

Abstract
Seasonal and novel influenza infections have the potential to cause worldwide pandemics. In order to properly treat infected patients and to limit its spread, a rapid, accurate and automatic influenza diagnostic tool needs to be developed. This study therefore presents a new integrated microfluidic system for the rapid detection of influenza infections. It integrated a suction-type, pneumatic-driven microfluidic control module, a magnetic bead-based fluorescent immunoassay (FIA) and an end-point optical detection module. This new system can successfully distinguish between influenza A and B using a single chip test within 15 min automatically, which is faster than existing devices. By utilizing the micromixers to thoroughly wash out the sputum-like mucus, this microfluidic system could be used for the diagnosis of clinical specimens and reduced the required sample volume to 40 μL. Furthermore, the results of diagnostic assays from 86 patient specimens have demonstrated that this system has 84.8 % sensitivity and 75.0 % specificity. This developed system may provide a powerful platform for the fast screening of influenza infections.
Electronic supplementary material
The online version of this article (doi:10.1007/s10544-013-9753-0) contains supplementary material, which is available to authorized users.
Keywords: Microfluidics, Magnetic bead, Rapid detection, Influenza, Clinical specimen
Introduction
Viral infectious diseases have caused a few worldwide pandemics. For instance, notorious viral infections include acquired immunodeficiency syndrome (AIDS) which is caused by the human immunodeficiency virus (HIV), severe acute respiratory syndrome (SARS) which is caused by the SARS coronavirus (SARS-CoV) and influenza due to the influenza virus. Even hepatic cancer has been proven to be related to a hepatitis B virus (HBV) infection. Among them, the influenza virus is a type of rapidly transmitted virus, infecting the upper respiratory tract and sometimes causing acute viral respiratory illness. Unlike most viruses, influenza infects all-age groups and recurs in many former patients. Furthermore, influenza epidemics show abrupt and rapid infections throughout a population with a high infection rate, especially in school-aged children.
Influenza viruses are enveloped ribonucleic acid (RNA) viruses and can be generally classified into three types, namely influenza viruses A, B and C. The majority of respiratory diseases in human beings are caused by influenza viruses A and B. The influenza virus A can be further subtyped by two different expressed surface proteins, hemagglutinin (HA) and neuraminidase (NA). Various subtypes of influenza virus A infections have caused severe pandemics. For example, a H1N1 pandemic happened in 1918 and is commonly known as the Spanish Flu (Johnson and Mueller 2002), a H2N2 pandemic in 1957 is known as the Asian Flu (Kawaoka et al. 1989) and a H3N2 pandemic in 1968 is known as the Hong Kong Flu (Both et al. 1983). Most new subtypes of influenza virus emerge and can lead to high mortality pandemics, such as the Hong Kong Flu in 1968–1969 which caused about one million deaths worldwide (Horimoto and Kawaoka 2005). Additionally, in early 2009, the novel influenza A (H1N1) virus, containing RNAs from human, pig and avian flu viruses (Medina and Garcia-Sastre 2011), emerged and made people realized that even a well-known seasonal influenza virus could develop into another unpredictable serious disease. For public health, it is therefore important to develop a rapid and accurate diagnostic method (or kit) for preventing the worldwide spread of these viral strains.
There are four main types of influenza diagnostic tests: virus culture, serological detection, viral nucleic acid detection and viral antigen detection. A virus culture is classically accepted as a “gold-standard” for the diagnosis of an influenza virus. However, it is a very time-consuming and labor-intensive process, often requiring 2 to 14 days for well-trained personnel (Kaiser et al. 1999; Woo et al. 1997). Serological detection methods for the diagnosis of influenza infection are traditionally conducted by performing a complement-binding reaction (CBR), a hemagglutination inhibition (HI) test, an enzyme immunoassay (EIA) or a neutralization test (Cox and Subbarao 1999). It requires less time for the treatment of clinical samples, when compared to a virus culture, thus making serological diagnosis methods more valuable for epidemiological research or for the clarification of influenza infection outbreaks (Espmark and Salenstedt 1961). Alternatively, molecular diagnosis based on a reverse transcription polymerase chain reaction (RT-PCR) is a very sensitive method with a high specificity. Nevertheless, polymerase chain reaction (PCR) is the most expensive method for influenza diagnosis and the diagnosis time can take from a couple of hours to up to 1 day (Atmar et al. 1996; Poon et al. 2009; McGeer 2009).
Methods of virus antigen detection include the immunofluorescence test (IFT) or the well-known enzyme-linked immunosorbent assay (ELISA), which is most commonly used by hospital medical departments for clinical diagnosis or in the research laboratory. ELISA can be performed faster than the other three types of influenza diagnostic tests (Zheng et al. 1990; Siavashi et al. 2005). However, the relatively time-consuming and labor-intensive protocols which require highly skilled operators are always the drawbacks of these diagnostic methods. Recently, microfluidic systems have been demonstrated as a rapid and automatic detection platform for diagnosis. For example, the on-chip fluorescence imaging for ELISA using a charge-coupled device (CCD) camera and a microscope for detection was reported to successfully analyze target immunoglobulin M (IgM) at 17-nM concentrations (Eteshola and Balberg 2004). Furthermore, a surface-modified microfluidic system to perform rapid ELISA was demonstrated for the detection of the dengue virus in 30 min (Weng et al. 2011a). Recently, bead-based immunoassays have further been integrated into microfluidic systems to enhance their performance. Because of the high surface-to-volume ratio and the fact that the surface of these microbeads can be selectively modified with a variety of detection targets, the sensitivity and applicability of these bead-based immunoassays can be greatly improved. These microbeads can be further easily manipulated, sorted, and concentrated in the microfluidic systems. Therefore, more bead-based assays have been reported recently on microfluidic systems (Vignali 2000; Choi et al. 2001; Fernández-Lafuente and Guisán 2006; Yang et al. 2008; Liu et al. 2009). In this study, rapid detection of influenza virus infection using an integrated microfluidic system will be explored.
Currently, many rapid diagnostic kits for influenza infections have already been developed and applied in hospitals by clinicians for treatment decisions and infection control practices, such as BDTM Directigen EZ Flu A+B test (Becton Dickinson, Franklin Lakes, NJ, USA), BinaxNOW Influenza A&B (Binax, Inc., Scarborough, Maine) and QuickVue Influenza A+B Test (Quidel Corporation, San Diego, California). Previous studies with rapid test kits showed that the sensitivity of the broad influenza tests ranged from 25 to 100 % and the range of selectivity were from 51 to 100 % (Gavin and Thomson 2003). However, some tests showed extremely low sensitivities of only 10 % but with a selectivity of 100 % (Hurt et al. 2007). Moreover, recent research regarding the detection of the influenza A/H1N1 virus using clinical specimens have shown sensitivity over a broad range from 10 to 69 % (Drexler et al. 2009; Faix et al. 2009; Ginocchio et al. 2009; Vasoo et al. 2009; Tsao et al. 2011). Most of the existing rapid tests for influenza infections do not show reliable detection sensitivity in clinical or laboratory tests. In order to accurately diagnose and treat influenza infected patients efficiently, a well-designed microfluidic system is thus in great need.
