JP, Mellencamp MA, Mouzaki D, Matika O, Waddington D, Drew TW, Bishop SC, Archibald AL. Host inhibits replication of European porcine reproductive and respiratory syndrome virus in macrophages by altering differential regulation of type-I interferon transcriptional response. Immunogenetics. 2011;63:437–448. doi: 10.1007/s00251-011-0518-8. [DOI] [PubMed] [Google Scholar]
- Altay G, Emmert-Streib F. Inferring the conservative causal core of gene regulatory networks. BMC Syst Biol. 2010;4:132. doi: 10.1186/1752-0509-4-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anselmo A, Flori L, Jaffrezic F, Rutigliano T, Cecere M, Cortes-Perez N, Lefevre F, Rogel-Gaillard C, Giuffra E. Co-expression of host and viral microRNAs in porcine dendritic cells infected by the pseudorabies virus. PLoS One. 2011;6:e17374. doi: 10.1371/journal.pone.0017374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arceo ME, Ernst CW, Lunney JK, Choi I, Raney NE, Huang T, Tuggle CK, Rowland RR, Steibel JP. Characterizing differential individual response to porcine reproductive and respiratory syndrome virus infection through statistical and functional analysis of gene expression. Front Genet. 2012;3:321. doi: 10.3389/fgene.2012.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badaoui B, Tuggle CK, Hu Z, Reecy JM, Ait-Ali T, Anselmo A, Botti S. Pig immune response to general stimulus and to porcine reproductive and respiratory syndrome virus infection: a meta-analysis approach. BMC Genomics. 2013;14:220. doi: 10.1186/1471-2164-14-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badaoui B, Rutigliano T, Anselmo A, Vanhee M, Nauwynck H, Giuffra E, Botti S. RNA-sequence analysis of primary alveolar macrophages after in vitro infection with porcine reproductive and respiratory syndrome virus strains of differing virulence. PLoS One. 2014;9:e91918. doi: 10.1371/journal.pone.0091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao WB, Ye L, Pan ZY, Zhu J, Du ZD, Zhu GQ, Huang XG, Wu SL. Microarray analysis of differential gene expression in sensitive and resistant pig to Escherichia coli F18. Anim Genet. 2012;43:525–534. doi: 10.1111/j.1365-2052.2011.02287.x. [DOI] [PubMed] [Google Scholar]
- Bao H, Kommadath A, Plastow GS, Tuggle CK, le Guan L, Stothard P. MicroRNA buffering and altered variance of gene expression in response to Salmonella infection. PLoS One. 2014;9:e94352. doi: 10.1371/journal.pone.0094352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H, Kommadath A, Tuggle CK, Plastow GS, Stothard P, Guan LL (unpublished) MicroRNA regulation in response to Salmonella infection in pigs [DOI] [PMC free article] [PubMed]
- Bates JS, Petry DB, Eudy J, Bough L, Johnson RK. Differential expression in lung and bronchial lymph node of pigs with high and low responses to infection with porcine reproductive and respiratory syndrome virus. J Anim Sci. 2008;86:3279–3289. doi: 10.2527/jas.2007-0685. [DOI] [PubMed] [Google Scholar]
- Bauersachs S, Wolf E. Transcriptome analyses of bovine, porcine and equine endometrium during the pre-implantation phase. Anim Reprod Sci. 2012;134:84–94. doi: 10.1016/j.anireprosci.2012.08.015. [DOI] [PubMed] [Google Scholar]
- Bearson SM, Allen HK, Bearson BL, Looft T, Brunelle BW, Kich JD, Tuggle CK, Bayles DO, Alt D, Levine UY, Stanton TB. Profiling the gastrointestinal microbiota in response to Salmonella: low versus high Salmonella shedding in the natural porcine host. Infect Genet Evol. 2013;16:330–340. doi: 10.1016/j.meegid.2013.03.022. [DOI] [PubMed] [Google Scholar]
- Birney E. The making of ENCODE: lessons for big-data projects. Nature. 2012;489:49–51. doi: 10.1038/489049a. [DOI] [PubMed] [Google Scholar]
- Boddicker N, Waide EH, Rowland RR, Lunney JK, Garrick DJ, Reecy JM, Dekkers JC. Evidence for a major QTL associated with host response to porcine reproductive and respiratory syndrome virus challenge. J Anim Sci. 2012;90:1733–1746. doi: 10.2527/jas.2011-4464. [DOI] [PubMed] [Google Scholar]
- Boddicker NJ, Garrick DJ, Rowland RR, Lunney JK, Reecy JM, Dekkers JC. Validation and further characterization of a major quantitative trait locus associated with host response to experimental infection with porcine reproductive and respiratory syndrome virus. Anim Genet. 2013;45:48–58. doi: 10.1111/age.12079. [DOI] [PubMed] [Google Scholar]
- Borca MV, Gudmundsdottir I, Fernandez-Sainz IJ, Holinka LG, Risatti GR. Patterns of cellular gene expression in swine macrophages infected with highly virulent classical swine fever virus strain Brescia. Virus Res. 2008;138:89–96. doi: 10.1016/j.virusres.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Chen H, Li C, Fang M, Zhu M, Li X, Zhou R, Li K, Zhao S. Understanding Haemophilus parasuis infection in porcine spleen through a transcriptomics approach. BMC Genomics. 2009;10:64. doi: 10.1186/1471-2164-10-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Ai H, Ren J, Li W, Li P, Qiao R, Ouyang J, Yang M, Ma J, Huang L. A global view of porcine transcriptome in three tissues from a full-sib pair with extreme phenotypes in growth and fat deposition by paired-end RNA sequencing. BMC Genomics. 2011;12:448. doi: 10.1186/1471-2164-12-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lunney JK, Cheng L, Li X, Cao J, Zhu M, Zhao S. Porcine S100A8 and S100A9: molecular characterizations and crucial functions in response to Haemophilus parasuis infection. Dev Comp Immunol. 2011;35:490–500. doi: 10.1016/j.dci.2010.11.017. [DOI] [PubMed] [Google Scholar]
