

Abstract
Historic and contemporary host ecology and evolutionary dynamics have profound impacts on viral diversity, virulence, and associated disease emergence. Bats have been recognized as reservoirs for several emerging viral pathogens, and are unique among mammals in their vagility, potential for long-distance dispersal, and often very large, colonial populations. We investigate the relative influences of host ecology and population genetic structure for predictions of viral richness in relevant reservoir species. We test the hypothesis that host geographic range area, distribution, population genetic structure, migratory behavior, International Union for Conservation of Nature and Natural Resources (IUCN) threat status, body mass, and colony size, are associated with known viral richness in bats. We analyze host traits and viral richness in a generalized linear regression model framework, and include a correction for sampling effort and phylogeny. We find evidence that sampling effort, IUCN status, and population genetic structure correlate with observed viral species richness in bats, and that these associations are independent of phylogeny. This study is an important first step in understanding the mechanisms that promote viral richness in reservoir species, and may aid in predicting the emergence of viral zoonoses from bats.
Keywords: Chiroptera, emerging infectious disease, IUCN, population structure, sampling effort, viral richness
Introduction
Emerging infectious diseases (EIDs) are mostly viral and zoonotic in origin (Taylor et al., 2001; Wolfe et al., 2007) and a growing number are discovered each year (Woolhouse et al., 2008). Several studies have undertaken large-scale analyses to determine risk factors associated with EIDs (Taylor et al., 2001; Jones et al., 2008), and have identified host range, as well as pathogen taxonomy and molecular characteristics as the most important (Cleaveland et al., 2001; Woolhouse and Gowtage-Sequeria, 2005; Holmes and Drummond, 2007; Pulliam and Dushoff, 2009). Molecular characteristics of viruses that likely contribute to emergence in novel hosts include physical or biochemical features, e.g., the ability to replicate in the cytoplasm (Pulliam and Dushoff, 2009), and evolutionary potential, e.g., rapid mutation rates and processes such as recombination or reassortment (Holmes and Drummond, 2007). Additionally, viruses that utilize highly conserved cell receptors (e.g., rabies virus) may have higher probabilities of successful cross-species transmission and emergence (Holmes and Drummond, 2007). Despite the importance of intrinsic pathogen characteristics, host life history, ecology, and spatial structure also contribute to the emergence, persistence, and intensity of zoonotic outbreaks (Bolzoni et al., 2007; Webb et al., 2007; Plowright et al., 2008).
As viruses may be relatively unstable outside their hosts, the dynamics and frequency of intra- or interspecific host interactions will largely affect encounter probabilities related to disease emergence (Pulliam, 2008). The structure of wildlife populations across the landscape also has strong implications for infectious disease dynamics and emergence (Real and Biek, 2007). Major developments have been made in theoretical (Rand et al., 1995; Boots and Sasaki, 1999, 2000; Haraguchi and Sasaki, 2000; Boots et al., 2004; Cross et al., 2005; Webb et al., 2007) and experimental (Bull et al., 1991; Messenger et al., 1999; Kerr et al., 2006; Boots and Mealor, 2007) studies linking population structure, disease dynamics, and pathogen evolution, with particular advances in our understanding of how contact networks regulate pathogen persistence and virulence. Evidence from natural systems has validated the ecological theories on spatial infection processes (Ewald, 1991; Herre, 1993; de Roode et al., 2008), and recent studies have highlighted the importance of contact and transmission heterogeneities in modeling disease prevalence and control strategies (Barlow, 1996; Lloyd-Smith et al., 2005; Donnelly et al., 2006; May, 2006; Bolzoni et al., 2007; Bohm et al., 2009).
Another key factor in predicting the emergence of zoonoses is the number of viruses, or viral richness, found in natural reservoir species (Wolfe et al., 2005). This largely unknown, natural diversity of pathogens in wildlife hosts has been termed the “zoonotic pool” (Morse, 1993). The fields of phylogenetics, population genetics, and biogeography have led to key advancements in our understanding of host–pathogen coevolution in relation to pathogen diversity (Pybus et al., 2000; Grenfell et al., 2004), and numerous studies have documented coevolution between pathogens and their hosts (Smith, 1996; Asikainen et al., 2000; Gaunt et al., 2001; Nieberding et al., 2004; Criscione et al., 2005; Biek et al., 2006; Dragoo et al., 2006; Criscione and Blouin, 2007; Liu et al., 2008). However, we know little about which factors are most important in creating and sustaining pathogen richness within reservoir hosts. Comparative approaches are needed that test for patterns of pathogen richness across multiple factors for groups of potential reservoir host species.
Few studies have simultaneously examined phylogenetic and socioecological factors affecting parasite richness, and the majority have focused on primates. Davies and Pedersen (2008) found that host geographic range overlap was a significant determinant of viral communities in primates, although phylogenetic relatedness was also an important predictor of richness for other taxonomic groups of parasites in their analysis. A separate study found a significant correlation between viral species richness, host geographic range area, and population density across primates when corrections for phylogeny and sampling effort were included (Nunn et al., 2003). A similar study also found a correlation between viral richness, host phylogenetic diversity, and host geographic range overlap (Nunn et al., 2004). In a more recent study, threatened primates harbored lower parasite species richness compared to nonthreatened hosts, although this effect was not significant for viruses when other host socioecological traits were included (Altizer et al., 2007). Ezenwa (2004) found that group size and social behavior were important correlates with parasite diversity in African bovids. Gregory (1990) found positive correlations between helminth richness and geographic range, but no associations with host socioecological traits, in Holarctic waterfowl. Across studies, host population density, geographic range area, and phylogeny emerge as consistent predictors of parasite richness. All of these studies also report highly significant positive correlations between parasite richness and sampling effort, which suggests that parasite richness has been largely underestimated for many vertebrate hosts.
Bats (Order Chiroptera), which comprise nearly 1200 species, have been linked as natural reservoir hosts to a rapidly growing number of EIDs (Calisher et al., 2006). This includes several viruses that cause severe disease in humans: Ebola (Leroy et al., 2005) and Marburg (Swanepoel et al., 2007) filoviruses, SARS and related coronaviruses (Li et al., 2005; Dominguez et al., 2007; Muller et al., 2007; Carrington et al., 2008; Gloza-Rausch et al., 2008; Tong et al., 2009), as well as Nipah (Johara et al., 2001), Hendra (Halpin et al., 2000) and related paramyxoviruses (Drexler et al., 2009). It has recently been suggested that two human cases of acute respiratory illness in Malaysia may be linked to reoviruses of bat origin (Chua et al., 2007; Chua et al., 2008). Furthermore, evidence supports the association of bats with lyssavirus infection in humans globally (Lumio et al., 1986; Allworth et al., 1996; Warner et al., 1999; Favi et al., 2002; Messenger et al., 2002; Fooks et al., 2003; da Rosa et al., 2006; van Thiel et al., 2008). The importance of bat-borne zoonoses has only recently been appreciated, and no studies to date have examined factors that may explain the large diversity of viruses in the Order Chiroptera. Bats exhibit a wide range of morphological, ecological, and behavioral characteristics, and similarly, are characterized by highly variable patterns of genetic population structure (Nowak, 1999; Burland and Worthington-Wilmer, 2001). These features make the Order Chiroptera ideal for testing hypotheses regarding patterns of host–pathogen coevolution and viral richness.
Many hypotheses have been suggested for the association of bats and EIDs, some of which include high species diversity, long life spans, capacity for long-distance dispersal, dense roosting aggregations (colony size) for many species, the use of torpor and hibernation, unique immunology, and spatial population structure (Messenger et al., 2003a; Calisher et al., 2006). While all of these factors are likely to be important, we investigate the significance of colony size, migratory ability, population genetic structure, geographic range area and distribution (continental vs. island), International Union for Conservation of Nature and Natural Resources (IUCN) threatened status, and body mass in generalized linear models of viral richness across Chiroptera, while accounting for sampling effort and phylogeny. We hypothesize that larger colony sizes will be positively correlated with viral richness, as a larger number of susceptible individuals in a population will permit greater viral establishment and persistence (Anderson and May, 1986). We also hypothesize that stronger population structure will be positively associated with viral richness, due to coevolutionary forces as a driver of within-lineage pathogen diversity and the importance of metapopulation structure in invasion and enzootic infection processes (Webb et al., 2007). As island taxa tend to have more restricted geographic ranges, and may be more prone to extinction, we predict that species with an island distribution will have fewer viruses than bats with a continental range distribution. Threatened bat species may have fewer viruses than nonthreatened taxa because parasites may be more likely to go extinct in smaller fragmented host populations (May and Anderson, 1979). However, threatened species may be more susceptible to multihost pathogens, potentially as a result of reduced immunological diversity, stress, and poor nutritional status of hosts across fragmented landscapes (Lyles and Dobson, 1993; Messenger et al., 2003a). Migratory species are expected to harbor more viruses than nonmigratory species as they sample more diverse habitats and may be subject to greater interspecific contact. Larger hosts with greater body mass may harbor greater viral richness, as has been suggested from studies in primates (Nunn et al., 2003). Although we explore associations of life history, genetics, and ecology with viral infection in bats, these factors may predict infection but not disease in bats. It has been suggested that bats may not develop clinical disease from infection with certain viral pathogens, such as filoviruses, henipaviruses, and coronaviruses; however, clinical signs of disease are observed following infection with other viral pathogens such as lyssaviruses. This is, to our knowledge, the first meta-analyses to quantitatively investigate the effect of these explanatory variables on viral richness in bats.
Methods
Data Collection
Population genetic structure data were gathered from published literature by searching Science Citation Index Expanded and ISI Zoological Record for the years 1985–2009 with various combinations of keywords: population, structure, genetic*, bat*, mammal*, and Chiroptera. Additional references not identified during keyword searches were collected from published reviews (Burland and Worthington-Wilmer, 2001), literature cited, and the Digital Dissertation Database. Population genetic data for 54 species of bats were compiled. Representative nuclear allelic FST (Wright, 1951) or mitochondrial haplotypic φST (Excoffier et al., 1992) estimates of population structure from each species were calculated separately as the mean of all pairwise FST or φST values in a study, and taken across multiple studies for the same species when available. A global mean value was also obtained by averaging FST and φST when available. Thus, the global FST value for each bat species summarizes data from single or multiple, mitochondrial and nuclear, empirical estimates. The advantage of FST over direct measures of genetic diversity such as nucleotide diversity is that, as a ratio that describes the proportion of population genetic subdivision, it is largely independent from the mutation process so that data from different molecular markers can be easily compared (Pannell and Charlesworth, 2000). This comparability, along with its common use, makes FST the most appropriate statistic for comparative analyses of population structure across studies using different molecular markers. As previous studies have suggested potential correlations between population structure indices and geographic sampling distance (Burland and Worthington-Wilmer, 2001), the average geographic sampling distance (km) between populations was calculated from empirical studies to test for this bias.
Bat species were assigned as being migratory or nonmigratory based on definitions presented in Fleming and Eby (2003) and data from previous ecological studies (including capture-mark-recapture data) or published reviews (McCracken, 1987; Burland and Worthington-Wilmer, 2001; Fleming and Eby, 2003). If a species had populations that were both migratory and nonmigratory, e.g., Tadarida brasiliensis (McCracken and Gassel, 1997), then the species was coded as migratory. Average colony size was recorded from the Mammalian Species account for a species when available, or the IUCN Red List database, or reconciled from both sources. The distribution of a species was coded from geographic range data presented in the IUCN Red List database (IUCN, 2009), and was either “continental” or “island.” If a species distribution included continental and island areas, assignment reflects the majority of the geographic range. IUCN Red List status was obtained directly from the database (IUCN, 2009). Body mass data were gathered from Norberg and Rayner (1987), and we used geographic range area estimates from Jones et al. (2003).