Advancements in microfluidics have attracted considerable interest and made substantial impacts on the development of compact, in-vitro diagnosis devices. Furthermore, the nucleoprotein (NP) were used to define the influenza serotypes A, B and C and showed much more stable than HA and NA. Thus a specific monoclonal antibody (mAbs) to influenza NP was useful for viral detection (de Boer et al. 1990). For instance, our group has reported an integrated microfluidic system for the rapid detection of the influenza A virus and successfully combined a three-dimensional (3D) magnetic-bead-based FIA and an optical detection module. It demonstrated that an efficient micromixer could enhance the interaction between the targets and surface modified magnetic beads (Lien et al. 2011). However, it could only perform one detection at a time and only influenza A virus detection was demonstrated. Based on our previous results, a microfluidic system equipped with four detection regions has been developed in this study. Different modified specific mAbs and fluorescence dye for influenza A and B can lead to successful detection within 15 min. Furthermore, 86 patient specimens have been tested. Experimental data showed that 84.8 % sensitivity and 75.0 % specificity can be achieved.
Materials and methods
Working principle and experimental procedure
A magnetic bead-based FIA has been developed, characterized, and applied for detection of influenza infections in a microfluidic system. For this diagnostic process, a newly custom-developed mAb (detailed in section 2.3) was first tested in the microfluidic system using optical detection. The developed mAb was directly tagged with a fluorescent dye that can offer a straightforward method to detect the antigen in patient samples. By utilizing this direct conjugated-R-Phycoerythrin (PE) developed mAb, detection of the influenza virus present in clinical specimens was feasible with fewer incubation steps and furthermore secondary antibody cross-reactions can be avoided.
A schematic illustration of the assay used in this study is schematically shown in Fig. 1. The clinical specimen, the washing buffer, the positive/negative developed mAbs and the specific mouse anti-influenza NP-A mAb-PE or anti-NP-B mAb-PE were first loaded into the sample loading chamber, washing buffer chambers, positive/negative developing mAb chambers and the influenza A/or B developing mAb chambers, respectively. Next, the magnetic beads coated with anti-NP-A/or B mAb were loaded into the detection and incubation chambers. The test sample was transported from the sample loading chamber to the incubation chambers by the transportation units. After transport of the test sample, an initial incubation process was engaged to mix the clinical sample with surface modified-mAbs magnetic beads for 5 min to capture influenza viral particles, as shown in Fig. 1a. After incubation, viral particles were attached onto the mAbs-modified magnetic beads forming bead-virus complexes. As shown in Fig. 1b, the magnetic bead-virus complexes were purified and collected with a magnet, and then the vacuum source, driven by the digital microfluidic control module, suctioned away all other unwanted particles, debris or mucous in the testing samples. Then the washing buffer was transported from the washing buffer chambers into the incubation chambers and gently mixing for 10 s to remove all the non-specific bound materials with the intention that the magnetic bead-virus complexes were completely purified from the clinical samples. Then, as shown in Fig. 1c, the positive/negative custom-developed mAbs and the anti-NP-A/or B mAb-PE were first loaded into the positive/negative mAb chambers and the anti-NP-A/or NP-B mAb-PE chambers, which were then transported into the incubation/detection chambers and incubated with the bead-virus complexes for 5 min. Then the magnetic bead-virus-mAb-PE complexes were formed. Because of the direct-conjugated PE fluorescent dye used in this study, there was no need to inject extra dyes. This automatic purification, collection and buffer washing processes were all driven by the vacuum source connected to the microfluidic control module. Finally, after completely washing out all the unbound mAb-PE, the magnetic bead-virus-mAb-PE complexes were concentrated on the button of the detection chamber where the fluorescence signal was excited and detected by the peripheral optical detection module, as shown in Fig. 1d, Likewise, the working process of positive and negative control testing is similar to virus testing by using different development Abs. The schematic illustration for the positive and negative testing can be found in Supplemental Information (SI) Figure 1. Furthermore, all the on-chip experimental procedures about the immunomagnetic bead-based microfluidic assay, including the reaction sample volume, the reaction time and the operating conditions of this microfluidic system are listed in Table 1.
Fig. 1.
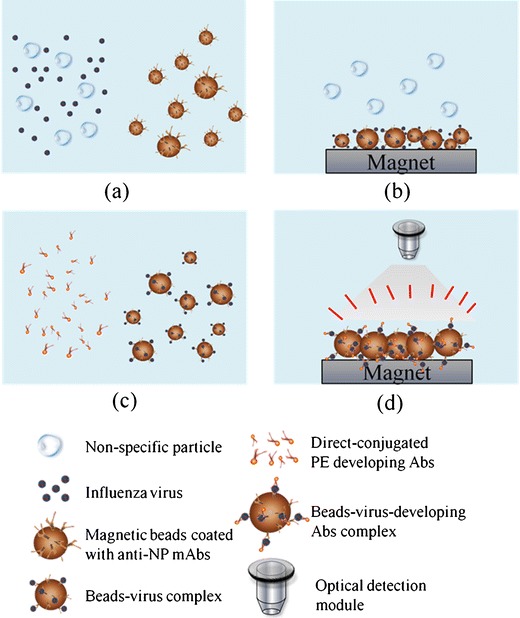
A schematic diagram of the immunomagnetic bead-based process used in this study. a Mix clinical sample and surface-modified mAbs magnetic beads in the mixing chamber, b collect the magnetic bead-virus complexes by using a magnet placed underneath the chip and wash out the non-specific materials, c transport the anti-NP-A/or B mAb-PE into the mixing chamber and mix with the magnetic bead-virus complexes, d collect the magnetic bead-virus-mAb-PE complexes by using the magnet, wash and detect the signals with optical detection module
Table 1.