- Choi I, Bao H, Kommadath A, Hosseini A, Sun X, Meng Y, Stothard P, Plastow GS, Tuggle CK, Reecy JM, Fritz-Waters E, Abrams SM, Lunney JK, Guan LL. Increasing gene discovery and coverage using RNA-seq of globin RNA reduced porcine blood samples. BMC Genomics. 2014;15(1):954. doi: 10.1186/1471-2164-15-954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinar MU, Islam MA, Uddin MJ, Tholen E, Tesfaye D, Looft C, Schellander K. Evaluation of suitable reference genes for gene expression studies in porcine alveolar macrophages in response to LPS and LTA. BMC Res Notes. 2012;5:107. doi: 10.1186/1756-0500-5-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinar MU, Islam MA, Proll M, Kocamis H, Tholen E, Tesfaye D, Looft C, Schellander K, Uddin MJ. Evaluation of suitable reference genes for gene expression studies in porcine PBMCs in response to LPS and LTA. BMC Res Notes. 2013;6:56. doi: 10.1186/1756-0500-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado-Romero M, Arce C, Ramirez-Boo M, Carvajal A, Garrido JJ. Quantitative analysis of the immune response upon Salmonella typhimurium infection along the porcine intestinal gut. Vet Res. 2010;41:23. doi: 10.1051/vetres/2009072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JL, Becares M, Sola I, Oliveros JC, Enjuanes L, Zuniga S. Alphacoronavirus protein 7 modulates host innate immune response. J Virol. 2013;87:9754–9767. doi: 10.1128/JVI.01032-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang Y, Lachance C, Wang Y, Gagnon CA, Savard C, Segura M, Grenier D, Gottschalk M. Transcriptional approach to study porcine tracheal epithelial cells individually or dually infected with swine influenza virus and Streptococcus suis. BMC Vet Res. 2014;10:86. doi: 10.1186/1746-6148-10-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HD. Comparative assessment of the pig, mouse, and human genomes: a structural and functional analysis of genes involved in immunity. In: McAnulty PA, Dayan A, Hastings KH, Ganderup N-C, editors. The minipig in biomedical research. Boca Raton: CRC Press; 2011. pp. 321–341. [Google Scholar]
- Dawson HD, Loveland JE, Pascal G, Gilbert JG, Uenishi H, Mann KM, Sang Y, Zhang J, Carvalho-Silva D, Hunt T, Hardy M, Hu Z, Zhao SH, Anselmo A, Shinkai H, Chen C, Badaoui B, Berman D, Amid C, Kay M, Lloyd D, Snow C, Morozumi T, Cheng RP, Bystrom M, Kapetanovic R, Schwartz JC, Kataria R, Astley M, Fritz E, Steward C, Thomas M, Wilming L, Toki D, Archibald AL, Bed’Hom B, Beraldi D, Huang TH, Ait-Ali T, Blecha F, Botti S, Freeman TC, Giuffra E, Hume DA, Lunney JK, Murtaugh MP, Reecy JM, Harrow JL, Rogel-Gaillard C, Tuggle CK. Structural and functional annotation of the porcine immunome. BMC Genomics. 2013;14:332. doi: 10.1186/1471-2164-14-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Greeff A, Benga L, Wichgers Schreur PJ, Valentin-Weigand P, Rebel JM, Smith HE. Involvement of NF-kappaB and MAP-kinases in the transcriptional response of alveolar macrophages to Streptococcus suis. Vet Microbiol. 2010;141:59–67. doi: 10.1016/j.vetmic.2009.07.031. [DOI] [PubMed] [Google Scholar]
- Dvorak CM, Hirsch GN, Hyland KA, Hendrickson JA, Thompson BS, Rutherford MS, Murtaugh MP. Genomic dissection of mucosal immunobiology in the porcine small intestine. Physiol Genomics. 2006;28:5–14. doi: 10.1152/physiolgenomics.00104.2006. [DOI] [PubMed] [Google Scholar]
- Edfors-Lilja I, Wattrang E, Marklund L, Moller M, Andersson-Eklund L, Andersson L, Fossum C. Mapping quantitative trait loci for immune capacity in the pig. J Immunol. 1998;161:829–835. [PubMed] [Google Scholar]
- Eisley C, Fritz-Waters E, Choi I, Koltes J, Boddicker N, Reecy J, Lunney J, Carpenter S, Tuggle C, Liu P, Dekkers J (2014) Analysis of gene expression in a region associated with host response to porcine reproductive and respiratory syndrome virus challenge. In: Plant & Animal Genome 22, Poster P592, San Diego
- Endale Ahanda ML, Fritz ER, Estelle J, Hu ZL, Madsen O, Groenen MA, Beraldi D, Kapetanovic R, Hume DA, Rowland RR, Lunney JK, Rogel-Gaillard C, Reecy JM, Giuffra E. Prediction of altered 3’- UTR miRNA-binding sites from RNA-Seq data: the swine leukocyte antigen complex (SLA) as a model region. PLoS One. 2012;7:e48607. doi: 10.1371/journal.pone.0048607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteve-Codina A, Kofler R, Palmieri N, Bussotti G, Notredame C, Perez-Enciso M. Exploring the gonad transcriptome of two extreme male pigs with RNA-seq. BMC Genomics. 2011;12:552. doi: 10.1186/1471-2164-12-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbairn L, Kapetanovic R, Sester DP, Hume DA. The mononuclear phagocyte system of the pig as a model for understanding human innate immunity and disease. J Leukoc Biol. 2011;89:855–871. doi: 10.1189/jlb.1110607. [DOI] [PubMed] [Google Scholar]
- Fairbairn L, Kapetanovic R, Beraldi D, Sester DP, Tuggle CK, Archibald AL, Hume DA. Comparative analysis of monocyte subsets in the pig. J Immunol. 2013;190:6389–6396. doi: 10.4049/jimmunol.1300365. [DOI] [PubMed] [Google Scholar]
- Ferrarini M, Moretto M, Ward JA, Surbanovski N, Stevanovic V, Giongo L, Viola R, Cavalieri D, Velasco R, Cestaro A, Sargent DJ. An evaluation of the PacBio RS platform for sequencing and de novo assembly of a chloroplast genome. BMC Genomics. 2013;14:670. doi: 10.1186/1471-2164-14-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flori L, Rogel-Gaillard C, Cochet M, Lemonnier G, Hugot K, Chardon P, Robin S, Lefevre F. Transcriptomic analysis of the dialogue between Pseudorabies virus and porcine epithelial cells during infection. BMC Genomics. 2008;9:123. doi: 10.1186/1471-2164-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman TC, Ivens A, Baillie JK, Beraldi D, Barnett MW, Dorward D, Downing A, Fairbairn L, Kapetanovic R, Raza S, Tomoiu A, Alberio R, Wu C, Su AI, Summers KM, Tuggle CK, Archibald AL, Hume DA. A gene expression atlas of the domestic pig. BMC Biol. 2012;10:90. doi: 10.1186/1741-7007-10-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo RC, Munoz PM, de Miguel MJ, Marin CM, Labairu J, Revilla M, Blasco JM, Gortazar C, de la Fuente J. Gene expression changes in spleens of the wildlife reservoir species, Eurasian wild boar (Sus scrofa), naturally infected with Brucella suis biovar 2. J Genet Genomics. 2010;37:725–736. doi: 10.1016/S1673-8527(09)60090-4. [DOI] [PubMed] [Google Scholar]
