

Abstract
Background
Autophagy has been reported to play a pivotal role on the replication of various RNA viruses. In this study, we investigated the role of autophagy on hepatitis C virus (HCV) RNA replication and demonstrated anti-HCV effects of an autophagic proteolysis inhibitor, chloroquine.
Methods
Induction of autophagy was evaluated following the transfection of HCV replicon to Huh-7 cells. Next, we investigated the replication of HCV subgenomic replicon in response to treatment with lysosomal protease inhibitors or pharmacological autophagy inhibitor. The effect on HCV replication was analyzed after transfection with siRNA of ATG5, ATG7 and light-chain (LC)-3 to replicon cells. The antiviral effect of chloroquine and/or interferon-α (IFNα) was evaluated.
Results
The transfection of HCV replicon increased the number of autophagosomes to about twofold over untransfected cells. Pharmacological inhibition of autophagic proteolysis significantly suppressed expression level of HCV replicon. Silencing of autophagy-related genes by siRNA transfection significantly blunted the replication of HCV replicon. Treatment of replicon cells with chloroquine suppressed the replication of the HCV replicon in a dose-dependent manner. Furthermore, combination treatment of chloroquine to IFNα enhanced the antiviral effect of IFNα and prevented re-propagation of HCV replicon. Protein kinase R was activated in cells treated with IFNα but not with chloroquine. Incubation with chloroquine decreased degradation of long-lived protein leucine.
Conclusion
The results of this study suggest that the replication of HCV replicon utilizes machinery involving cellular autophagic proteolysis. The therapy targeted to autophagic proteolysis by using chloroquine may provide a new therapeutic option against chronic hepatitis C.
Keywords: Autophagy, Autophagosome, HCV replicon, Chloroquine
Introduction
The genome of HCV, a member of the family Flaviviridae, consists of a positive-sense single-stranded RNA. Peg-interferon/ribavirin combination therapy, which is the most effective therapy against HCV infection, is effective in around 50% for genotype 1 and 80% for genotypes 2 and 3 [1–3], however, many people cannot tolerate the serious side effects and are resistant to Peg-interferon/ribavirin combination therapy. Difficulties in eradicating HCV are attributable to the limited number of treatment options against HCV [4, 5]. Therefore, the search for novel therapeutic agents remains a strong aspiration.
Autophagy is an evolutionarily conserved cellular pathway in which the cytoplasm and organelles are engulfed within double-membraned vesicles, known as autophagosomes. While cellular autophagy is thought to be in preparation for the turnover and recycling of cellular constituents [6–8], this process has been proposed as a mechanism of virus replication complex formation in positive-stranded RNA viruses including poliovirus, equine arteritis virus and coronavirus [9–12]. In these viruses, the replication complexes consist of double membrane vesicles in the cytoplasm, suggestive of an autophagosome origin [9, 12]. Recently, it was reported that transfection of HCV replicon induced autophagy [11]. Additionally, Sir et al. [13] demonstrated that the suppression of autophagy inhibited the replication of HCV. These findings suggested that the autophagy plays a pivotal role in HCV replication.
Chloroquine, which is widely used for the treatment of malaria, is a well-established inhibitor of autophagic proteolysis which acts by inhibiting acidification of lysosomes and endosomes [14]. It has been reported that chloroquine exerts direct antiviral effects on several RNA viruses including coronaviruses, flaviviruses and human immunodeficiency virus (HIV) [8, 15–17]. Moreover, clinical studies have demonstrated the safety, tolerability, and efficacy of chloroquine in the antiviral treatment of HIV infection [18, 19]. Here, we have demonstrated that autophagic proteolysis plays a pivotal role on HCV replication, moreover, the inhibition of autophagic proteolytic pathways can constitute an effective new therapeutic target against HCV.
Materials and methods
Cell culture and treatment
Huh-7 cells were stably transfected with HCV replicon expressing chimeric protein of firefly luciferase and neomycin phosphotransferase [20, 21]. They were cultured in Dulbecco’s modified essential medium (DMEM) (Sigma, St. Louis, MO) supplemented with 10% foetal bovine serum (FBS) at 37°C under 5% CO2. To maintain cell lines carrying the HCV replicon, G418 (Wako, Osaka, Japan) was added to the medium at a final concentration 500 μg/ml.