Details of the experimental protocol performed on the microfluidic chip
| Step | Operating process | Sample volume | On-chip operating condition | Reaction time |
|---|---|---|---|---|
| I | Load the mAb-conjugated magnetic beads into the sample loading chamber | 10 μL | ||
| Load clinical specimen into the sample loading chamber | 80 μL | |||
| Load the washing buffer into the washing buffer chamber | 100 μL | |||
| Load the positive/negative developed mAbs into the positive/negative developing mAb chambers | 20 μL | |||
| Load the anti-NP-A/or B mAb-PE into the influenza A/or B developing mAb chambers | 20 μL | |||
| II | Mix the sample with the mAb-magnetic beads by using the micro-mixer | −70 kPa and 4.0 Hz for the micro-mixer | 5 min | |
| III | Attach the permanent magnet onto the bottom surface of the mixing chamber to attract the well-mixed magnetic complexes. | 30 s | ||
| Suck all the other substances in the biological solution away into the waste chamber using the vacuum pump | −70 kPa for the vacuum pump | 5 s | ||
| Pump the washing buffer into the mixing chamber and re-suspend the magnetic complexes in 20 μL | 20 μL | −70 kPa for sample transport unit | ||
| IV | Attach the permanent magnet onto the bottom surface of the mixing chamber to attract the re-suspended magnetic complexes | 30 s | ||
| Suck all the other substances in the sample away into the waste chamber using the vacuum pump | −70 kPa for the vacuum pump | 5 s | ||
| V | Pump the anti-NP-A/or B mAb-PE into the mixing chamber | 20 μL | −70 kPa for sample transport unit | |
| Mix the magnetic complexes with anti-NP-A/or B mAb-PE by using the micro-mixer. | −70 kPa and 4.0 Hz for the micro-mixer | 5 min | ||
| VI | Attach the permanent magnet onto the bottom surface of the mixing chamber to attract the well-mixed magnetic complexes | 30 s | ||
| Suck all the other substances in the biological solution away into the waste chamber using the vacuum pump | −70 kPa for the vacuum pump | 5 s | ||
| Pump the washing buffer into the mixing chamber and re-suspend the magnetic complexes in 20 μL | 20 μL | −70 kPa for sample transport unit | ||
| VII | Attach the permanent magnet onto the bottom surface of the mixing chamber to attract the re-suspended magnetic complexes | 30 s | ||
| Suck all the other substances in the bio-sample away into the waste chamber using the vacuum pump | −70 kPa for the vacuum pump | 5 s | ||
| VIII | Attach the permanent magnet onto the bottom surface of the mixing chamber to attract the re-suspended magnetic complexes | 30 s | ||
| Suck all the other substances in the bio-sample away into the waste chamber by the vacuum pump | −70 kPa for the vacuum pump | 5 s | ||
| IX | Pump the PBS buffer into the mixing chamber to complete the FIA process | 10 μL | −70 kPa for sample transport unit | |
| Analyze the optical signals of the magnetic complexes by utilizing the optical detection module | 5 s |
Chip design and fabrication
In order to apply this integrated microfluidic system for clinical diagnosis in patients, this chip is composed of four regions which have the positive control, the negative control, and detection of influenza A and influenza B. The test references (positive and negative controls) are the critical indexes for a clinical trial. Therefore, this type of “four-in-one” multiplexed detection area design will allow technicians in a clinical laboratory to use a single chip to confirm if the patient is infected or not. Thus, this chip design has the potential to work as a rapid test kit for influenza.
The integrated microfluidic chip consisted of four layers including three layers of polydimethylsiloxane (PDMS) structures and one glass substrate, as schematically shown in Fig. 2a. Two of the three PDMS layers were thin layers with a thickness of 150 μm. One of them was a liquid channel layer (indicated as purple color layer in Fig. 2a), and another was used for air barriers and gates (green color layer in Fig. 2a) because the multiplexed detection area required many interlaced air-control channels. This integrated microfluidic system worked by utilizing one thick layer for air channels (blue color layer in Fig. 2a). Figure 2b shows a photograph of the developed microfluidic chip. The dimensions of the chip were measured to be 6.7 cm × 5.5 cm. In addition, this microfluidic chip was composed of several micro-devices such as normally-closed microvalves (Yang et al. 2009a), suction-type sample transportation units (Weng et al. 2011b) and vortex-type micromixers (Yang et al. 2009b) which were all integrated into the system to automate the entire assay process. The normally-closed microvalves were integrated with suction-type sample transportation units, thus the pumping rate could be significantly improved. The working principle of the sample transportation units can be briefly described as follows. First, the normally-closed PDMS membranes were deflected upwards by the vacuum force in the air channels and the liquid samples were drawn into the central transportation unit PDMS membrane. Second, the PDMS membrane of the left normally-closed microvalve was released as the right microvalve was deflected and opened such that the sample could be pushed forward. Finally, all the PDMS membranes of the central transportation unit and right normally-closed microvalve were released to push the liquid sample into the right channel. Furthermore, the vortex-type micromixers were also driven by the vacuum force to mix the liquid samples with the magnetic microbeads. The PDMS membranes of the micromixer were deflected on and released sequentially, and then the vortex-type flow force would be generated in the circular micromixer chamber (the control systems will be described in section 2.5). Figure 2c shows a schematic illustration of the microfluidic chip, which was composed of four detection regions for the positive control, the negative control, influenza A and influenza B, respectively.
Fig. 2.
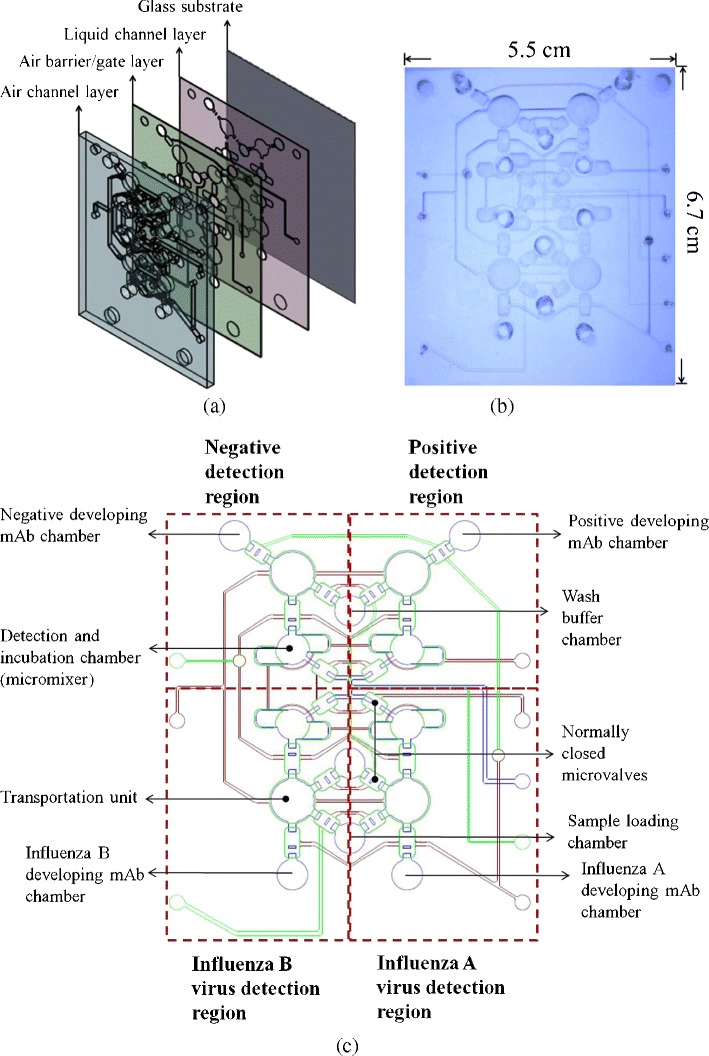
a An exploded view of the chip consisting of three PDMS layers and one glass substrate; the liquid channel layer, the air barrier/gate layer and the air channel layer are indicated with purple, green, and blue colors, respectively; b A photograph of the microfluidic chip with dimensions of 6.7 cm × 5.5 cm; c A schematic illustration of the microfluidic chip containing several normally-closed microvalves, four suction-type sample transportation units and four vortex-type micromixers for both sample incubation and detection
The fabrication of the microfluidic system utilized a computer numerical control (CNC) machining process and a PDMS replication process. In brief, the micro-structured master molds on a polymethylmethacrylate (PMMA) plate was first formed by a CNC machine (EGX-400, Roland Inc., Japan) with a 0.5-mm drill bit. The rotational rate and feed speed of the drill bit were 26,000 rpm and 7 mm/s, respectively. After the PMMA master molds were fabricated, PDMS (Sylgard 184A/B, Sil-More Industrial Ltd., USA) casting process was used to replicate inversed images, thus forming three PDMS layers (Wang and Lee 2006). Finally, the three PDMS layers and a glass substrate were bonded together by an oxygen plasma treatment to form the integrated microfluidic chip. Details of the microfabrication procedure can be referenced in our previous work (Yang et al. 2008).
