

Abstract
The appearance of single cell microorganisms on earth dates back to more than 3.5 billion years ago, ultimately leading to the development of multicellular organisms approximately 3 billion years later. The evolutionary burst of species diversity and the “struggle for existence”, as proposed by Darwin, generated a complex host defense system. Host survival during infection in vital organs, such as the lung, requires a delicate balance between host defense, which is essential for the detection and elimination of pathogens and host tolerance, which is critical for minimizing collateral tissue damage. Whereas the cellular and molecular mechanisms of host defense against many invading pathogens have been extensively studied, our understanding of host tolerance as a key mechanism in maintaining host fitness is extremely limited. This may also explain why current therapeutic and preventive approaches targeting only host defense mechanisms have failed to provide full protection against severe infectious diseases, including pulmonary influenza virus and Mycobacterium tuberculosis infections. In this review, we aim to outline various host strategies of resistance and tolerance for effective protection against acute or chronic pulmonary infections.
Keywords: Host resistance, Host tolerance, Pulmonary infections, Tuberculosis, Influenza virus
Nothing in biology makes sense except in the light of evolution.
Theodosius Dobzhnansky
Introduction
For a long time, immunologists considered host defense as the hallmark of immunity, through the detection and destruction of pathogens. However, we now recognize that the host may also provide protection against infections by tolerating them and controlling tissue damage caused either directly via pathogen-derived toxins or indirectly by the immune response. Host tolerance was initially studied in plants (Kover and Schaal 2002; Ayres and Schneider 2012) and then in Drosophila (Ayres et al. 2008; Ayres and Schneider 2008, 2009) and the concept has recently been introduced in animals (Raberg et al. 2007; Read et al. 2008; Schneider and Ayres 2008; Raberg et al. 2009; Medzhitov et al. 2012; Fig. 1).
Fig. 1.
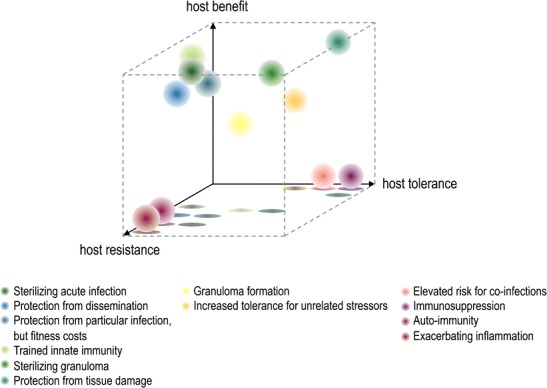
Three-dimensional representation of host resistance and host tolerance and their beneficial impact on the organism. Each dot represents a major type of immune response in its general placement between host resistance and host tolerance and its contribution to the host benefit. Contribution to the host benefit can vary because of host and pathogen factors. The different placements are not mutually exclusive and the transition is fluid
During an infection, the host protects itself by two major mechanisms, namely resistance and tolerance and these mechanisms are not mutually exclusive. Resistance mechanisms are typically associated with a decrease in microbial burden through innate mechanisms, including pathogen detection by various sensors, such as Toll-like receptors (TLR), NOD-like receptors and C-type lectins, or phagocytosis and neutralization by macrophages and adaptive mechanisms via the killing of infected cells by T cell-mediated immunity. Although the host’s resistance response is essential in controlling the infection and preventing further dissemination, it is frequently associated with significant fitness costs, as it also induces tissue damage. For instance, lung infections often result in mucus production, increased airway permeability, alterations in vascular function, and thereby, impaired lung function. Therefore, a balance between decreasing the microbial burden and restricting tissue damage is required. In this vein, the collateral damage caused by the resistive immune response can be dampened by host tolerance. Disease tolerance is defined as the mechanisms that “decrease host susceptibility to tissue damage, or other fitness costs caused by pathogens or by the immune response” (Medzhitov et al. 2012). Common tolerance mechanisms include the activation of the stress response to eliminate reactive oxygen species (ROS) and the secretion of anti-inflammatory cytokines such as interleukin-10 (IL-10) and transforming growth factor-β (TGF-β). An additional major tolerance mechanism is the tissue repair response, which has been classically associated with the production of type 2 cytokines (e.g., IL-4 and IL-13) that share a STAT6-dependent signaling pathway (Martinez et al. 2009) and the induction of alternatively activated macrophages (AAMφ).
Pulmonary infections are usually associated with alterations in vascular function and increased permeability of the airways, which lead to lung dysfunction. For example, influenza A virus (IAV) induces type 2 cytokines such as IL-33, IL-4 and IL-13 early during infection, driving goblet cell hyperplasia, mucus production and airway hyperreactivity (Chang et al. 2011). However, production of these cytokines upon viral clearance is associated with the restoration of lung function and tissue remodeling (Monticelli et al. 2011). Alternatively, slow growing pathogens such as Mycobacterium tuberculosis (Mtb) may exploit type 2 cytokine production for its own benefit to favor the invasion of the lungs and to establish a chronic infection (Heitmann et al. 2014). Mechanistically, IL-4 and IL-13 have been shown to stimulate macrophage fusion and giant cell formation (Helming and Gordon 2007), leading to granulomas, a hallmark of chronic tuberculosis (TB) and an important mechanism of immune evasion and chronicity. Pairing the formation of granulomas with the increased lifespan of AAMφ may provide an important reservoir for Mtb to facilitate bacterial persistence. Although some groups have found that the inhibition of type 2 cytokine production via antibody blockade or deletion of the IL-4/13 signaling pathway confers enhanced resistance to Mtb infection (Buccheri et al. 2007; Roy et al. 2008), this topic remains controversial (Jung et al. 2002). Undoubtedly though, during pulmonary infection, both resistance to the pathogen, by mounting an effective immune response and tolerance of its presence to control immunopathology have a direct impact on host protection and fitness. In this review, we explore the cost-benefit trade-offs of the immune response and immune-mediated pathology with a particular focus on respiratory infections.
Host resistance in pulmonary infections
Most acute infections in the lungs cause excessive tissue damage that needs to be rapidly controlled and repaired for host survival. During an acute pulmonary infection, the pathogen burden and the magnitude of the immune response that will ultimately determine the extent of the tissue damage are directly correlated. Thus, the extent of the initial resistance mechanisms is an important determinant of host tolerance to infection.
Both pulmonary epithelial and immune cells express a wide range pattern-recognition receptors (PRRs) that identify pathogen-associated molecular patterns (PAMPs) originating from the invading pathogen and of damage-associated molecular patterns (DAMPs) released from infected cells (Holt et al. 2008; Takeuchi and Akira 2009; Braciale et al. 2012). In 1989, Charles Janeway brilliantly predicted the importance of PRRs in the recognition of pathogens by macrophages and their potential link to adaptive immunity (Janeway 1989). This hypothesis led to an explosion of research in the field of innate immunity, beginning with the discovery of the Toll pathway in anti-microbial defense in the plant (Whitham et al. 1994) and the fruit fly (Drosophila melanogaster; Lemaitre et al. 1996). This work was followed by Medzhitov and Janeway describing the first mammalian Toll-like receptor (TLR4; Medzhitov et al. 1997) that recognizes lipopolysaccharide (LPS) on gram-negative bacteria (Poltorak et al. 1998). In the last 15 years, more than 13 TLR family members have been discovered that localize to the cell surface or endosome and that recognize diverse bacterial or viral components. Whereas several TLRs detect conserved PAMPs present on bacteria (e.g., LPS, flagellin, or peptidoglycans), a PRR that directly recognizes viruses has yet to be identified. As most structural components of viruses are derived from the host, recognition is necessarily limited to viral nucleic acids. Thus, TLRs involved in sensing viruses are unsurprisingly localized to the endosome and detect extracellular viral particles by stripping and exposing their genome (RNA/DNA) via endosomal acidification. In addition to TLR family members that have evolved to recognize microbial components in the extracellular microenvironment, there are also cytosolic PRRs including Nod-like receptors (NLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) and others that play a crucial role in recognition of PAMPs within infected cells (Kumar et al. 2011). The evolution of this second layer of PRRs is essential for host survival as the role of TLRs in host defense against intracellular pathogens is less pronounced. Indeed, NLRs are highly conserved (Rast et al. 2006) and crucial in the recognition of various intracellular bacteria including Mtb (Guirado et al. 2013). For example, NOD2 is an NLR family member that recognizes the bacterial peptidoglycan fragment muramyl dipeptide (MDP) and activates nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways (Inohara et al. 2005). Macrophages from NOD2-deficient mice or humans are impaired in cytokine production (e.g., TNF-α and IL-12) following Mtb infection (Ferwerda et al. 2005; Divangahi et al. 2008). In addition, NOD2-deficient mice are more susceptible to the late stages of Mtb infection (Gandotra et al. 2007; Divangahi et al. 2008) emphasizing a potential role for NOD2 in adaptive immunity (Divangahi et al. 2008; Philpott et al. 2014). Whereas most bacteria express an N-acetylated form of MDP, mycobacteria produce an N-glycolylated form, converted via N-acetyl muramic acid hydroxylase activity. N-glycolylated MDP is more potent in inducing NOD2-mediated innate and adaptive immune responses (Coulombe et al. 2009; Behr and Divangahi 2015). Thus, PAMP localization and post-transcriptional modifications can diversify the outcome of the immune response.