Luciferase assay
Luciferase activities were quantified to evaluate the replication of HCV replicon by a luminometer (Lumat LB9507; Berthold, Germany) using a Bright-Glo Luciferase Assay System (Promega, Madison, WI). Assays were performed in triplicate, and the results were expressed as mean ± SD as percents of controls.
Cell viability assay
The viability of cells was assessed by WST-1 assay. Cells were cultured in 96-well plates at 5 × 103/well for 24 h, and then treated with 3-methyladenine [22] (10 mM), mixture of E64d (1 μg/ml) and pepstatin A [23] (1 μg/ml), and chloroquine (10−6–10−3 M) for 18 h. Cell proliferation reagent WST-1 (Roche, Swiss) was added to each well, and the cells were incubated for another 1 h at 37°C. The absorbance was measured against a background control by microplates reader (SPECTRA max 340PC, Molecular Devices, Sunnyvale, CA) at 450 nm. The reference wavelength was 650 nm.
Inhibition of autophagy and replication of HCV replicon
Cells were treated with 3-methyladenine (10 mM) or mixture of E64d (1 μg/ml) and pepstatin A (1 μg/ml), chloroquine (10−7–10−3 M), interferon (IFN)α (100 U/ml) for 18 h, the levels of replication of HCV replicon were assessed by luciferase assay. Moreover, cells were cultured with chloroquine (10−5 M) and/or IFNα (100 U/ml) for 7 days, then continued to incubate without drugs for another 21 days. Replication levels of HCV replicon were determined by luciferase assay at 7th and 21st days from cessation of drugs.
Identification of autophagosomes
Naïve Huh7 cells, Huh7/Rep-Feo cells, and Huh7/Rep-Feo treated with IFNα for 14 days were seeded on 30 mm dishes and incubated for 48 h. In addition, Huh7/Rep-Feo cells were treated with chloroquine (10−5 M) for 18 h. Cells were prefixed with 2% glutaraldehyde, post-fixed with 1% osmic acid, dehydrated in graded ethanol, embedded in resin, and cut into sections on an ultramicrotome. The cells were analyzed by a transmission electron microscope (Hitachi H7100, Japan). The number of autolysosomes in 100 μm2 of cytoplasm was counted by using transmission electron microscopy.
Small interfering RNA knockdown of ATG5, 7, LC-3
A combination of four chemically synthesized siRNA duplex molecules targeted to the human ATG5, 7, LC-3α, LC-3β mRNA sequence (Dharmacon, Lafayette, CO) was transiently transfected (final concentration 50 nM) into Huh7/Rep-Feo cells using a transfection reagent (Dharmacon, Lafayette, CO). siRNA targeted to enhanced green fluorescence protein was used as a control. Forty-eight hours after transfection, levels of HCV replication were analyzed by luciferase assay.
Western blot analysis
Twenty-five micrograms of total cell lysates were subjected to SDS/PAGE on a 10% gradient gel and electrophoretically transferred onto polyvinylidene fluoride membranes. After blocking with 5% non-fat dry milk in Tris-buffered saline, membranes were incubated with primary rabbit monoclonal antibody against Phospho-protein kinase R (P-PKR) (Cell Signaling Technology, Danvers, MA) or light-chain 3 (LC3), followed by a secondary horseradish peroxidase (HRP)-conjugated anti-rabbit IgG antibody (Cell Signaling Technology, Danvers, MA). Subsequently, specific bands were visualized using ECL detection kit (Amersham Pharmacia Biotech, Midland, ON, Canada).