Materials and the magnetic bead-coating process
This microfluidic system showed its specificity to capture different types of influenza virus by utilizing the specific mouse anti-NP-A mAb (H16L-10-4R5 cell line (HB-65), ATCC, USA) and mouse anti-NP-B mAb (influenza B nucleoprotein (B017), GeneTex, USA), respectively. They were conjugated onto the surface of magnetic beads to form anti-NP-A/or B mAb-conjugated magnetic beads. Furthermore, the process for directly conjugating the PE fluorescent dye to these custom-developed mAbs was conducted by a commercial kit-EasyLink R-Phycoerythrin Conjugation Kit (Abcam, UK). Alexa Fluor® 488 Goat Anti-Mouse IgG (H+L) (Invitrogen, USA) was used as the positive custom-developed mAbs and anti-Core Dengue virus2 (DV2) antibody-PE (provided by the Microbiology and Immunology Laboratory, NCKU) was used as the negative custom-developed mAbs, which also conjugate with the PE fluorescent dye by using the EasyLink R-Phycoerythrin Conjugation Kit. Therefore, two types of influenza virus can be accurately detected optically.
Magnetic beads (stock concentration = 4 × 108 beads/mL, beads diameter = 4.5 μm, Dynabeads® M-450 Epoxy, Invitrogen, USA) with surfaces covered with epoxy groups were treated with the anti-NP-A/or B mAb. The epoxy group on the surface of the beads were used to couple to specific mAb. The magnetic beads were conjugated to mAb via the following process: 5 μg of anti-NP-A/or B mAbs were conjugated with 2 μL of magnetic beads (1 × 107 beads) in a phosphate-buffered saline (1 × PBS, Invitrogen, USA) solution with a total volume of 1,000 μL for overnight incubation (more than 16 h) at 4 °C. Then the mAb-coated magnetic beads were washed, purified and re-suspended in 1,000 μL of PBS. In order to prevent non-specific binding in the subsequent immunological steps, the mAb-coated magnetic beads were blocked by using a blocking solution of (1xPBS with 1 % (w/v) bovine serum albumin (BSA, Sigma, USA)) at 4 °C for 2 h. Finally, the mAb-coated magnetic beads were stored at 4 °C in 1 × PBS with 0.1 % (w/v) BSA until used in the FIA processes.
Virus particles, clinical specimens and clinical laboratory tests
Influenza A virus types, specifically influenza A/H1 (subtype of H1N1, 97N510H1) and influenza A/H3 (subtype of H3N2, 90N225H3), were used to verify the performance of the integrated microfluidic system. The initial titers of the virus stock were 128 hemagglutin units (HAU) and the titer determination method of HA testing can be found in SI section II (Vyas and Shulman 1970). The assay for influenza B virus detection used viral strain-94N399IB and the initial titers of the type B virus stock were also 128 HAU. All the tested influenza viral strains were infected in Mardin-Darby canine kidney (MDCK) cells, which were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen, USA) with 2 μg/mL of Trypsin (TPCK treated from bovine pancreas, Sigma-Aldrich, USA) at 37 °C for 2–4 days, then the propagated virus was harvested from the fluid medium by using 10,000 rpm centrifuge at 4 °C (Sugiura et al. 1972). After harvesting from the culture medium supernatant, the viruses were frozen at −80 °C until testing. Four times serial dilutions of the influenza viruses were used to determine the limit of detection (LOD). They were diluted with 4 °C-cloded 1 × PBS buffer to form various viral concentrations ranging from 1:128 with the 4−2 dilution (8 HAU/50 μL) to 1:128 with the 4−8 dilution (0.002 HAU/50 μL). In addition, two other different types of viruses including, DV serotype 2/PL046 (1.25 × 106 plaque-forming unit (PFU)/mL) and enterovirus (EV) 71/4643 (2.5 × 106 PFU/mL) were employed for the verification of the selectivity of the immunomagnetic bead-based microfluidic assay (provided and viral inactivated by the Microbiology and Immunology Laboratory, NCKU). The HA testing was used to determine the HAU titers at room temperature in a 96-well V plate (NUNC, Thermo Fisher Scientific, Roskilde, Denmark). With serial dilutions of the virus twice (25 μL) in PBS, viruses were incubated with 25 μL of 0.5 % suspension from human type O red blood cells. Agglutination was determined after the settlement of the sample for 1 h, and the HAU titers were expressed as the reciprocal of the highest dilution which caused complete agglutination (HAU/50 μL).
Clinical specimens were obtained from the Microbiology and Immunology Laboratory of NCKU and the Department of Pathology of NCKU Hospital. The specimens used in this study had been tested previously using the rapid test BDTM Directigen EZ Flu A+B test (Becton Dickinson, Franklin Lakes, NJ USA.). These specimens were stored at −80 °C until tested. A total of 86 clinical samples were collected, 20 of which were known negative samples and the other 66 samples were known positive samples. For comparison, in this study, parts of the clinical samples were analyzed with the BD Directigen test (the experimental process is shown in the SI, Figure 2 and Table 1) and the integrated microfluidic chip. Furthermore, bio-statistical analysis was performed and both the receiver-operator characteristic (ROC) curves and Youden’s index were used to decide the threshold from positive or negative signals (Wians 2009) (as shown in Supplemental Figure 3 and Table 2 in the SI).
Experimental setup and optical detection
A digital controller (8051 microcontroller, model AT89C51 24PC, ATMEL, USA), a custom-made, programmable control system, electro-magnetic valves (EMVs, SD70M-6BG-32, SMC, Japan) and an external vacuum pump (UN-90V, UNI-CROWN Inc., Taiwan) were integrated with the microfluidic system for automating the diagnosis process. This control system was used to automatically drive all the microfluidic components, including the normally-closed microvalves, the micromixers and the sample transportation unit. The driving frequencies of the EMVs were regulated such that the pumping rate of the sample transportation unit can be precisely controlled. The red ink was transported from the loading chambers to the detection chambers to calibrate the pumping rate. Detailed information about this measurement process can be found in our previous work (Lien et al. 2011). In brief, the volume of loaded red ink was known and then the working volume in each pumping cycle was measured consequently. Furthermore, the mixing performance was recorded by a high-speed CCD camera (MC1311, Mikrotron, Germany), coupled with a high-speed image acquisition interface card (INSPECTA-5, Mikrotron, Germany) and a fluorescence microscope (E-400, Nikon, Japan). The captured images were then analyzed by digital imaging techniques to calculate the mixing index (Yang et al. 2009b).
