

Abstract
Background
Pancreatic digestive enzymes present in meconium might be responsible for meconium-induced lung injury. The local Renin Angiotensin System plays an important role in lung injury and inflammation. Particularly, angiotensin converting enzyme-2 (ACE-2) has been identified as a protective lung enzyme against the insult. ACE-2 converts pro-apoptotic Angiotensin II to anti-apoptotic Angiotensin 1–7. However, the effect of meconium on ACE-2 has never been studied before.
Objective
To study the effect of meconium on ACE-2, and whether inhibition of proteolytic enzymes present in the meconium reverses its effects on ACE-2.
Methods
Alveolar epithelial A549 cells were exposed to F-12 medium, 2.5% meconium, meconium + a protease inhibitor cocktail (PIc) and PIc alone for 16 h. At the end of incubation, apoptosis was measured with a nuclear fragmentation assay and cell lysates were collected for ACE-2 immunoblotting and enzyme activity.
Results
Meconium caused a fourfold increase in apoptotic nuclei (p < 0.001). The pro-apoptotic effect of meconium can be reversed by PIc. Meconium reduced ACE-2 enzyme activity by cleaving ACE-2 into a fragment detected at ~ 37 kDa by immunoblot. PIc prevented the degradation of ACE-2 and restored 50% of ACE-2 activity (p < 0.05).
Conclusion
These data suggest that meconium causes degradation of lung protective ACE-2 by proteolytic enzymes present in meconium, since the effects of meconium can be reversed by PIc.
Keywords: Meconium aspiration syndrome, Angiotensin-converting enzyme 2, Renin-angiotensin system, Neonatal lung injury
Introduction
Despite improved obstetric care in last several decades, meconium aspiration syndrome (MAS) remains a leading cause of respiratory distress in term neonates. None of the interventions (oropharyngeal, nasopharyngeal, and tracheal suctioning) after delivery has proven effective in preventing MAS [1]. Meconium aspiration is characterized by initial obstruction of the airways resulting in ventilation-perfusion mismatch and hypoxemia. Within a few hours of exposure to meconium, a severe inflammatory response in the lungs is initiated [2]. Recent experimental data using animal models have indicated the presence of non-inflammatory cell death, apoptosis, in meconium-contaminated lungs [3]. Still, the potential pro-inflammatory and pro-apoptotic action of meconium in the pathogenesis of MAS is not well understood.
The role of the Renin Angiotensin System (RAS) is well established in neonatal lung injury and inflammation [4]. RAS is thought to be a systemic cascade and plays an important role in fluid balance and blood pressure regulation [5]. However, many studies support the pivotal role of tissue specific RAS in injury and repair [6–11]. A subset of these studies supports the existence of an intrinsic angiotensin system in lung alveolar epithelial cells (AECs) and demonstrates that activation of this local angiotensin system plays a major role in lung injury [12]. Angiotensinogen gets converted to Angiotensin I (ANG I) via renin. ANG I is then converted to ANG II by angiotensin converting enzyme (ACE). ANG II induces apoptosis in AEC’s through angiotensin type 1 receptor (AT1R). In contrast, ANG 1–7 is antiapoptotic, anti-inflammatory, antifibrotic and vasodilatory and acts through the “Mas receptor.” The main function of ACE-2 is to convert the octapeptide ANG II to the heptapeptide ANG 1–7, thereby counterbalancing ACE activity [13].
Human meconium consists of toxic substances such as pancreatic enzymes, bile pigments and desquamated cells and is considered to be responsible for meconium-induced lung injury [14]. Ivanov et al. [15] showed that meconium causes intense lung epithelial cell detachment and that the effect of meconium on AECs can be reversed by adding a protease inhibitor cocktail (PIc) to the meconium. Therefore, we hypothesized that pancreatic proteolytic digestive enzymes present in the meconium cause downregulation of ACE-2 and that its effect can be reversed by inhibiting proteolytic enzymes.
Methods and Materials
Meconium Collection, Storage and Preparation
First-pass meconium was collected from random anonymous healthy babies without a history of MSAF in the full-term nursery at Sparrow Hospital, Lansing, Michigan. The meconium samples were divided into 1 g aliquots and kept at − 20 °C until use. On the day of experiment, meconium was prepared and sterilized as described before [15]. Meconium samples were collected from four individual babies. We did not pool samples.