Protein degradation assay
Long lived protein is mainly degraded by autophagy [24]. Cells were incubated with Williams’ E/10% FBS containing 0.5 μCi/ml [14C]leucine for 24 h to label long-lived proteins. Cells were washed with Williams’ E/10% FBS containing 10−5 M of unlabeled leucine and incubated with the medium for 2 h to allow degradation of short-lived proteins and minimize the incorporation of labeled leucine. The cells were then washed with phosphate-buffered saline (PBS) and incubated at 37°C with Williams’ E/10% FBS in the presence or absence of chloroquine (10−5 M). After 4 h, aliquots of the medium were taken and a one-tenth volume of 100% trichloroacetic acid was added to each aliquot. The mixtures were centrifuged at 12,000g for 5 min, and the acid-soluble radioactivity was determined using a liquid scintillation counter. At the end of the experiment, the cultures were washed twice with PBS, and 1 ml of cold trichloroacetic acid was added to fix the cell proteins. The fixed cell monolayers were washed with trichloroacetic acid and dissolved in 1 ml of 1 N NaOH at 37°C. Radioactivity in an aliquot of 1 N NaOH was determined by liquid scintillation counting. The percentage of protein degradation was calculated according to published procedures [25].
Statistical analysis
Differences were compared using ANOVA. Basically P values less than 0.05 were considered as statistically significant.
Results
The inhibition of autophagy suppressed replication of HCV replicon
We counted numbers of autophagosome and autolysosome in cells transduced with HCV replicon Rep-Feo by using electron microscopy. Double membrane vesicles with the morphology of autophagosomes were identified at 2.3 vacuoles/cells in naïve Huh-7 cells, while transfection of HCV replicon increased the number of vacuoles to about fourfold over untransfected Huh-7 cells (Fig. 1a, b). Subsequent treatment of the cells with IFNα (100 U/ml) for 14 days to eliminate HCV replicon substantially decreased the autophagolysosome in cytoplasm of Huh7/Rep-Feo cells (Fig. 1a, b). These observations suggested that HCV replicon induces formation of autophagosomes. To clarify the role of autophagy on the replication of HCV, Huh7/Rep-Feo cells were treated with 3-methyladenine (10 mM) or a mixture of E64d (10 μg/ml) and pepstatin A (10 μg/ml) which inhibited autophagic protein degradation. Replication level of HCV replicon in cells was increased to about twofold after 18 h in control media, however incubation with 3-methyladenine completely blunted increases in replication of HCV replicon. Treatment with 3-methyladenine decreased the number of autophagosomes to about 19% of Huh7/Rep-Feo cells. Furthermore co-incubation with E64d and pepstatin A decreased replication of HCV replicon to about 66% of control (Fig. 2a). Next, WST-1 assay was performed to check the cytotoxicity of these drugs. Treatment with 3-methyladenine or a mixture of E64d and pepstatin A did not affect cell viability (Fig. 2b). To clarify the role of autophagy induction in the replication of HCV, we suppressed the induction of autophagy by silencing autophagy-related genes (ATG5, ATG7, LC-3α and LC-3β) by siRNA transfection. Silencing of autophagy-related genes reduced the replication of HCV replicon to about 70% of control (Fig. 2c). Transfection with siRNA of autophagy related genes decreased the number of autophagosomes to about 30% of control. These results indicated that autophagy plays a pivotal role in replication of HCV.
Fig. 1.
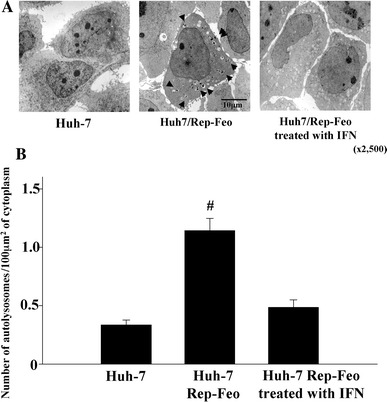
Expression of autophagy is changed by presence or absence of HCV replicon. a Naïve Huh7 cells, Huh7/Rep-Feo cells, and Huh7/Rep-Feo treated with IFNα for 14 days were seeded on 30 mm dishes and incubated for 48 h. The cells were analyzed by a transmission electron microscopy. Autophagosomes (arrow heads) were detected by transmission electron microscopy. b The number of autolysosomes in 100 μm2 of cytoplasm was counted by using transmission electron microscopy
Fig. 2.