Particularly relevant to anti-viral immunity are RLRs, such as RIG-I and melanoma differentiated-associated gene 5 (MDA5), which detect viral single-stranded RNA/short double-stranded RNA (<300 bp) and long double-stranded RNA (>1000 bp), respectively, in infected cells (Iwasaki and Pillai 2014). Additionally, mitochondrial antiviral signaling protein (MAVS), an essential adaptor protein downstream of RIG-I/MDA5, activates NF-κB and interferon (IFN) regulatory factor 3 (IRF3)-dependent signaling pathways and is localized to the mitochondrial outer membrane (Seth et al. 2005). Recognition of a virus by these sensors potently induces type I IFN. Type I IFN is a critical mediator of host resistance to viral infection, as it rapidly induces more than 300 known IFN-stimulated genes (ISG) within infected and neighboring cells rapidly to restrict viral replication (Doly et al. 1998; Schneider et al. 2014). Viruses have also evolved several mechanisms to inhibit RIG-I/MDA5 signaling and to paralyze antiviral responses. For instance, the NS1 protein expressed by IAV inhibits RIG-I signaling, hinting at the crucial role of RLRs in innate immunity to viral infections. Furthermore, type I IFN also promotes the cytotoxic activity of CD8+ T cells by inducing granzyme B expression (Kohlmeier et al. 2010), highlighting a role for type I IFN in orchestrating adaptive immunity. Apart from type I IFN, type II IFN (IFN-γ) and type III IFN (IFN-λ) have also been shown to induce overlapping sets of ISG, which promote viral clearance (Der et al. 1998). Interestingly, influenza PB1-F2 is encoded by an alternate reading frame within the viral polymerase PB1 gene segment and selectively targets the mitochondria of macrophages but not epithelial cells, to inhibit type I IFN production and induce apoptosis, enhancing IAV virulence in mice (Chen et al. 2001; McAuley et al. 2007). Moreover, non-pathogenic influenza viruses engineered to express the PB1 gene from virulent H5N1 or 1918 pandemic influenza strains become highly pathogenic in mice (McAuley et al. 2007, 2010). Mechanistically, PB1-F2 interacts with the permeability transition pore (PTP) complex, containing the adenine nucleotide translocator 3 (ANT3) and voltage-dependent anion channel 1 (VDAC1), both of which have been implicated in mitochondrial-dependent apoptosis (Varga et al. 2011). Recently, we have shown that NLR-family member NLRX1 interacts with PB1-F2 in the mitochondria to disarm its pro-apoptotic effect and to promote the capacity of macrophages to produce type I IFN (Jaworska et al. 2014). It will be interesting to discover other viral or bacterial proteins with similar functions. In this regard, part of the Mtb genome called Region of Difference 1 (RD1), which encompasses a system essential for the secretion of bacterial proteins including Early Secretory Antigenic Target 6 (ESAT6), has the ability to target mitochondria in macrophages and to induce mitochondrial inner membrane potential dissipation, leading to necrosis (Welin et al. 2011) and increased bacterial invasiveness (Hsu et al. 2003). However, the role of NLRX1 in Mtb infection and its potential interaction with ESAT6 remains to be determined.
Host tolerance in acute pulmonary infection
Although immune cells are the critical component of host resistance to infection, the integrity of structural cells (e.g., lung parenchymal epithelial cells) is essential in mitigating tissue damage and promoting host tolerance. While a robust host resistance may result in the complete elimination of the pathogen, collateral tissue damage caused by such a response must be controlled in order to avoid jeopardizing host survival. Several mechanisms contribute to host tolerance:
PRRs: Because the cost of tissue damage is high and may result in the permanent loss of physiological function, repair mechanisms must be initiated from the onset of the infection. Interestingly, the overall effects of single nucleotide polymorphisms in genes encoding TLRs or their signaling components appear to have only a modest effect on host resistance to infectious diseases, thus suggesting a potentially important role in host tolerance (Neagos et al. 2015). Matzinger (2002) initially suggested that the activation of TLRs via DAMPs would serve as an early “alarm signal” of tissue damage for the initiation of the repair process. For example, during Pseudomonas aeruginosa infection, high-mobility group box 1 (HMGB1) has been demonstrated to contribute to the inflammation in the lungs of patients suffering from cystic fibrosis (CF) through recognition by TLR2 and TLR4. Indeed, the neutralization of HMGB1 in a mouse model of Pseudomonas aeruginosa infection dampens lung inflammation (Entezari et al. 2012). Furthermore, C-type lectin receptors (CLRs) can function as PPRs by recognizing glycan structures expressed by various pathogens (Davicino et al. 2011). For instance, in macrophages, galactose-type lectin-1 recognizes galactose and/or its monosaccharide derivative on Klebsiella pneumonia and orchestrates the immune response in the lungs by triggering the recruitment of neutrophils (Jondle et al. 2016). However, conversely, HMGB1 recognition through TLR4 and the receptor for advanced glycated end products (RAGE) on bronchial epithelial cells promotes extracellular matrix synthesis and wound repair (Ojo et al. 2015). Thus, the initial recognition of PAMPs via PRRs in immune cells initiates host resistance, whereas the recognition of DAMPs via PRRs in structural cells may potentially initiate host tolerance by activating tissue repair mechanisms at a very early stage of infection.
Stress response: The cellular stress response has evolved to provide rapid metabolic adaptation to environmental changes including oxygen, glucose, cellular redox, or local ADP/ATP concentrations. This adaptation is required for the maintenance of tissue integrity and for functionality during infection (Soares et al. 2014). During pulmonary infection with IAV or respiratory syncytial virus (RSV), large amounts of ROS are produced by neutrophils and macrophages; this creates oxidative stress in the surrounding tissue. The actin-anchored protein Keap-1 senses the oxidative stress and liberates the transcription factor Nrf2 to induce multiple proteins to scavenge free radicals, eliminate damaged proteins, metabolize oxidized membrane lipids, and repair damaged DNA (Thimmulappa et al. 2006; Kensler et al. 2007; Garofalo et al. 2013). Stress response pathways are complex and have been extensively reviewed with regard to disease tolerance elsewhere (Soares et al. 2014).
Th2 response: Type 2 immunity has been extensively studied in the context of host resistance to metazoan parasites and a remarkable overlap occurs between pathways regulating host resistance to parasites and tissue repair (Allen and Maizels 2011). Type 2 immunity is characterized by Th2 cells secreting IL-4, IL-5, IL-10 and IL-13 and host Th2 immunity has been proposed to have evolved mainly to control tissue damage in the host resulting from parasitic infections (e.g. worms; Allen and Sutherland 2014). Additionally, type 2 cytokines, including IL-10, are produced by regulatory T cells (Treg) and effector CD4+ or CD8+ T cells in the lungs of humans and mice following infection with IAV or RSV (Sun et al. 2009, 2011; Palmer et al. 2010). Blockade of IL-10 signaling by using an IL-10R neutralizing antibody during sublethal IAV infection results in increased morbidity and mortality in mice. Similarly, IL-10-deficient mice are more susceptible to Th17-mediated immunopathology following a challenge with a lethal dose of IAV (McKinstry et al. 2009). However, the role of IL-10 during RSV infection is less clear. Although some studies have demonstrated that IL-10 reduces immunopathology by decreasing T cell responses in mice, other groups have observed that the overexpression of IL-10 augments pathology (Loebbermann et al. 2012; Sun et al. 2013). TGF-β is another important cytokine whose role in resistance versus tolerance has been evaluated in a mouse model of acute IAV infection. TGF-β is expressed in an inactive form by most cell types and needs to be cleaved to become activated (Khalil 1999). Interestingly, IAV neuraminidase can also cleave TGF-β into its active form during infection (Schultz-Cherry and Hinshaw 1996). Whereas some groups have demonstrated that an in vivo blockade of TGF-β increases the mortality of mice infected with the 2009 H1N1 or H5N1 IAV without affecting viral titres, others have observed that the overexpression of the protein by the injection of plasmid DNA reduces inflammation but impairs viral clearance of a H3N1 virus (Williams et al. 2005; Carlson et al. 2010). Collectively, these studies highlight the importance of a balance between host resistance and tolerance mechanisms in a pathogen-specific manner.