Cell Preparation
The human lung adenocarcinoma A549 cell line was grown, maintained, and handled according to the supplier’s manual in Ham’s F12 medium (ATCC, Manassas, VA), with 10% fetal bovine serum (Gibco, Grand Island, NY), as described earlier [12]. Data from our laboratory showed that ACE-2 expression is cell cycle dependent and maximum at postconfluent (quiescent) densities [16]. Therefore, we used cells 5 days postconfluence for ACE-2 protein experiments. In contrast, we used A549 cells at ~ 90% confluence for ACE-2 enzyme activity assay due to previous reports of interference of ACE-2 activity with higher ACE-2 protein [17].
Treatment of Cells
All subculture experiments were performed in serum-free medium. Proprietary PIc (Roche, Nutley, NJ) was diluted in 2.5% meconium and serum-free media according to manufacturer’s recommendations. The composition of the proprietary PIc is not available in the manufacturer’s manual. However, it inhibits the effect of pancreatic enzymes of our interest, e.g., trypsin and chymotrypsin. The cells were exposed to serum free medium (control), 2.5% meconium, 2.5% meconium and PIc and PIc alone for 16 h.
At the end of the incubation period, cells were assayed for ACE-2 by immunoblotting for reactive protein and for ACE-2 enzyme activity.
Nuclear Fragmentation Assay
Apoptotic cells were detected by nuclear fragmentation assay using propidium iodide, after enzymatic digestion of ethanol-fixed cells with DNase-free RNase in PBS containing 5 µg/ml propidium iodide, as previously described [18, 19]. During fixation with 70% ethanol, detached cells were retained by centrifugation of the 24-well culture plates. Cells with discrete nuclear fragments with condensed chromatin were counted as apoptotic using epifluorescence microscopy. Apoptotic cells were scored over a minimum of three separate microscopic fields from each of at least six culture vessels per treatment group.
Western Blotting
After performing a protein assay using the BCA method, 45 µg of protein lysate was loaded in each well of 10% Tris HCL polyacrylamide gels, and separated by SDS-PAGE, in 10× Tris/glycine/SDS buffer. Proteins were transferred to Polyvinylidene Difluoride “PVDF” blotting membrane and blocked by 5% nonfat dry milk in 0.1% tween 20 in Tris-buffered saline.
Western blot analysis was performed using polyclonal antibody against C-terminal amino acids of Human ACE2 (Abcam Catalog Ab15348). PVDF membrane was incubated with ACE-2 antibody for 16 h at + 4 °C. β-actin (Cell Signaling Technology, Danvers, MA) was used to normalize the assay. Bands were visualized by HRP-conjugated goat anti-rabbit antibody using enhanced chemiluminescence detection by standard film techniques.
ACE-2 Enzyme Assay
After the treatment, A549 cells were harvested in ice-cold complete Tris–HCl buffer (pH 6.5), 1 × Complete PIc EDTA-free and lisinopril (50 µg/l; Sigma-Aldrich). Lisinopril was added to the buffer to block the ACE activity. In a half-area black 96-well microtiter plate, the fluorogenic peptide substrate for ACE-2, MCA-APK (Dnp) (Enzo Life Sciences, Farmingdale, NY) was added at a final concentration of 10 µmol/l to 30 µl cell lysate (in a total volume of 50 µl) on ice. DX600 (AnaSpec Inc., Fremont, CA), at a final concentration of 10 µmol/l, a competitive inhibitor of ACE-2, was added to half of the wells to compare enzymatic ACE-2 activity inhibition. The plate was warmed to room temperature, and the fluorescence was read on a plate reader (310/20 nm excitation and 420/50 nm emission) in a FL600 Microplate Fluorescence Reader (BioTek, Burlington, VT) for 30 min. ACE-2 activity in the culture medium was corrected for the cell protein amounts on the culture dishes.
Statistics
All data are shown as mean ± SEM. Group comparisons were evaluated by one-way analysis of variance (ANOVA). When the overall ANOVA was significant, comparisons between the groups were made using Student–Newman–Keul’s post hoc test. For comparisons involving two groups, Student’s t test was used. P < 0.05 was considered significant.