- Galindo RC, Ayllon N, Smrdel KS, Boadella M, Beltran-Beck B, Mazariegos M, Garcia N, de la Lastra JM, Avsic-Zupanc T, Kocan KM, Gortazar C, de la Fuente J. Gene expression profile suggests that pigs (Sus scrofa) are susceptible to Anaplasma phagocytophilum but control infection. Parasit Vectors. 2012;5:181. doi: 10.1186/1756-3305-5-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Flori L, Lecardonnel J, Esquerre D, Hu ZL, Teillaud A, Lemonnier G, Lefevre F, Oswald IP, Rogel-Gaillard C. Transcriptome analysis of porcine PBMCs after in vitro stimulation by LPS or PMA/ionomycin using an expression array targeting the pig immune response. BMC Genomics. 2010;11:292. doi: 10.1186/1471-2164-11-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Wahlberg P, Marthey S, Esquerre D, Jaffrezic F, Lecardonnel J, Hugot K, Rogel-Gaillard C. Analysis of porcine MHC using microarrays. Vet Immunol Immunopathol. 2012;148:78–84. doi: 10.1016/j.vetimm.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Gao L, Guo XK, Wang L, Zhang Q, Li N, Chen XX, Wang Y, Feng WH. MicroRNA 181 suppresses porcine reproductive and respiratory syndrome virus (PRRSV) infection by targeting PRRSV receptor CD163. J Virol. 2013;87:8808–8812. doi: 10.1128/JVI.00718-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Nicolas O, Quereda JJ, Gomez-Laguna J, Salguero FJ, Carrasco L, Ramis G, Pallares FJ. Cytokines transcript levels in lung and lymphoid organs during genotype 1 Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) infection. Vet Immunol Immunopathol. 2014;160:26–40. doi: 10.1016/j.vetimm.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Gardy JL, Lynn DJ, Brinkman FS, Hancock RE. Enabling a systems biology approach to immunology: focus on innate immunity. Trends Immunol. 2009;30:249–262. doi: 10.1016/j.it.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, James JJ, Maysuria M, Mitton JD, Oliveri P, Osborn JL, Peng T, Ratcliffe AL, Webster PJ, Davidson EH, Hood L, Dimitrov K. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- Genini S, Delputte PL, Malinverni R, Cecere M, Stella A, Nauwynck HJ, Giuffra E. Genome-wide transcriptional response of primary alveolar macrophages following infection with porcine reproductive and respiratory syndrome virus. J Gen Virol. 2008;89:2550–2564. doi: 10.1099/vir.0.2008/003244-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genini S, Malinverni R, Delputte PL, Fiorentini S, Stella A, Botti S, Nauwynck HJ, Giuffra E. Gene expression profiling of porcine alveolar macrophages after antibody-mediated cross-linking of Sialoadhesin (Sn, Siglec-1) J Recept Signal Transduct Res. 2008;28:185–243. doi: 10.1080/10799890802084226. [DOI] [PubMed] [Google Scholar]
- Giles CB, Girija-Devi R, Dozmorov MG, Wren JD. mirCoX: a database of miRNA-mRNA expression correlations derived from RNA-seq meta-analysis. BMC Bioinform. 2013;14(Suppl 14):S17. doi: 10.1186/1471-2105-14-S14-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go JT, Belisle SE, Tchitchek N, Tumpey TM, Ma W, Richt JA, Safronetz D, Feldmann H, Katze MG. 2009 pandemic H1N1 influenza virus elicits similar clinical course but differential host transcriptional response in mouse, macaque, and swine infection models. BMC Genomics. 2012;13:627. doi: 10.1186/1471-2164-13-627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucl Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryev DN, Cheranova DI, Heruth DP, Huang P, Zhang LQ, Rabb H, Ye SQ. Meta-analysis of molecular response of kidney to ischemia reperfusion injury for the identification of new candidate genes. BMC Nephrol. 2013;14:231. doi: 10.1186/1471-2369-14-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenen MA, Archibald AL, Uenishi H, Tuggle CK, Takeuchi Y, Rothschild MF, Rogel-Gaillard C, Park C, Milan D, Megens HJ, Li S, Larkin DM, Kim H, Frantz LA, Caccamo M, Ahn H, Aken BL, Anselmo A, Anthon C, Auvil L, Badaoui B, Beattie CW, Bendixen C, Berman D, Blecha F, Blomberg J, Bolund L, Bosse M, Botti S, Bujie Z, Bystrom M, Capitanu B, Carvalho-Silva D, Chardon P, Chen C, Cheng R, Choi SH, Chow W, Clark RC, Clee C, Crooijmans RP, Dawson HD, Dehais P, De Sapio F, Dibbits B, Drou N, Du ZQ, Eversole K, Fadista J, Fairley S, Faraut T, Faulkner GJ, Fowler KE, Fredholm M, Fritz E, Gilbert JG, Giuffra E, Gorodkin J, Griffin DK, Harrow JL, Hayward A, Howe K, Hu ZL, Humphray SJ, Hunt T, Hornshoj H, Jeon JT, Jern P, Jones M, Jurka J, Kanamori H, Kapetanovic R, Kim J, Kim JH, Kim KW, Kim TH, Larson G, Lee K, Lee KT, Leggett R, Lewin HA, Li Y, Liu W, Loveland JE, Lu Y, Lunney JK, Ma J, Madsen O, Mann K, Matthews L, McLaren S, Morozumi T, Murtaugh MP, Narayan J, Nguyen DT, Ni P, Oh SJ, Onteru S, Panitz F, Park EW, Park HS, Pascal G, Paudel Y, Perez-Enciso M, Ramirez-Gonzalez R, Reecy JM, Rodriguez-Zas S, Rohrer GA, Rund L, Sang Y, Schachtschneider K, Schraiber JG, Schwartz J, Scobie L, Scott C, Searle S, Servin B, Southey BR, Sperber G, Stadler P, Sweedler JV, Tafer H, Thomsen B, Wali R, Wang J, White S, Xu X, Yerle M, Zhang G, Zhang J, Zhao S, Rogers J, Churcher C, Schook LB. Analyses of pig genomes provide insight into porcine demography and evolution. Nature. 2012;491:393–398. doi: 10.1038/nature11622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross G, van der Meulen J, Snel J, van der Meer R, Kleerebezem M, Niewold TA, Hulst MM, Smits MA. Mannose-specific interaction of Lactobacillus plantarum with porcine jejunal epithelium. FEMS Immunol Med Microbiol. 2008;54:215–223. doi: 10.1111/j.1574-695X.2008.00466.x. [DOI] [PubMed] [Google Scholar]
- Guo XK, Zhang Q, Gao L, Li N, Chen XX, Feng WH. Increasing expression of MicroRNA 181 inhibits porcine reproductive and respiratory syndrome virus replication and has implications for controlling virus infection. J Virol. 2013;87:1159–1171. doi: 10.1128/JVI.02386-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He T, Feng G, Chen H, Wang L, Wang Y. Identification of host encoded microRNAs interacting with novel swine-origin influenza A (H1N1) virus and swine influenza virus. Bioinformation. 2009;4:112–118. doi: 10.6026/97320630004112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedegaard J, Skovgaard K, Mortensen S, Sorensen P, Jensen TK, Hornshoj H, Bendixen C, Heegaard PM. Molecular characterisation of the early response in pigs to experimental infection with Actinobacillus pleuropneumoniae using cDNA microarrays. Acta Vet Scand. 2007;49:11. doi: 10.1186/1751-0147-49-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeke L, Sharbati J, Pawar K, Keller A, Einspanier R, Sharbati S. Intestinal Salmonella typhimurium infection leads to miR-29a induced caveolin 2 regulation. PLoS One. 2013;8:e67300. doi: 10.1371/journal.pone.0067300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong LZ, Li J, Schmidt-Kuntzel A, Warren WC, Barsh GS. Digital gene expression for non-model organisms. Genome Res. 2011;21:1905–1915. doi: 10.1101/gr.122135.