Efferocytosis: Considering the high turnover of cells in our body under physiological conditions (∼ one million cells die/second), the removal of this cumbersome number of dying cells is essential for host survival. Recent studies have identified efferocytosis (from the Latin “to bury”) as a critical mechanism for the disposal of cell corpses, a mechanism that differs from classical phagocytosis (Ravichandran 2010). Interestingly, this disposal is mandatory in both host resistance and tolerance to infection. For instance, during Mtb infection, efferocytosis is a crucial promoter of macrophage bactericidal activity following the engulfment of Mtb-infected apoptotic cells (Martin et al. 2012). We have also recently demonstrated that efferocytosis by dendritic cells is required for cross-presentation to enhance adaptive immunity during Mtb infection (Tzelepis et al. 2015). TGF-β, together with IL-10 and prostaglandins, have furthermore been shown to be produced by dendritic cells following the efferocytosis of apoptotic cells in order to decrease lung inflammation (Voll et al. 1997; Fadok et al. 1998; Huynh et al. 2002). During IAV infection, alveolar macrophages (aMφ) are critical for the clearance of apoptotic bodies during the resolution phase of infection and thereby contribute importantly to the resolution of inflammation (Kosmider et al. 2012; Nelson et al. 2014). Similarly, the depletion of aMφ during Streptococcus pneumoniae infection is associated with a poor outcome, because of impaired apoptotic cell clearance (Knapp et al. 2003). The removal of these apoptotic bodies by aMφ has been shown to limit the release of cellular content that may further enhance the inflammation. This anti-inflammatory effect is mediated by the activation of the peroxisome proliferator-activated receptor γ (PPARγ) following the interaction of apoptotic cells with aMφ. It consequently contributes to the AAMφ macrophage phenotype to foster wound healing and the resolution of inflammation (Sica and Mantovani 2012; Mantovani et al. 2013; Novak and Koh 2013; von Knethen et al. 2013).
CD200 signaling pathway: Another mechanism promoting tolerance in the lungs is mediated by CD200R, which is expressed by aMφ and dampens their inflammatory state following the interaction with CD200 expressed by epithelial cells. Studies in CD200-null or CD200R−/− mice have demonstrated that the absence of this signaling pathway leads to the delayed resolution of inflammation and subsequently enhances host morbidity and mortality after IAV infection. The absence of CD200R-CD200 signaling has also been associated with the enhanced influx of both CD4+ and CD8+ T cells in the lungs, further augmenting lung immunopathology (Snelgrove et al. 2008; Rygiel et al. 2009). Whether this interesting lung tolerance strategy is relevant to other infections remains to be determined.
Innate lymphoid cells (ILCs): ILCs represent a family of cells that are of lymphoid origin and that do not express traditional lineage markers (e.g., CD3, CD19, CD11b). Based on the expression of cell surface markers and transcription factors, they are divided into three major subsets (ILC1-3), each with unique functions (Saenz et al. 2010; Spits and Di Santo 2011). During IAV infection, epithelial cells produce IL-33 to induce the secretion of amphiregulin by ILC2 to promote tissue repair mechanisms. Although the depletion of ILC2 does not impair host resistance to IAV, it significantly affects host tolerance as determined by the loss of airway epithelial integrity, decreased lung function, and impaired restoration of airway homeostasis and repair (Monticelli et al. 2011; Zaiss et al. 2015). In addition, a recent study demonstrated that multiple pathogenic strains of RSV induce the production of IL-13 (downstream of IL-33) by ILC2 in mice (Stier et al. 2016). Conversely, rhinovirus-infected epithelial cells have been shown to produce IL-33 to trigger the production of type 2 cytokines by ILC2, which subsequently promotes airway inflammation and exacerbates asthma (Jackson et al. 2014), suggesting a potentially dichotomous role for ILC2 in immunity to acute viral infections.
Although host tolerance is essential during primary infection, it may increase susceptibility to other pathogens. For example, the induction of a tolerant state in the lungs following IAV infection elevates vulnerability to subsequent bacterial infections (Didierlaurent et al. 2008). IAV infection has been shown to desensitize aMφ, rendering them irresponsive to subsequent TLR stimulation by inhibiting the nuclear translocation of NF-κB-p65 (Didierlaurent et al. 2008). This impaired TLR signaling allows invading bacteria to remain undetected by the innate immune response, promoting pathogenicity. Moreover, the induction of pulmonary tolerance through the CD200-CD200R axis has also been associated with increased susceptibility to secondary bacterial infections (Goulding et al. 2011). Similarly, infection with rhinovirus predisposes mice for subsequent infection with the bacterium Haemophilus influenzae by desensitizing aMφ and bronchial epithelial cells to TLR stimuli (Unger et al. 2012). Furthermore, mice stimulated intranasally with the TLR3 agonist poly I:C also exhibit more severe prognosis to secondary infection (Tian et al. 2012). Interestingly, in the case of IAV infection, this susceptibility appears to be mediated by type I IFN (Tian et al. 2012). Although the anti-viral role of type I IFN is well defined, its role in promoting secondary bacterial infections is less clear. However, the increase in host susceptibility to primary bacterial infections including Francisella tularensis (Freudenberg et al. 2002), Listeria monocytogenes (Fehr et al. 1997) and Mtb (Manca et al. 2005) via type I IFN induction is well documented, signifying a permissive role of type I IFN in secondary bacterial infection. We have previously reviewed this concept (Divangahi et al. 2015).
Inefficiency of host tolerance mechanisms in vital organs is catastrophic and often leads to mortality. The consequences of the replication of the pathogen and a robust immune response in the lungs are severe epithelial loss, increased airway resistance, diminished gas exchange and ultimately, respiratory failure. Highly pathogenic strains of IAV (e.g., 1918 Spanish H1N1 or H5N1), severe acute respiratory syndrome (SARS), or hantavirus infections can induce considerable inflammation in the lungs resulting in death (de Jong et al. 2006; Macneil et al. 2011; Gralinski and Baric 2015). The sequential events and factors contributing to such exacerbated immune responses and the breakdown of host tolerance processes are still not well understood. However, both host and pathogen factors have been shown to contribute to this effect. Indeed, pathogen-intrinsic characteristics, such as the expression of virulence factors, replication rate and tissue tropism, markedly affect the host response. Additionally, the host itself can express genes that will either favor resistance and survival to infection or predispose it to severe immunopathology (Keynan et al. 2013; Arcanjo et al. 2014; Charbonnel et al. 2014). For instance, IL-17A, IL-17F and TNF-α are rapidly secreted following IAV infection and animals deficient in these cytokines show reduced morbidity compared with wild-type animals (Szretter et al. 2007; Crowe et al. 2009). Interestingly, others have observed that TNF-α deficiency during IAV infection leads to increased immunopathology characterized by elevated cellular infiltration and cytokine production in the airways. Thus, depending on the phase of infection, TNF-α may play a dual role by also acting as a negative regulator of inflammatory responses to control immunopathology (Damjanovic et al. 2011). A delay in type I IFN production during SARS infection promotes the accumulation of inflammatory monocytes-macrophages in the lungs, which in turn leads to elevated cytokine and chemokine levels (cytokine storm), increased vascular leakage and decreased survival (Channappanavar et al. 2016). Thus, an understanding of the underlying cellular and molecular mechanisms involved in the induction and maintenance of host tolerance during pulmonary viral infection may provide new avenues for developing therapeutic approaches.