Results
Apoptosis in human A549 AECs was quantitated by fluorescence detection of nuclear fragmentation in alcohol-fixed cells stained with propidium iodide (Fig. 1a). Figure 1b shows meconium-induced apoptosis of AECs in a concentration-dependent manner. Scoring of fragmented nuclei revealed dose-dependent apoptosis of AEC that reached statistical significance beginning at 2.5% meconium, whereas the effect of 0.5% meconium was indistinguishable from control values. Therefore, we used 2.5% meconium for all other experiments.
Fig. 1.

Nuclear fragmentation in human A549 AECs in response to meconium. a Cells exhibiting nuclear fragmentation with propidium iodide (arrowhead) were scored as apoptotic as compared to normal morphologic nuclei (white arrow). Magnification 200 ×. The scale bar is 100 µm. b Meconium induces nuclear fragmentation in a dose-dependent manner (2.5–20%). Bars are means ± SE of six observations per treatment. A significant increase in number of apoptotic nuclei as compared to control (C); *p < 0.05, **p < 0.01 versus C
As shown in Fig. 2, meconium causes a fourfold increase in apoptotic nuclei as compared to control. Addition of the PIc to the meconium completely blocked the meconium-induced apoptosis of A549 cells (p < 0.0001), PIc alone was not different from control.
Fig. 2.

Meconium induces apoptosis in human A549 AECs. A significant increase in percentage of apoptotic nuclei in meconium-treated cells as compared to control was noted. Its effect can be reversed by adding PIc to the meconium. Bars are means ± SE of at least six observations; *P < 0.001 meconium versus C, M + PI and PI alone
We next studied the effect of meconium on ACE-2 in A549 cells. Figure 3 shows proteolytic cleavage of ACE-2 by human meconium. Immunoreactive ACE-2, which appears at ~ 100 kDa by Western blot in A549 AECs decreased nonsignificantly after treating cells with meconium (Fig. 3a). Nevertheless, the cleaved portion of ACE-2 at ~ 37 kDa is significantly increased in meconium-treated cells as compared to control (twofold increase, p < 0.01) by densitometry. This degraded ACE-2 (at ~ 37 kDa) was only found in meconium-treated cells (Fig. 3b). The degraded ACE-2 at ~ 37 kDa by Western blot has been detected in an LPS-induced rat model of lung injury [20]. We did not observe a statistically significant decrease in intact ACE-2 at ~ 100 kDa by densitometry. This might be due to abundance of ACE-2 protein expression in A549 cells after 5 days postconfluence. (We used cells 5 days postconfluence to maximize ACE-2 expression in our experiments.) At the same time, generation of a small quantity of cleaved ACE-2 in meconium-treated cells as compared to other groups did reach statistical significance.
Fig. 3.
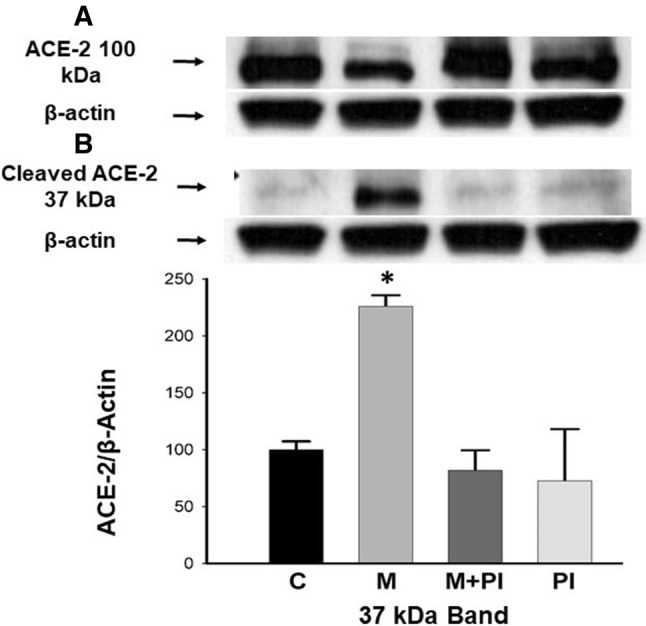
Meconium proteolytically cleaves ACE-2 in A549 AECs. a ACE-2 protein that appears at ~ 100 kDa decreased slightly in meconium-treated cells as compared to control, meconium + PI and PI alone. b The cleaved portion of ACE-2 at ~ 37 kDa increased in meconium-treated cells. *P < 0.05 versus C, M + PI and PI alone. Bars are means ± SE of at least three observations over two experiments (n = 4 babies)
Table 1 shows that meconium-treated cells have increased cleaved ACE-2 at ~ 37 kDa as compared to control cells by densitometry (Western blot). We collected meconium from normal healthy term neonates without the history of meconium-stained amniotic fluid and studied the effect of meconium on ACE-2 individually. There was a wide variation in production of cleaved ACE-2 by meconium from each baby as compared to control (control vs meconium, p < 0.05, paired t test). This variation might explain the heterogeneity in the clinical presentation of MAS.