Inhibition of autophagy suppressed replication of HCV replicon. a Cells were treated with 3-methyladenine (3-MA) (10 mM) or a mixture of E64d (1 μg/ml) and pepstatin A (Pep) (1 μg/ml) for 18 h, the levels of replication of HCV replicon were assessed by luciferase assay. b Cells were treated with 3-methyladenine (10 mM), mixture of E64d (1 μg/ml) and pepstatin A (1 μg/ml) for 18 h. Cell proliferation reagent WST-1 was added to each well, and the cells were incubated for 1 more hour at 37°C. The absorbance was measured against a background control by microplates reader at 450 nm. The reference wavelength was 650 nm. c A combination of four chemically synthesized siRNA duplex molecules targeted to the human ATG5, 7, LC-3α, LC-3β mRNA sequence was transiently transfected into Huh7/Rep-Feo cells using a transfection reagent. siRNA targeted to enhanced green fluorescence protein was used as the control. Forty-eight hours after transfection, levels of HCV replication were analyzed by luciferase assay
Chloroquine inhibits the replication of HCV replicon
Next, we evaluated the anti-HCV effect of chloroquine, which is a lysosomotropic agent that raises intralysosomal pH and impairs autophagic protein degradation. To assess the effects of chloroquine on the intracellular replication of the HCV replicon, Huh7/Rep-Feo cells were cultured with various concentrations of chloroquine in the medium. The replication of the HCV replicon was increased to about twofold within 18 h in the control media, however, which was suppressed by chloroquine in a dose-dependent manner (Fig. 3a). Next, cytotoxicity of chloroquine was analysed by WST-1 assays. Huh7/Rep-Feo cells treated with chloroquine showed no significant effect on cell viability in doses of lower than 10−5 M (Fig. 3b). However, incubation with 10−4 M of chloroquine reduced the cell viability to 25% of control. On the basis of the toxicity curve, the IC50 of the drug was calculated to be 3.6 × 10−5 M (Fig. 3c). The average EC50 of chloroquine was calculated as 2.2 × 10−7 M (Fig. 3c). The replication of HCV replicon was suppressed to nearly 40% of control at 10−5 M of chloroquine, which did not affect cell viability. These data indicated that chloroquine efficiently inhibited the replication of HCV replicon in the absence of toxic effect to cells at the concentration of 10−5 M. Accordingly, we used 10−5 M of chloroquine for the following study.
Fig. 3.
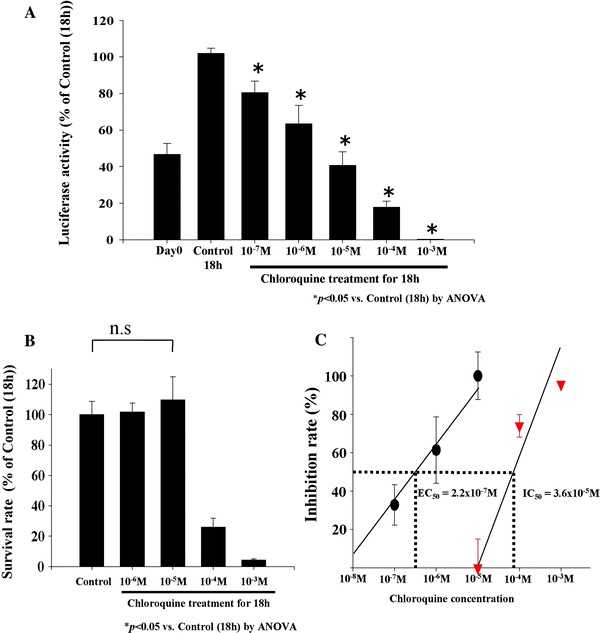
Effect of chloroquine on inhibition of HCV replication and cell viability. a Effect of chloroquine on replication of HCV replicon. Huh-7 Rep/Feo cells were seeded in 48-well plate and incubated with chloroquine (10−7–10−3 M) for 18 h. Replication levels of HCV replicon were determined by luciferase assay. Values are shown as percentages of the control cells. [*P < 0.05 vs. control (18 h) by ANOVA]. b Effect of chloroquine on proliferation of Huh-7 Rep/Feo cell lines in vitro. Cells seeded in 96-well plates were treated with 10−6 to 10−3M of chloroquine. After 18 h, effects on cell proliferation were determined by WST-1 assay. [*P < 0.05 vs. control (18 h) by ANOVA]. c Calculation of EC50 and IC50. Concentration of chloroquine inhibiting 50% of the replication of HCV replicon is showed as EC50. IC50 is the concentration of chloroquine which inhibits 50% of the cell proliferation of Huh-7 Rep/Feo cells
Next, we conducted the following assay to determine the synergistic inhibitory effect of chloroquine to IFNα on HCV replication. Treatment with chloroquine for 18 h resulted in a significant decrease of HCV replicon to about 40% of control. On the other hand, incubation with IFNα for 18 h inhibited the replication of HCV replicon to the levels about 15% of controls as expected. However, co-incubation with 100U/ml of IFNα and 10−5 M of chloroquine further decreased HCV replication significantly (Fig. 4a).
