

Abstract
A rapid, accurate, specific, repeatable and robust HPTLC method for the determination of lycorine in different Amaryllidaceae plant extracts is presented in this work. No article related to the HPTLC determination of lycorine in plant extracts has been reported in literature. Lycorine, a common alkaloid of family Amaryllidaceae, moreover, there have been some recent reports which reveal the interaction of lycorine with DNA and tRNA. It has, therefore, been to the interest of phytochemists to determine the content of this alkaloid in Amaryllidaceaous plants.
Keywords: Thin layer chromatography, Amaryllidaceae plant extracts, Lycorine, Narcissus ‘Breath of Spring’
Introduction
Lycorine, the most frequent Amaryllidaceae alkaloid, has proven to have a wide spectrum of biological activities including antiviral, cytotoxic, antimalarial and anti-inflammatory activities [1–10]. Moreover, there have been some recent reports which reveal the interaction of lycorine with DNA and tRNA [11, 12], and recently it has been identified as anti-SARS-CoV component [13]. It has, therefore, been to the interest of phytochemists to determine the content of this alkaloid in Amaryllidaceaous plants.
Different analytical techniques have been described for the qualitative and quantitative determination of lycorine in various parts of different Amaryllidaceae plants including (capillary gas chromatography-mass spectrometry) CGC-MS [14], spectrophotometric [15, 16], fluorimetric [16] techniques and reversed-phase high-performance liquid chromatography [17]. The aim of the present work is to an develop accurate, specific, repeatable and robust high performance thin layer chromatographic (HPTLC) method for the determination of lycorine in plant extracts.
Experimental
Instrumentation
Sample solutions for HPTLC analyses were applied by means of a Camag (Wilmington, NC, USA) Linomat IV automated spray-on band applicator. Zones were quantified by linear scanning at λ = 368 nm with a Camag TLC Scanner 3 with a deuterium source in the fluorescence mode, slit dimension settings of length 6 and width 0.1, monochromator bandwidth 20 nm, a scanning rate of 10 mm s−1. The peak areas of chromatograms were determined using CATS TLC software (version 4.X).
Chemicals
HPTLC analyses were performed on Merck (Darmstadt, Germany) 20 × 10 cm HPTLC silica gel 60F254 (0.25 mm) plates. Lycorine was supplied by Sigma, Aldrich, Germany.
Plant Material
Narcissus ‘Breath of Spring’ was collected in October 2005 (flowering stage), cultivated in Alexandria, Egypt. A voucher sample is deposited in the department of Pharmacognosy, Faculty of Pharmacy, Alexandria, Egypt.
Quantitative Determination of Lycorine in Plant Extracts
Preparation of Standard Solutions
Standard lycorine of 2.0 mg were accurately weighed, quantitatively transferred into a 10 mL volumetric flask, dissolved in methanol and the volume was adjusted with the same solvent.
Sample Preparation
Fresh bulbs (10 g) of Narcissus ‘Breath of Spring’ were chopped into small pieces and exhaustively extracted using methanol (0.5 L × 3). The solvent was evaporated under reduced pressure producing the crude extract (0.1 g). The crude extract was re-dissolved in a mixture of chloroform:methanol 3:1, filtered, transferred quantitatively to a 10 mL volumetric flask, adjusted to volume with methanol and shaken to mix thoroughly.
Preparation of Calibration Graphs
As recommended by the international conference on harmonization of drugs (ICH) guidelines [18, 19], a calibration curve was established using six analyte concentrations (1.0, 2.0, 3.0, 4.0, 5.0 and 6.0 μL zone−1) of the TLC standard applied in duplicate, representing 0.2–1.2 μg of lycorine. For routine analytical procedures, a three-point calibration curve within this range was used, produced by applying duplicate 2.0, 4.0 and 6.0 μL (0.4, 0.8, 1.2 μg) of the HPTLC standard on each plate. Standard solutions were applied in the form of bands on pre-coated HPTLC silica gel plates 60 F-254 (20 × 10 cm with 250 μm thickness) by means of Linomat IV automated spray-on band applicator. The volumes applied for routine analysis were duplicate 2.0, 4.0 and 6.0 μL of the TLC lycorine standard (0.4, 0.8 and 1.2 μg) and duplicate 2 μL aliquots of sample solution. The mobile phase consisted of chloroform: methanol (9:1) and 25 mL of mobile phase were used per development. Ascending development of the plates was carried out in 20 × 20 cm Camag HPTLC twin trough chamber saturated with the mobile phase. The optimized chamber saturation time for the mobile phase was 30 min at room temperature. Plates were developed to a distance of 7 cm beyond the origin. The development time was 11 min. After development, the plates were air-dried for 5 min. Densitometric scanning was performed on Camag TLC scanner III in the reflectance-fluorescence mode at 368 nm and operated by CATS TLC software (version 4.X) and using the optical filter K400. The source of radiation utilized was deuterium lamp emitting a continuous UV spectrum between 190 and 400 nm. The slit dimension was kept at 6 × 0.1 mm.