We then searched the literature and compiled records for all known viruses occurring in the 54 bat species with available population structure data. Viral information was available for 33 of 54 (61%) species for which we also had population structure estimates. The majority of the viral associations were obtained from previously published reviews (Messenger et al., 2003a; Calisher et al., 2006; Wong et al., 2007), but additional literature searches on PubMed and Web of Science were conducted to identify recent events. When possible, we also recorded the method of viral detection from the original papers cited. Typical methods of detection included mouse inoculation or cell culture for viral isolation, although alternate methods included the fluorescent antibody test (FAT) or polymerase chain reaction (PCR) to detect viral antigen from organs or tissues of bats. One exclusion criterion was employed in our database, and this was for records of arbovirus detection through serology only. As bats are likely to be incidental hosts to most arboviruses, and positive serological results may be nonspecific in differentiating viruses with noted cross-reactivity (e.g., genus Flavivirus), serological detection of arbovirus antibodies was not considered as evidence of a viral association. All viral records were considered unique for subsequent analysis with the exception of bat gammaherpesviruses (BatGHV), where BatGHV-1, 2, 4, 6, and 7 were considered one viral species, and the phylogenetically distant BatGHV-3 was considered to be a separate unique virus (Wibbelt et al., 2007).
Previous studies have demonstrated that sampling effort is likely to be uneven across species, and can confound interpretations with other biological variables of interest. To account for variation in sampling effort across species, we searched the ISI Zoological Record from 1945 to 2009 for each bat species. The number of citations per species was recorded, and used as an index of sampling effort (Gregory, 1990; Nunn et al., 2004, 2005; Altizer et al., 2007). Only bat species with one or more known viral associations were included in our dataset (n = 33), as we found that sampling effort was a highly significant predictor of the presence or absence of viral records across all taxa (n = 54; r 2 = 0.56; P < 0.0001).
Statistical Analysis
In the full dataset for bats with population structure and viral information (n = 33), the global FST statistic averaged nuclear and mitochondrial indices (n = 8), or represented a single nuclear (n = 17) or mitochondrial (n = 8) statistic. Generalized linear models of the viral richness data included migration, distribution, and IUCN status as binomial categorical predictors, and colony size, population genetic structure, body mass, geographic range area, and sampling effort as covariates. Covariates were log-transformed to satisfy normality criteria. There were no significant covariate interactions in our analyses, and our limited sample sizes precluded us from testing across multiple levels for all categorical interactions. A mixed stepwise variable selection procedure was employed in JMP v.7.0.1 (SAS Institute, Inc., Cary, NC) to identify optimal models, with the cutoff value for variables to enter and leave set at α = 0.20. As we had incomplete information for the geographic range area of all taxa (28 of 33), the stepwise procedure was run with and without this variable included for optimal model selection. The Egyptian fruit bat (Rousettus aegyptiacus) had the most viral records (n = 8), was identified as an outlier in the generalized linear model testing (Studentized residual >3), and was omitted from subsequent analyses. The best model and subsets identified from the stepwise procedure were then analyzed in the generalized linear framework using a normal distribution and identity link function. For categorical levels, means and standard deviation of viral richness estimates are presented.
Phylogenetic Correction
The best model and subsets were reanalyzed to test for phylogenetic dependence, as traits among related species may be more similar due to common ancestry and should not be considered independent in comparative analyses (Felsenstein, 1985; Harvey and Pagel, 1991). A generalized least squares (GLS) framework (Grafen, 1989; Martins and Hansen, 1997; Garland and Ives, 2000), using the APE package (Paradis et al., 2004) in R (The R Foundation for Statistical Computing, http://www.R-project.org) and supplementary code (Duncan et al., 2007), was used to estimate λ, a parameter that tests whether the evolution of phenotypic traits are independent of phylogenetic relationships among taxa. As comparative sequence data were not available for all of the bats with viral information, a mammalian supertree with branch length information was used as a source tree for the phylogenetic correlation matrix (Bininda-Emonds et al., 2007). After excluding one outlier, 29 of the 32 taxa of interest were represented in the mammalian supertree, and a pruned phylogeny with branch lengths was extracted (Fig. 1a). The remaining (three) taxa not found in the supertree were manually added to the pruned phylogeny in a way that minimized assumptions of relationships (i.e., branch length = 0) (Fig. 1b). A correlation matrix was computed from the expected variances and covariances of shared branch lengths between taxa, assuming a model of Brownian motion (Martins and Hansen, 1997; Garland and Ives, 2000). The PGLS function (Duncan et al., 2007) was used to find the maximum likelihood estimate of λ, as well as estimated coefficients and confidence intervals on regression predictor variables. The parameter λ varies between 0 and 1, with zero values indicating independent phenotypic trait evolution with respect to phylogeny, and values close to one indicating dependence between trait evolution and phylogeny (Pagel, 1999; Freckleton et al., 2002). A null distribution of λ was also created, through 1000 randomizations of the model and data, to evaluate the significance of the observed estimate. Akaike information criterion scores corrected for small sample sizes (AICc) were used to identify the best model among subsets (Burnham and Anderson, 2002).
Figure 1.

a The phylogeny extracted from the mammalian supertree (with branch lengths) for 29 of 32 taxa (Bininda-Emonds et al., 2007). b The phylogeny in which three additional taxa were added (highlighted in bold).
Results
One hundred unique viral records were obtained for 33 bat species (Table 1). The majority of the viral records, 31% (31 of 100), were from the Family Rhabdoviridae and most of these were lyssaviruses (i.e., rabies and related viruses). Viruses from the Flaviviridae and Paramyxoviridae were the second and third most frequent, accounting for 13% (13 of 100) and 12% (12 of 100) of associations, respectively. Each bat species was associated with 1–8 viruses, and the mean number of viral records per bat species was 3.03 ± 1.83 (Fig. 2). Average sampling distance was a marginal predictor of global FST in a univariate regression (n = 31, P = 0.06), and was not included in subsequent analyses.
Table 1.
Bats with Available Population Genetic Structure and Viral Richness Data
| Family | Genus | Species | IUCNa | Migratory | Rangeb | Colony size | Global FST | No. of publications | No. of viruses | FST references | Virus references |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Vespertilionidae | Antrozous | pallidus | LC | Y | C | 60 | 0.244 | 175 | 1 | (Weyandt and Van Den Bussche, 2007) | (Burns et al., 1956) |
| Phyllostomidae | Artibeus | jamaicensis | LC | N | I | 10 | 0.013 | 271 | 4 | (Ortega et al., 2003; Carstens et al., 2004) | (Downs et al., 1963; Calisher et al., 1971; Price and Everard, 1977; Aguilar-Setien et al., 2008) |
| Vespertilionidae | Corynorhinus | townsendii | LC | N | C | 50 | 0.081 | 50 | 1 | (Piaggio and Perkins, 2005), [Piaggio, personal communication] | (Blanton et al., 2007) |
| Pteropodidae | Cynopterus | brachyotis | LC | N | I | 8 | 0.074 | 105 | 5 | (Peterson and Heaney, 1993; Heaney et al., 2005; Campbell et al., 2006) | (Salaun et al., 1974; Johara et al., 2001; Calisher et al., 2006) |
| Pteropodidae | Cynopterus | sphinx | LC | N | C | 36 | 0.164 | 180 | 3 | (Storz et al., 2001; Campbell et al. 2006) | (Pavri and Singh, 1968; Kelkar et al., 1981; Reynes et al., 2004) |
| Phyllostomidae | Desmodus | rotundus | LC | N | C | 60 | 0.468 | 400 | 2 | (Wilkinson, 1985; Martins et al., 2007) | (Burns et al., 1956; Correa-Giron et al., 1972; Seymour et al., 1978) |
| Pteropodidae | Eidolon | helvum | NT | Y | C | 500,000 | 0.153 | 142 | 5 | (Juste et al., 2000) | (Kemp et al., 1988; Hayman et al., 2008; Kuzmin et al., 2008; Markotter et al., 2008; Razafindratsimandresy et al., 2009; Tong et al., 2009) |
| Pteropodidae | Eonycteris | spelaea | LC | N | I | 5000 | 0.120 | 71 | 4 | (Maharadatunkamsi et al. 2003) | (Johara et al., 2001; Lumlertdacha et al., 2005; Calisher et al., 2006) |
| Vespertilionidae | Eptesicus | fuscus | LC | N | C | 50 | 0.464 | 841 | 5 | (Turmelle, 2002), [Turmelle, personal communication] | (Constantine, 1970; Trimarchi and Debbie, 1977; CDC, 2000; Dominguez et al., 2007) |
| Pteropodidae | Macroglossus | minimus | LC | N | I | 1 | 0.105 | 36 | 1 | (Heaney et al., 2005) | (Calisher et al., 2006) |
| Vespertilionidae | Miniopterus | schreibersii | NT | Y | C | 5000 | 0.620 | 251 | 6 | (Miller-Butterworth et al., 2003) | (Sulkin et al., 1970; Serra-Cobo et al., 2002; Botvinkin et al., 2003; Poon et al., 2005; Konstantinov et al., 2006; Markotter et al., 2008) |
| Vespertilionidae | Myotis | daubentonii | LC | N | C | 100 | 0.017 | 306 | 2 | (Ngamprasertwong et al., 2008) | (Whitby et al., 2000; Gloza-Rausch et al., 2008) |
| Vespertilionidae | Myotis | myotis | LC | N | C | 5000 | 0.310 | 753 | 3 | (Castella et al., 2000, 2001; Ruedi and Castella, 2003; Ruedi et al., 2008) | (Serra-Cobo et al., 2002; Amengual et al., 2007; Wibbelt et al., 2007) |
| Vespertilionidae | Myotis | nattereri | LC | N | C | 275 | 0.017 | 367 | 4 | (Rivers et al., 2005) | (Serra-Cobo et al., 2002; Wibbelt et al., 2007) |
| Vespertilionidae | Myotis | septentrionalis | LC | N | C | 30 | 0.002 | 129 | 1 | (Arnold, 2007) | (Blanton et al., 2007) |
| Vespertilionidae | Nyctalus | noctula | LC | Y | C | 100 | 0.020 | 574 | 5 | (Petit and Mayer, 1999) | (L’vov et al., 1973, 1979; Muller et al., 2004; Wibbelt et al., 2007) |
| Phyllostomidae | Phyllostomus | hastatus | LC | N | C | 55 | 0.031 | 131 | 1 | (McCracken and Bradbury, 1981) | (Constantine, 1970) |
| Vespertilionidae | Pipistrellus | pipistrellus | LC | Y | C | 50 | 0.044 | 715 | 5 | (Racey et al., 2007) | (Muller et al., 2004; Calisher et al., 2006; Cui et al., 2007; Wibbelt et al., 2007) |
| Vespertilionidae | Pipistrellus | pygmaeus | LC | Y | I | 250 | 0.024 | 130 | 1 | (Racey et al., 2007) | (Gloza-Rausch et al., 2008) |
| Vespertilionidae | Plecotus | auritus | LC | N | C | 50 | 0.019 | 568 | 2 | (Burland et al., 1999) | (Muller et al., 2004; Wibbelt et al., 2007) |
| Pteropodidae | Pteropus | alecto | LC | Y | C | 5000 | 0.023 | 86 | 4 | (Webb and Tidemann, 1996) | (Fraser et al., 1996; Halpin et al., 1996; Speare et al., 1997; Philbey et al., 1998; Halpin et al., 2000, 2007) |
| Pteropodidae | Pteropus | hypomelanus | LC | N | I | 250 | 0.882 | 48 | 4 | (Olival, 2008) | (Chua et al., 2001, 2002; Johara et al., 2001; Arguin et al., 2002; Pritchard et al., 2006) |
| Pteropodidae | Pteropus | poliocephalus | NT* | Y | C | 25,000 | 0.014 | 175 | 4 | (Webb and Tidemann, 1996) | (Gard and Marshall, 1973; Halpin et al., 1996, 2000; Philbey et al., 1998; Barrett et al., 2005) |
| Pteropodidae | Pteropus | scapulatus | LC | Y | C | 200,000 | 0.028 | 92 | 2 | (Sinclair et al., 1996) | (Halpin et al., 1996; Speare et al., 1997) |
| Pteropodidae | Pteropus | vampyrus | NT | Y | I | 5000 | −0.006 | 58 | 1 | (Olival, 2008) | (Johara et al., 2001) |
| Rhinolophidae | Rhinolophus | ferrumequinum | LC | N | C | 125 | 0.102 | 735 | 4 | (Rossiter et al., 2000) | (Kim et al., 1994; Serra-Cobo et al., 2002; Li et al., 2005; Calisher et al., 2006; Cui et al., 2007; Shi and Hu, 2008) |
| Rhinolophidae | Rhinolophus | pusillus | LC | N | C | 1000 | 0.088 | 110 | 2 | (Yoshino et al., 2008) | (Sulkin et al., 1970; Iwasaki et al., 2004) |
| Pteropodidae | Rousettus | aegyptiacus | LC | N | C | 5000 | 0.493 | 249 | 8 | (Juste et al., 1996) | (Kalunda et al., 1986; Wellenberg et al., 2002; Calisher et al., 2006; McKnight et al., 2006; Rector et al., 2006; Muller et al., 2007; Swanepoel et al., 2007; Towner et al., 2007; Markotter et al., 2008) |
| Pteropodidae | Rousettus | amplexicaudatus | LC | N | I | 2000 | 0.100 | 47 | 1 | (Heaney et al., 2005) | (Arguin et al., 2002) |
| Vespertilionidae | Scotophilus | kuhlii | LC | N | I | 500 | 0.070 | 23 | 4 | (Hisheh et al., 2004) | (Johara et al., 2001; Arguin et al., 2002; Reynes et al., 2004; Calisher et al., 2006; Cui et al., 2007) |
| Molossidae | Tadarida | brasiliensis | LC | Y | C | 500,000 | 0.070 | 412 | 3 | (McCracken and Gassel, 1997; Russell et al., 2005) | (Enright et al., 1955; Burns et al., 1956; Burns and Farinacci 1956; Constantine and Woodall, 1964; Sulkin et al., 1966) |
| Phyllostomidae | Uroderma | bilobatum | LC | N | C | 30 | 0.010 | 47 | 1 | (Meyer et al., 2009) | (Seymour et al., 1978) |
| Vespertilionidae | Vespertilio | murinus | LC | Y | C | 100 | 0.071 | 229 | 1 | (Safi et al., 2007) | (Hutson, 2004) |
aIUCN status is least concern (LC) or near-threatened/vulnerable (NT) (*Pteropus poliocephalus is listed as vulnerable)
bRange distribution is continental (C) or island (I)
Figure 2.