Fig. 4.

Combination effect of chloroquine with IFNα on HCV replication. a Huh-7 Rep/Feo cells were treated with chloroquine (10−5 M) and/or IFNα (100 U/ml) for 18 h. Values are shown as percentages of the control cells [*P < 0.05 vs. control (18 h) by ANOVA, **P < 0.05 vs. IFNα treatment group by ANOVA]. b Assessment of re-propagation of HCV replicon after long term treatment of chloroquine and/or IFNα. Huh-7 Rep/Feo was incubated with chloroquine (10−5 M) and/or IFNα (100 U/ml) for 7 days, then drugs were removed from the medium and incubation continued for another 21 days. Luciferase assay was performed at the 7th and 21st days from cessation of drugs. Values are shown as percentages of the control cells [*P < 0.05 vs. IFNα treatment group (day 28) by ANOVA]
To determine whether long-term chloroquine treatment inhibits post-treatment re-propagation of HCV replicon, we followed up luciferase activity of the cells at the 7th and 21st days after 7 days of treatment with chloroquine and/or IFNα (Fig. 4b). In HCV replicon cells treated by chloroquine, luciferase activities recovered to 53 and 88% on 7 and 21 days after cessation of treatment. In cells that were treated by IFNα, luciferase activity maintained background level for 7 days post-treatment. However, it reappeared in 21 days. In sharp contrast, co-incubation with IFNα and chloroquine for 7 days suppressed HCV replication for the extensive period up to 21 days, even in the absence of these drugs (Fig. 4b).
Anti-HCV effect of chloroquine independent of IFN signaling pathway
IFN-inducible double-stranded RNA-activated protein kinase R (PKR) plays a key antiviral role against hepatitis C virus [26, 27]. To elucidate the mechanisms of the inhibitory effect of chloroquine on HCV replication, phosphorylated PKR (P-PKR) was evaluated by western blotting analysis. P-PKR was detectable in cells treated with IFNα after 24 h; this increase in P-PKR expression peaked at 24 h after IFNα treatment and was reduced at 48 h (Fig. 5a). In contrast, P-PKR was not observed in cells treated with chloroquine at any time point.
Fig. 5.