Table 1.
Difference in production of cleaved ACE-2 ~ 37 kDa by individual baby’s meconium (by densitometry), p value < 0.05; paired t test
| Baby | Cleaved ACE-2/β-actin 37 kDa Mean ± SEM |
p Value | |
|---|---|---|---|
| Control | Meconium | ||
| 1 | 0.43 ± 0.03 | 0.96 ± 0.04 | |
| 2 | 0.53 ± 0.03 | 0.84 ± 0.09 | |
| 3 | 0.45 ± 0.14 | 1.31 ± 0.23 | |
| 4 | 0.46 ± 0.11 | 1.26 ± 0.15 | |
| Mean | 0.47 ± 0.05 | 1.12 ± 0.10 | 0.0005 |
In the next step, we studied the effect of meconium-induced ACE-2 cleavage on ACE-2 enzymatic activity. Figure 4 shows a reduction of 60% in ACE-2 enzymatic activity in meconium-treated cells as compared to control (p < 0.0001). However, adding the PIc to the meconium partially but significantly reversed the effect of meconium on ACE-2 enzyme activity [60 ± 7 (C) vs 23 ± 11 (M) vs 44 ± 6 (M + PIc)]. PIc alone had no effect on ACE-2 enzyme activity (data not shown). For each treatment group, the enzymatic activity detected by fluorogenic peptide substrate was essentially eliminated by the addition of peptide DX600, a competitive inhibitor of ACE-2 [21]. 85% of ACE-2 activity was inhibited by DX600, suggesting a key role for ACE 2 specific enzyme activity rather than other enzymes capable of cleaving the substrate.
Fig. 4.
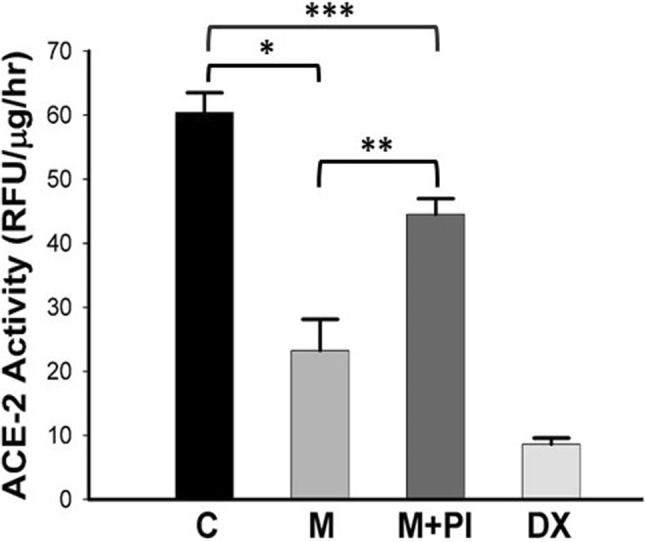
Meconium reduces ACE-2 activity in A549 cells. Note the essentially complete inhibition of ACE-2 activity by DX600. Bars are means ± SE of at least six observations. *P < 0.0001 versus C, **p < 0.001 versus M + PI and ***p < 0.05 C versus M + PI
Discussion
MAS is still a major cause of morbidity and mortality in near term and term neonates, particularly in developing countries. However, the pathophysiology of MAS is not completely understood. The bulk of evidence implies that toxic substances present in the meconium cause lung AEC detachment, surfactant inactivation, apoptosis and lung injury. Nevertheless, attempts to prevent meconium-induced lung injury remain largely unsuccessful. Previous studies from our laboratory showed that apoptosis is regulated by the local RAS in AECs and that protective lung enzyme ACE-2 is downregulated in experimental lung injury [12, 13]. To our knowledge, the role of ACE-2 in meconium-induced lung injury has never been studied before.