Host tolerance in chronic pulmonary infection
Although host tolerance aims to reduce or control tissue damage inflicted by acute infection, it may also be a powerful defense strategy for host survival in chronic infections. This adaptation strategy is a reflection of the co-evolutionary dynamics of host–pathogen interactions (Best et al. 2014). The best example of this unique interaction between a pathogen and human is perhaps Mtb, as humans are the only reservoir for this bacterium (Comas et al. 2010). Exposure to Mtb leads to two broad outcomes: elimination of the bacteria or its persistence. It has long been recognized that, even among close household contacts of TB cases and despite ample exposure, nearly half of the exposed individuals are negative for the tuberculin skin test (TST), which demonstrates that they are disease-free (Morrison et al. 2008). This finding indicates that some people are naturally resistant to Mtb, because of an efficient immune response (Cobat et al. 2009). However, if Mtb is not eliminated, the pathogen can persist in a quiescent or latent state and typically, the individual develops latent tuberculosis infection (LTBI). Although one third of the world population is infected with Mtb and approximately 1.5 million people die from this pathogen each year (Barry et al. 2009), only 5–15% of individuals with LTBI progress (over months to years) to active TB (Vynnycky and Fine 1997). The specific mechanisms leading to this protection are still elusive in the vast majority of Mtb-infected people but several experimental studies in small mammals (mice, guinea pigs, rabbits) and non-human primates have significantly contributed to identifying the importance of early, probably innate, immune events during primary infection (Orme et al. 2015).
The importance of innate immune mechanisms is reflected by its remarkable existence and diversity at almost every level of the evolutionary tree of life. Macrophages are a particularly ancient cellular compartment of innate immunity and are the dominant cell type that Mtb infects. The route of entry of Mtb is mainly via the respiratory tract through the inhalation of bacteria and it passages to the lower respiratory tract, where it encounters aMφ. Mtb is internalized by macrophages via a vacuolar structure called a phagosome. Unlike organisms that are destroyed when the phagosome fuses with the lysosome, Mtb actively blocks phagosomal maturation, ensuring its survival in the phagocytic compartment (Podinovskaia et al. 2013). Next, through an ESX-1 mediated process, Mtb disrupts the phagosomal membrane and translocates into the cytosol (Houben et al. 2012). The advantages for the bacterium being delivered into the cytosol are a matter of ongoing investigation but one possibility is that the activation of the cytosolic surveillance pathway results in the induction of type I IFN, which is beneficial for Mtb (Pandey et al. 2009; Manzanillo et al. 2012). Although type I IFN is a major component of antiviral host defense mechanisms (Coulombe et al. 2014) and most viruses contain genes to block the type I IFN pathway, Mtb expresses genes (Stanley et al. 2007) to activate the type I IFN pathway to promote bacterial growth (Manca et al. 2001; Mayer-Barber et al. 2014). Importantly, a type I IFN gene signature has been directly linked to active human TB (Berry et al. 2010). Thus, Mtb has evolved into a parasite of the intracellular milieu of macrophages, where it not only survives but replicates in a naturally hostile environment. This allows Mtb access to the lung interstitium to initiate granuloma formation, which is the early stage of chronic infection. However, how Mtb is translocated from the airways into the parenchyma for the progression of infection is unknown. Do Mtb-infected macrophages migrate through pneumocytes? Or alternatively, do free bacteria directly infect pneumocytes and reach the lung interstitial tissue where they are phagocytosed by interstitial macrophages? As the success of Mtb in establishing chronic infection is dictated by the initial actions of pulmonary macrophages, early inhibition of macrophage activation and eventually cell death are critical for Mtb survival and replication. Experimental studies of a variety of pathogens have shown that the fate of pulmonary macrophages (i.e., the type of cell death) is critical not only for the innate response to infection (Divangahi et al. 2009; Singh et al. 2012) but also for the ensuing adaptive immune response to Mtb infection (Schaible et al. 2003; Behar et al. 2010; Divangahi et al. 2010, 2015; Coulombe et al. 2014; Tzelepis et al. 2015).
Once the primary infection is established, inflammatory monocytes transport Mtb to pulmonary lymph nodes and transfer Mtb antigens to classic dendritic cells for T cell priming (Samstein et al. 2013). T cell responses are essential in immunity to Mtb infection by restricting bacterial growth. Conversely, through various mechanisms, Mtb actively delays initial T cell priming and their trafficking into the lung (Chackerian et al. 2002; Wolf et al. 2008). HIV infection is a clear risk factor for the progression from Mtb infection to disease, because of the significant reduction of CD4+ T cells. However, for the purposes of vaccination, whether increased T cell responses above the population norm provide better protection is unclear. Indeed, recent studies in the experimental murine model of TB have shown that unleashing CD4+ T cell responses in a PD1-dependent manner leads to reduced protection and enhanced mortality (Aubert et al. 2011; Barber et al. 2011). Thus, an understanding of the regulatory mechanisms involved in immunity to TB is fundamental for generating a strong host defense to hinder bacterial growth, while maintaining host tolerance.
One can hypothesize that, during Mtb infection, the initiation of granuloma formation represents the transition of host defense mechanisms from resistance to tolerance. Granulomas have long been thought of as a critical component of host protective immunity to Mtb infection (Davis et al. 2002). However, they have recently been shown also to be beneficial to pathogens (Kaplan et al. 2003; Hunter 2011; Davis and Ramakrishnan 2009; Cronan et al. 2016). For instance, Mtb requires complex granuloma formation at various stages of disease progression for a more efficient spread within individuals (Hunter 2011). The extracellular location of bacteria in chronic granulomas is associated with highly elevated replication rates (Kaplan et al. 2003). In contrast, immunocompromised individuals usually only display poorly formed granulomas and have lower extracellular bacterial burdens (Hunter 2011). Interestingly, a recent study demonstrated that immune responses are geographically segregated, with the center of the granuloma being pro-inflammatory, while the surrounding tissue is anti-inflammatory (Marakalala et al. 2016). In addition, Mtb can initiate a type I IFN response, which has been directly linked to the recruitment of a unique myeloid population (CD11b+F4/80+Gr1int) to the nascent granuloma; this population is highly permissive for Mtb infection (Antonelli et al. 2010).
Similar to monocytes and macrophages, the protective role of T cells during Mtb infection has recently been scrutinized. Conventionally, the identification of immunodominant Mtb antigens for the generation of a repertoire of Mtb-specific T cells was thought to be the foundation for T cell-mediated protective immunity and, hence, an effective vaccine-based strategy against TB. However, despite inducing enhanced T-cell-mediated responses, one such vaccine has failed to improve protection in a human trial (Tameris et al. 2013). After more than half a century of BCG vaccination, we still do not know the precise way in which BCG provides protection in children and to what extent this protection is mediated via CD4+ T cells. An emerging hypothesis is that BCG protection is mainly mediated through innate immune pathways, as reviewed elsewhere (Blankley et al. 2014). In addition, the finding that Mtb genes involved in the production of immunodominant CD4+ T cell antigens are hyper-conserved suggests that Mtb may paradoxically benefit from antigen-specific CD4+ T cell activation in humans (Comas et al. 2010). This theory derives further indirect support from the HIV-TB syndemic: whereas HIV is clearly a risk factor for individual progression from Mtb infection to disease, HIV/AIDS is negatively associated with contagion (Corbett et al. 2006). Furthermore, the risk of active TB is enhanced during the early stage of HIV infection, when the number of CD4+ T cells is still in the normal range (Sonnenberg et al. 2005). Together, these observations argue that Mtb depends on the elaboration of a T-cell-mediated immune response for the development of pathology that enables it to be transmitted to other humans.
Concluding remarks
Whereas the concept of host tolerance has been well established in plant biology, an appreciation of this phenomenon in the animal kingdom is just emerging. This delay is mainly attributable to the dominant conventional concept of host resistance to infectious diseases. The cellular and regulatory mechanisms of host tolerance appear to be pathogen-specific, which is mainly reflected by the mode of pathogen transmission. For instance, during an IAV pandemic (e.g., with the 1918 Spanish strain), the success of the influenza virus depends on rapid replication and early transmission. This kinetic of infection ultimately generates a dysregulated immune response, in terms of both the intensity and the duration, which leads to a breakdown of host tolerance. Although this massive immune response completely eliminates the pathogen, host survival will still be significantly compromised in the absence of tolerance. In sharp contrast to the influenza virus, Mtb survival depends on host tolerance. Death from tuberculosis was initially known as “consumption” as this chronic infection causes dramatic cachexia (wasting). Interestingly, a study examining Mycobacterium marinum infection in the fruit fly Drosophila melanogaster showed that the increased mortality is independent of bacterial load and is mediated by altered host metabolism and increased body wasting (Lazzaro and Galac 2006). This result indicates that, in the absence of tolerance, host resistance is not sufficient to control the infection. Therefore, when human resistance mechanisms fail to reduce the fitness of Mtb and to eliminate the bacteria during the early phase of infection, the host alters its defense strategy from antagonism to symbiosis, which leads to tolerance of the bacteria and chronic, if not lifelong, infection. However, unraveling the cellular and molecular mechanisms involved in the regulation of host tolerance is essential for a better understanding of the pathogenesis of any infectious disease.