In the optical detection process, a photo-multiplier tube (PMT, R928, Hamamatsu, Japan), a mercury lamp (MODEL C-SHG1, Nikon, Japan) and a set of optical components including one collimation lens, one objective lens (Nikon LU Plan 10×/0.30 A, Nikon, Japan) and three fluorescence filters (Nikon G-2A, Nikon, Japan) for performing the microfluidic FIA were used. The results from conventional bench-top systems, including a large-scale shaker (INTELLI-MIXER, ELMI Ltd, Latvia) and a permanent magnetic concentrator (DynaMagTM-2, Invitrogen, USA) using a manual preparation protocol were used for comparison while using the same optical detection module. In brief, testing viruses were incubated with anti-NP-A/B mAb coated beads in a larger-scale shaker for 60 min. After manual washing process with 200 μL pipetman (RAININ, Anachem Ltd., United Kingdom), anti-NP-A/or B mAb-PE was used to incubate with bead-virus complexes for another 60 min in the shaker. Finally, the washed bead-virus-anti-NP-PE complexes were used for optical analysis, which used the same system as the one for the microfluidic system detection (Lien et al. 2011). A mercury lamp was used to excite the fluorescent both Alexa 488 and PE dye, and then the induced fluorescent signal was first collected through an excitation band-pass (BP) filter (535/25BP, Nikon, Japan). Next, the emitted fluorescence signals from the PE dye were passed through a dichroic mirror with a 565-nm cut-off wavelength (Nikon, Japan) and other signals were filtered out by using a 590 long-pass (LP) barrier filter (Nikon, Japan). Only signals with wavelengths greater than 590 nm could pass through and be detected by the PMT. Similar process with different filters was applied for Alexa 488 fluorescent dye. The excitation filter was 460/95BP. A dichroic mirror with a 505-nm cut-off wavelength was used and then other signals were filtered by a 510/50BP barrier filter (Nikon, Japan).
Results and discussion
Characterization of the microfluidic system
Before the integrated microfluidic system was used to detect influenza viruses, each microfluidic device including the micromixer and the micro-transportation unit was first characterized. As shown in Fig. 2c, the incubation/detection chambers were composed of micromixers which were the key units of this microfluidic chip. Therefore, the normalized concentration profile of these four air-channel-connected micromixers was first measured, as shown in Fig. 3a, where the normalization location (D+) was measured across the center of the micromixer chamber (X-X’ line). The mixing period, the applied negative pressure and the working frequency regulated by the EMVs in this test were 2 s, −60 kPa and 2.0 Hz, respectively. As shown in this figure, the micromixer can mix the red ink and deionized water completely within a short period of time. Four different micromixers integrated in this chip show very similar performance. The optimal working conditions for the micromixers were then explored by measuring the mixing performance at various driving frequencies (0.5, 1.0, 2.0 and 4.0 Hz) under −70 kPa, which was the negative pressure provided by the vacuum pump. As shown in Fig. 3b, when the driving frequency was 4.0 Hz, the mixing index could be as high as 96 % after 1 s of mixing. Note that the mixing index (σ) is defined as follows to characterize the mixing performance (Erickson and Li 2002; Yang et al. 2009b):
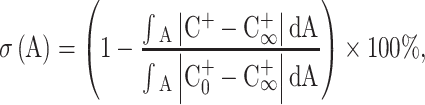 |
1 |
which σ(A) is the mixing index for the normalized concentration (C+) distributing in the area of the micromixer chamber (A).  is the normalized initial condition, as
is the normalized initial condition, as 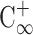 is the completely mixed condition with normalization (
is the completely mixed condition with normalization (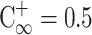 ). σ = 0 % means that sample was completely unmixed. When the sample was completely mixed, the σ was 100 %. The optimal operating conditions for the micromixer for influenza diagnosis were with a 4.0 Hz driving frequency under −70 kPa applied air pressure.
). σ = 0 % means that sample was completely unmixed. When the sample was completely mixed, the σ was 100 %. The optimal operating conditions for the micromixer for influenza diagnosis were with a 4.0 Hz driving frequency under −70 kPa applied air pressure.
Fig. 3.
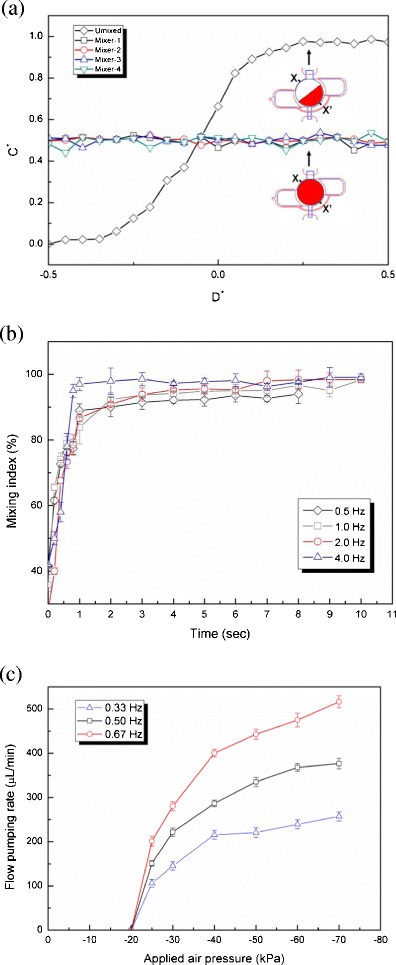
a Normalized concentrations along the X-X’ line across the center of the micromixer for the four integrated vortex-type micromixers; b The relationship between the mixing time and mixing index while operated at different driving frequencies. A mixing index of 96 % is achieved in 1 s while mixing at 4.0 Hz frequency and −70 kPa; c The relationship between the applied negative pressure and the flow pumping rate. Note that when the transportation frequency is set as 0.67 Hz (1.5 s/cycle), the flow pumping rate can be as high as 516.5 ± 13.5 μL/min at −70 kPa vacuum pressure
Next, the sample transportation unit was also characterized to determine the relationship between the applied negative pressure and the flow pumping rate, as shown in Fig. 3c. Because the transportation process required three steps to transport the liquid sample, reducing the working time of each step increased the flow pumping rate. It also increased with the applied pressure. At a negative applied pressure of −70 kPa, the flow pumping rate was as high as 516.5 ± 13.5 μL/min at a driving frequency of 0.67 Hz. The working frequency of 0.67 Hz corresponds to the case where each step only takes 0.5 s to transport the liquid samples. Therefore, a complete cycle by the transportation unit to transfer a sample was 1.5 s. As a result, the subsequent influenza detections were conducted with a 0.67 Hz (1.5 s/cycle) driving frequency under −70 kPa applied air pressure for liquid transportation.