The number of viruses associated with bats, for species with known population structure estimates and at least one viral association (n = 33).
Correlates of Viral Richness
The mixed stepwise variable selection procedure converged on a model including IUCN status, distribution, log-transformed global FST, and log-transformed sampling effort as covariates. The stepwise procedure omitted migratory status and log-transformed average colony size as model predictors (P > 0.2). The multivariate model explained 37% of the viral richness data (Likelihood Ratio
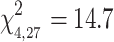 , P = 0.005). IUCN status was a significant predictor of viral richness among bats (P = 0.02), and near-threatened or vulnerable bats had higher mean viral richness (4.00 ± 2.16) compared to bats of least concern (2.71 ± 1.51) (Fig. 3a). Global FST was positively associated with viral richness (P = 0.03) (Fig. 3b), as was sampling effort (P = 0.007) (Fig. 3c). Species range distribution (i.e., island vs. continental) was not a significant predictor of viral richness (P = 0.12). A reduced multivariate model excluding distribution as a predictor explained 32% of the viral richness data (Likelihood Ratio
, P = 0.005). IUCN status was a significant predictor of viral richness among bats (P = 0.02), and near-threatened or vulnerable bats had higher mean viral richness (4.00 ± 2.16) compared to bats of least concern (2.71 ± 1.51) (Fig. 3a). Global FST was positively associated with viral richness (P = 0.03) (Fig. 3b), as was sampling effort (P = 0.007) (Fig. 3c). Species range distribution (i.e., island vs. continental) was not a significant predictor of viral richness (P = 0.12). A reduced multivariate model excluding distribution as a predictor explained 32% of the viral richness data (Likelihood Ratio
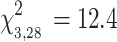 , P = 0.006) (Table 2). In the reduced model, all three predictors significantly explained variation in viral richness (P < 0.03).
, P = 0.006) (Table 2). In the reduced model, all three predictors significantly explained variation in viral richness (P < 0.03).
Figure 3.

The three significant predictor variables from the best generalized linear model. Panels show the relationship between IUCN threat status (a), global FST (b), sampling effort (c), and viral richness across bat species (n = 32). Each data point represents one bat species in the linear regressions. NT/VU, near-threatened/vulnerable; LC, least concern.
Table 2.
A Summary of the Models and Scores
| Model | Predictors | LogLik | AICc | K | I | Rank | Lambda |
|---|---|---|---|---|---|---|---|
| 2 | iucn, log (fst all), log (no. pubs) | −54.17 | 123.7 | 6 | 0.0 | 1 | 0 |
| 1 | iucn, distribution, log (fst all), log (no. pubs) | −52.99 | 124.7 | 7 | 0.9 | 2 | 0 |
| 3 | iucn, log (no. pubs) | −56.42 | 125.2 | 5 | 1.5 | 3 | 0 |
| 5 | log (fst all), log (no. pubs) | −56.49 | 125.3 | 5 | 1.6 | 4 | 0.02 |
| 4 | iucn, log (fst all) | −56.70 | 125.7 | 5 | 2.0 | 5 | 0 |
The maximum likelihood estimate of phylogenetic signal in the full or reduced models was λ = 0 (Table 2), indicating that patterns of viral richness are independent from the phylogenetic relationships among bats in this study. Effect tests for parameters in the full or reduced models were similar when compared to the generalized linear models without any phylogenetic correction. The reduced model provided the best explanation of the data (Table 2). The randomization of viral richness trait values across taxa to obtain a null distribution of λ revealed that we cannot reject that our observed value is outside of the null distribution (P = 0.22), indicating a lack of power. To test the estimation procedure, a dummy phenotypic data set was created with trait values skewed to match the phylogeny, and an estimate of λ = 1 was obtained (data not shown).
Discussion
Zoonotic disease emergence is a multipart process which includes: the build-up of risk factors during a “pre-spillover” period; a spillover event—when an infected individual from a reservoir population transmits the disease to a new host (Power and Mitchell, 2004); a period of local transmission in the new host that may or may not die out depending on characteristics of the pathogen and host population; and, a potentially self-sustaining transmission period within the new host (Anderson and May, 1986). Risk factors that exist in the “pre-spillover” period can be environmental, demographic, or evolutionary; these can be viewed as existing along an eco-evolutionary continuum. Successful cross-species transmission is usually the result of repeated spillover events, i.e., “viral chatter” (Wolfe et al., 2005). Standing pathogen diversity in reservoir populations can be critically important for promoting the evolutionary flexibility of pathogens, and likelihood of successful spillover and emergence in a new host (Morse, 1993). Thus, the taxonomic diversity of pathogens in hosts and the genetic diversity of each respective pathogen may both be important predictors of spillover success, or emergence. Mechanisms that promote diversity on either level may increase the likelihood of emergence.
Impact of Genetic Spatial Structure
Population genetic structure and sampling effort were correlated with viral richness in bats. Although a positive linear fit best described the relationship between population genetic structure and viral richness, compared to other polynomial models (data not shown), it is possible that population structure may have a nonlinear impacts on epizootiology and subsequent viral evolution within a host. Modeling, coupled with empirical observations, has shown that the spatial structure of host populations can influence the likelihood of disease establishment (Rodriguez and Torres-Sorando, 2001), rate of pathogen spread (Caraco et al., 2002), duration of epidemics (Park et al., 2002), as well as pathogen virulence (Boots et al., 2004). Extremely low levels of population genetic structure predict a homogeneously mixing population, potentially leading to viral epidemics that rapidly burn out without establishing an enzootic state. This is supported by metapopulation models that have shown an increased extinction probability in cases with increasing migration rates and moderate pathogen virulence (Hess, 1996). In hosts that exhibit extremely high levels of population structure, characterized by little migration between demes and small effective populations, localized disease extinction may preclude viral establishment. However, point estimates of population genetic structure may not capture all migration or fission–fusion dynamics that impact epizootiology, as this one statistic generally describes population substructure that relates to mating probabilities. Furthermore, the duration of incubation and infectious periods has equal importance for predicting the potential reproductive capabilities of a pathogen in any population, structured or not. While we did not examine the genetic diversity within host-specific viral lineages, the epizootiology of lyssaviruses in bats suggests that moderate levels of host population structure can positively influence viral genetic diversity within hosts (Smith, 1996; Messenger et al., 2003b; Davis et al., 2005; Hughes et al., 2005; Franka et al., 2006; Velasco-Villa et al., 2006). Bats with increasing population genetic structure are positively associated with viral richness in this study. However, the current host sampling excludes several families, hundreds of species, and may not be representative for all bats. Furthermore, it will be important to distinguish between pathogens that are directly transmitted or vector-borne, and those that infect single or multiple hosts, as additional data become available. The influence of host population genetic structure may be negligible for vector-borne or multihost viruses, where pathogen distribution and richness may be determined by spatial structure of vectors and all competent hosts. Low sample sizes in the current study precluded partitioned statistical treatment for all possible transmission strategies. There is also evidence that many other factors, such as host immune response, may promote lineage diversity within pathogens (Grenfell et al., 2004). However, the spatial genetic structure of hosts can serve as a starting point for investigating pathogen distribution, diversification, and spatial control strategies.
Impacts of Host Ecology and Social Behavior
Theoretical modeling of epizootic and enzootic processes predicts that larger populations generally will increase the likelihood of viral establishment (Anderson and May, 1979); however, host colony size was not correlated with viral richness across bats in this study. Bat colony sizes can be extremely variable across species (Kunz, 1982; O’Shea and Bogan, 2003), and are known to be seasonally dynamic in response to variation in prey and habitat resource availability and life history. There is evidence that Brazilian free-tailed bats (Tadarida brasiliensis) migrate from Mexico to track seasonal insect abundance in conjunction with formation of large maternity colonies in the southwestern United States (McCracken, 2003). The straw-colored fruit bat (Eidolon helvum) also exhibits seasonal fluctuations in colony size correlated with temporal and geographic variation in fruit availability across sub-Saharan Africa (Richter and Cumming, 2006). Colony sizes of the large flying fox (Pteropus vampyrus) in Malaysia fluctuate dramatically over time, and dispersal behavior may reflect variation in fruit availability and hunting pressure (Epstein et al., 2009). Although generalized predictions can be made from long-term summary statistics on host aggregation, seasonality also plays a critical role in epizootiological processes and disease emergence (Altizer et al., 2006). In our search of the literature for species-specific colony size estimates, there were frequent references to seasonal and gender variation in gregarious behavior. Seasonal variation in colony size may be more important than typical colony size per se in terms of understanding the importance of contact networks for viral emergence and persistence in bat hosts. Irrespective of potential intra-annual variation, the typical colony sizes of individual host species may also be less important when considering vector-borne and multihost pathogens.
Host geographic range may be an important determinant of pathogen richness, as larger geographic ranges encompass a greater diversity of ecosystems, faunal communities, and may lead to greater spatial population substructure. Geographic range area was not a significant predictor of viral richness among bats in this study, but other studies have highlighted that host geographic range may be an important predictor of parasite richness (Gregory, 1990; Nunn et al., 2003). Smaller geographic ranges have been linked to increased extinction risk across bats (Jones et al., 2003), and may similarly govern parasite dynamics associated with such hosts. However, a study across primates found evidence that host relatedness and the presence of conspecific range overlap were significant predictors of parasite community similarity across taxa (Davies and Pedersen, 2008). Given recent implication of bats in EID spillover events (Halpin et al., 2000; Johara et al., 2001; Leroy et al., 2005; Li et al., 2005; Swanepoel et al., 2007), additional studies on pathogen community similarity that include multiple pathogen taxonomic groups and tests of phylogenetic dependence across bats are needed. Although several socioecological variables were not significant predictors of viral richness across bats in this study, it would be prudent to search more exhaustively among known viral hosts, without limiting the search to species with characterized genetic spatial structure.