Acknowledgements
This work was supported by the Canadian Institute of Health Research (CIHR) Foundation Grant (FDN-143273) to M.D., who also holds a CIHR New Investigator Award and by the Fonds de la Recherche du Quebec-Sante (FRQS). I.M. is a recipient of the American Association of Immunologists Careers in Immunology Awards. E.K. is a recipient of funds from the German Research Foundation (DFG).
Footnotes
Isabelle Meunier and Eva Kaufmann contributed equally to this work.
References
- Allen JE, Maizels RM. Diversity and dialogue in immunity to helminths. Nat Rev Immunol. 2011;11:375–388. doi: 10.1038/nri2992. [DOI] [PubMed] [Google Scholar]
- Allen JE, Sutherland TE. Host protective roles of type 2 immunity: parasite killing and tissue repair, flip sides of the same coin. Semin Immunol. 2014;26:329–340. doi: 10.1016/j.smim.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli LR, Gigliotti Rothfuchs A, Goncalves R, Roffe E, Cheever AW, Bafica A, Salazar AM, Feng CG, Sher A. Intranasal poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest. 2010;120:1674–1682. doi: 10.1172/JCI40817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcanjo AC, Mazzocco G, de Oliveira SF, Plewczynski D, Radomski JP. Role of the host genetic variability in the influenza A virus susceptibility. Acta Biochim Pol. 2014;61:403–419. [PubMed] [Google Scholar]
- Aubert RD, Kamphorst AO, Sarkar S, Vezys V, Ha SJ, Barber DL, Ye L, Sharpe AH, Freeman GJ, Ahmed R. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc Natl Acad Sci U S A. 2011;108:21182–21187. doi: 10.1073/pnas.1118450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres JS, Schneider DS. A signaling protease required for melanization in Drosophila affects resistance and tolerance to infections. PLoS Biol. 2008;6:e305. doi: 10.1371/journal.pbio.0060305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres JS, Schneider DS. The role of anorexia in resistance and tolerance to infection in Drosophila. PLoS Biol. 2009;7:e1000150. doi: 10.1371/journal.pbio.1000150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres JS, Schneider DS. Tolerance of infections. Annu Rev Immunol. 2012;30:271–294. doi: 10.1146/annurev-immunol-020711-075030. [DOI] [PubMed] [Google Scholar]
- Ayres JS, Freitag N, Schneider DS. Identification of Drosophila mutants altering defense of and endurance to Listeria monocytogenes infection. Genetics. 2008;178:1807–1815. doi: 10.1534/genetics.107.083782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber DL, Mayer-Barber KD, Feng CG, Sharpe AH, Sher A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J Immunol. 2011;186:1598–1607. doi: 10.4049/jimmunol.1003304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry CE, 3rd, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, Schnappinger D, Wilkinson RJ, Young D. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol. 2009;7:845–855. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar SM, Divangahi M, Remold HG. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat Rev Microbiol. 2010;8:668–674. doi: 10.1038/nrmicro2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr MA, Divangahi M. Freund’s adjuvant, NOD2 and mycobacteria. Curr Opin Microbiol. 2015;23:126–132. doi: 10.1016/j.mib.2014.11.015. [DOI] [PubMed] [Google Scholar]
- Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O’Garra A. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best A, White A, Boots M. The coevolutionary implications of host tolerance. Evolution. 2014;68:1426–1435. doi: 10.1111/evo.12368. [DOI] [PubMed] [Google Scholar]
- Blankley S, Berry MP, Graham CM, Bloom CI, Lipman M, O’Garra A. The application of transcriptional blood signatures to enhance our understanding of the host response to infection: the example of tuberculosis. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130427. doi: 10.1098/rstb.2013.0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braciale TJ, Sun J, Kim TS. Regulating the adaptive immune response to respiratory virus infection. Nat Rev Immunol. 2012;12:295–305. doi: 10.1038/nri3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buccheri S, Reljic R, Caccamo N, Ivanyi J, Singh M, Salerno A, Dieli F. IL-4 depletion enhances host resistance and passive IgA protection against tuberculosis infection in BALB/c mice. Eur J Immunol. 2007;37:729–737. doi: 10.1002/eji.200636764. [DOI] [PubMed] [Google Scholar]
- Carlson CM, Turpin EA, Moser LA, O’Brien KB, Cline TD, Jones JC, Tumpey TM, Katz JM, Kelley LA, Gauldie J, Schultz-Cherry S. Transforming growth factor-beta: activation by neuraminidase and role in highly pathogenic H5N1 influenza pathogenesis. PLoS Pathog. 2010;6:e1001136. doi: 10.1371/journal.ppat.1001136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes initiation of T-cell immunity. Infect Immun. 2002;70:4501–4509. doi: 10.1128/IAI.70.8.4501-4509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, Dekruyff RH, Umetsu DT. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12:631–638. doi: 10.1038/ni.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, Perlman S. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. 2016;19:181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbonnel N, Pages M, Sironen T, Henttonen H, Vapalahti O, Mustonen J, Vaheri A. Immunogenetic factors affecting susceptibility of humans and rodents to hantaviruses and the clinical course of hantaviral disease in humans. Viruses. 2014;6:2214–2241. doi: 10.3390/v6052214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O’Neill R, Schickli J, Palese P, Henklein P, Bennink JR, Yewdell JW. A novel influenza A virus mitochondrial protein that induces cell death. Nat Med. 2001;7:1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- Cobat A, Gallant CJ, Simkin L, Black GF, Stanley K, Hughes J, Doherty TM, Hanekom WA, Eley B, Jais JP, Boland-Auge A, van Helden P, Casanova JL, Abel L, Hoal EG, Schurr E, Alcais A. Two loci control tuberculin skin test reactivity in an area hyperendemic for tuberculosis. J Exp Med. 2009;206:2583–2591. doi: 10.1084/jem.20090892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, Ernst JD, Gagneux S. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet. 2010;42:498–503. doi: 10.1038/ng.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett EL, Marston B, Churchyard GJ, De Cock KM. Tuberculosis in sub-Saharan Africa: opportunities, challenges, and change in the era of antiretroviral treatment. Lancet. 2006;367:926–937. doi: 10.1016/S0140-6736(06)68383-9. [DOI] [PubMed] [Google Scholar]
- Coulombe F, Divangahi M, Veyrier F, Leseleuc L de, Gleason JL, Yang Y, Kelliher MA, Pandey AK, Sassetti CM, Reed MB, Behr MA (2009) Increased NOD2-mediated recognition of N-glycolyl muramyl dipeptide. J Exp Med 206:1709–1716 [DOI] [PMC free article] [PubMed]
- Coulombe F, Jaworska J, Verway M, Tzelepis F, Massoud A, Gillard J, Wong G, Kobinger G, Xing Z, Couture C, Joubert P, Fritz JH, Powell WS, Divangahi M. Targeted prostaglandin E2 inhibition enhances antiviral immunity through induction of type I interferon and apoptosis in macrophages. Immunity. 2014;40:554–568. doi: 10.1016/j.immuni.2014.02.013. [DOI] [PubMed] [Google Scholar]
- Cronan MR, Beerman RW, Rosenberg AF, Saelens JW, Johnson MG, Oehlers SH, Sisk DM, Smith KLJ, Medvitz NA, Miller SE, Trinh LA, Fraser SE, Madden JF, Turner J, Stout JE, Lee S, Tobin DM (2016) Macrophage epithelial reprogramming underlies mycobacterial granuloma formation and promotes infection. Immunity 45(4):861–876 [DOI] [PMC free article] [PubMed]
- Crowe CR, Chen K, Pociask DA, Alcorn JF, Krivich C, Enelow RI, Ross TM, Witztum JL, Kolls JK. Critical role of IL-17RA in immunopathology of influenza infection. J Immunol. 2009;183:5301–5310. doi: 10.4049/jimmunol.0900995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damjanovic D, Divangahi M, Kugathasan K, Small CL, Zganiacz A, Brown EG, Hogaboam CM, Gauldie J, Xing Z. Negative regulation of lung inflammation and immunopathology by TNF-alpha during acute influenza infection. Am J Pathol. 2011;179:2963–2976. doi: 10.1016/j.ajpath.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davicino RC, Eliçabe RJ, Di Genaro MS, Rabinovich GA. Coupling pathogen recognition to innate immunity through glycan-dependent mechanisms. Int Immunopharmacol. 2011;11:1457–1463. doi: 10.1016/j.intimp.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Davis JM, Ramakrishnan L (2009) The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 136(1):37–49 [DOI] [PMC free article] [PubMed]