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TH, Uthe JJ, Bearson SM, Demirkale CY, Nettleton D, Knetter S, Christian C, Ramer-Tait AE, Wannemuehler MJ, Tuggle CK. Distinct peripheral blood RNA responses to Salmonella in pigs differing in Salmonella shedding levels: intersection of IFNG, TLR and miRNA pathways. PLoS One. 2011;6:e28768. doi: 10.1371/journal.pone.0028768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulst M, Smits M, Vastenhouw S, de Wit A, Niewold T, van der Meulen J. Transcription networks responsible for early regulation of Salmonella-induced inflammation in the jejunum of pigs. J Inflamm (Lond) 2013;10:18. doi: 10.1186/1476-9255-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MA, Cinar MU, Uddin MJ, Tholen E, Tesfaye D, Looft C, Schellander K. Expression of toll-like receptors and downstream genes in lipopolysaccharide-induced porcine alveolar macrophages. Vet Immunol Immunopathol. 2012;146:62–73. doi: 10.1016/j.vetimm.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Islam MA, Uddin MJ, Tholen E, Tesfaye D, Looft C, Schellander K, Cinar MU. Age-related changes in phagocytic activity and production of pro-inflammatory cytokines by lipopolysaccharide stimulated porcine alveolar macrophages. Cytokine. 2012;60:707–717. doi: 10.1016/j.cyto.2012.08.011. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Zhou X, Michal JJ, Wu XL, Zhang L, Zhang M, Ding B, Liu B, Manoranjan VS, Neill JD, Harhay GP, Kehrli ME, Jr, Miller LC. Reactomes of porcine alveolar macrophages infected with porcine reproductive and respiratory syndrome virus. PLoS One. 2013;8:e59229. doi: 10.1371/journal.pone.0059229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapetanovic R, Fairbairn L, Beraldi D, Sester DP, Archibald AL, Tuggle CK, Hume DA. Pig bone marrow-derived macrophages resemble human macrophages in their response to bacterial lipopolysaccharide. J Immunol. 2012;188:3382–3394. doi: 10.4049/jimmunol.1102649. [DOI] [PubMed] [Google Scholar]
- Kapetanovic R, Fairbairn L, Downing A, Beraldi D, Sester DP, Freeman TC, Tuggle CK, Archibald AL, Hume DA. The impact of breed and tissue compartment on the response of pig macrophages to lipopolysaccharide. BMC Genomics. 2013;14:581. doi: 10.1186/1471-2164-14-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knetter S (2013) Characterizing the porcine immune response to an environmental and pathogenic challenge: swine barn dust and Salmonella infection. Graduate Theses and Dissertations. Paper 13142
- Knetter S, Bearson S, Huang T, Kurkiewicz D, Schroyen M, Nettleton D, Berman D, Cohen V, Lunney J, Ramer-Tait A, Wannemuehler M, Tuggle C (2014) Salmonella enterica serovar Typhimurium-infected pigs with different shedding levels exhibit distinct clinical, peripheral cytokine and transcriptomic immune response phenotypes. Innate Immun (in press) [DOI] [PubMed]
- Koltes JE, Fritz-Waters E, Hu Z, Reecy JM (2014) Livestock EpiDB: an integrated epigenetics and gene expression database and discovery resource. In: Plant and animal genome 22, workshop session W025, San Diego
- Kommadath A, Bao H, Arantes AS, Plastow GS, Tuggle CK, Bearson SM, Guan LL, Stothard P. Gene co-expression network analysis identifies porcine genes associated with variation in Salmonella shedding. BMC Genomics. 2014;15:452. doi: 10.1186/1471-2164-15-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucl Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansson E, Osterlund T, Gunnarsson L, Arne G, Larsson DG, Nerman O. A novel method for cross-species gene expression analysis. BMC Bioinform. 2013;14:70. doi: 10.1186/1471-2105-14-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn A, Luthi-Carter R, Delorenzi M. Cross-species and cross-platform gene expression studies with the Bioconductor-compliant R package ‘annotationTools’. BMC Bioinform. 2008;9:26. doi: 10.1186/1471-2105-9-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, Chen Y, DeSalvo G, Epstein C, Fisher-Aylor KI, Euskirchen G, Gerstein M, Gertz J, Hartemink AJ, Hoffman MM, Iyer VR, Jung YL, Karmakar S, Kellis M, Kharchenko PV, Li Q, Liu T, Liu XS, Ma L, Milosavljevic A, Myers RM, Park PJ, Pazin MJ, Perry MD, Raha D, Reddy TE, Rozowsky J, Shoresh N, Sidow A, Slattery M, Stamatoyannopoulos JA, Tolstorukov MY, White KP, Xi S, Farnham PJ, Lieb JD, Wold BJ, Snyder M. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lastra JM, Galindo RC, Gortazar C, Ruiz-Fons F, Aranaz A, de la Fuente J. Expression of immunoregulatory genes in peripheral blood mononuclear cells of European wild boar immunized with BCG. Vet Microbiol. 2009;134:334–339. doi: 10.1016/j.vetmic.2008.08.026. [DOI] [PubMed] [Google Scholar]
- Ledger TN, Pinton P, Bourges D, Roumi P, Salmon H, Oswald IP (2004) Development of a macroarray to specifically analyze immunological gene expression in swine. Clin Diagn Lab Immunol 11:691–698 [DOI] [PMC free article] [PubMed]
- Lee SM, Schommer SK, Kleiboeker SB. Porcine reproductive and respiratory syndrome virus field isolates differ in in vitro interferon phenotypes. Vet Immunol Immunopathol. 2004;102:217–231. doi: 10.1016/j.vetimm.2004.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Xia Y, Gu Y, Zhang K, Lang Q, Chen L, Guan J, Luo Z, Chen H, Li Y, Li Q, Li X, Jiang AA, Shuai S, Wang J, Zhu Q, Zhou X, Gao X. MicroRNAome of porcine pre- and postnatal development. PLoS One. 2010;5:e11541. doi: 10.1371/journal.pone.0011541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Zhang A, Chen B, Teng L, Wang Y, Chen H, Jin M. Response of swine spleen to Streptococcus suis infection revealed by transcription analysis. BMC Genomics. 2010;11:556. doi: 10.1186/1471-2164-11-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhou H, Wen Z, Wu S, Huang C, Jia G, Chen H, Jin M. Transcription analysis on response of swine lung to H1N1 swine influenza virus. BMC Genomics. 2011;12:398. doi: 10.1186/1471-2164-12-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Liu S, Wang Y, Deng F, Yan W, Yang K, Chen H, He Q, Charreyre C, Audoneet JC. Transcription analysis of the porcine alveolar macrophage response to porcine circovirus type 2. BMC Genomics. 2013;14:353. doi: 10.1186/1471-2164-14-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS. Getting started in tiling microarray analysis. PLoS Comput Biol. 2007;3:1842–1844. doi: 10.1371/journal.pcbi.0030183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HC, Hicks JA, Trakooljul N, Zhao SH. Current knowledge of microRNA characterization in agricultural animals. Anim Genet. 2010;41:225–231. doi: 10.1111/j.1365-2052.2009.01995.x. [DOI] [PubMed] [Google Scholar]