IUCN threat status, which combines data on population sizes, distribution, and geographic range area, was a significant predictor of viral richness across bats in this study, whereas individual contributing factors were nonsignificant. In this study, bats that were near-threatened (n = 3) or vulnerable (n = 1) had higher viral richness compared to bats of least concern (n = 28). This result contrasts a previous study in primates (Altizer et al., 2007), where threatened taxa harbored lower parasite richness compared to nonthreatened taxa. Although the categories of threat status under comparison were not identical compared to the Altizer et al. (2007) study, the current study was also much more limited in taxonomic scope for both hosts and parasites under consideration. Although the data suggest that near-threatened or vulnerable bats may be more susceptible to viral infection compared to bats of least concern, the current result warrants caution given the uneven and limited sampling across IUCN threat categories.
Although significant links between phylogeny and parasite richness have been demonstrated in other studies (Nunn et al., 2004), there was no evidence of phylogenetic dependence for patterns of viral richness in this study. Familial monophyly across bats was well supported for the phylogeny used in this study (Bininda-Emonds et al., 2007). However, small sample sizes and the lack of phylogenetic resolution within and among bat genera may have lessened our power to detect dependence between phylogenetic relatedness and patterns of viral richness, as demonstrated by our inability to reject that our estimate of λ was not nested within the null distribution. It is also possible that viral richness traits may not be evolving under the model of Brownian motion that was employed in our test (Freckleton et al., 2002). Sampling that is more broadly representative of chiropteran diversity may allow for more powerful tests of phylogenetic dependence for trait evolution.
Limitations of a Biogeographic Approach
Although estimates of FST provide information on historic dispersal and social organization of reservoir species over relatively large geographic or temporal scales, studies of emerging zoonoses and wildlife EIDs would benefit from collecting fine-scale, or real-time spatial data on host movement. Few studies have focused on this, but examples include studies of the reservoir hosts for chronic wasting disease (Conner and Miller, 2004), canine pathogens (Riley et al., 2004), the use of satellite telemetry to quantify migration activity for flying fox reservoirs of Nipah virus (Epstein et al., 2009), and use of contact transmitters on domestic animals and wildlife (Bohm et al., 2009). It is often logistically difficult to collect these data, but the combination of population genetic data with direct measures of host dispersal and contact have the potential to more accurately predict the spatial patterns of host–pathogen dynamics across the landscape (Hess et al., 2004; Real and Biek, 2007; Olival, 2008). Thus, biogeographic inferences can make important macroecological predictions on pathogen richness and epizootiological processes, but control strategies for zoonotic disease will also require attention to multihost interactions, as well as local geographic and temporal scales.
Future Directions
As this study was only able to include a small fraction of bat species (33 of 1200) for which both population genetic and viral richness data were available, it also highlights how little we know about this diverse group of animals that are key players in the health of ecosystems and emergence of infectious diseases globally. Viral richness estimates in this study surely underestimate the real number of viruses associated with bats, in part due to a lack of population structure estimates for the overwhelming majority of bat species and also incomplete disease surveillance. Models fit to the temporal emergence of viruses suggests that, even for human pathogens, the process of discovery is far from complete (Woolhouse et al., 2008). A number of recently discovered viruses, in addition to many other previously published associations, could not be included in our data set because they were found in species for which we did not have population genetic structure data or other host socioecological variables. This points to a number of considerations for future studies. First, more bat species should be examined using population genetic markers, and efforts should be made to collect samples for genetic work at the same time that animals are captured and handled during viral surveillance or telemetry studies. Second, more standardized methods for detecting viruses in wildlife reservoirs are needed. The majority of studies target limited taxonomic breadth in viral surveillance efforts, and often serology alone can be problematic due to cross-reactivity (Hanlon et al., 2005; Hayman et al., 2008). Novel methods, such as multiplex PCR and DNA microarrays, in addition to diverse collaborative research teams, have the capacity to revolutionize the information gained through viral surveillance efforts and will surely advance our understanding of the processes of pathogen richness, coinfection, and emerging bat zoonoses. Lastly, this study highlights the importance of species-specific evolutionary and life-history characteristics for predicting viral richness in wildlife, and this should be examined for other taxonomic groups. This approach could greatly aid pathogen discovery by focusing sampling efforts to host species with potentially greater viral richness and, hence, greater likelihood of harboring novel zoonotic pathogens.
Acknowledgments
The authors thank the International Society of Biogeography, S. Scheiner, and K. Smith, for organizing and supporting the Biogeography of Disease Symposium and its participants. This research was supported in part by two NIH/NSF “Ecology of Infectious Diseases” awards (R01-TW05869 and 0430418). AST thanks G. McCracken, the Department of Ecology and Evolutionary Biology at the University of Tennessee, and the Rabies Team at the Centers for Disease Control and Prevention. KJO thanks the Department of Ecology, Evolution, and Environmental Biology at Columbia University and the Sackler Institute for Comparative Genomics at the American Museum of Natural History. AST and KJO were supported by US Environmental Protection Agency Science-To-Achieve-Results (STAR) fellowships. The authors also thank D. Streicker, B. Fitzpatrick, and J. Fordyce for statistical advice, and two anonymous reviewers for comments that improved this manuscript. The ideas presented in this study are those of the authors only, and do not necessarily represent the views of the funding agency or institutions.
References
- Aguilar-Setien A, Romero-Almaraz ML, Sanchez-Hernandez C, Figueroa R, Juarez-Palma LP, Garcia-Flores MM, et al. Dengue virus in Mexican bats. Epidemiology and Infection. 2008;136:1678–1683. doi: 10.1017/S0950268808000460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allworth A, Murray K, Morgan J. A human case of encephalitis due to a lyssavirus recently identified in fruit bats. Communicable Diseases Intelligence. 1996;20:504. [Google Scholar]
- Altizer S, Dobson A, Hosseini P, Hudson P, Pascual M, Rohani P. Seasonality and the dynamics of infectious diseases. Ecology Letters. 2006;9:467–484. doi: 10.1111/j.1461-0248.2005.00879.x. [DOI] [PubMed] [Google Scholar]
- Altizer S, Nunn CL, Lindenfors P. Do threatened hosts have fewer parasites? A comparative study in primates. Journal of Animal Ecology. 2007;76:304–314. doi: 10.1111/j.1365-2656.2007.01214.x. [DOI] [PubMed] [Google Scholar]
- Amengual B, Bourhy H, Lopez-Roig M, Serra-Cobo J. Temporal dynamics of European bat lyssavirus type 1 and survival of Myotis myotis bats in natural colonies. PLoS ONE. 2007;2:e566. doi: 10.1371/journal.pone.0000566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RM, May RM. Population biology of infectious diseases: part I. Nature. 1979;280:361–367. doi: 10.1038/280361a0. [DOI] [PubMed] [Google Scholar]
- Anderson RM, May RM. The invasion, persistence and spread of infectious diseases within animal and plant communities. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences. 1986;314:533–570. doi: 10.1098/rstb.1986.0072. [DOI] [PubMed] [Google Scholar]
- Arguin PM, Murray-Lillibridge K, Miranda ME, Smith JS, Calaor AB, Rupprecht CE. Serologic evidence of lyssavirus infections among bats, the Philippines. Emerging Infectious Diseases. 2002;8:258–262. doi: 10.3201/eid0803.010330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold BD. Population structure and sex-biased dispersal in the forest dwelling vespertilionid bat, Myotis septentrionalis. American Midland Naturalist. 2007;157:374–384. doi: 10.1674/0003-0031(2007)157[374:PSASDI]2.0.CO;2. [DOI] [Google Scholar]
- Asikainen K, Hanninen T, Henttonen H, Niemimaa J, Laakkonen J, Andersen HK, et al. Molecular evolution of Puumala hantavirus in Fennoscandia: phylogenetic analysis of strains from two recolonization routes, Karelia and Denmark. Journal of General Virology. 2000;81:2833–2841. doi: 10.1099/0022-1317-81-12-2833. [DOI] [PubMed] [Google Scholar]
- Barlow ND. The ecology of wildlife disease control: simple models revisited. Journal of Applied Ecology. 1996;33:303–314. doi: 10.2307/2404752. [DOI] [Google Scholar]
- Barrett J, Rodwell B, Lunt R, Rupprecht C, Field H, Smith G, et al. (2005) Australian bat lyssavirus: observations of natural and experimental infection in bats. In: Wildlife Disease Association International Conference, Cairns, Queensland, Australia, 149 pp
- Biek R, Drummond AJ, Poss M. A virus reveals population structure and recent demographic history of its carnivore host. Science. 2006;311:538–541. doi: 10.1126/science.1121360. [DOI] [PubMed] [Google Scholar]
- Bininda-Emonds OR, Cardillo M, Jones KE, MacPhee RD, Beck RM, Grenyer R, et al. The delayed rise of present-day mammals. Nature. 2007;446:507–512. doi: 10.1038/nature05634. [DOI] [PubMed] [Google Scholar]
- Blanton JD, Hanlon CA, Rupprecht CE. Rabies surveillance in the United States during 2006. Journal of the American Veterinary Medical Association. 2007;231:540–556. doi: 10.2460/javma.231.4.540. [DOI] [PubMed] [Google Scholar]
- Bohm M, Hutchings MR, White PC. Contact networks in a wildlife-livestock host community: identifying high-risk individuals in the transmission of bovine TB among badgers and cattle. PLoS One. 2009;4:e5016. doi: 10.1371/journal.pone.0005016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolzoni L, Real L, De Leo G. Transmission heterogeneity and control strategies for infectious disease emergence. PLoS One. 2007;2:e747. doi: 10.1371/journal.pone.0000747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boots M, Hudson PJ, Sasaki A. Large shifts in pathogen virulence relate to host population structure. Science. 2004;303:842–844. doi: 10.1126/science.1088542. [DOI] [PubMed] [Google Scholar]
- Boots M, Mealor M. Local interactions select for lower pathogen infectivity. Science. 2007;315:1284–1286. doi: 10.1126/science.1137126. [DOI] [PubMed] [Google Scholar]