- Davis JM, Clay H, Lewis JL, Ghori N, Herbomel P, Ramakrishnan L. Real-time visualization of mycobacterium-macrophage interactions leading to initiation of granuloma formation in zebrafish embryos. Immunity. 2002;17:693–702. doi: 10.1016/S1074-7613(02)00475-2. [DOI] [PubMed] [Google Scholar]
- Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didierlaurent A, Goulding J, Patel S, Snelgrove R, Low L, Bebien M, Lawrence T, Rijt LS van, Lambrecht BN, Sirard JC, Hussell T (2008) Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J Exp Med 205:323–329 [DOI] [PMC free article] [PubMed]
- Divangahi M, Mostowy S, Coulombe F, Kozak R, Guillot L, Veyrier F, Kobayashi KS, Flavell RA, Gros P, Behr MA. NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity. J Immunol. 2008;181:7157–7165. doi: 10.4049/jimmunol.181.10.7157. [DOI] [PubMed] [Google Scholar]
- Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, Fortune S, Behar SM, Remold HG. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat Immunol. 2009;10:899–906. doi: 10.1038/ni.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat Immunol. 2010;11:751–758. doi: 10.1038/ni.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divangahi M, King IL, Pernet E. Alveolar macrophages and type I IFN in airway homeostasis and immunity. Trends Immunol. 2015;36:307–314. doi: 10.1016/j.it.2015.03.005. [DOI] [PubMed] [Google Scholar]
- Doly J, Civas A, Navarro S, Uze G. Type I interferons: expression and signalization. Cell Mol Life Sci. 1998;54:1109–1121. doi: 10.1007/s000180050240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entezari M, Weiss DJ, Sitapara R, Whittaker L, Wargo MJ, Li J, Wang H, Yang H, Sharma L, Phan BD, Javdan M, Chavan SS, Miller EJ, Tracey KJ, Mantell LL. Inhibition of high-mobility group box 1 protein (HMGB1) enhances bacterial clearance and protects against Pseudomonas aeruginosa pneumonia in cystic fibrosis. Mol Med. 2012;18:477–485. doi: 10.2119/molmed.2012.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr T, Schoedon G, Odermatt B, Holtschke T, Schneemann M, Bachmann MF, Mak TW, Horak I, Zinkernagel RM. Crucial role of interferon consensus sequence binding protein, but neither of interferon regulatory factor 1 nor of nitric oxide synthesis for protection against murine listeriosis. J Exp Med. 1997;185:921–931. doi: 10.1084/jem.185.5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferwerda G, Girardin SE, Kullberg BJ, Le Bourhis L, Jong DJ de, Langenberg DM, Crevel R van, Adema GJ, Ottenhoff TH, Van der Meer JW, Netea MG (2005) NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog 1:279–285 [DOI] [PMC free article] [PubMed]
- Freudenberg MA, Merlin T, Kalis C, Chvatchko Y, Stubig H, Galanos C. Cutting edge: a murine, IL-12-independent pathway of IFN-gamma induction by gram-negative bacteria based on STAT4 activation by Type I IFN and IL-18 signaling. J Immunol. 2002;169:1665–1668. doi: 10.4049/jimmunol.169.4.1665. [DOI] [PubMed] [Google Scholar]
- Gandotra S, Schnappinger D, Monteleone M, Hillen W, Ehrt S. In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nat Med. 2007;13:1515–1520. doi: 10.1038/nm1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garofalo RP, Kolli D, Casola A. Respiratory syncytial virus infection: mechanisms of redox control and novel therapeutic opportunities. Antioxid Redox Signal. 2013;18:186–217. doi: 10.1089/ars.2011.4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding J, Godlee A, Vekaria S, Hilty M, Snelgrove R, Hussell T. Lowering the threshold of lung innate immune cell activation alters susceptibility to secondary bacterial superinfection. J Infect Dis. 2011;204:1086–1094. doi: 10.1093/infdis/jir467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gralinski LE, Baric RS. Molecular pathology of emerging coronavirus infections. J Pathol. 2015;235:185–195. doi: 10.1002/path.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guirado E, Schlesinger LS, Kaplan G. Macrophages in tuberculosis: friend or foe. Semin Immunopathol. 2013;35:563–583. doi: 10.1007/s00281-013-0388-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitmann L, Abad Dar M, Schreiber T, Erdmann H, Behrends J, McKenzie AN, Brombacher F, Ehlers S, Holscher C. The IL-13/IL-4Ralpha axis is involved in tuberculosis-associated pathology. J Pathol. 2014;234:338–350. doi: 10.1002/path.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helming L, Gordon S. Macrophage fusion induced by IL-4 alternative activation is a multistage process involving multiple target molecules. Eur J Immunol. 2007;37:33–42. doi: 10.1002/eji.200636788. [DOI] [PubMed] [Google Scholar]
- Holt PG, Strickland DH, Wikstrom ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol. 2008;8:142–152. doi: 10.1038/nri2236. [DOI] [PubMed] [Google Scholar]
- Houben D, Demangel C, Ingen J van, Perez J, Baldeon L, Abdallah AM, Caleechurn L, Bottai D, Zon M van, Punder K de, Laan T van der, Kant A, Bossers-de Vries R, Willemsen P, Bitter W, Soolingen D van, Brosch R, Wel N van der, Peters PJ (2012) ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol 14:1287–1298 [DOI] [PubMed]
- Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, Eisenberg D, Russell RG, Derrick SC, Collins FM, Morris SL, King CH, Jacobs WR., Jr The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci U S A. 2003;100:12420–12425. doi: 10.1073/pnas.1635213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter RL. Pathology of post primary tuberculosis of the lung: an illustrated critical review. Tuberculosis (Edinb) 2011;91:497–509. doi: 10.1016/j.tube.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI0211638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inohara N, Chamaillard M, McDonald C, Nunez G. NOD-LRR proteins: role in host–microbial interactions and inflammatory disease. Annu Rev Biochem. 2005;74:355–383. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. 2014;14:315–328. doi: 10.1038/nri3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, Jerico D-R, Telcian AG, Nikonova A, Zhu J, Aniscenko J, Gogsadze L, Bakhsoliani E, Traub S, Dhariwal J, Porter J, Hunt D, Hunt T, Stanciu LA, Khaitov M, Bartlett NW, Edwards MR, Kon OM, Mallia P, Papadopoulos NG, Akdis CA, Westwick J, Edwards MJ, Cousins DJ, Walton RP, Johnston SL. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med. 2014;190:1373–1382. doi: 10.1164/rccm.201406-1039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway CA., Jr Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54:1–13. doi: 10.1101/SQB.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- Jaworska J, Coulombe F, Downey J, Tzelepis F, Shalaby K, Tattoli I, Berube J, Rousseau S, Martin JG, Girardin SE, McCullers JA, Divangahi M. NLRX1 prevents mitochondrial induced apoptosis and enhances macrophage antiviral immunity by interacting with influenza virus PB1-F2 protein. Proc Natl Acad Sci U S A. 2014;111:E2110–E2119. doi: 10.1073/pnas.1322118111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jondle CN, Sharma A, Simonson TJ, Larson B, Mishra BB, Sharma J. Macrophage galactose-type lectin-1 deficiency is associated with increased neutrophilia and hyperinflammation in gram-negative pneumonia. J Immunol. 2016;196:3088–3096. doi: 10.4049/jimmunol.1501790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong MD de, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC, Qui PT, Cam BV, Ha do Q, Guan Y, Peiris JS, Chinh NT, Hien TT, Farrar J (2006) Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 12:1203–1207 [DOI] [PMC free article] [PubMed]