Mullinger and Palasi reported trypsin and chymotrypsin activities in meconium on day 1 and showed that enzymatic activity increases in first 3 days of life [22]. We used a PIc which caused near complete inhibition of trypsin, chymotrypsin and pancreatic metalloprotease enzyme activities. We demonstrated that meconium-induced lung AEC apoptosis by nuclear fragmentation assay in human A549 cells is reversed by inhibiting the activities of fetal proteolytic enzymes. Ivanov et al. [15] has shown that meconium causes A549 cell detachment and the detached A549 cells were alive (using the acridine orange staining method) in contrast to our results demonstrating apoptosis after centrifugation of detached cells. It is important to point out that the acridine orange staining method is not a reliable method for detection of cell apoptosis, particularly for in vitro studies. We used the nuclear fragmentation assay which is considered the gold standard for measuring apoptosis in in vitro studies [23–25]. It has been shown that nuclear fragmentation is a crucial event in apoptosis [26]. The morphology of this event is strikingly similar in different cell types [26, 27]. Previous studies have shown that protease inhibitors can prevent cell apoptosis in response to various noxious stimuli by nuclear fragmentation assay method [28]. This method of detecting apoptosis has been validated in previous publications from our laboratory [18, 19, 29] by simultaneously using other methods (In Situ End Labelling and anti-caspase 3 immunolabeling) in human A549 cells. This observation is in agreement with studies by Zagariya and co-workers, who have demonstrated lung AEC detachment and apoptosis in meconium-induced lung injury [30].
Rosenfeld et al. showed that exposure to meconium induces AT1 receptor expression and further that losartan, an antagonist for the AT1 receptor, attenuates meconium-induced AEC apoptosis in newborn rabbit lung [31]. An extensive body of literature has shown that the ACE/ANGII/AT1 axis promotes lung injury and is counteracted by the ACE-2/ANG1-7/Mas axis. Interestingly, previous attempts to downregulate ACE/ANGII/AT1 axis by using captopril and losartan have produced mixed results in human and animal studies [32]. In particular, ACE2 has been identified as an essential receptor for SARS coronavirus infections, as well as a protective molecule against lethal lung failure in SARS [33]. Imai et al. showed the severe acute lung injury in ACE-2 knock-out mice and symptoms of acute lung injury can be rescued by a recombinant ACE-2 protein [34]. Intriguingly, ACE2 localization was mapped to the apical surface of epithelial cells in the lungs [35]. This leads to our hypothesis that proteolytic enzymes in the meconium causes cleavage of ACE-2 present on the surface of AECs.
To our knowledge, this is the first study reporting proteolytic cleavage of ACE-2 by human meconium in human A549 cells. There is an increased cleaved portion of ACE-2 ~ 37 kDa in meconium-treated cells as compared to control. Also, there is a reduced ACE-2 activity in meconium-treated cells, and adding PIc to the meconium partially reverses the effect of meconium on ACE-2 activity. Previously, Wosten-van Asperen et al. [20] have shown ACE-2 degradation and decreased ACE-2 activity by LPS induce lung injury in a rat model of acute respiratory distress syndrome. We have observed a similar effect in our study. Interestingly, there are reports of the presence of RAS in the GI tract, particularly on the intestinal brush border [21]. However, we did not observe ACE-2 activity in collected meconium. It is becoming increasingly apparent that the proteolytic cleavage of cell surface proteins is an important mechanism regulating their expression and function. We speculate that meconium induces lung epithelial cell apoptosis by proteolytically cleaving ACE-2. However, the mechanism by which this protective axis prevents lung injury is still an active area of research.
As mentioned in Table 1, the proteolytic cleavage of ACE-2 is a consistent finding but there is a considerable variation in the effect of meconium on ACE-2 cleavage in each individual baby. We speculate that this effect is due to individual variation in the amounts of proteolytic enzymes present in each meconium sample. This could also explain the partial reversal (50%) of ACE-2 enzyme activity by PIc. It is possible that that particular baby had a very high amount of proteolytic enzymes in the meconium; therefore, protease inhibitor could only partially inhibit pancreatic enzymes which led to only 50% restoration of ACE-2 enzyme activity. This observed effect may provide an insight into the clinical spectrum of MAS.