- Boots M, Sasaki A. ‘Small worlds’ and the evolution of virulence: infection occurs locally and at a distance. Proceedings of the Royal Society of London. Series B: Biological Sciences. 1999;266:1933–1938. doi: 10.1098/rspb.1999.0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boots M, Sasaki A. The evolutionary dynamics of local infection and global reproduction in host-parasite interactions. Ecology Letters. 2000;3:181–185. doi: 10.1046/j.1461-0248.2000.00139.x. [DOI] [Google Scholar]
- Botvinkin AD, Poleschuk EM, Kuzmin IV, Borisova TI, Gazaryan SV, Yager P, et al. Novel lyssaviruses isolated from bats in Russia. Emerging Infectious Diseases. 2003;9:1623–1625. doi: 10.3201/eid0912.030374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull JJ, Molineux IJ, Rice WR. Selection of benevolence in a host-parasite system. Evolution. 1991;45:875–882. doi: 10.2307/2409695. [DOI] [PubMed] [Google Scholar]
- Burland TM, Barratt EM, Beaumont MA, Racey PA (1999) Population genetic structure and gene flow in a gleaning bat, Plecotus auritus. Proceedings of the Royal Society of London. Series B: Biological Sciences 266:975–980
- Burland TM, Worthington-Wilmer JW. Seeing in the dark: molecular approaches to the study of bat populations. Biological Reviews. 2001;76:389–409. doi: 10.1017/S1464793101005747. [DOI] [PubMed] [Google Scholar]
- Burnham KP, Anderson DR. Model Selection and Multimodel Inference. 2. New York: Springer; 2002. [Google Scholar]
- Burns KF, Farinacci CJ. Virus of bats antigenically related to St. Louis encephalitis. Science. 1956;123:227. doi: 10.1126/science.123.3189.227. [DOI] [PubMed] [Google Scholar]
- Burns KF, Farinacci CF, Murnane VC. Insectivorous bats naturally infected with rabies in the southwestern United States. American Journal of Public Health. 1956;46:1089–1097. doi: 10.2105/AJPH.46.9.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calisher CH, Chappell WA, Maness KS, Lord RD, Sudia WD. Isolations of Nepuyo virus strains from Honduras, 1967. American Journal of Tropical Medicine and Hygiene. 1971;20:331–337. doi: 10.4269/ajtmh.1971.20.331. [DOI] [PubMed] [Google Scholar]
- Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clinical Microbiology Reviews. 2006;19:531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell P, Schneider CJ, Adnan AM, Zubaid A, Kunz TH. Comparative population structure of Cynopterus fruit bats in peninsular Malaysia and southern Thailand. Molecular Ecology. 2006;15:29–47. doi: 10.1111/j.1365-294X.2005.02769.x. [DOI] [PubMed] [Google Scholar]
- Caraco T, Glavanakov S, Chen G, Flaherty JE, Ohsumi TK, Szymanski BK. Stage-structured infection transmission and a spatial epidemic: a model for Lyme disease. American Naturalist. 2002;160:348–359. doi: 10.1086/341518. [DOI] [PubMed] [Google Scholar]
- Carrington CVF, Foster JE, Zhu CH, Zhang JX, Smith GJD, Thompson N, et al. Detection and phylogenetic analysis of group 1 coronaviruses in South American bats. Emerging Infectious Diseases. 2008;14:1890–1893. doi: 10.3201/eid1412.080642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstens BC, Sullivan J, Davalos LM, Larsen PA, Pedersen SC. Exploring population genetic structure in three species of Lesser Antillean bats. Molecular Ecology. 2004;13:2557–2566. doi: 10.1111/j.1365-294X.2004.02250.x. [DOI] [PubMed] [Google Scholar]
- Castella V, Ruedi M, Excoffier L. Contrasted patterns of mitochondrial and nuclear structure among nursery colonies of the bat Myotis myotis. Journal of Evolutionary Biology. 2001;14:708–720. doi: 10.1046/j.1420-9101.2001.00331.x. [DOI] [Google Scholar]
- Castella V, Ruedi M, Excoffier L, Ibanez C, Arlettaz R, Hausser J. Is the Gibraltar strait a barrier to gene flow for the bat Myotis myotis (Chiroptera: Vespertilionidae)? Molecular Ecology. 2000;9:1761–1772. doi: 10.1046/j.1365-294x.2000.01069.x. [DOI] [PubMed] [Google Scholar]
- CDC Update: West Nile virus activity—Northeastern United States, 2000. Morbidity and Mortality Weekly Report. 2000;49:820–822. [PubMed] [Google Scholar]
- Chua KB, Crameri G, Hyatt A, Yu M, Tompang MR, Rosli J, et al. A previously unknown reovirus of bat origin is associated with an acute respiratory disease in humans. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:11424–11429. doi: 10.1073/pnas.0701372104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua KB, Koh CL, Hooi PS, Wee KF, Khong JH, Chua BH, et al. Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes and Infection. 2002;4:145–151. doi: 10.1016/S1286-4579(01)01522-2. [DOI] [PubMed] [Google Scholar]
- Chua KB, Voon K, Crameri G, Tan HS, Rosli J, McEachern JA, et al. Identification and characterization of a new orthoreovirus from patients with acute respiratory infections. PLoS ONE. 2008;3:e3803. doi: 10.1371/journal.pone.0003803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua KB, Wang LF, Lam SK, Crameri G, Yu M, Wise T, et al. Tioman virus, a novel paramyxovirus isolated from fruit bats in Malaysia. Virology. 2001;283:215–229. doi: 10.1006/viro.2000.0882. [DOI] [PubMed] [Google Scholar]
- Cleaveland S, Laurenson MK, Taylor LH. Diseases of humans and their domestic mammals: pathogen characteristics, host range and the risk of emergence. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences. 2001;356:991–999. doi: 10.1098/rstb.2001.0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner MM, Miller MW. Movement patterns and spatial epidemiology of a prion disease in mule deer population units. Ecological Applications. 2004;14:1870–1881. doi: 10.1890/03-5309. [DOI] [Google Scholar]
- Constantine DG. Bats in relation to the health, welfare, and economy of man. In: Wimsatt WA, editor. Biology of Bats. New York: Academic Press; 1970. pp. 319–449. [Google Scholar]
- Constantine DG, Woodall DF. Latent infection of Rio Bravo virus in salivary glands of bats. Public Health Reports. 1964;79:1033–1039. [PMC free article] [PubMed] [Google Scholar]
- Correa-Giron P, Calisher CH, Baer GM. Epidemic strain of Venezuelan equine encephalomyelitis virus from a vampire bat captured in Oaxaca, Mexico, 1970. Science. 1972;175:546–547. doi: 10.1126/science.175.4021.546. [DOI] [PubMed] [Google Scholar]
- Criscione CD, Blouin MS. Parasite phylogeographical congruence with salmon host evolutionarily significant units: implications for salmon conservation. Molecular Ecology. 2007;16:993–1005. doi: 10.1111/j.1365-294X.2006.03220.x. [DOI] [PubMed] [Google Scholar]
- Criscione CD, Poulin R, Blouin MS. Molecular ecology of parasites: elucidating ecological and microevolutionary processes. Molecular Ecology. 2005;14:2247–2257. doi: 10.1111/j.1365-294X.2005.02587.x. [DOI] [PubMed] [Google Scholar]
- Cross PC, Lloyd-Smith JO, Johnson PLF, Getz WM. Duelling timescales of host movement and disease recovery determine invasion of disease in structured populations. Ecology Letters. 2005;8:587–595. doi: 10.1111/j.1461-0248.2005.00760.x. [DOI] [Google Scholar]
- Cui J, Han N, Streicker D, Li G, Tang X, Shi Z, et al. Evolutionary relationships between bat coronaviruses and their hosts. Emerging Infectious Diseases. 2007;13:1526–1532. doi: 10.3201/eid1310.070448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Rosa ES, Kotait I, Barbosa TF, Carrieri ML, Brandao PE, Pinheiro AS, et al. Bat-transmitted human rabies outbreaks, Brazilian Amazon. Emerging Infectious Diseases. 2006;12:1197–1202. doi: 10.3201/1208.050929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies TJ, Pedersen AB. Phylogeny and geography predict pathogen community similarity in wild primates and humans. Proceedings of the Royal Society of London. Series B: Biological Sciences. 2008;275:1695–1701. doi: 10.1098/rspb.2008.0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis PL, Holmes EC, Larrous F, Van der Poel WH, Tjornehoj K, Alonso WJ, et al. Phylogeography, population dynamics, and molecular evolution of European bat lyssaviruses. Journal of Virology. 2005;79:10487–10497. doi: 10.1128/JVI.79.16.10487-10497.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Roode JC, Yates AJ, Altizer S (2008) Virulence-transmission trade-offs and population divergence in virulence in a naturally occurring butterfly parasite. Proceedings of the National Academy of Sciences of the United States of America 105:7489–7494 [DOI] [PMC free article] [PubMed]
- Dominguez SR, O’Shea TJ, Oko LM, Holmes KV. Detection of group 1 coronaviruses in bats in North America. Emerging Infectious Diseases. 2007;13:1295–1300. doi: 10.3201/eid1309.070491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly CA, Woodroffe R, Cox DR, Bourne FJ, Cheeseman CL, Clifton-Hadley RS, et al. Positive and negative effects of widespread badger culling on tuberculosis in cattle. Nature. 2006;439:843–846. doi: 10.1038/nature04454. [DOI] [PubMed] [Google Scholar]
- Downs WG, Anderson CR, Spence L, Aitken TH, Greenhall AH. Tacaribe virus, a new agent isolated from Artibeus bats and mosquitoes in Trinidad, West Indies. American Journal of Tropical Medicine and Hygiene. 1963;12:640–646. doi: 10.4269/ajtmh.1963.12.640. [DOI] [PubMed] [Google Scholar]
- Dragoo JW, Lackey JA, Moore KE, Lessa EP, Cook JA, Yates TL. Phylogeography of the deer mouse (Peromyscus maniculatus) provides a predictive framework for research on hantaviruses. Journal of General Virology. 2006;87:1997–2003. doi: 10.1099/vir.0.81576-0. [DOI] [PubMed] [Google Scholar]
- Drexler JF, Corman VM, Gloza-Rausch F, Seebens A, Annan A, Ipsen A, et al. Henipavirus RNA in African bats. PLoS ONE. 2009;4:e6367. doi: 10.1371/journal.pone.0006367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan RP, Forsyth DM, Hone J. Testing the metabolic theory of ecology: allometric scaling exponents in mammals. Ecology. 2007;88:324–333. doi: 10.1890/0012-9658(2007)88[324:TTMTOE]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Enright JB, Sadler WW, Moulton JE, Constantine D. Isolation of rabies virus from an insectivorous bat (Tadarida mexicana) in California. Proceedings of the Society for Experimental Biology and Medicine. 1955;89:94–96. doi: 10.3181/00379727-89-21725. [DOI] [PubMed] [Google Scholar]
- Epstein JH, Olival KJ, Pulliam JRC, Smith CS, Westrum J, Hughes T, et al. Management of Pteropus vampyrus, a hunted migratory species with a multinational home-range. Journal of Applied Ecology. 2009;46:991–1002. doi: 10.1111/j.1365-2664.2009.01699.x. [DOI] [Google Scholar]
- Ewald PW. Waterborne transmission and the evolution of virulence among gastrointestinal bacteria. Epidemiology and Infection. 1991;106:83–119. doi: 10.1017/S0950268800056478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes—application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezenwa VO. Host social behavior and parasitic infection: a multifactorial approach. Behavioral Ecology. 2004;15:446–454. doi: 10.1093/beheco/arh028. [DOI] [Google Scholar]
- Favi M, de Mattos CA, Yung V, Chala E, Lopez LR, de Mattos CC. First case of human rabies in Chile caused by an insectivorous bat virus variant. Emerging Infectious Diseases. 2002;8:79–81. doi: 10.3201/eid0801.010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Phylogenies and the comparative method. American Naturalist. 1985;125:1–15. doi: 10.1086/284325. [DOI] [PubMed] [Google Scholar]
- Fleming TH, Eby P. Ecology of bat migration. In: Kunz TH, Fenton B, editors. Bat Ecology. Chicago: The University of Chicago Press; 2003. pp. 156–208. [Google Scholar]