- Liu M, Fang L, Tan C, Long T, Chen H, Xiao S. Understanding Streptococcus suis serotype 2 infection in pigs through a transcriptional approach. BMC Genomics. 2011;12:253. doi: 10.1186/1471-2164-12-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Huang J, Yang S, Zhao Y, Xiang A, Cao J, Fan B, Wu Z, Zhao J, Zhao S, Zhu M. Whole blood transcriptome comparison of pigs with extreme production of in vivo dsRNA-induced serum IFN-a. Dev Comp Immunol. 2014;44:35–43. doi: 10.1016/j.dci.2013.11.008. [DOI] [PubMed] [Google Scholar]
- Loveday EK, Svinti V, Diederich S, Pasick J, Jean F. Temporal- and strain-specific host microRNA molecular signatures associated with swine-origin H1N1 and avian-origin H7N7 influenza A virus infection. J Virol. 2012;86:6109–6122. doi: 10.1128/JVI.06892-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveland JE, Gilbert JG, Griffiths E, Harrow JL. Community gene annotation in practice. Database (Oxford) 2012;2012:bas009. doi: 10.1093/database/bas009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Fu WX, Luo YR, Ding XD, Zhou JP, Liu Y, Liu JF, Zhang Q. Genome-wide association study for T lymphocyte subpopulations in swine. BMC Genomics. 2012;13:488. doi: 10.1186/1471-2164-13-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunney JK. Advances in swine biomedical model genomics. Int J Biol Sci. 2007;3:179–184. doi: 10.7150/ijbs.3.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Belisle SE, Mosier D, Li X, Stigger-Rosser E, Liu Q, Qiao C, Elder J, Webby R, Katze MG, Richt JA. 2009 pandemic H1N1 influenza virus causes disease and upregulation of genes related to inflammatory and immune responses, cell death, and lipid metabolism in pigs. J Virol. 2011;85:11626–11637. doi: 10.1128/JVI.05705-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach N, Gao Y, Lemonnier G, Lecardonnel J, Oswald IP, Estelle J, Rogel-Gaillard C. The peripheral blood transcriptome reflects variations in immunity traits in swine: towards the identification of biomarkers. BMC Genomics. 2013;14:894. doi: 10.1186/1471-2164-14-894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JA, Wang Z. Next-generation transcriptome assembly. Nat Rev Genet. 2011;12:671–682. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- Martins RP, Collado-Romero M, Arce C, Lucena C, Carvajal A, Garrido JJ. Exploring the immune response of porcine mesenteric lymph nodes to Salmonella enterica serovar Typhimurium: an analysis of transcriptional changes, morphological alterations and pathogen burden. Comp Immunol Microbiol Infect Dis. 2013;36:149–160. doi: 10.1016/j.cimid.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Martins RP, Lorenzi V, Arce C, Lucena C, Carvajal A, Garrido JJ. Innate and adaptive immune mechanisms are effectively induced in ileal Peyer’s patches of Salmonella typhimurium infected pigs. Dev Comp Immunol. 2013;41:100–104. doi: 10.1016/j.dci.2013.04.020. [DOI] [PubMed] [Google Scholar]
- McDaneld TG. MicroRNA: mechanism of gene regulation and application to livestock. J Anim Sci. 2009;87:E21–E28. doi: 10.2527/jas.2008-1303. [DOI] [PubMed] [Google Scholar]
- Mellencamp MA, Galina-Pantoja L, Gladney CD, Torremorell M. Improving pig health through genomics: a view from the industry. Dev Biol (Basel) 2008;132:35–41. doi: 10.1159/000317142. [DOI] [PubMed] [Google Scholar]
- Meurens F, Summerfield A, Nauwynck H, Saif L, Gerdts V. The pig: a model for human infectious diseases. Trends Microbiol. 2012;20:50–57. doi: 10.1016/j.tim.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LC, Fleming D, Arbogast A, Bayles DO, Guo B, Lager KM, Henningson JN, Schlink SN, Yang HC, Faaberg KS, Kehrli ME., Jr Analysis of the swine tracheobronchial lymph node transcriptomic response to infection with a Chinese highly pathogenic strain of porcine reproductive and respiratory syndrome virus. BMC Vet Res. 2012;8:208. doi: 10.1186/1746-6148-8-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LC, Jiang Z, Sang Y, Harhay GP, Lager KM. Evolutionary characterization of pig interferon-inducible transmembrane gene family and member expression dynamics in tracheobronchial lymph nodes of pigs infected with swine respiratory disease viruses. Vet Immunol Immunopathol. 2014;159:180–191. doi: 10.1016/j.vetimm.2014.02.015. [DOI] [PubMed] [Google Scholar]
- Mockler TC, Chan S, Sundaresan A, Chen H, Jacobsen SE, Ecker JR. Applications of DNA tiling arrays for whole-genome analysis. Genomics. 2005;85:1–15. doi: 10.1016/j.ygeno.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008;92:255–264. doi: 10.1016/j.ygeno.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Nfon CK, Leung A, Smith G, Embury-Hyatt C, Kobinger G, Weingartl HM. Immunopathogenesis of severe acute respiratory disease in Zaire ebolavirus-infected pigs. PLoS One. 2013;8:e61904. doi: 10.1371/journal.pone.0061904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewold TA, Kerstens HH, van der Meulen J, Smits MA, Hulst MM (2005) Development of a porcine small intestinal cDNA micro-array: characterization and functional analysis of the response to enterotoxigenic E. coli. Vet Immunol Immunopathol 105:317–329 [DOI] [PubMed]
- Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, Habib N, Yosef N, Chang CY, Shay T, Frampton GM, Drake AC, Leskov I, Nilsson B, Preffer F, Dombkowski D, Evans JW, Liefeld T, Smutko JS, Chen J, Friedman N, Young RA, Golub TR, Regev A, Ebert BL. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144:296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton JB, Rowland RR, Yoo D, Chang KO. Modulation of CD163 receptor expression and replication of porcine reproductive and respiratory syndrome virus in porcine macrophages. Virus Res. 2009;140:161–171. doi: 10.1016/j.virusres.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Perez-Montarelo D, Hudson NJ, Fernandez AI, Ramayo-Caldas Y, Dalrymple BP, Reverter A. Porcine tissue-specific regulatory networks derived from meta-analysis of the transcriptome. PLoS One. 2012;7:e46159. doi: 10.1371/journal.pone.0046159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzulo AA, Tang XX, Hoegger MJ, Alaiwa MH, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, Banfi B, Horswill AR, Stoltz DA, McCray PB, Jr, Welsh MJ, Zabner J. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487:109–113. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilcher C, Jones C, Schroyen M, Severin A, Patience J, Tuggle C, Koltes JE. Gene expression profiling of longissimus dorsi and adipose tissue in pigs with differing post-weaning growth rate. Champaign: American Society of Animal Science Des Moines; 2014. [Google Scholar]
- Podolska A, Anthon C, Bak M, Tommerup N, Skovgaard K, Heegaard PM, Gorodkin J, Cirera S, Fredholm M. Profiling microRNAs in lung tissue from pigs infected with Actinobacillus pleuropneumoniae. BMC Genomics. 2012;13:459. doi: 10.1186/1471-2164-13-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Cinar MU, Fan H, Proll M, Tesfaye D, Tholen E, Looft C, Holker M, Schellander K, Uddin MJ (2014) Comparison of the innate immune responses of porcine monocyte-derived dendritic cells and splenic dendritic cells stimulated with LPS. Innate Immun [DOI] [PubMed]
- Ramakrishnan R, Qin J, Jones RC, Weaver LS. Integrated fluidic circuits (IFCs) for digital PCR. Methods Mol Biol. 2013;949:423–431. doi: 10.1007/978-1-62703-134-9_27. [DOI] [PubMed] [Google Scholar]
- Robic A, Feve K, Larzul C, Billon Y, van Son M, Liaubet L, Sarry J, Milan D, Grindflek E, Bidanel JP, Riquet J. Expression levels of 25 genes in liver and testis located in a QTL region for androstenone on SSC7q1.2. Anim Genet. 2011;42:662–665. doi: 10.1111/j.1365-2052.2011.02195.x. [DOI] [PubMed] [Google Scholar]
- Rojas M, Parker RE, Thorn N, Corredor C, Iyer SS, Bueno M, Mroz L, Cardenes N, Mora AL, Stecenko AA, Brigham KL. Infusion of freshly isolated autologous bone marrow derived mononuclear cells prevents endotoxin-induced lung injury in an ex vivo perfused swine model. Stem Cell Res Ther. 2013;4:26. doi: 10.1186/scrt174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JW, Prather RS. Transgenics and modern reproductive technologies. In: Rothschild MF, Ruvinsky A, editors. The genetics of the pig. Iowa City: Iowa State University; 2011. pp. 242–262. [Google Scholar]
- Rowland RR, Lunney J, Dekkers J. Control of porcine reproductive and respiratory syndrome (PRRS) through genetic improvements in disease resistance and tolerance. Front Genet. 2012;3:260. doi: 10.3389/fgene.2012.00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang Y, Brichalli W, Rowland RR, Blecha F. Genome-wide analysis of antiviral signature genes in porcine macrophages at different activation statuses. PLoS One. 2014;9:e87613. doi: 10.1371/journal.pone.0087613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaria V, Hariharan M, Maiti S, Pillai B, Brahmachari SK. Host-virus interaction: a new role for microRNAs. Retrovirology. 2006;3:68. doi: 10.1186/1742-4690-3-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwerk C, Adam R, Borkowski J, Schneider H, Klenk M, Zink S, Quednau N, Schmidt N, Stump C, Sagar A, Spellerberg B, Tenenbaum T, Koczan D, Klein-Hitpass L, Schroten H. In vitro transcriptome analysis of porcine choroid plexus epithelial cells in response to Streptococcus suis: release of pro-inflammatory cytokines and chemokines. Microbes Infect. 2011;13:953–962. doi: 10.1016/j.micinf.2011.05.012. [DOI] [PubMed] [Google Scholar]
- Searle SM, Gilbert J, Iyer V, Clamp M. The otter annotation system. Genome Res. 2004;14:963–970. doi: 10.1101/gr.1864804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serao NV, Matika O, Kemp RA, Harding JC, Bishop SC, Plastow GS, Dekkers JC. Genetic analysis of reproductive traits and antibody response in a PRRS outbreak herd. J Anim Sci. 2014;92:2905–2921. doi: 10.2527/jas.2014-7821. [DOI] [PubMed] [Google Scholar]
- Sharbati S, Friedlander MR, Sharbati J, Hoeke L, Chen W, Keller A, Stahler PF, Rajewsky N, Einspanier R. Deciphering the porcine intestinal microRNA transcriptome. BMC Genomics. 2010;11:275. doi: 10.1186/1471-2164-11-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay T, Kang J. Immunological genome project and systems immunology. Trends Immunol. 2013;34:602–609. doi: 10.1016/j.it.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen-Orr SS, Tibshirani R, Khatri P, Bodian DL, Staedtler F, Perry NM, Hastie T, Sarwal MM, Davis MM, Butte AJ. Cell type-specific gene expression differences in complex tissues. Nat Methods. 2010;7:287–289. doi: 10.1038/nmeth.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalsky RL, Cullen BR. Viruses, microRNAs, and host interactions. Annu Rev Microbiol. 2010;64:123–141. doi: 10.1146/annurev.micro.112408.134243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovgaard K, Mortensen S, Boye M, Hedegaard J, Heegaard PM (2010) Hepatic gene expression changes in pigs experimentally infected with the lung pathogen Actinobacillus pleuropneumoniae as analysed with an innate immunity focused microarray. Innate Immun 16:343–353 [DOI] [PubMed]
- Skovgaard K, Cirera S, Vasby D, Podolska A, Breum SO, Durrwald R, Schlegel M, Heegaard PM. Expression of innate immune genes, proteins and microRNAs in lung tissue of pigs infected experimentally with influenza virus (H1N2) Innate Immun. 2013;19:531–544. doi: 10.1177/1753425912473668. [DOI] [PubMed] [Google Scholar]
- Smith SH, Wilson AD, Van Ettinger I, MacIntyre N, Archibald AL, Ait-Ali T. Down-regulation of mechanisms involved in cell transport and maintenance of mucosal integrity in pigs infected with Lawsonia intracellularis. Vet Res. 2014;45:55. doi: 10.1186/1297-9716-45-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits M, Schokker D. Host–pathogen interactions. In: te Pas MFW, Woelders H, Bannink A, editors. Systems biology and livestock science. New York: Wiley; 2011. pp. 247–276. [Google Scholar]
- Solinhac R, Mompart F, Martin P, Robelin D, Pinton P, Iannuccelli E, Lahbib-Mansais Y, Oswald IP, Yerle-Bouissou M (2011) Transcriptomic and nuclear architecture of immune cells after LPS activation. Chromosoma 120:501–520 [DOI] [PubMed]