Sample Assay
Aliquots (4.0 μL) of the prepared sample solution were subjected to HPTLC analysis as described above under preparation of calibration graph for HPTLC.
Validation
A 5 mL aliquot of sample solution of known previously determined concentration (1.3 mg mL−1) was mixed with 5 mL of standard lycorine solution (0.4 mg mL−1) to give mix. 1 and another 5 mL aliquot of the same sample solution was mixed with 5 ml of standard lycorine solution (0.8 mg mL−1) to give mix. 2. The original and the two fortified sample solutions (mix. 1 and 2) were analyzed on the same plate by application of triplicate 4.0, 3.0 and 2.0 μL volumes, respectively, in addition to the three standards described earlier for routine analyses which were applied in duplicate.
Results and Discussion
Experimental conditions, such as mobile phase composition, scan mode, scan speed and wavelength of detection were optimized to provide accurate and precise results. Development with the mobile phase described above on the HPTLC silica gel layers produced compact, flat, fluorescent bands of lycorine (R F 0.3) when viewed under a 368 nm UV light.
The relationship between the peak areas and the amount of substance applied showed linearity over the range 0.2–1.2 μg spot−1. The regression equation data, correlation coefficient (r-value) and other statistical parameters are listed in Table 1.
Table 1.
Linear regression data for the calibration curve (n = 6)
| Lycorine | |
|---|---|
| Linearity range (μg spot−1) | 0.2–1.2 |
| Intercept (a ± SD) | 357.33 ± 0.48 |
| Slope (b ± SD) | 1464.3 ± 2.1 |
| Correlation coefficient (r ± SD) | 0.99964 ± 0.000137 |
| Sy/xcovariance of x and y | 170.8333 |
| Sastandard deviation of x | 0.374166 |
| Sbstandard deviation of y | 548.0834 |
| Standard error | 16.466 |
| LOD (μg spot−1) | 0.00108 |
| LOQ (μg spot−1) | 0.0032 |
The three-point calibration was repeated many times and was also found to have a linear regression correlation coefficient of 0.99972.
Validation
The scan-densitogram obtained from a representative sample, Fig. 1, showed selective baseline separation between lycorine and other components in the sample.
Fig. 1.
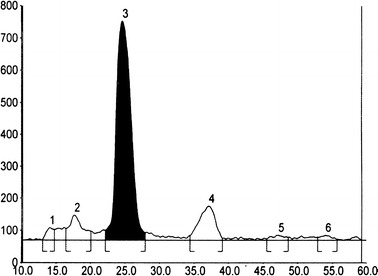
HPTLC scan-densitogram showing the separation of lycorine [3] from other components in the sample at 368 nm
The proposed method was applied for the determination of lycorine in the concentration range of 50–150% of the working concentrations. Limit of detection (LOD) and limit of quantitation (LOQ) were experimentally verified by diluting known concentrations of standard lycorine solutions. The results are shown in Table 1.
In accordance with the ICH guideline [18, 19], precision was determined by independent repeated analysis of six different lycorine standard solutions and repeated analysis of a homogenous sample (5.19 μg spot−1). The assay was repeated in the same day for intra-day precision and on different days to obtain inter-day precision. The percentage of relative standard deviation (RSD%) was calculated and the results are shown in Tables 2 and 3. The results shown in the tables meet the acceptance criterion for RSD% specified by the ICH, which is a precision of less than 2%.
Table 2.