Selectivity of anti-NP-A/or B mAb-PE
In order to test the specificity of the PE-conjugated anti A/B mAbs, two other types of viruses were used as comparison samples, including DV-2/PL046 at a concentration of 1.25 × 106 PFU/mL and EV-71/4643 at 2.5 × 106 PFU/mL. As mentioned previously, influenza A (both H1N1 and H3N2) and influenza B viruses were tested at the stock viral titer of 128 HAU/50 μL. First, the anti-NP-A mAb-PE was used to detect five different strains of viruses and the results are shown in Fig. 4. The anti-NP-A mAb-PE demonstrates its excellent selectivity to both H1N1 and H3N2 influenza viruses with extremely high optical signals, 2223.5 ± 256.2 and 2331.1 ± 169.1 mV (n = 3, p = 0.001 < 0.01), respectively, but no significant reaction with the other three viruses, as indicated by detection signals close to the negative control level (HB, DV, EV and NC were 387.1 ± 15, 313.2 ± 43, 322.7 ± 20 and 412.2 ± 31 mV, respectively, n = 3, p > 0.05). As the anti-NP-B mAb-PE were applied to detect the same five virus strains and the results are also shown in Fig. 4. Similarly, anti-NP-B mAb-PE showed its excellent selectivity with a strong signal of 2098.3 ± 166 mV (n = 3, p = 0.001 < 0.01) for the sample of influenza B virus while the other four viruses appeared no differently than the negative control signals (A/H1, A/H3, DV, EV and NC were 447.1 ± 22, 423.5 ± 11, 306.1 ± 46, 311.2 ± 39 and 400.2 ± 54 mV, respectively, n = 3, p > 0.05). With the results shown in Fig. 4, the anti-NP-A/or B mAb-PE has a high selectivity to their respective target viruses, indicating that both of them are suitable for accurate detection of influenza infections. The details of the statistical analysis could be found in the SI Table 3.
Fig. 4.
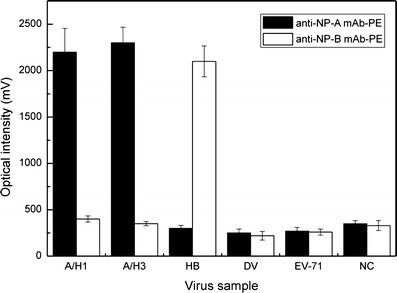
Selectivity of immunomagnetic bead-based FIA with directly conjugated PE anti-NP antibody for detection of influenza A or B infections. Testing viruses contain Influenza A virus H1N1 (97N510H1), H3N2 (90N225H3), influenza B virus (94N399IB), dengue virus (2/PL046) and enterovirus (71/4643). Black bars (■) show that the anti-NP-A mAb-PE can be used specifically for A/H1 and A/H3 (p = 0.001 < 0.01). White bars (□) shows that the anti-NP-B mAb-PE can be used specifically for HB (p = 0.001 < 0.01)
Limit of detection of the integrated microfluidic system
After verifying the selectivity of the anti-NP-A/or B mAb-PE, it was crucial to explore the LOD of the developed assay by utilizing this integrated microfluidic system. Purified viral particles were used to test the microfluidic chip. For comparison, the same assay was conducted by manually performing the protocol using bench-top equipment. Figure 5a-1 shows a series of microscopic fluorescent images of the magnetic complexes with different concentrations of influenza A viruses. At higher viral concentrations, the fluorescent intensity of PE was observed to be stronger. For comparison, the optical signals from the on-chip sample and manually-prepared sample were calculated and shown in Fig. 5a-2. The LOD for influenza A was measured to be 128 × 4−6 HAU/50 μL (about 0.031 HAU/50 μL) with an optical signal of 462.5 ± 47.7 mV (n = 3, p = 0.011 < 0.05) when utilizing the microfluidic system, which was four times more sensitive than that from the manual protocol (about 128 × 4−5 HAU/50 μL with an optical signal of 446.2 ± 37.7 mV (n = 3, p = 0.015 < 0.05)). Note that the signal for the negative control is around 336.2 ± 27.7 mV. As the concentration of the influenza virus A increased, the optical signals would increase accordingly.
Fig. 5.
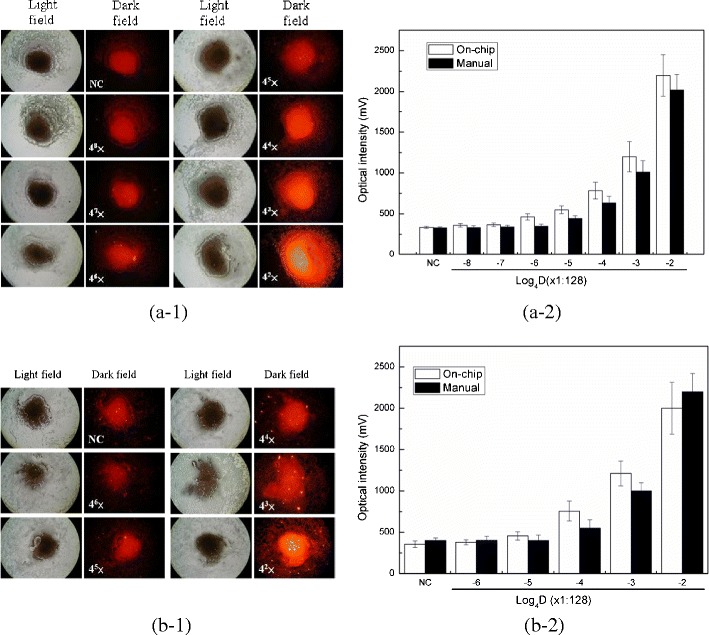
Limit of detection for influenza A and B viruses by using the integrated microfluidic system. a-1 A series of images of the magnetic complexes with different viral concentrations of influenza virus A when operated by the microfluidic assay. The detection concentrations are from 8.0, 2.0, 0.50, 0.125, 0.031, 0.008 to 0.002 HAU with 4-fold serial dilutions; a-2 Optical signals from the detection of influenza virus A at different concentrations. White bars (□) shows that the microfluidic assay has a LOD measured as 0.031 HAU (p = 0.011 < 0.05), while black bars (■) shows that the manual assay only has a LOD measured as 0.125 HAU (p = 0.015 < 0.05); b-1 A series of images of the magnetic complexes with different viral concentrations of influenza virus B. The detection concentrations are from 8.0, 2.0, 0.50, 0.125 to 0.031 with 4-fold serial dilutions b-2 Optical signals from the detection of influenza virus B at different concentrations. White bars (□) shows that the microfluidic assay has a LOD measured as 0.125 HAU (p = 0.002 < 0.01), while black bars (■) shows that the manual assay only has a LOD measured as 0.5 HAU (p = 0.008 < 0.01)
Similarly, influenza B viral particles were also tested in this microfluidic system. As shown in Fig. 5b-1, the detection of influenza virus B was successfully realized and the LOD was measured to be 128 × 4−5 HAU/50 μL (about 0.125 HAU/50 μL) with an optical signal of 443.6 ± 31.7 mV (n = 3, p = 0.028 < 0.05). Figure 5b-2 shows a comparison of the LOD between the on-chip and the manual operation. Again, the on-chip assay had better performance than the manual one (about 128 × 4−4 HAU/50 μL with an optical signal of 532.5 ± 54.4 mV). Note that the signal for the negative control is around 356.2 ± 32.3 mV (n = 3, p = 0.008 < 0.01). The LOD of influenza virus A and B were shown to be lower with the integrated microfluidic system than from manual operation of bench-top equipment. For further comparison, the optical signals of the various viruses shown in Fig. 4 were taken as background noise to test the LOD of influenza virus A and B in Fig. 5 with statistical analysis. The details of the statistical analysis of Influenza A could be found in the SI Table 4 and SI Table 5.