- Sorg D, Danowski K, Korenkova V, Rusnakova V, Kuffner R, Zimmer R, Meyer HH, Kliem H. Microfluidic high-throughput RT-qPCR measurements of the immune response of primary bovine mammary epithelial cells cultured from milk to mastitis pathogens. Animal. 2013;7:799–805. doi: 10.1017/S1751731112002315. [DOI] [PubMed] [Google Scholar]
- Spurgeon SL, Jones RC, Ramakrishnan R. High throughput gene expression measurement with real time PCR in a microfluidic dynamic array. PLoS One. 2008;3:e1662. doi: 10.1371/journal.pone.0001662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steibel JP, Wysocki M, Lunney JK, Ramos AM, Hu ZL, Rothschild MF, Ernst CW. Assessment of the swine protein-annotated oligonucleotide microarray. Anim Genet. 2009;40:883–893. doi: 10.1111/j.1365-2052.2009.01928.x. [DOI] [PubMed] [Google Scholar]
- Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, Hanfland RA, Wohlford-Lenane C, Dohrn CL, Bartlett JA, Nelson GAT, Chang EH, Taft PJ, Ludwig PS, Estin M, Hornick EE, Launspach JL, Samuel M, Rokhlina T, Karp PH, Ostedgaard LS, Uc A, Starner TD, Horswill AR, Brogden KA, Prather RS, Richter SS, Shilyansky J, McCray PB, Jr., Zabner J, Welsh MJ (2010) Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med 2: 29ra31 [DOI] [PMC free article] [PubMed]
- Suzuki S, Iwamoto M, Saito Y, Fuchimoto D, Sembon S, Suzuki M, Mikawa S, Hashimoto M, Aoki Y, Najima Y, Takagi S, Suzuki N, Suzuki E, Kubo M, Mimuro J, Kashiwakura Y, Madoiwa S, Sakata Y, Perry AC, Ishikawa F, Onishi A. Il2rg gene-targeted severe combined immunodeficiency pigs. Cell Stem Cell. 2012;10:753–758. doi: 10.1016/j.stem.2012.04.021. [DOI] [PubMed] [Google Scholar]
- Szeto CY, Lin CH, Choi SC, Yip TT, Ngan RK, Tsao GS, Li Lung M. Integrated mRNA and microRNA transcriptome sequencing characterizes sequence variants and mRNA-microRNA regulatory network in nasopharyngeal carcinoma model systems. FEBS Open Bio. 2014;4:128–140. doi: 10.1016/j.fob.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Consortium Project (2011) Standards, Guidelines and Best Practices for RNA-Seq
- The ENCODE Consortium Project A user’s guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timoneda O, Nunez-Hernandez F, Balcells I, Munoz M, Castello A, Vera G, Perez LJ, Egea R, Mir G, Cordoba S, Rosell R, Segales J, Tomas A, Sanchez A, Nunez JI. The role of viral and host microRNAs in the Aujeszky’s disease virus during the infection process. PLoS One. 2014;9:e86965. doi: 10.1371/journal.pone.0086965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas A, Fernandes LT, Sanchez A, Segales J. Time course differential gene expression in response to porcine circovirus type 2 subclinical infection. Vet Res. 2010;41:12. doi: 10.1051/vetres/2009060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuggle CK, Wang Y, Couture O. Advances in swine transcriptomics. Int J Biol Sci. 2007;3:132–152. doi: 10.7150/ijbs.3.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuggle CK, Bearson SM, Uthe JJ, Huang TH, Couture OP, Wang YF, Kuhar D, Lunney JK, Honavar V. Methods for transcriptomic analyses of the porcine host immune response: application to Salmonella infection using microarrays. Vet Immunol Immunopathol. 2010;138:280–291. doi: 10.1016/j.vetimm.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuggle CK, Towfic F, Honavar VG. Introduction to systems biology for animal scientists. In: te Pas MFW, Woelders H, Bannink A, editors. Systems biology and livestock science. New York: Wiley; 2011. pp. 1–30. [Google Scholar]
- Uddin MJ, Cinar MU, Grosse-Brinkhaus C, Tesfaye D, Tholen E, Juengst H, Looft C, Wimmers K, Phatsara C, Schellander K. Mapping quantitative trait loci for innate immune response in the pig. Int J Immunogenet. 2011;38:121–131. doi: 10.1111/j.1744-313X.2010.00985.x. [DOI] [PubMed] [Google Scholar]
- Uddin MJ, Nuro-Gyina PK, Islam MA, Tesfaye D, Tholen E, Looft C, Schellander K, Cinar MU. Expression dynamics of Toll-like receptors mRNA and cytokines in porcine peripheral blood mononuclear cells stimulated by bacterial lipopolysaccharide. Vet Immunol Immunopathol. 2012;147:211–222. doi: 10.1016/j.vetimm.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin MJ, Kaewmala K, Tesfaye D, Tholen E, Looft C, Hoelker M, Schellander K, Cinar MU. Expression patterns of porcine Toll-like receptors family set of genes (TLR1-10) in gut-associated lymphoid tissues alter with age. Res Vet Sci. 2013;95:92–102. doi: 10.1016/j.rvsc.2013.01.027. [DOI] [PubMed] [Google Scholar]
- Uthe JJ, Wang Y, Qu L, Nettleton D, Tuggle CK, Bearson SM. Correlating blood immune parameters and a CCT7 genetic variant with the shedding of Salmonella enterica serovar Typhimurium in swine. Vet Microbiol. 2009;135:384–388. doi: 10.1016/j.vetmic.2008.09.074. [DOI] [PubMed] [Google Scholar]
- Valdmanis PN, Roy-Chaudhuri B, Kim HK, Sayles LC, Zheng Y, Chuang CH, Caswell DR, Chu K, Zhang Y, Winslow MM, Sweet-Cordero EA, Kay MA (2013) Upregulation of the microRNA cluster at the Dlk1-Dio3 locus in lung adenocarcinoma. Oncogene. doi:10.1038/onc.2013.523 [DOI] [PMC free article] [PubMed]
- Van Reeth K, Labarque G, Nauwynck H, Pensaert M. Differential production of proinflammatory cytokines in the pig lung during different respiratory virus infections: correlations with pathogenicity. Res Vet Sci. 1999;67:47–52. doi: 10.1053/rvsc.1998.0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters EM, Wolf E, Whyte JJ, Mao J, Renner S, Nagashima H, Kobayashi E, Zhao J, Wells KD, Critser JK, Riley LK, Prather RS. Completion of the swine genome will simplify the production of swine as a large animal biomedical model. BMC Med Genomics. 2012;5:55. doi: 10.1186/1755-8794-5-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Qu L, Uthe JJ, Bearson SM, Kuhar D, Lunney JK, Couture OP, Nettleton D, Dekkers JC, Tuggle CK. Global transcriptional response of porcine mesenteric lymph nodes to Salmonella enterica serovar Typhimurium. Genomics. 2007;90:72–84. doi: 10.1016/j.ygeno.2007.03.018. [DOI] [PubMed] [Google Scholar]