- Jung YJ, LaCourse R, Ryan L, North RJ. Evidence inconsistent with a negative influence of T helper 2 cells on protection afforded by a dominant T helper 1 response against Mycobacterium tuberculosis lung infection in mice. Infect Immun. 2002;70:6436–6443. doi: 10.1128/IAI.70.11.6436-6443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan G, Post FA, Moreira AL, Wainwright H, Kreiswirth BN, Tanverdi M, Mathema B, Ramaswamy SV, Walther G, Steyn LM, Barry CE, 3rd, Bekker LG. Mycobacterium tuberculosis growth at the cavity surface: a microenvironment with failed immunity. Infect Immun. 2003;71:7099–7108. doi: 10.1128/IAI.71.12.7099-7108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Keynan Y, Malik S, Fowke KR. The role of polymorphisms in host immune genes in determining the severity of respiratory illness caused by pandemic H1N1 influenza. Public Health Genom. 2013;16:9–16. doi: 10.1159/000345937. [DOI] [PubMed] [Google Scholar]
- Khalil N. TGF-beta: from latent to active. Microbes Infect. 1999;1:1255–1263. doi: 10.1016/S1286-4579(99)00259-2. [DOI] [PubMed] [Google Scholar]
- Knapp S, Leemans JC, Florquin S, Branger J, Maris NA, Pater J, Rooijen N van, Poll T van der (2003) Alveolar macrophages have a protective antiinflammatory role during murine pneumococcal pneumonia.Am J Respir Crit Care Med 167:171–179 [DOI] [PubMed]
- Knethen A von, Sha LK, Kuchler L, Heeg AK, Fuhrmann D, Heide H, Wittig I, Maier TJ, Steinhilber D, Brune B (2013) 5-Lipoxygenase contributes to PPARgamma activation in macrophages in response to apoptotic cells. Cell Signal 25:2762–2768 [DOI] [PubMed]
- Kohlmeier JE, Cookenham T, Roberts AD, Miller SC, Woodland DL. Type I interferons regulate cytolytic activity of memory CD8(+) T cells in the lung airways during respiratory virus challenge. Immunity. 2010;33:96–105. doi: 10.1016/j.immuni.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmider B, Messier EM, Janssen WJ, Nahreini P, Wang J, Hartshorn KL, Mason RJ. Nrf2 protects human alveolar epithelial cells against injury induced by influenza A virus. Respir Res. 2012;13:43. doi: 10.1186/1465-9921-13-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kover PX, Schaal BA. Genetic variation for disease resistance and tolerance among Arabidopsis thaliana accessions. Proc Natl Acad Sci U S A. 2002;99:11270–11274. doi: 10.1073/pnas.102288999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- Lazzaro BP, Galac MR. Disease pathology: wasting energy fighting infection. Curr Biol. 2006;16:R964–R965. doi: 10.1016/j.cub.2006.10.015. [DOI] [PubMed] [Google Scholar]
- Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/S0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- Loebbermann J, Schnoeller C, Thornton H, Durant L, Sweeney NP, Schuijs M, O’Garra A, Johansson C, Openshaw PJ. IL-10 regulates viral lung immunopathology during acute respiratory syncytial virus infection in mice. PLoS ONE. 2012;7:e32371. doi: 10.1371/journal.pone.0032371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macneil A, Nichol ST, Spiropoulou CF. Hantavirus pulmonary syndrome. Virus Res. 2011;162:138–147. doi: 10.1016/j.virusres.2011.09.017. [DOI] [PubMed] [Google Scholar]
- Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, Barry CE, 3rd, Freedman VH, Kaplan G. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc Natl Acad Sci U S A. 2001;98:5752–5757. doi: 10.1073/pnas.091096998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, Barry C, Kaplan G. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res. 2005;25:694–701. doi: 10.1089/jir.2005.25.694. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229:176–185. doi: 10.1002/path.4133. [DOI] [PubMed] [Google Scholar]
- Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe. 2012;11:469–480. doi: 10.1016/j.chom.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marakalala MJ, Raju RM, Sharma K, Zhang YJ, Eugenin EA, Prideaux B, Daudelin IB, Chen PY, Booty MG, Kim JH, Eum SY, Via LE, Behar SM, Barry CE, 3rd, Mann M, Dartois V, Rubin EJ. Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat Med. 2016;22:531–538. doi: 10.1038/nm.4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CJ, Booty MG, Rosebrock TR, Nunes-Alves C, Desjardins DM, Keren I, Fortune SM, Remold HG, Behar SM. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe. 2012;12:289–300. doi: 10.1016/j.chom.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei W, Yuan X, Zhang G, Cai Y, Babu S, Catalfamo M, Salazar AM, Via LE, Barry CE, 3rd, Sher A. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. 2014;511:99–103. doi: 10.1038/nature13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley JL, Hornung F, Boyd KL, Smith AM, McKeon R, Bennink J, Yewdell JW, McCullers JA. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe. 2007;2:240–249. doi: 10.1016/j.chom.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley JL, Chipuk JE, Boyd KL, Van De Velde N, Green DR, McCullers JA. PB1-F2 proteins from H5N1 and 20 century pandemic influenza viruses cause immunopathology. PLoS Pathog. 2010;6:e1001014. doi: 10.1371/journal.ppat.1001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinstry KK, Strutt TM, Buck A, Curtis JD, Dibble JP, Huston G, Tighe M, Hamada H, Sell S, Dutton RW, Swain SL. IL-10 deficiency unleashes an influenza-specific Th17 response and enhances survival against high-dose challenge. J Immunol. 2009;182:7353–7363. doi: 10.4049/jimmunol.0900657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;335:936–941. doi: 10.1126/science.1214935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, Kubota M, Turner D, Diamond JM, Goldrath AW, Farber DL, Collman RG, Wherry EJ, Artis D. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. 2011;12:1045–1054. doi: 10.1038/ni.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison J, Pai M, Hopewell PC. Tuberculosis and latent tuberculosis infection in close contacts of people with pulmonary tuberculosis in low-income and middle-income countries: a systematic review and meta-analysis. Lancet Infect Dis. 2008;8:359–368. doi: 10.1016/S1473-3099(08)70071-9. [DOI] [PubMed] [Google Scholar]
- Neagos J, Standiford TJ, Newstead MW, Zeng X, Huang SK, Ballinger MN. Epigenetic regulation of tolerance to Toll-like receptor ligands in alveolar epithelial cells. Am J Respir Cell Mol Biol. 2015;53:872–881. doi: 10.1165/rcmb.2015-0057OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson B, Zhou X, White M, Hartshorn K, Takahashi K, Kinane TB, Anandaiah A, Koziel H. Recombinant human mannose-binding lectin dampens human alveolar macrophage inflammatory responses to influenza A virus in vitro. J Leukoc Biol. 2014;95:715–722. doi: 10.1189/jlb.0313161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak ML, Koh TJ. Phenotypic transitions of macrophages orchestrate tissue repair. Am J Pathol. 2013;183:1352–1363. doi: 10.1016/j.ajpath.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo OO, Ryu MH, Jha A, Unrhu H, Halayko AJ. High-mobility group box 1 promotes extracellular matrix synthesis and wound repair in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2015;309:154–1366. doi: 10.1152/ajplung.00054.2015. [DOI] [PubMed] [Google Scholar]
- Orme IM, Robinson RT, Cooper AM. The balance between protective and pathogenic immune responses in the TB-infected lung. Nat Immunol. 2015;16:57–63. doi: 10.1038/ni.3048. [DOI] [PubMed] [Google Scholar]
- Palmer EM, Holbrook BC, Arimilli S, Parks GD, Alexander-Miller MA. IFNgamma-producing, virus-specific CD8+ effector cells acquire the ability to produce IL-10 as a result of entry into the infected lung environment. Virology. 2010;404:225–230. doi: 10.1016/j.virol.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey AK, Yang Y, Jiang Z, Fortune SM, Coulombe F, Behr MA, Fitzgerald KA, Sassetti CM, Kelliher MA (2009) NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog 5:e1000500 [DOI] [PMC free article] [PubMed]
- Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol. 2014;14:9–23. doi: 10.1038/nri3565. [DOI] [PubMed] [Google Scholar]
- Podinovskaia M, Lee W, Caldwell S, Russell DG. Infection of macrophages with Mycobacterium tuberculosis induces global modifications to phagosomal function. Cell Microbiol. 2013;15:843–859. doi: 10.1111/cmi.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Raberg L, Sim D, Read AF. Disentangling genetic variation for resistance and tolerance to infectious diseases in animals. Science. 2007;318:812–814. doi: 10.1126/science.1148526. [DOI] [PubMed] [Google Scholar]