Our study is limited by small sample size, and the fact that it used adult human lung AECs. However, our laboratory has demonstrated the presence of ACE-2 in neonatal mouse lung as well as in fetal lung fibroblasts [36, 37]. Therefore, it is plausible that a similar effect of meconium on fetal and neonatal lungs would be observed. Also, we have not examined the direct effect of downregulation of ACE-2 and meconium-induced apoptosis in this study. Nevertheless, our laboratory has previously shown that ACE-2 downregulation leads to apoptosis of human A549 cells, MLE12 cells and fetal fibroblast IMR 90 cells in response to various noxious stimuli, e.g., bleomycin [18], hyperoxia [36] and hypoxia [37]. Therefore, it is safe to assume that meconium-induced human A549 cells apoptosis is due to downregulation of ACE-2.
Previously, various studies have shown AEC apoptotic induction by meconium, and it was therefore theorized that pharmacological inhibition of apoptotic factors may provide potential therapeutic targets for MAS. Additional experimental studies have shown the activation of the local RAS system mediated apoptosis in response to meconium [30, 31]. This led to our speculation that maintaining high levels of tissue ACE-2 by prevention of its cleavage, and/or replacement of the protein through administration of biologically active enzyme, may hold potential as a therapeutic strategy for treating MAS. In support of this theory, a recent pilot clinical trial of GSK2586881, a recombinant form of human angiotensin-converting enzyme 2 (rhACE-2), was performed in adults with acute respiratory distress syndrome [38].
In the current study, we have further established the role of pulmonary RAS activation in meconium-induced lung injury. This apoptotic effect is presumably due to reduction in ACE-2 activity caused by proteolytic cleavage of the lung protective enzyme ACE-2 by pancreatic digestive enzymes present in the meconium. Since it is well known that lung injury can be abrogated by ACE-2, the demonstration of a direct effect of meconium on ACE-2 in human A549 cells has significant implications. The findings from this preliminary report suggest the ACE-2 mediated RAS modification may be a viable therapeutic option for treatment of MAS.
Acknowledgements
We thank Dr. Amal T. Abdul-Hafez for helping in designing and executing the experiments. We also thank Sparrow Hospital (Lansing, MI) Newborn Unit nursing staff for helping in collecting the meconium for the study.
Funding
This work was supported by a grant from the Fellowship Research Fund of Sparrow Hospital, Lansing, MI (to C.G).
Compliance with Ethical Standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
The study protocol was approved by the Sparrow Hospital and Michigan State University institutional review board.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Gandhi CK. Management of meconium-stained newborns in the delivery room. Neonatal Netw NN. 2018;37(3):141–148. doi: 10.1891/0730-0832.37.3.141. [DOI] [PubMed] [Google Scholar]
- 2.Tyler DC, Murphy J, Cheney FW. Mechanical and chemical damage to lung tissue caused by meconium aspiration. Pediatrics. 1978;62(4):454–459. [PubMed] [Google Scholar]
- 3.Zagariya A, Bhat R, Chari G, Uhal B, Navale S, Vidyasagar D. Apoptosis of airway epithelial cells in response to meconium. Life Sci. 2005;76(16):1849–1858. doi: 10.1016/j.lfs.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 4.Gandhi C, Uhal BD. Roles of the angiotensin system in neonatal lung injury and disease. JSM Atheroscler. 2016;1(3):1014. [PMC free article] [PubMed] [Google Scholar]
- 5.Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM. Classical renin-angiotensin system in kidney physiology. Compr Physiol. 2014;4(3):1201–1228. doi: 10.1002/cphy.c130040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Nakatani T, Tsujinoue H, Fukui H. Angiotensin-II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology. 2001;34(4 Pt 1):745–750. doi: 10.1053/jhep.2001.28231. [DOI] [PubMed] [Google Scholar]