Diagnosis of influenza infections using clinical specimens
According to the aforementioned experimental conditions and promising characterization results from the integrated microfluidic system, clinical specimens were next tested. The clinical specimens, however, were more complicated than purified viral particles, containing additional components such as debris, sputum, mucous or ever other types of microorganisms. First, the ability of the microfluidic system to wash out the nonspecific materials in the clinical samples was confirmed. A comparison between the optical signals of the negative clinical samples and those signals from the on-chip and manually operated diagnosis with on-chip or manual operation, is shown in Fig. 6. The optical images from the manual operation showed more debris and sputum-like mucus than the on-chip operations. This was because the microfluidic chip washed out the debris more efficiently and thus reduced the background noise caused by debris or sputum-like mucus. Parts of the negative specimens showed more mucous than expected, causing mAB-PE to become attached around these nonspecific materials and this resulted in a strong optical signal from the manually operated assay. In contrast, when operated with the microfluidic system, the micromixers could wash the specimens thoroughly and reduced the nonspecific signals when dealing with clinical specimens. Note that the entire process only takes 15 min, which is much faster than manually operation (180 min).
Fig. 6.
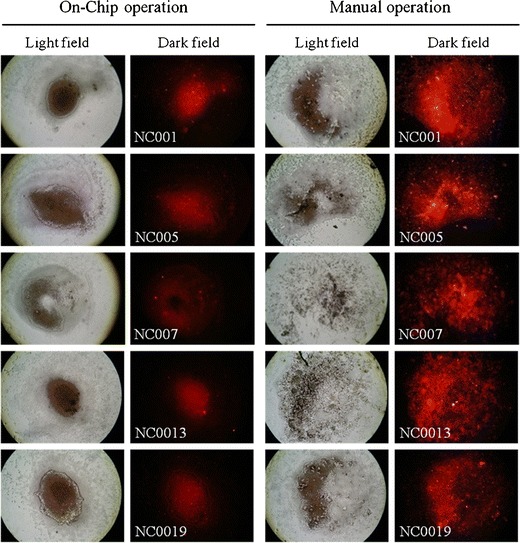
Comparison of the optical images with the processed results of clinical specimens using the manual and the on-chip operation. The images of the magnetic beads in on-chip operation show more concentrated beads than the ones obtained by the manual operation
Sixty-six positive specimens and 20 negative specimens were tested using the integrated microfluidic system. Table 2 shows the summary of the test results. A sensitivity of 84.8 % and a specificity of 75.0 % were verified. For comparison, parts of the same clinical specimens were also analyzed with the rapid test set, BD Directigen EZ Flu A+B test, which displayed a sensitivity of 60.9 % and a specificity of 100 % with 200 μL of clinical specimens (Supplemental Table 1). These results showed that the microfluidic system had better detection sensitivity than the traditional rapid test kit. In addition, by utilizing this integrated microfluidic system, each reaction only consumed 40-μL of clinical specimens (as shown in Table 1). This would be more advantageous in the diagnosis of rare clinical specimens. Furthermore, the entire diagnosis protocol was performed automatically within 15 min. Although the specificity of the developed assay is inferior to the rapid test kits, the results have shown that the developed microfluidic chip could detect clinical samples successfully with reasonable specificity and a high sensitivity.
Table 2.
Test results for influenza infections in clinical specimens
| NCKU hospital clinical reports | |||
|---|---|---|---|
| Positive | Negative | ||
| Integrated microfluidic system | Tested positive | 56 | 5 |
| Tested negative | 10 | 15 | |
Sensitivity = 56/(56 + 10) = 0.848; specificity = 15/(5 + 15) = 0.750; positive predictive value (pPV) = 56/(56 + 5) = 0.918; negative predictive value (nPV) = 15/(10 + 15) = 0.600
Conclusion
A new integrated microfluidic system was demonstrated for the diagnosis of both types of influenza A and B viral infections within 15 min by utilizing specific anti-NP-A/or B mAbs and the direct-conjugated PE fluorescent dye. The system was designed to be equipped with four detection regions, including a positive test, a negative test, influenza A test and influenza B test, enabling automatic viral diagnosis feasible on a single chip. Furthermore, the characterized micro-devices could wash the clinical samples more cleanly, thus reducing the noise level in the signal. Finally, 86 patient specimens were tested with the resulting sensitivity of 84.8 % and a specificity of 75.0 %. This indicated that this developed platform may provide a powerful tool for the rapid detection of various types of influenza infections.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(DOC 2400 kb)
Acknowledgments
The authors would like to thank the National Science Council in Taiwan (NSC 100-2120-M-007-014) and the “Towards A World-class University” Project for financial support of this study.