Precision of the HPTLC method for the determination of lycorine (repeatability)
| Experiment number | Intra-day precision | Inter-day precision | ||||
|---|---|---|---|---|---|---|
| Concentration (μg spot−1) | Mean ± SD | RSD (%) | Concentration (μg spot−1) | Mean ± SD | RSD % | |
| 1 |
0.215 0.219 0.22 |
0.218 ± 0.0027 | 1.21 |
0.205 0.211 0.208 |
0.003 ± 0.208 | 1.44 |
| 2 |
0.419 0.420 0.411 |
0.4167 ± 0.005 | 0.98 |
0.403 0.409 0.412 |
0.408 ± 0.0046 | 1.12 |
| 3 |
0.628 0.620 0.615 |
0.621 ± .0066 | 1.05 |
0.602 0.608 0.615 |
0.6083 ± 0.006 | 1.07 |
| 4 |
0.805 0.813 0.823 |
0.814 ± 0.0092 | 1.1 |
0.811 0.807 0.826 |
0.81467 ± 0.011 | 1.22 |
| 5 |
1.028 1.015 1.037 |
1.0267 ± 0.01 | 1.07 |
1.055 1.081 1.074 |
1.07 ± 0.013 | 1.25 |
| 6 |
1.215 1.203 1.225 |
0.011 ± 1.214 | 0.907 |
1.215 1.227 1.202 |
1.21467 ± 0.012 | 1.03 |
Table 3.
Results of replicate analysis of a homogenous sample by HPTLC method
| Measurement | Concentration (μg spot−1) |
|---|---|
| 1 | 5.19 |
| 2 | 5.2 |
| 3 | 5.22 |
| 4 | 5.15 |
| 5 | 5.17 |
| 6 | 5.11 |
| Mean | 5.1733 |
| SD | 0.0393 |
| RSD% | 0.76 |
The accuracy of the method was validated by a standard addition analysis. The sample solutions were spiked with two different, known, concentrations of lycorine. The results of the experiments are presented in Table 4.
Table 4.
Results of the standard addition experiments
| Concentration of lycorine in sample (mg mL−1) | Concentration of lycorine added (mg mL−1) | Concentration of lycorine found in mixture (mg mL−1)a | Recovery (%) ± SD | RSD(%) | |
|---|---|---|---|---|---|
| 1 | 1.3 | 0.4 | 0.83 | 97.6 ± 1.094 | 1.1 |
| 2 | 1.3 | 0.8 | 1.03 | 98.08 ± 1.43 | 1.4 |
| 3 | 1.3 | 1.2 | 1.23 | 98.4 ± 1.23 | 1.25 |
a Each concentration is the average of three determinations
Robustness tests examine the effect of the operational parameters on the analysis results. By introducing small changes in the mobile phase composition, the effects on the results were examined. Mobile phases having different composition like (CHCl3:MeOH 9:1, CHCl3:MeOH 8.5:1.5 and EtOAc:MeOH 19:1) were tried. The amount of mobile phase was varied in the range of ±5%. Time from spotting to chromatography and from chromatography to scanning was varied from 0, 10, 20, to 40 min. Robustness of the method was carried out at three concentration levels 1, 3 and 5 μg spot−1. Results are shown in Table 5.
Table 5.
Robustness testing
| Parameter | SDa | RSD (%)a |
|---|---|---|
| Mobile phase composition | 0.0073 | 0.96 |
| Amount of mobile phase | 0.0054 | 0.65 |
| Time from spotting to chromatography | 0.0049 | 0.58 |
| Time from chromatography to scanning | 0.0054 | 0.65 |
a Average of three concentrations 1, 3 and 5 μg spot−1
In conclusion it has been demonstrated above that validation data for the new quantitative HPTLC method meet the acceptance criteria for accuracy, precision, linearity, detection and quantification limits set by ICH [18, 19]. The precision and accuracy values of the proposed HPTLC method compare favourably with those obtained with HPLC method [17] (the detection limit was 5 ng, recovery was 97.0% compared to the suggested HPTLC method which showed a limit of detection of 1.1 ng and 98% recovery) and CGC-MS (where only good peak resolution was obtained while LOD and recovery are not mentioned) [14] for determination of lycorine in plants extracts. The proposed HPTLC method is simpler, faster, more accurate and precise than the spectrophotometric [15, 16] and fluorimetric [16] methods.