- 7.Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006;86(3):747–803. doi: 10.1152/physrev.00036.2005. [DOI] [PubMed] [Google Scholar]
- 8.Chen LN, Yang XH, Nissen DH, Chen YY, Wang LJ, Wang JH, Gao JL, Zhang LY. Dysregulated renin-angiotensin system contributes to acute lung injury caused by hind-limb ischemia-reperfusion in mice. Shock (Augusta GA) 2013;40(5):420–429. doi: 10.1097/SHK.0b013e3182a6953e. [DOI] [PubMed] [Google Scholar]
- 9.Liu L, Qiu HB, Yang Y, Wang L, Ding HM, Li HP. Losartan, an antagonist of AT1 receptor for angiotensin II, attenuates lipopolysaccharide-induced acute lung injury in rat. Arch Biochem Biophys. 2009;481(1):131–136. doi: 10.1016/j.abb.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 10.Marshall RP. The pulmonary renin-angiotensin system. Curr Pharm Des. 2003;9(9):715–722. doi: 10.2174/1381612033455431. [DOI] [PubMed] [Google Scholar]
- 11.Zhang H, Sun GY. LPS induces permeability injury in lung microvascular endothelium via AT(1) receptor. Arch Biochem Biophys. 2005;441(1):75–83. doi: 10.1016/j.abb.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 12.Wang R, Alam G, Zagariya A, Gidea C, Pinillos H, Lalude O, Choudhary G, Oezatalay D, Uhal BD. Apoptosis of lung epithelial cells in response to TNF-alpha requires angiotensin II generation de novo. J Cell Physiol. 2000;185(2):253–259. doi: 10.1002/1097-4652(200011)185:2<253::AID-JCP10>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Molina-Molina M, Abdul-Hafez A, Uhal V, Xaubet A, Uhal BD. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;295(1):L178–L185. doi: 10.1152/ajplung.00009.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antonowicz I, Shwachman H. Meconium in health and in disease. Adv Pediatr. 1979;26:275–310. [PubMed] [Google Scholar]
- 15.Ivanov VA, Gewolb IH, Uhal BD. A new look at the pathogenesis of the meconium aspiration syndrome: a role for fetal pancreatic proteolytic enzymes in epithelial cell detachment. Pediatr Res. 2010;68(3):221–224. doi: 10.1203/PDR.0b013e3181ebd4c3. [DOI] [PubMed] [Google Scholar]
- 16.Uhal BD, Dang M, Dang V, Llatos R, Cano E, Abdul-Hafez A, Markey J, Piasecki CC, Molina-Molina M. Cell cycle dependence of ACE-2 explains downregulation in idiopathic pulmonary fibrosis. Eur Respir J. 2013;42(1):198–210. doi: 10.1183/09031936.00015612. [DOI] [PubMed] [Google Scholar]
- 17.Xiao F, Burns KD. Measurement of angiotensin converting enzyme 2 activity in biological fluid (ACE2) Methods Mol Biol (Clifton NJ) 2017;1527:101–115. doi: 10.1007/978-1-4939-6625-7_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, Zhang H, Soledad-Conrad V, Zhuang J, Uhal BD. Bleomycin-induced apoptosis of alveolar epithelial cells requires angiotensin synthesis de novo. Am J Physiol Lung Cell Mol Physiol. 2003;284(3):L501–L507. doi: 10.1152/ajplung.00273.2002. [DOI] [PubMed] [Google Scholar]
- 19.Uhal BD, Joshi I, Hughes WF, Ramos C, Pardo A, Selman M. Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am J Physiol. 1998;275(6 Pt 1):L1192–L1199. doi: 10.1152/ajplung.1998.275.6.L1192. [DOI] [PubMed] [Google Scholar]
- 20.Wosten-van Asperen RM, Lutter R, Specht PA, Moll GN, van Woensel JB, van der Loos CM, van Goor H, Kamilic J, Florquin S, Bos AP. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1–7) or an angiotensin II receptor antagonist. J Pathol. 2011;225(4):618–627. doi: 10.1002/path.2987. [DOI] [PubMed] [Google Scholar]
- 21.Garg M, Angus PW, Burrell LM, Herath C, Gibson PR, Lubel JS. Review article: the pathophysiological roles of the renin-angiotensin system in the gastrointestinal tract. Aliment Pharmacol Ther. 2012;35(4):414–428. doi: 10.1111/j.1365-2036.2011.04971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mullinger M, Palasi M. Tryptic and chymotryptic activity of stools of newborn infants. Pediatrics. 1966;38(4):657–659. [PubMed] [Google Scholar]