- Raberg L, Graham AL, Read AF. Decomposing health: tolerance and resistance to parasites in animals. Philos Trans R Soc Lond B Biol Sci. 2009;364:37–49. doi: 10.1098/rstb.2008.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rast JP, Smith LC, Loza-Coll M, Hibino T, Litman GW. Genomic insights into the immune system of the sea urchin. Science. 2006;314:952–956. doi: 10.1126/science.1134301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med. 2010;207:1807–1817. doi: 10.1084/jem.20101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read AF, Graham AL, Raberg L. Animal defenses against infectious agents: is damage control more important than pathogen control. PLoS Biol. 2008;6:e4. doi: 10.1371/journal.pbio.1000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy E, Brennan J, Jolles S, Lowrie DB. Beneficial effect of anti-interleukin-4 antibody when administered in a murine model of tuberculosis infection. Tuberculosis (Edinb) 2008;88:197–202. doi: 10.1016/j.tube.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Rygiel TP, Rijkers ES, Ruiter T de, Stolte EH, Valk M van der, Rimmelzwaan GF, Boon L, Loon AM van, Coenjaerts FE, Hoek RM, Tesselaar K, Meyaard L (2009) Lack of CD200 enhances pathological T cell responses during influenza infection. J Immunol 183:1990–1996 [DOI] [PubMed]
- Saenz SA, Noti M, Artis D. Innate immune cell populations function as initiators and effectors in Th2 cytokine responses. Trends Immunol. 2010;31:407–413. doi: 10.1016/j.it.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Samstein M, Schreiber HA, Leiner IM, Susac B, Glickman MS, Pamer EG. Essential yet limited role for CCR2(+) inflammatory monocytes during Mycobacterium tuberculosis-specific T cell priming. eLife. 2013;2:e01086. doi: 10.7554/eLife.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaible UE, Winau F, Sieling PA, Fischer K, Collins HL, Hagens K, Modlin RL, Brinkmann V, Kaufmann SH. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat Med. 2003;9:1039–1046. doi: 10.1038/nm906. [DOI] [PubMed] [Google Scholar]
- Schneider DS, Ayres JS. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol. 2008;8:889–895. doi: 10.1038/nri2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz-Cherry S, Hinshaw VS. Influenza virus neuraminidase activates latent transforming growth factor beta. J Virol. 1996;70:8624–8629. doi: 10.1128/jvi.70.12.8624-8629.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V, Jamwal S, Jain R, Verma P, Gokhale R, Rao KV. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe. 2012;12:669–681. doi: 10.1016/j.chom.2012.09.012. [DOI] [PubMed] [Google Scholar]
- Snelgrove RJ, Goulding J, Didierlaurent AM, Lyonga D, Vekaria S, Edwards L, Gwyer E, Sedgwick JD, Barclay AN, Hussell T. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat Immunol. 2008;9:1074–1083. doi: 10.1038/ni.1637. [DOI] [PubMed] [Google Scholar]
- Soares MP, Gozzelino R, Weis S. Tissue damage control in disease tolerance. Trends Immunol. 2014;35:483–494. doi: 10.1016/j.it.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Sonnenberg P, Glynn JR, Fielding K, Murray J, Godfrey-Faussett P, Shearer S. How soon after infection with HIV does the risk of tuberculosis start to increase? A retrospective cohort study in South African gold miners. J Infect Dis. 2005;191:150–158. doi: 10.1086/426827. [DOI] [PubMed] [Google Scholar]
- Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. 2011;12:21–27. doi: 10.1038/ni.1962. [DOI] [PubMed] [Google Scholar]
- Stanley SA, Johndrow JE, Manzanillo P, Cox JS. The type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol. 2007;178:3143–3152. doi: 10.4049/jimmunol.178.5.3143. [DOI] [PubMed] [Google Scholar]
- Stier MT, Bloodworth MH, Toki S, Newcomb DC, Goleniewska K, Boyd KL, Quitalig M, Hotard AL, Moore ML, Hartert TV, Zhou B, McKenzie AN, Peebles RS., Jr Respiratory syncytial virus infection activates IL-13-producing group 2 innate lymphoid cells through thymic stromal lymphopoietin. J Allergy Clin Immunol. 2016;138:814–824. doi: 10.1016/j.jaci.2016.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Madan R, Karp CL, Braciale TJ. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med. 2009;15:277–284. doi: 10.1038/nm.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Cardani A, Sharma AK, Laubach VE, Jack RS, Muller W, Braciale TJ. Autocrine regulation of pulmonary inflammation by effector T-cell derived IL-10 during infection with respiratory syncytial virus. PLoS Pathog. 2011;7:e1002173. doi: 10.1371/journal.ppat.1002173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Cornell TT, LeVine A, Berlin AA, Hinkovska-Galcheva V, Fleszar AJ, Lukacs NW, Shanley TP. Dual role of interleukin-10 in the regulation of respiratory syncitial virus (RSV)-induced lung inflammation. Clin Exp Immunol. 2013;172:263–279. doi: 10.1111/cei.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szretter KJ, Gangappa S, Lu X, Smith C, Shieh WJ, Zaki SR, Sambhara S, Tumpey TM, Katz JM. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J Virol. 2007;81:2736–2744. doi: 10.1128/JVI.02336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, Shea JE, McClain JB, Hussey GD, Hanekom WA, Mahomed H, McShane H. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet. 2013;381:1021–1028. doi: 10.1016/S0140-6736(13)60177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Xu F, Lung WY, Meyerson C, Ghaffari AA, Cheng G, Deng JC. Poly I:C enhances susceptibility to secondary pulmonary infections by gram-positive bacteria. PLoS ONE. 2012;7:e41879. doi: 10.1371/journal.pone.0041879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzelepis F, Verway M, Daoud J, Gillard J, Hassani-Ardakani K, Dunn J, Downey J, Gentile ME, Jaworska J, Sanchez AM, Nedelec Y, Vali H, Tabrizian M, Kristof AS, King IL, Barreiro LB, Divangahi M. Annexin1 regulates DC efferocytosis and cross-presentation during Mycobacterium tuberculosis infection. J Clin Invest. 2015;125:752–768. doi: 10.1172/JCI77014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger BL, Faris AN, Ganesan S, Comstock AT, Hershenson MB, Sajjan US. Rhinovirus attenuates non-typeable Hemophilus influenzae-stimulated IL-8 responses via TLR2-dependent degradation of IRAK-1. PLoS Pathog. 2012;8:e1002969. doi: 10.1371/journal.ppat.1002969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga ZT, Ramos I, Hai R, Schmolke M, Garcia-Sastre A, Fernandez-Sesma A, Palese P. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 2011;7:e1002067. doi: 10.1371/journal.ppat.1002067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- Vynnycky E, Fine PE. The natural history of tuberculosis: the implications of age-dependent risks of disease and the role of reinfection. Epidemiol Infect. 1997;119:183–201. doi: 10.1017/S0950268897007917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welin A, Eklund D, Stendahl O, Lerm M. Human macrophages infected with a high burden of ESAT-6-expressing M. tuberculosis undergo caspase-1- and cathepsin B-independent necrosis. PLoS ONE. 2011;6:e20302. doi: 10.1371/journal.pone.0020302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitham S, Dinesh-Kumar SP, Choi D, Hehl R, Corr C, Baker B. The product of the tobacco mosaic virus resistance gene N: similarity to toll and the interleukin-1 receptor. Cell. 1994;78:1101–1115. doi: 10.1016/0092-8674(94)90283-6. [DOI] [PubMed] [Google Scholar]
- Williams AE, Humphreys IR, Cornere M, Edwards L, Rae A, Hussell T. TGF-beta prevents eosinophilic lung disease but impairs pathogen clearance. Microbes Infect. 2005;7:365–374. doi: 10.1016/j.micinf.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, Ernst JD. Initiation of the adaptive response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not in the lungs. J Exp Med. 2008;205:105–115. doi: 10.1084/jem.20071367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaiss DM, Gause WC, Osborne LC, Artis D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity. 2015;42:216–226. doi: 10.1016/j.immuni.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]