- Fooks AR, McElhinney LM, Pounder DJ, Finnegan CJ, Mansfield K, Johnson N, et al. Case report: isolation of a European bat lyssavirus type 2a from a fatal human case of rabies encephalitis. Journal of Medical Virology. 2003;71:281–289. doi: 10.1002/jmv.10481. [DOI] [PubMed] [Google Scholar]
- Franka R, Constantine DG, Kuzmin I, Velasco-Villa A, Reeder SA, Streicker D, et al. A new phylogenetic lineage of rabies virus associated with western pipistrelle bats (Pipistrellus hesperus) Journal of General Virology. 2006;87:2309–2321. doi: 10.1099/vir.0.81822-0. [DOI] [PubMed] [Google Scholar]
- Fraser GC, Hooper PT, Lunt RA, Gould AR, Gleeson LJ, Hyatt AD, et al. Encephalitis caused by a lyssavirus in fruit bats in Australia. Emerging Infectious Diseases. 1996;2:327–331. doi: 10.3201/eid0204.960408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freckleton RP, Harvey PH, Pagel M. Phylogenetic analysis and comparative data: a test and review of evidence. American Naturalist. 2002;160:712–726. doi: 10.1086/343873. [DOI] [PubMed] [Google Scholar]
- Gard GP, Marshall ID. Nelson Bay virus. A novel reovirus. Archiv Für Die Gesamte Virusforschung. 1973;43:34–42. doi: 10.1007/BF01249346. [DOI] [PubMed] [Google Scholar]
- Garland T, Ives AR. Using the past to predict the present: confidence intervals for regression equations in phylogenetic comparative methods. American Naturalist. 2000;155:346–364. doi: 10.1086/303327. [DOI] [PubMed] [Google Scholar]
- Gaunt MW, Sall AA, de Lamballerie X, Falconar AKI, Dzhivanian TI, Gould EA. Phylogenetic relationships of flaviviruses correlate with their epidemiology, disease association and biogeography. Journal of General Virology. 2001;82:1867–1876. doi: 10.1099/0022-1317-82-8-1867. [DOI] [PubMed] [Google Scholar]
- Gloza-Rausch F, Ipsen A, Seebens A, Gottsche M, Panning M, Felix Drexler J, et al. Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerging Infectious Diseases. 2008;14:626–631. doi: 10.3201/eid1404.071439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafen A. The phylogenetic regression. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences. 1989;326:119–157. doi: 10.1098/rstb.1989.0106. [DOI] [PubMed] [Google Scholar]
- Gregory RD. Parasites and host geographic range as illustrated by waterfowl. Functional Ecology. 1990;4:645–654. doi: 10.2307/2389732. [DOI] [Google Scholar]
- Grenfell BT, Pybus OG, Gog JR, Wood JLN, Daly JM, Mumford JA, et al. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303:327–332. doi: 10.1126/science.1090727. [DOI] [PubMed] [Google Scholar]
- Halpin K, Hyatt AD, Plowright RK, Epstein JH, Daszak P, Field HE, et al. Emerging viruses: coming in on a wrinkled wing and a prayer. Clinical Infectious Diseases. 2007;44:711–717. doi: 10.1086/511078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin K, Young P, Field H. Identification of likely natural hosts for equine morbillivirus. Communicable Diseases Intelligence. 1996;20:476. [Google Scholar]
- Halpin K, Young PL, Field HE, Mackenzie JS. Isolation of Hendra virus from pteropid bats: a natural reservoir of Hendra virus. Journal of General Virology. 2000;81:1927–1932. doi: 10.1099/0022-1317-81-8-1927. [DOI] [PubMed] [Google Scholar]
- Hanlon CA, Kuzmin IV, Blanton JD, Weldon WC, Manangan JS, Rupprecht CE. Efficacy of rabies biologics against new lyssaviruses from Eurasia. Virus Research. 2005;111:44–54. doi: 10.1016/j.virusres.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Haraguchi Y, Sasaki A. The evolution of parasite virulence and transmission rate in a spatially structured population. Journal of Theoretical Biology. 2000;203:85–96. doi: 10.1006/jtbi.1999.1065. [DOI] [PubMed] [Google Scholar]
- Harvey PH, Pagel MD. The Comparative Method in Evolutionary Biology. Oxford, UK: Oxford University Press; 1991. [Google Scholar]
- Hayman DT, Suu-Ire R, Breed AC, McEachern JA, Wang L, Wood JL, et al. Evidence of henipavirus infection in West African fruit bats. PLoS ONE. 2008;3:e2739. doi: 10.1371/journal.pone.0002739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaney LR, Walsh JS, Peterson AT. The roles of geological history and colonization abilities in genetic differentiation between mammalian populations in the Philippine archipelago. Journal of Biogeography. 2005;32:229–247. doi: 10.1111/j.1365-2699.2004.01120.x. [DOI] [Google Scholar]
- Herre EA. Population-structure and the evolution of virulence in nematode parasites of fig wasps. Science. 1993;259:1442–1445. doi: 10.1126/science.259.5100.1442. [DOI] [PubMed] [Google Scholar]
- Hess G. Disease in metapopulation models: implications for conservation. Ecology. 1996;77:1617–1632. doi: 10.2307/2265556. [DOI] [Google Scholar]
- Hess GR, Randolph SE, Arneberg P, Chemini C, Furlanello C, Harwood J, et al. Spatial aspects of disease dynamics. In: Hudson PJ, Rizzoli A, Grenfell B, Heesterbeek JAP, Dobson A, et al., editors. The Ecology of Wildlife Diseases. Oxford, UK: Oxford University Press; 2004. pp. 102–118. [Google Scholar]
- Hisheh S, How RA, Suyanto A, Schmitt LH. Implications of contrasting patterns of genetic variability in two vespertilionid bats from the Indonesian archipelago. Biological Journal of the Linnean Society. 2004;83:421–431. doi: 10.1111/j.1095-8312.2004.00401.x. [DOI] [Google Scholar]
- Holmes EC, Drummond AJ. The evolutionary genetics of viral emergence. Current Topics in Microbiology and Immunology. 2007;315:51–66. doi: 10.1007/978-3-540-70962-6_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes GJ, Orciari LA, Rupprecht CE. Evolutionary timescale of rabies virus adaptation to North American bats inferred from the substitution rate of the nucleoprotein gene. Journal of General Virology. 2005;86:1467–1474. doi: 10.1099/vir.0.80710-0. [DOI] [PubMed] [Google Scholar]
- Hutson AM (2004) Occurrence, distribution and incidence of lyssavirus in bats. Scottish Natural Heritage Commissioned Report No. 063
- IUCN (2009) IUCN Red List of Threatened Species. Available: http://www.iucnredlist.org [accessed March 1, 2009]
- Iwasaki T, Inoue S, Tanaka K, Sato Y, Morikawa S, Hayasaka D, et al. Characterization of Oita virus 296/1972 of Rhabdoviridae isolated from a horseshoe bat bearing characteristics of both lyssavirus and vesiculovirus. Archives of Virology. 2004;149:1139–1154. doi: 10.1007/s00705-003-0271-x. [DOI] [PubMed] [Google Scholar]
- Johara MY, Field H, Rashdi AM, Morrissy C, Heide Bvd, Rosta P, et al. Nipah virus infection in bats (Order Chiroptera) in Peninsular Malaysia. Emerging Infectious Diseases. 2001;7:439–441. doi: 10.3201/eid0703.010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, et al. Global trends in emerging infectious diseases. Nature. 2008;451:990–993. doi: 10.1038/nature06536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KE, Purvis A, Gittleman JL. Biological correlates of extinction risk in bats. American Naturalist. 2003;161:601–614. doi: 10.1086/368289. [DOI] [PubMed] [Google Scholar]
- Juste J, Ibanez C, Machordom A. Morphological and allozyme variation of Eidolon helvum (Mammalia: Megachiroptera) in the islands of the Gulf of Guinea. Biological Journal of the Linnean Society. 2000;71:359–378. doi: 10.1111/j.1095-8312.2000.tb01262.x. [DOI] [Google Scholar]
- Juste JB, Machordom A, Ibanez C. Allozyme variation of the Egyptian rousette (Rousettus aegyptiacus) in the Gulf of Guinea. Biochemical Systematics and Ecology. 1996;24:499–508. doi: 10.1016/0305-1978(96)00055-5. [DOI] [Google Scholar]
- Kalunda M, Mukwaya LG, Mukuye A, Lule M, Sekyalo E, Wright J, et al. Kasokero virus: a new human pathogen from bats (Rousettus aegyptiacus) in Uganda. American Journal of Tropical Medicine and Hygiene. 1986;35:387–392. doi: 10.4269/ajtmh.1986.35.387. [DOI] [PubMed] [Google Scholar]
- Kelkar SD, Kadam SS, Banerjee K. Haemagglutination inhibition antibodies against influenza virus in bats. Indian Journal of Medical Research. 1981;74:147–152. [PubMed] [Google Scholar]
- Kemp GE, Le Gonidec G, Karabatsos N, Rickenbach A, Cropp CB. Ife: a new African orbivirus isolated from Eidolon helvum bats captured in Nigeria, Cameroon and the Central African Republic. Bulletin de la Société de Pathologie Exotique et de Ses Filiales. 1988;81:40–48. [PubMed] [Google Scholar]
- Kerr B, Neuhauser C, Bohannan BJM, Dean AM. Local migration promotes competitive restraint in a host-pathogen ‘tragedy of the commons’. Nature. 2006;442:75–78. doi: 10.1038/nature04864. [DOI] [PubMed] [Google Scholar]
- Kim GR, Lee YT, Park CH. A new natural reservoir of hantavirus: isolation of hantaviruses from lung tissues of bats. Archives of Virology. 1994;134:85–95. doi: 10.1007/BF01379109. [DOI] [PubMed] [Google Scholar]
- Konstantinov OK, Diallo M, Inapogui A, Bah A, Camara S. The mammals of guinea as reservoirs and carriers of arboviruses. Meditsinskaia Parazitologiia i Parazitarnye Bolezni. 2006;1:34–39. [PubMed] [Google Scholar]
- Kunz TH. Roosting ecology of bats. In: Kunz TH, editor. Ecology of Bats. New York: Plenum Publishing; 1982. pp. 1–55. [Google Scholar]
- Kuzmin IV, Niezgoda M, Franka R, Agwanda B, Markotter W, Beagley JC, et al. Lagos bat virus in Kenya. Journal of Clinical Microbiology. 2008;46:1451–1461. doi: 10.1128/JCM.00016-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L’vov DK, Easterday B, Hinshow W, Dandurov IV, Arkhipov PN. Isolation of strains of the Hong Kong complex (H3N2) influenza virus from Nyctalus noctula bats in Kazakhstan. Voprosy Virusologii. 1979;4:338–341. [PubMed] [Google Scholar]
- L’vov DK, Karas FR, Timofeev EM, Tsyrkin YM, Vargina SG, Veselovskaya OV, et al. Issyk-Kul virus, a new arbovirus isolated from bats and Argas (Carios) Vespertilionis (Latr., 1802) in Kirghiz S.S.R. Archiv Für Die Gesamte Virusforschung. 1973;42:207–209. doi: 10.1007/BF01270841. [DOI] [PubMed] [Google Scholar]
- Leroy EM, Kumulungui B, Pourrut X, Rouquet P, Hassanin A, Yaba P, et al. Fruit bats as reservoirs of Ebola virus. Nature. 2005;438:575–576. doi: 10.1038/438575a. [DOI] [PubMed] [Google Scholar]
- Li WD, Shi ZL, Yu M, Ren WZ, Smith C, Epstein JH, et al. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- Liu WM, Worobey M, Li YY, Keele BF, Bibollet-Ruche F, Guo YY, et al. Molecular ecology and natural history of simian foamy virus infection in wild-living chimpanzees. PLoS Pathogens. 2008;4:e1000097. doi: 10.1371/journal.ppat.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Smith JO, Schreiber SJ, Kopp PE, Getz WM. Superspreading and the effect of individual variation on disease emergence. Nature. 2005;438:355–359. doi: 10.1038/nature04153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumio J, Hillbom M, Roine R, Ketonen L, Haltia M, Valle M, et al. Human rabies of bat origin in Europe. Lancet. 1986;1:378. doi: 10.1016/S0140-6736(86)92336-6. [DOI] [PubMed] [Google Scholar]
- Lumlertdacha B, Boongird K, Wanghongsa S, Wacharapluesadee S, Chanhome L, Khawplod P, et al. Survey for bat lyssaviruses, Thailand. Emerging Infectious Diseases. 2005;11:232–236. doi: 10.3201/eid1102.040691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyles AM, Dobson AP. Infectious disease and intensive management: population dynamics, threatened hosts, and their parasites. Journal of Zoo and Wildlife Medicine. 1993;24:315–326. [Google Scholar]