Conclusion
The described method is suitable for routine use by manufacturers for product quality control. It is simpler than HPLC and faster because up to six samples (applied in duplicate singly with a minimum of three standard concentrations) can be analyzed on each plate rather than performing sequential injection of the samples and standards in HPLC. Cost of solvent purchase and disposal is very low because no more than 15 mL of mobile phase for development is required in the chamber trough containing the plate, and an additional 10 mL for vapor saturation in the other trough. The processing of samples and standards together at the same time (in-system calibration) leads to improved reproducibility and accuracy.
References
- 1.Carrasco L, Fresno M, Vazquez D. FEBS Lett. 1975;52:236–239. doi: 10.1016/0014-5793(75)80813-1. [DOI] [PubMed] [Google Scholar]
- 2.Trost B, Pulley S. J Am Chem Soc. 1995;117:10143–10144. doi: 10.1021/ja00145a038. [DOI] [Google Scholar]
- 3.Akgun H, Hudlicky T. Tetrahedron Lett. 1999;40:3081–3084. doi: 10.1016/S0040-4039(99)00434-7. [DOI] [Google Scholar]
- 4.Hudson S. J Am Chem Soc. 1925;47:265–268. doi: 10.1021/ja01678a039. [DOI] [Google Scholar]
- 5.Carey F, Sundberg R (2000) Advanced organic chemistry, Part A: Structure and Mechanism. 4th edn. Plenum Publishers, p 451
- 6.Liu J, Hu WX, He LF, Ye M, Li Y. FEBS Letters. 2004;578(3):245–250. doi: 10.1016/j.febslet.2004.10.095. [DOI] [PubMed] [Google Scholar]
- 7.Yui S, Mikami M, Mimaki Y, Sashida Y, Yamazaki M. Yakugaku Zasshi. 2001;121(2):167–171. doi: 10.1248/yakushi.121.167. [DOI] [PubMed] [Google Scholar]
- 8.Sener B, Orhan I, Satayavivad J. Phytother Res. 2003;17(10):1220–1223. doi: 10.1002/ptr.1346. [DOI] [PubMed] [Google Scholar]
- 9.Hohmann J, Forgo P, Molnar J, Wolfard K, Molnar A, Thalhammer T, Mathe I, Sharples D. Planta Med. 2002;68(5):454–457. doi: 10.1055/s-2002-32068. [DOI] [PubMed] [Google Scholar]
- 10.Satoru Y, Masaaki M., Mikio K, Masatoshi Y. Immunopharmacology. 1998;40(2):151–162. doi: 10.1016/S0162-3109(98)00040-X. [DOI] [PubMed] [Google Scholar]
- 11.Hohmann J, Forgo P, Molnár J, Wolfard K, Molnár A, Thalhammer T, Máthe I, Sharples D. Planta Med. 2002;68(5):454–457. doi: 10.1055/s-2002-32068. [DOI] [PubMed] [Google Scholar]
- 12.Karadeniz H, Gulmez B, Sahinci F, Erdem A, Kaya GI, Unver N, Kivcak B, Ozsoz M. J Pharm Biomed Anal. 2003;33(2):295–302. doi: 10.1016/S0731-7085(03)00283-8. [DOI] [PubMed] [Google Scholar]
- 13.Li SY, Chen C, Zhang HQ, Guo HY, Wang H, Wang L, Zhang X, Hua SN, Yu J, Xiao PG, Li RS, Tan X. Antiviral Res. 2005;67(1):18–23. doi: 10.1016/j.antiviral.2005.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berkov S, Pavlov A, Ilieva M, Burrus M, Popov S, Stanilova M. Phytochem Anal. 2005;16(2):98–103. doi: 10.1002/pca.824. [DOI] [PubMed] [Google Scholar]
- 15.Makhkamova AU, Safonova EV. Chem Nat Comp (English Translation) 1995;30(4):529–530. [Google Scholar]
- 16.El-Din AS, Korany M, Abou-Donia A, Sabry NN. Acta Pharm Jugosl. 1983;33(2):143–147. [Google Scholar]
- 17.Evidente A, Iasiello I, Randazzo G. J Chromatogr. 1983;281:362–366. doi: 10.1016/S0021-9673(01)87900-0. [DOI] [Google Scholar]
- 18.ICH-Guidelines Q 2A, (CPMP/ICH/381/95)
- 19.ICH-Guidelines Q 2B, Validation (CPMP/ICH/281/95)