- Wang Y, Couture OP, Qu L, Uthe JJ, Bearson SM, Kuhar D, Lunney JK, Nettleton D, Dekkers JC, Tuggle CK. Analysis of porcine transcriptional response to Salmonella enterica serovar Choleraesuis suggests novel targets of NFkappaB are activated in the mesenteric lymph node. BMC Genomics. 2008;9:437. doi: 10.1186/1471-2164-9-437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Luo YR, Fu WX, Lu X, Zhou JP, Ding XD, Liu JF, Zhang Q. Genome-wide association studies for hematological traits in swine. Anim Genet. 2012;44:34–43. doi: 10.1111/j.1365-2052.2012.02366.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu C, Fang Y, Liu X, Li W, Liu S, Liu Y, Charreyre C, Audonnet JC, Chen P, He Q. Transcription analysis on response of porcine alveolar macrophages to Haemophilus parasuis. BMC Genomics. 2012;13:68. doi: 10.1186/1471-2164-13-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei N, Pang W, Wang Y, Xiong Y, Xu R, Wu W, Zhao C, Yang G. Knockdown of PU.1 mRNA and AS lncRNA regulates expression of immune-related genes in zebrafish Danio rerio. Dev Comp Immunol. 2014;44:315–319. doi: 10.1016/j.dci.2014.01.015. [DOI] [PubMed] [Google Scholar]
- Wilkinson JM, Sargent CA, Galina-Pantoja L, Tucker AW. Gene expression profiling in the lungs of pigs with different susceptibilities to Glasser’s disease. BMC Genomics. 2010;11:455. doi: 10.1186/1471-2164-11-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson JM, Dyck MK, Dixon WT, Foxcroft GR, Dhakal S, Harding JC. Transcriptomic analysis identifies candidate genes and functional networks controlling the response of porcine peripheral blood mononuclear cells to mitogenic stimulation. J Anim Sci. 2012;90:3337–3352. doi: 10.2527/jas.2012-5167. [DOI] [PubMed] [Google Scholar]
- Wu Z, Irizarry RA (2004) Preprocessing of oligonucleotide array data. Nat Biotechnol 22:656–658; author reply 658 [DOI] [PubMed]
- Wu YQ, Chen DJ, He HB, Chen DS, Chen LL, Chen HC, Liu ZF. Pseudorabies virus infected porcine epithelial cell line generates a diverse set of host microRNAs and a special cluster of viral microRNAs. PLoS One. 2012;7:e30988. doi: 10.1371/journal.pone.0030988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki M, Chen H, Steibel JP, Kuhar D, Petry D, Bates J, Johnson R, Ernst CW, Lunney JK. Identifying putative candidate genes and pathways involved in immune responses to porcine reproductive and respiratory syndrome virus (PRRSV) infection. Anim Genet. 2012;43:328–332. doi: 10.1111/j.1365-2052.2011.02251.x. [DOI] [PubMed] [Google Scholar]
- Xiao S, Jia J, Mo D, Wang Q, Qin L, He Z, Zhao X, Huang Y, Li A, Yu J, Niu Y, Liu X, Chen Y. Understanding PRRSV infection in porcine lung based on genome-wide transcriptome response identified by deep sequencing. PLoS One. 2010;5:e11377. doi: 10.1371/journal.pone.0011377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Mo D, Wang Q, Jia J, Qin L, Yu X, Niu Y, Zhao X, Liu X, Chen Y. Aberrant host immune response induced by highly virulent PRRSV identified by digital gene expression tag profiling. BMC Genomics. 2010;11:544. doi: 10.1186/1471-2164-11-544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie SS, Li XY, Liu T, Cao JH, Zhong Q, Zhao SH. Discovery of porcine microRNAs in multiple tissues by a Solexa deep sequencing approach. PLoS One. 2011;6:e16235. doi: 10.1371/journal.pone.0016235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KC, Yamada KA, Patel AY, Topkara VK, George I, Cheema FH, Ewald GA, Mann DL, Nerbonne JM (2014) Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNA in failing human heart and remodeling with mechanical circulatory support. Circulation. doi:10.1161/ciculationaha.113.003863 [DOI] [PMC free article] [PubMed]
- Ye L, Su X, Wu Z, Zheng X, Wang J, Zi C, Zhu G, Wu S, Bao W. Analysis of differential miRNA expression in the duodenum of Escherichia coli F18-sensitive and -resistant weaned piglets. PLoS One. 2012;7:e43741. doi: 10.1371/journal.pone.0043741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Zheng L, Li Y, Li C, Ma C, Li X, Hao P. A cross-species analysis method to analyze animal models’ similarity to human’s disease state. BMC Syst Biol. 2012;6(Suppl 3):S18. doi: 10.1186/1752-0509-6-S3-S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JF, Zhang SJ, Jafer O, Furlong RA, Chausiaux OE, Sargent CA, Zhang GH, Affara NA. Global transcriptional response of pig brain and lung to natural infection by Pseudorabies virus. BMC Microbiol. 2009;9:246. doi: 10.1186/1471-2180-9-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Hopwood P, Abrams CC, Downing A, Murray F, Talbot R, Archibald A, Lowden S, Dixon LK (2006) Macrophage transcriptional responses following in vitro infection with a highly virulent African swine fever virus isolate. J Virol 80:10514–10521 [DOI] [PMC free article] [PubMed]
- Zhang H, Guo X, Nelson E, Christopher-Hennings J, Wang X. Porcine reproductive and respiratory syndrome virus activates the transcription of interferon alpha/beta (IFN-alpha/beta) in monocyte-derived dendritic cells (Mo-DC) Vet Microbiol. 2012;159:494–498. doi: 10.1016/j.vetmic.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao SH, Recknor J, Lunney JK, Nettleton D, Kuhar D, Orley S, Tuggle CK. Validation of a first-generation long-oligonucleotide microarray for transcriptional profiling in the pig. Genomics. 2005;86:618–625. doi: 10.1016/j.ygeno.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Zhao M, Liu XD, Li XY, Chen HB, Jin H, Zhou R, Zhu MJ, Zhao SH. Systems infection biology: a compartmentalized immune network of pig spleen challenged with Haemophilus parasuis. BMC Genomics. 2013;14:46. doi: 10.1186/1471-2164-14-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Zhai S, Zhou X, Lin P, Jiang T, Hu X, Jiang Y, Wu B, Zhang Q, Xu X, Li JP, Liu B. Molecular characterization of transcriptome-wide interactions between highly pathogenic porcine reproductive and respiratory syndrome virus and porcine alveolar macrophages in vivo. Int J Biol Sci. 2011;7:947–959. doi: 10.7150/ijbs.7.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Z, Cui H, Li M, Peng X, Zhu L, Zhang M, Ma J, Xu Z, Gan M, Deng J, Li X, Fang J. Transcriptional profiling of swine lung tissue after experimental infection with Actinobacillus pleuropneumoniae. Int J Mol Sci. 2013;14:10626–10660. doi: 10.3390/ijms140510626. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.