- Maharadatunkamsi, Hisheh S, Kitchener DJ, Schmitt LH (2003) Relationships between morphology, genetics, and geography in the cave fruit bat Eonycteris spelaea (Dobson, 1871) from Indonesia. Biological Journal of the Linnean Society 79:511–522
- Markotter W, Van Eeden C, Kuzmin IV, Rupprecht CE, Paweska JT, Swanepoel R, et al. Epidemiology and pathogenicity of African bat lyssaviruses. Developments in Biologicals. 2008;131:317–325. [PubMed] [Google Scholar]
- Martins EP, Hansen TF. Phylogenies and the comparative method: a general approach to incorporating phylogenetic information into the analysis of interspecific data. American Naturalist. 1997;149:646–667. doi: 10.1086/286013. [DOI] [Google Scholar]
- Martins FM, Ditchfield AD, Meyer D, Morgante JS. Mitochondrial DNA phylogeography reveals marked population structure in the common vampire bat, Desmodus rotundus (Phyllostomidae) Journal of Zoological Systematics and Evolutionary Research. 2007;45:372–378. doi: 10.1111/j.1439-0469.2007.00419.x. [DOI] [Google Scholar]
- May RM. Network structure and the biology of populations. Trends in Ecology and Evolution. 2006;21:394–399. doi: 10.1016/j.tree.2006.03.013. [DOI] [PubMed] [Google Scholar]
- May RM, Anderson RM. Population biology of infectious diseases: part II. Nature. 1979;280:455–461. doi: 10.1038/280455a0. [DOI] [PubMed] [Google Scholar]
- McCracken GF. Genetic structure of bat social groups. In: Fenton B, Racey PA, Rayner JMV, editors. Recent Advances in the Study of Bats. Cambridge, UK: Cambridge University Press; 1987. pp. 281–298. [Google Scholar]
- McCracken GF (2003) Estimates of population sizes in summer colonies of Brazilian free-tailed bats (Tadarida brasiliensis). USGS/BRD/ITR-2003-003, U.S. Geological Survey
- McCracken GF, Bradbury JW. Social organization and kinship in the polygynous bat Phyllostomus hastatus. Behavioral Ecology and Sociobiology. 1981;8:11–34. doi: 10.1007/BF00302840. [DOI] [Google Scholar]
- McCracken GF, Gassel MF. Genetic structure in migratory and nonmigratory populations of Brazilian free-tailed bats. Journal of Mammalogy. 1997;78:348–357. doi: 10.2307/1382888. [DOI] [Google Scholar]
- McKnight CA, Wise AG, Maes RK, Howe C, Rector A, Van Ranst M, et al. Papillomavirus-associated basosquamous carcinoma in an Egyptian fruit bat (Rousettus aegyptiacus) Journal of Zoo and Wildlife Medicine. 2006;37:193–196. doi: 10.1638/05-101.1. [DOI] [PubMed] [Google Scholar]
- Messenger SL, Molineux IJ, Bull JJ. Virulence evolution in a virus obeys a trade-off. Proceedings of the Royal Society of London. Series B: Biological Sciences. 1999;266:397–404. doi: 10.1098/rspb.1999.0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messenger SL, Smith JS, Rupprecht CE. Emerging epidemiology of bat-associated cryptic cases of rabies in humans in the United States. Clinical Infectious Diseases. 2002;35:738–747. doi: 10.1086/342387. [DOI] [PubMed] [Google Scholar]
- Messenger S, Rupprecht C, Smith J. Bats, emerging virus infections, and the rabies paradigm. In: Kunz TH, Fenton MB, editors. Bat Ecology. Chicago: The University of Chicago Press; 2003. pp. 622–679. [Google Scholar]
- Messenger SL, Smith JS, Orciari LA, Yager PA, Rupprecht CE. Emerging pattern of rabies deaths and increased viral infectivity. Emerging Infectious Diseases. 2003;9:151–154. doi: 10.3201/eid0902.020083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer CF, Kalko EK, Kerth G. Small-scale fragmentation effects on local genetic diversity in two phyllostomid bats with different dispersal abilities in Panama. Biotropica. 2009;41:95–102. doi: 10.1111/j.1744-7429.2008.00443.x. [DOI] [Google Scholar]
- Miller-Butterworth C, Jacobs D, Harley E. Strong population substructure is correlated with morphology and ecology in a migratory bat. Nature. 2003;424:187–191. doi: 10.1038/nature01742. [DOI] [PubMed] [Google Scholar]
- Morse SS. Emerging Viruses. New York: Oxford University Press; 1993. [Google Scholar]
- Muller MA, Paweska JT, Leman PA, Drosten C, Grywna K, Kemp A, et al. Coronavirus antibodies in African bat species. Emerging Infectious Diseases. 2007;13:1367–1370. doi: 10.3201/eid1309.070342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller T, Cox J, Peter W, Schafer R, Johnson N, McElhinney LM, et al. Spill-over of European bat lyssavirus type 1 into a stone marten (Martes foina) in Germany. Journal of Veterinary Medicine, B: Infectious Diseases and Veterinary Public Health. 2004;51:49–54. doi: 10.1111/j.1439-0450.2003.00725.x. [DOI] [PubMed] [Google Scholar]
- Ngamprasertwong T, Mackie IJ, Racey PA, Piertney SB. Spatial distribution of mitochondrial and microsatellite DNA variation in Daubenton’s bat within Scotland. Molecular Ecology. 2008;17:3243–3258. doi: 10.1111/j.1365-294X.2008.03845.x. [DOI] [PubMed] [Google Scholar]
- Nieberding C, Morand S, Libois R, Michaux JR (2004) A parasite reveals cryptic phylogeographic history of its host. Proceedings of the Royal Society of London. Series B: Biological Sciences 271:2559–2568 [DOI] [PMC free article] [PubMed]
- Norberg UM, Rayner JMV. Ecological morphology and flight in bats (Mammalia, Chiroptera)—wing adaptations, flight performance, foraging strategy and echolocation. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences. 1987;316:337–419. doi: 10.1098/rstb.1987.0030. [DOI] [Google Scholar]
- Nowak RM. Walker’s Mammals of the World. 6. Baltimore: Johns Hopkins University Press; 1999. [Google Scholar]
- Nunn CL, Altizer S, Jones KE, Sechrest W. Comparative tests of parasite species richness in primates. American Naturalist. 2003;162:597–614. doi: 10.1086/378721. [DOI] [PubMed] [Google Scholar]
- Nunn CL, Altizer S, Sechrest W, Jones KE, Barton RA, Gittleman JL. Parasites and the evolutionary diversification of primate clades. American Naturalist. 2004;164(Suppl 5):S90–S103. doi: 10.1086/424608. [DOI] [PubMed] [Google Scholar]
- Nunn CL, Altizer SM, Sechrest W, Cunningham AA. Latitudinal gradients of parasite species richness in primates. Diversity and Distributions. 2005;11:249–256. doi: 10.1111/j.1366-9516.2005.00160.x. [DOI] [Google Scholar]
- O’Shea TJ, Bogan MA (2003) Monitoring trends in bat populations of the United States and territories: problems and prospects. USGS/BRD/ITR-2003-003, U.S. Geological Survey
- Olival KJ (2008) Population genetic structure and phylogeography of Southeast Asian flying foxes: implications for conservation and disease ecology. Thesis. Columbia University, New York
- Ortega J, Maldonado JE, Wilkinson GS, Arita HT, Fleischer RC. Male dominance, paternity, and relatedness in the Jamaican fruit-eating bat (Artibeus jamaicensis) Molecular Ecology. 2003;12:2409–2415. doi: 10.1046/j.1365-294X.2003.01924.x. [DOI] [PubMed] [Google Scholar]
- Pagel M. Inferring the historical patterns of biological evolution. Nature. 1999;401:877–884. doi: 10.1038/44766. [DOI] [PubMed] [Google Scholar]
- Pannell JR, Charlesworth B. Effects of metapopulation processes on measures of genetic diversity. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences. 2000;355:1851–1864. doi: 10.1098/rstb.2000.0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- Park AW, Gubbins S, Gilligan CA. Extinction times for closed epidemics: the effects of host spatial structure. Ecology Letters. 2002;5:747–755. doi: 10.1046/j.1461-0248.2002.00378.x. [DOI] [Google Scholar]
- Pavri KM, Singh KR. Kyasanur forest disease virus infection in the frugivorous bat, Cynopterus sphinx. Indian Journal of Medical Research. 1968;56:1202–1204. [PubMed] [Google Scholar]
- Peterson AT, Heaney LR. Genetic differentiation in Philippine bats of the genera Cynopterus and Haplonycteris. Biological Journal of the Linnean Society. 1993;49:203–218. doi: 10.1111/j.1095-8312.1993.tb00900.x. [DOI] [Google Scholar]
- Petit E, Mayer F. Male dispersal in the noctule bat (Nyctalus noctula): where are the limits? Proceedings of the Royal Society of London. Series B: Biological Sciences. 1999;266:1717–1722. doi: 10.1098/rspb.1999.0837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philbey AW, Kirkland PD, Ross AD, Davis RJ, Gleeson AB, Love RJ, et al. An apparently new virus (family Paramyxoviridae) infectious for pigs, humans, and fruit bats. Emerging Infectious Diseases. 1998;4:269–271. doi: 10.3201/eid0402.980214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piaggio AJ, Perkins SL. Molecular phylogeny of North American long-eared bats (Vespertilionidae: Corynorhinus); inter- and intraspecific relationships inferred from mitochondrial and nuclear DNA sequences. Molecular Phylogenetics and Evolution. 2005;37:762–775. doi: 10.1016/j.ympev.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Plowright RK, Field HE, Smith C, Divljan A, Palmer C, Tabor G, et al. Reproduction and nutritional stress are risk factors for Hendra virus infection in little red flying foxes (Pteropus scapulatus) Proceedings of the Royal Society of London. Series B: Biological Sciences. 2008;275:861–869. doi: 10.1098/rspb.2007.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon LL, Chu DK, Chan KH, Wong OK, Ellis TM, Leung YH, et al. Identification of a novel coronavirus in bats. Journal of Virology. 2005;79:2001–2009. doi: 10.1128/JVI.79.4.2001-2009.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power AG, Mitchell CE. Pathogen spillover in disease epidemics. American Naturalist. 2004;164:S79–S89. doi: 10.1086/424610. [DOI] [PubMed] [Google Scholar]
- Price JL, Everard CO. Rabies virus and antibody in bats in Grenada and Trinidad. Journal of Wildlife Diseases. 1977;13:131–134. doi: 10.7589/0090-3558-13.2.131. [DOI] [PubMed] [Google Scholar]
- Pritchard LI, Chua KB, Cummins D, Hyatt A, Crameri G, Eaton BT, et al. Pulau virus; a new member of the Nelson Bay orthoreovirus species isolated from fruit bats in Malaysia. Archives of Virology. 2006;151:229–239. doi: 10.1007/s00705-005-0644-4. [DOI] [PubMed] [Google Scholar]
- Pulliam JR, Dushoff J. Ability to replicate in the cytoplasm predicts zoonotic transmission of livestock viruses. Journal of Infectious Diseases. 2009;199:565–568. doi: 10.1086/596510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulliam JRC. Viral host jumps: moving toward a predictive framework. EcoHealth. 2008;5:80–91. doi: 10.1007/s10393-007-0149-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pybus OG, Rambaut A, Harvey PH. An integrated framework for the inference of viral population history from reconstructed genealogies. Genetics. 2000;155:1429–1437. doi: 10.1093/genetics/155.3.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racey PA, Barratt EM, Burland TM, Deaville R, Gotelli D, Jones G, et al. Microsatellite DNA polymorphism confirms reproductive isolation and reveals differences in population genetic structure of cryptic pipistrelle bat species. Biological Journal of the Linnean Society. 2007;90:539–550. doi: 10.1111/j.1095-8312.2007.00746.x. [DOI] [Google Scholar]
- Rand DA, Keeling M, Wilson HB (1995) Invasion, stability and evolution to criticality in spatially extended, artificial host-pathogen ecologies. Proceedings of the Royal Society of London. Series B: Biological Sciences 259:55–63