Nomenclature
- 3D
Three-dimensional
- AIDS
Acquired immunodeficiency syndrome
- BP
Band-pass
- BSA
Bovine serum albumin
- C+
Normalized concentration
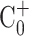
Normalized initial condition
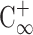
Normalized completed mixed condition
- CCD
Charge-coupled device
- CBR
Complement-binding reaction
- CNC
Computer numerical control
- D+
Normalized circular coordinate
- DV
Dengue virus
- EIA
Enzyme immunoassay
- ELISA
Enzyme-linked immunosorbent assay
- EMVs
Electromagnetic valves
- EV
Enterovirus
- FIA
Fluorescent immunoassay
- Flu
Influenza
- HA
Hemagglutinin
- HAU
Hemagglutin unit
- HBV
Hepatitis B virus
- HI
Hemagglutination inhibition
- HIV
Human immunodeficiency virus
- IFT
Immunofluorescence test
- IgG
Immunoglobulin G
- IgM
Immunoglobulin M
- LOD
Limit of detection
- LP
Long-pass
- mAb
Monoclonal antibody
- MDCK
Mardin-Darby canine kidney
- NA
Neuraminidase
- NP
Nucleoprotein
- PBS
Phosphate-buffered saline
- PCR
Polymerase chain reaction
- PDMS
Polydimethylsiloxane
- PE
R-Phycoerythrin
- PFU
Plaque-forming unit
- PMMA
Polymethylmethacrylate
- PMT
Photo-multiplier tube
- RNA
Ribonucleic acid
- ROC
Receiver-operator characteristic
- RT-PCR
Reverse-transcription polymerase chain reaction
- SARS
Severe acute respiratory syndrome
- SARS-CoV
Severe acute respiratory syndrome-associated coronavirus
- σ
Mixing index
- UV
Ultraviolet
- X-X’ line
Center of the micromixer chamber
References
- Atmar RL, Baxter BD, Dominguez EA, Taber LH. J. Clin. Microbiol. 1996;34(10):2604–2606. doi: 10.1128/jcm.34.10.2604-2606.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Both GW, Sleigh MJ, Cox NJ, Kendal AP. J. Virol. 1983;48(1):52–60. doi: 10.1128/jvi.48.1.52-60.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JW, Oh KW, Han A, Wijayawardhana CA, Lannes C, Bhansali S, Schlueter KT, Heineman WR, Halsall HB, Nevin JH, Henderson HT, Ahn CH. Biomed. Microdevices. 2001;3(3):191–200. doi: 10.1023/A:1011490627871. [DOI] [Google Scholar]
- Cox NJ, Subbarao K. Lancet. 1999;354(9186):1277–1282. doi: 10.1016/S0140-6736(99)01241-6. [DOI] [PubMed] [Google Scholar]
- de Boer GF, Back W, Osterhaus ADME. Arch. Virol. 1990;115(1–2):47–61. doi: 10.1007/BF01310622. [DOI] [PubMed] [Google Scholar]
- Drexler JF, Helmer A, Kirberg H, Reber U, Panning M, Müller M, Höfling K, Matz B, Drosten C, Eis-Hübinger AM. Emerg. Infect. Dis. 2009;15(10):1662–1664. doi: 10.3201/eid1510.091186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson D, Li D. Langmuir. 2002;18(5):1883–1892. doi: 10.1021/la015646z. [DOI] [Google Scholar]
- Espmark JA, Salenstedt CR. Arch. Gesamte. Virusforsch. 1961;11(1):64–72. doi: 10.1007/BF01241248. [DOI] [PubMed] [Google Scholar]
- E. Eteshola, M. Balberg, Biomed Microdecives. 6(1) 7–9 (2004) [DOI] [PubMed]
- Faix DJ, Sherman SS, Waterman SH. N. Engl. J. Med. 2009;361(7):728–729. doi: 10.1056/NEJMc0904264. [DOI] [PubMed] [Google Scholar]
- Fernández-Lafuente R, Guisán JM. Biosens. Bioelectron. 2006;21(8):1574–1580. doi: 10.1016/j.bios.2005.07.017. [DOI] [PubMed] [Google Scholar]
- Gavin P, Thomson RB., Jr Clin. Appl. Immunol. Rev. 2003;4(3):151–172. doi: 10.1016/S1529-1049(03)00064-3. [DOI] [Google Scholar]
- Ginocchio CC, Zhang F, Manji R, Arora S, Bornfreund M, Falk L, Lotlikar M, Kowerska M, Becker G, Korologos D, de Geronimo M, Crawford JM. J. Clin. Virol. 2009;45(3):191–195. doi: 10.1016/j.jcv.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horimoto T, Kawaoka Y. Nat. Rev. Microbiol. 2005;3(8):591–600. doi: 10.1038/nrmicro1208. [DOI] [PubMed] [Google Scholar]
- Hurt AC, Alexander R, Hibbert J, Deed N, Barr IG. J. Clin. Virol. 2007;39(2):132–135. doi: 10.1016/j.jcv.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Johnson NP, Mueller J. Bull. Hist. Med. 2002;76(1):105–115. doi: 10.1353/bhm.2002.0022. [DOI] [PubMed] [Google Scholar]
- Kaiser L, Briones MS, Hayden FG. J. Clin. Virol. 1999;14(3):191–197. doi: 10.1016/S1386-6532(99)00058-X. [DOI] [PubMed] [Google Scholar]
- Kawaoka Y, Krauss S, Webster RG. J. Virol. 1989;63(11):4603–4608. doi: 10.1128/jvi.63.11.4603-4608.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien KY, Hung LY, Huang TB, Tsai YC, Lei HY, Lee GB. Biosens. Bioelectron. 2011;26(9):3900–3907. doi: 10.1016/j.bios.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CJ, Lien KY, Weng CY, Shin JW, Chang TY, Lee GB. Biomed. Microdevices. 2009;11(2):339–350. doi: 10.1007/s10544-008-9240-1. [DOI] [PubMed] [Google Scholar]
- McGeer AJ. Clin. Infect. Dis. 2009;48(Suppl. 1):S14–S19. doi: 10.1086/591852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina RA, Garcia-Sastre A. Nat. Rev. Microbiol. 2011;9(8):590–603. doi: 10.1038/nrmicro2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon LLM, Chan KH, Smith GJ, Leung CS, Guan Y, Yuen KY, Peiris JS. Clin. Chem. 2009;55(8):1555–1558. doi: 10.1373/clinchem.2009.130229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siavashi MR, Taherkhani H, Rezaei K, Deligani MRR, Assmar M. Iran. Biomed. J. 2005;9(2):91–94. [Google Scholar]
- Sugiura A, Tobita K, Kilbourne ED. J. Virol. 1972;10(4):639–647. doi: 10.1128/jvi.10.4.639-647.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao KC, Kuo YB, Huang CG, Chau SW, Chan EC. J. Virol. Methods. 2011;173(2):387–389. doi: 10.1016/j.jviromet.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Vasoo S, Stevens J, Singh K. Clin. Infect. Dis. 2009;49(7):1090–1093. doi: 10.1086/644743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignali DA. J. Immunol. Methods. 2000;243(1–2):243–255. doi: 10.1016/S0022-1759(00)00238-6. [DOI] [PubMed] [Google Scholar]
- Vyas GN, Shulman NR. Science. 1970;170(3955):332–333. doi: 10.1126/science.170.3955.332. [DOI] [PubMed] [Google Scholar]
- Wang CH, Lee GB. J. Micromech. Microeng. 2006;16(2):341–348. doi: 10.1088/0960-1317/16/2/019. [DOI] [Google Scholar]
- Weng CH, Huang TB, Huang CC, Yeh CS, Lei HY, Lee GB. Biomed. Microdevices. 2011;13(3):585–595. doi: 10.1007/s10544-011-9529-3. [DOI] [PubMed] [Google Scholar]
- Weng CH, Lien KY, Yang SY, Lee GB. Microfluid. Nanofluid. 2011;10(2):301–310. doi: 10.1007/s10404-010-0669-1. [DOI] [Google Scholar]
- Wians FH. Lab. Med. 2009;40(2):105–113. [Google Scholar]
- Woo PC, Chiu SS, Seto WH, Peiris M. J. Clin. Microbiol. 1997;35(6):1579–1581. doi: 10.1128/jcm.35.6.1579-1581.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SY, Lien KY, Huang KJ, Lei HY, Lee GB. Biosens. Bioelectron. 2008;24(4):855–862. doi: 10.1016/j.bios.2008.07.019. [DOI] [PubMed] [Google Scholar]
- Yang SY, Lin JL, Lee GB. J. Micromech. Microeng. 2009;19(3):035020. doi: 10.1088/0960-1317/19/3/035020. [DOI] [Google Scholar]
- Yang YN, Hsiung SK, Lee GB. Microfluid. Nanofluid. 2009;6(6):823–833. doi: 10.1007/s10404-008-0356-7. [DOI] [Google Scholar]
- Zheng HJ, Tao ZH, Cheng WF, Piessens WF. Am.J.Trop. Med. Hyg. 1990;42(6):546–549. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 2400 kb)