- 23.Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284(5756):555–556. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- 24.Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006;1(3):1458–1461. doi: 10.1038/nprot.2006.238. [DOI] [PubMed] [Google Scholar]
- 25.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139(2):271–279. doi: 10.1016/0022-1759(91)90198-O. [DOI] [PubMed] [Google Scholar]
- 26.Dini L, Coppola S, Ruzittu MT, Ghibelli L. Multiple pathways for apoptotic nuclear fragmentation. Exp Cell Res. 1996;223(2):340–347. doi: 10.1006/excr.1996.0089. [DOI] [PubMed] [Google Scholar]
- 27.Oberhammer F, Fritsch G, Schmied M, Pavelka M, Printz D, Purchio T, Lassmann H, Schulte-Hermann R. Condensation of the chromatin at the membrane of an apoptotic nucleus is not associated with activation of an endonuclease. J Cell Sci. 1993;104(Pt 2):317–326. doi: 10.1242/jcs.104.2.317. [DOI] [PubMed] [Google Scholar]
- 28.Ghibelli L, Maresca V, Coppola S, Gualandi G. Protease inhibitors block apoptosis at intermediate stages: a compared analysis of DNA fragmentation and apoptotic nuclear morphology. FEBS Lett. 1995;377(1):9–14. doi: 10.1016/0014-5793(95)01284-2. [DOI] [PubMed] [Google Scholar]
- 29.Gopallawa I, Uhal BD. Angiotensin-(1–7)/mas inhibits apoptosis in alveolar epithelial cells through upregulation of MAP kinase phosphatase-2. Am J Physiol Lung Cell Mol Physiol. 2016;310(3):L240–L248. doi: 10.1152/ajplung.00187.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zagariya A, Bhat R, Uhal B, Navale S, Freidine M, Vidyasagar D. Cell death and lung cell histology in meconium aspirated newborn rabbit lung. Eur J Pediatr. 2000;159(11):819–826. doi: 10.1007/s004310000581. [DOI] [PubMed] [Google Scholar]
- 31.Rosenfeld CR, Zagariya AM, Liu XT, Willis BC, Fluharty S, Vidyasagar D. Meconium increases type 1 angiotensin II receptor expression and alveolar cell death. Pediatr Res. 2008;63(3):251–256. doi: 10.1203/PDR.0b013e318163a2b8. [DOI] [PubMed] [Google Scholar]
- 32.Huang L, Sexton DJ, Skogerson K, Devlin M, Smith R, Sanyal I, Parry T, Kent R, Enright J, Wu QL, Conley G, DeOliveira D, Morganelli L, Ducar M, Wescott CR, Ladner RC. Novel peptide inhibitors of angiotensin-converting enzyme 2. J Biol Chem. 2003;278(18):15532–15540. doi: 10.1074/jbc.M212934200. [DOI] [PubMed] [Google Scholar]
- 33.Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, Huan Y, Yang P, Zhang Y, Deng W, Bao L, Zhang B, Liu G, Wang Z, Chappell M, Liu Y, Zheng D, Leibbrandt A, Wada T, Slutsky AS, Liu D, Qin C, Jiang C, Penninger JM. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H, Crackower MA, Fukamizu A, Hui CC, Hein L, Uhlig S, Slutsky AS, Jiang C, Penninger JM. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiener RS, Cao YX, Hinds A, Ramirez MI, Williams MC. Angiotensin converting enzyme 2 is primarily epithelial and is developmentally regulated in the mouse lung. J Cell Biochem. 2007;101(5):1278–1291. doi: 10.1002/jcb.21248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oarhe CI, Dang V, Dang M, Nguyen H, Gopallawa I, Gewolb IH, Uhal BD. Hyperoxia downregulates angiotensin-converting enzyme-2 in human fetal lung fibroblasts. Pediatr Res. 2015;77(5):656–662. doi: 10.1038/pr.2015.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohamed TL, Nguyen HT, Abdul-Hafez A, Dang VX, Dang MT, Gewolb IH, Uhal BD. Prior hypoxia prevents downregulation of ACE-2 by hyperoxia in fetal human lung fibroblasts. Exp Lung Res. 2016;42(3):121–130. doi: 10.3109/01902148.2016.1157712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khan A, Benthin C, Zeno B, Albertson TE, Boyd J, Christie JD, Hall R, Poirier G, Ronco JJ, Tidswell M, Hardes K, Powley WM, Wright TJ, Siederer SK, Fairman DA, Lipson DA, Bayliffe AI, Lazaar AL. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Critic Care (Lond Engl) 2017;21(1):234. doi: 10.1186/s13054-017-1823-x. [DOI] [PMC free article] [PubMed] [Google Scholar]