- Razafindratsimandresy R, Jeanmaire EM, Counor D, Vasconcelos PF, Sall AA, Reynes JM. Partial molecular characterization of alphaherpesviruses isolated from tropical bats. Journal of General Virology. 2009;90:44–47. doi: 10.1099/vir.0.006825-0. [DOI] [PubMed] [Google Scholar]
- Real LA, Biek R. Spatial dynamics and genetics of infectious diseases on heterogeneous landscapes. Journal of the Royal Society Interface. 2007;4:935–948. doi: 10.1098/rsif.2007.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rector A, Mostmans S, Van Doorslaer K, McKnight CA, Maes RK, Wise AG, et al. Genetic characterization of the first chiropteran papillomavirus, isolated from a basosquamous carcinoma in an Egyptian fruit bat: the Rousettus aegyptiacus papillomavirus type 1. Veterinary Microbiology. 2006;117:267–275. doi: 10.1016/j.vetmic.2006.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynes JM, Molia S, Audry L, Hout S, Ngin S, Walston J, et al. Serologic evidence of lyssavirus infection in bats, Cambodia. Emerging Infectious Diseases. 2004;10:2231–2234. doi: 10.3201/eid1012.040459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter HV, Cumming GS. Food availability and annual migration of the straw-colored fruit bat (Eidolon helvum) Journal of Zoology. 2006;268:35–44. [Google Scholar]
- Riley SPD, Foley J, Chomel B. Exposure to feline and canine pathogens in bobcats and gray foxes in urban and rural zones of a National Park in California. Journal of Wildlife Diseases. 2004;40:11–22. doi: 10.7589/0090-3558-40.1.11. [DOI] [PubMed] [Google Scholar]
- Rivers NM, Butlin RK, Altringham JD. Genetic population structure of Natterer’s bats explained by mating at swarming sites and philopatry. Molecular Ecology. 2005;14:4299–4312. doi: 10.1111/j.1365-294X.2005.02748.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez DJ, Torres-Sorando L. Models of infectious diseases in spatially heterogeneous environments. Bulletin of Mathematical Biology. 2001;63:547–571. doi: 10.1006/bulm.2001.0231. [DOI] [PubMed] [Google Scholar]
- Rossiter SJ, Jones G, Ransome RD, Barratt EM. Genetic variation and population structure in the endangered greater horseshoe bat Rhinolophus ferrumequinum. Molecular Ecology. 2000;9:1131–1135. doi: 10.1046/j.1365-294x.2000.00982.x. [DOI] [PubMed] [Google Scholar]
- Ruedi M, Castella V. Genetic consequences of the ice ages on nurseries of the bat Myotis myotis: a mitochondrial and nuclear survey. Molecular Ecology. 2003;12:1527–1540. doi: 10.1046/j.1365-294X.2003.01828.x. [DOI] [PubMed] [Google Scholar]
- Ruedi M, Walter S, Fischer MC, Scaravelli D, Excoffier L, Heckel G. Italy as a major Ice Age refuge area for the bat Myotis myotis (Chiroptera: Vespertilionidae) in Europe. Molecular Ecology. 2008;17:1801–1814. doi: 10.1111/j.1365-294X.2008.03702.x. [DOI] [PubMed] [Google Scholar]
- Russell AL, Medellin RA, McCracken GF. Genetic variation and migration in the Mexican free-tailed bat (Tadarida brasiliensis mexicana) Molecular Ecology. 2005;14:2207–2222. doi: 10.1111/j.1365-294X.2005.02552.x. [DOI] [PubMed] [Google Scholar]
- Safi K, Konig B, Kerth G. Sex differences in population genetics, home range size and habitat use of the parti-colored bat (Vespertilio murinus, Linnaeus 1758) in Switzerland and their consequences for conservation. Biological Conservation. 2007;137:28–36. doi: 10.1016/j.biocon.2007.01.011. [DOI] [Google Scholar]
- Salaun JJ, Klein JM, Hebrard G. A new virus, Phnom-Penh bat virus, isolated in Cambodia from a short-nosed fruit bat, Cynopterus brachyotis angulatus Miller, 1898. Annales de Microbiologie. 1974;125:485–495. [PubMed] [Google Scholar]
- Serra-Cobo J, Amengual B, Abellan C, Bourhy H. European bat lyssavirus infection in Spanish bat populations. Emerging Infectious Diseases. 2002;8:413–420. doi: 10.3201/eid0804.010263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour C, Dickerman RW, Martin MS. Venezuelan encephalitis virus infection in neotropical bats. I. Natural infection in a Guatemalan enzootic focus. American Journal of Tropical Medicine and Hygiene. 1978;27:290–296. doi: 10.4269/ajtmh.1978.27.290. [DOI] [PubMed] [Google Scholar]
- Shi ZL, Hu ZH. A review of studies on animal reservoirs of the SARS coronavirus. Virus Research. 2008;133:74–87. doi: 10.1016/j.virusres.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair EA, Webb NJ, Marchant AD, Tidemann CR. Genetic variation in the little red flying-fox Pteropus scapulatus (Chiroptera: Pteropodidae): implications for management. Biological Conservation. 1996;76:45–50. doi: 10.1016/0006-3207(95)00086-0. [DOI] [Google Scholar]
- Smith JS. New aspects of rabies with emphasis on epidemiology, diagnosis, and prevention of the disease in the United States. Clinical Microbiology Reviews. 1996;9:166–176. doi: 10.1128/cmr.9.2.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speare R, Skerratt L, Foster R, Berger L, Hooper P, Lunt R, et al. Australian bat lyssavirus infection in three fruit bats from north Queensland. Communicable Diseases Intelligence. 1997;21:117–120. [PubMed] [Google Scholar]
- Storz JF, Bhat HR, Kunz TH. Genetic consequences of polygyny and social structure in an Indian fruit bat, Cynopterus sphinx. I. Inbreeding, outbreeding, and population subdivision. Evolution. 2001;55:1215–1223. doi: 10.1111/j.0014-3820.2001.tb00641.x. [DOI] [PubMed] [Google Scholar]
- Sulkin SE, Allen R, Miura T, Toyokawa K. Studies of arthropod-borne virus infections in chiroptera. VI. Isolation of Japanese B encephalitis virus from naturally infected bats. American Journal of Tropical Medicine and Hygiene. 1970;19:77–87. doi: 10.4269/ajtmh.1970.19.77. [DOI] [PubMed] [Google Scholar]
- Sulkin SE, Sims RA, Allen R. Isolation of St. Louis encephalitis virus from bats (Tadarida b. mexicana) in Texas. Science. 1966;152:223–225. doi: 10.1126/science.152.3719.223. [DOI] [PubMed] [Google Scholar]
- Swanepoel R, Smit SB, Rollin PE, Formenty P, Leman PA, Kemp A, et al. Studies of reservoir hosts for Marburg virus. Emerging Infectious Diseases. 2007;13:1847–1851. doi: 10.3201/eid1312.071115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor LH, Latham SM, Woolhouse MEJ. Risk factors for human disease emergence. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences. 2001;356:983–989. doi: 10.1098/rstb.2001.0888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong S, Conrardy C, Ruone S, Kuzmin IV, Guo X, Tao Y, et al. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerging Infectious Diseases. 2009;15:482–485. doi: 10.3201/eid1503.081013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towner JS, Pourrut X, Albarino CG, Nkogue CN, Bird BH, Grard G, et al. Marburg virus infection detected in a common African bat. PLoS ONE. 2007;2:e764. doi: 10.1371/journal.pone.0000764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimarchi C, Debbie JG. Naturally occurring rabies virus and neutralizing antibody in two species of insectivorous bats of New York State. Journal of Wildlife Diseases. 1977;13:366–369. doi: 10.7589/0090-3558-13.4.366. [DOI] [PubMed] [Google Scholar]
- Turmelle AS (2002) Phylogeography and population structure of the big brown bat (Eptesicus fuscus). Thesis. Boston University, Boston
- van Thiel PP, van den Hoek JA, Eftimov F, Tepaske R, Zaaijer HJ, Spanjaard L, et al. Fatal case of human rabies (Duvenhage virus) from a bat in Kenya: The Netherlands, December 2007. European Communicable Disease Bulletin. 2008;13:1–2. [PubMed] [Google Scholar]
- Velasco-Villa A, Orciari LA, Juarez-Islas V, Gomez-Sierra M, Padilla-Medina I, Flisser A, et al. Molecular diversity of rabies viruses associated with bats in Mexico and other countries of the Americas. Journal of Clinical Microbiology. 2006;44:1697–1710. doi: 10.1128/JCM.44.5.1697-1710.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner CK, Zaki SR, Shieh WJ, Whitfield SG, Smith JS, Orciari LA, et al. Laboratory investigation of human deaths from vampire bat rabies in Peru. American Journal of Tropical Medicine and Hygiene. 1999;60:502–507. doi: 10.4269/ajtmh.1999.60.502. [DOI] [PubMed] [Google Scholar]
- Webb NJ, Tidemann CR. Megachiroptera): evidence from genetic variation. Proceedings of the Royal Society of London. Series B: Biological Sciences. 1996;263:497–502. doi: 10.1098/rspb.1996.0075. [DOI] [PubMed] [Google Scholar]
- Webb SD, Keeling MJ, Boots M. Host-parasite interactions between the local and the mean-field: how and when does spatial population structure matter? Journal of Theoretical Biology. 2007;249:140–152. doi: 10.1016/j.jtbi.2007.06.013. [DOI] [PubMed] [Google Scholar]
- Wellenberg GJ, Audry L, Ronsholt L, van der Poel WH, Bruschke CJ, Bourhy H. Presence of European bat lyssavirus RNAs in apparently healthy Rousettus aegyptiacus bats. Archives of Virology. 2002;147:349–361. doi: 10.1007/s705-002-8324-3. [DOI] [PubMed] [Google Scholar]
- Weyandt SE, Van Den Bussche RA. Phylogeographic structuring and volant mammals: the case of the pallid bat (Antrozous pallidus) Journal of Biogeography. 2007;34:1233–1245. doi: 10.1111/j.1365-2699.2007.01690.x. [DOI] [Google Scholar]
- Whitby JE, Heaton PR, Black EM, Wooldridge M, McElhinney LM, Johnstone P. First isolation of a rabies-related virus from a Daubenton’s bat in the United Kingdom. Veterinary Record. 2000;147:385–388. doi: 10.1136/vr.147.14.385. [DOI] [PubMed] [Google Scholar]
- Wibbelt G, Kurth A, Yasmum N, Bannert M, Nagel S, Nitsche A, et al. Discovery of herpesviruses in bats. Journal of General Virology. 2007;88:2651–2655. doi: 10.1099/vir.0.83045-0. [DOI] [PubMed] [Google Scholar]
- Wilkinson GS. The social organization of the common vampire bat. 2. Mating system, genetic structure, and relatedness. Behavioral Ecology and Sociobiology. 1985;17:123–134. [Google Scholar]
- Wolfe ND, Daszak P, Kilpatrick AM, Burke DS. Bushmeat hunting, deforestation, and prediction of zoonotic disease. Emerging Infectious Disease. 2005;11:1822–1827. doi: 10.3201/eid1112.040789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe ND, Dunavan CP, Diamond J. Origins of major human infectious diseases. Nature. 2007;447:279–283. doi: 10.1038/nature05775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S, Lau S, Woo P, Yuen KY. Bats as a continuing source of emerging infections in humans. Reviews in Medical Virology. 2007;17:67–91. doi: 10.1002/rmv.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolhouse MEJ, Gowtage-Sequeria S. Host range and emerging and reemerging pathogens. Emerging Infectious Diseases. 2005;11:1842–1847. doi: 10.3201/eid1112.050997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolhouse MEJ, Howey R, Gaunt E, Reilly L, Chase-Topping M, Savill N. Temporal trends in the discovery of human viruses. Proceedings of the Royal Society of London. Series B: Biological Sciences. 2008;275:2111–2115. doi: 10.1098/rspb.2008.0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. The genetical structure of populations. Annals of Eugenics. 1951;15:323–354. doi: 10.1111/j.1469-1809.1949.tb02451.x. [DOI] [PubMed] [Google Scholar]
- Yoshino H, Armstrong KN, Izawa M, Yokoyama J, Kawata M. Genetic and acoustic population structuring in the Okinawa least horseshoe bat: are intercolony acoustic differences maintained by vertical maternal transmission? Molecular Ecology. 2008;17:4978–4991. doi: 10.1111/j.1365-294X.2008.03975.x. [DOI] [PubMed] [Google Scholar]