Chloroquine suppresses autophagic protein degradation, not interferon pathways. a Cells were treated with 10−5 M of chloroquine (CQ) or 100 U/ml of IFNα for 24–48 h. Phosphorylation of PKR was assessed by western blot analysis. GAPDH was used as loading control. b Ultrastructural analysis showing the effect of chloroquine on the number of autolysosomes. Huh-7/Rep-Feo cells were incubated with chloroquine for 18 h. Autolysosomes were identified as the double membrane vesicles (arrow heads) of cytoplasm in Huh-7 Rep/Feo. The number of autolysosomes in 100 μm2 of cytoplasm was counted by using transmission electron microscopy. Data represent mean ± SEM of individual preparations from pictures (*P < 0.05 vs. control by ANOVA). c Western blot analysis of LC3 in Huh7 Rep/Feo. The lysate of Huh7 Rep/Feo treated with chloroquine for 4–8 h were immunoblotted with LC3. GAPDH was used as loading control
Chloroquine blunts autophagic proteolysis in cells transfected with HCV replicon
It is reported that chloroquine disrupts lysosomal function, preventing effective autophagic protein degradation, leading to the accumulation of ineffective autophagosomes [28]. Therefore, we investigated if chloroquine led to the accumulation of autolysosomes as a result of suppression of proteolysis. We performed electron microscopic investigation to evaluate quantities of autophagosomes and autolysosomes. Ultrastructural analysis identified 0.94 ± 0.1 vacuoles/100 μm2 of autolysosomes in control cells; however, treatment with chloroquine increased the number of autolysosomes dramatically to about 13-fold over control (Fig. 5b). Furthermore, the molecular form of LC3 protein of the cells, which is a component of autophagosomes, was examined by western blot analysis to ensure that chloroquine treatment leads to the accumulation of autophagosomes and autolysosomes. As shown in Fig. 5c, immunopositive protein bands for LC3-I and LC3-II forms were clearly evident in control cells. After chloroquine treatment, LC3-II expression increased at 4 h (Fig. 5c) to about threefold over control without enhancing LC3-I expression, and at 8 h (Fig. 5c) LC3-II expression was further enhanced. Finally, we evaluated turnover of the long-lived protein leucine, which was mainly degraded by autophagy. Huh7/Rep-Feo cells were labeled with [14C]leucine for 24 h, and degradation of [14C]leucine in cells treated with or without chloroquine was measured. Chloroquine treatment decreased degradation of leucin to 76% of control, indicating that chloroquine blunts degradation of proteins via an autophagic pathway (Fig. 6). These results demonstrate that chloroquine-induced the accumulation of autolysosomes was due to disruption of autophagic proteolysis.
Fig. 6.

Turnover of long lived protein. Huh-7 Rep/Feo cells were labeled with [14C]leucin for 4 h, then degradation of long-lived protein in chloroquine treated cells was measured as described in Materials and Methods. The percentage of protein degradation was calculated by dividing the amount of acid-soluble radioactivity in the medium at that time by the amount of acid-precipitable radioactivity present in the cells at time zero. Data are mean ± SEM of value of triplicate in each group (*P < 0.05)
Discussion
Previous reports have disclosed that autophagy plays a pivotal role on the replication of several RNA viruses [10–12]. Our present results demonstrate that autophagy is induced by transfection of HCV replicon and is reduced by deletion of replicon due to IFNα (Fig. 1a, b). These results suggest that autophagy is induced in the presence of HCV replication in its host cells. However, the role of autophagy in the pathogenesis of HCV is largely unclear. We found that the inhibition of autophagosome formation and autophagic proteolysis blunt the replication of genotype 1b subgenomic HCV replicon (Fig. 2a, c). Sir et al. [13] reported that inhibition of autophagy also reduced the replication of the JFH1-based full length genotype 2a genome. Therefore, the utilization of autophagy on viral replication is shown by HCV strains across different genotypes.
On the other hand, not only a silencing of autophagic gene but also pharmacological inhibition of autophagic proteolysis possesses anti-HCV effects (Fig. 2a, c). However, treatment with both chloroquine and the mixture of E64d and pepstatin induced the accumulation of autophagosomes in cytoplasm. Therefore, it is likely that HCV does not utilize the double membrane structure as the localization of the viral replication formation. These results support the hypothesis that protein degradation due to autophagy is important for HCV replication.
Chloroquine is a well-known inhibitor of autophagic protein degradation and is often used as an anti-malarial agent. Moreover, the anti-viral effect of chloroquine on other RNA viruses has been already reported in clinical trials [15, 16]. In our results, chloroquine inhibits the intracellular replication of an HCV replicon in a dose-dependent manner (Fig. 3a). This antiviral effect of chloroquine was clearly not due to cytotoxic effects (Fig. 3b). Moreover, chloroquine possesses a synergistic effect with IFNα on HCV replication (Fig. 4a). Although IFNα possesses strong anti-HCV effects, re-propagation of HCV replicon was observed after 3 weeks following 7 days of treatment with IFNα. Interestingly, co-incubation with IFNα and chloroquine for 7 days prevented re-propagation of HCV replicon (Fig. 4b). Chloroquine is a lysosomal weak base that is known to affect acid vesicles leading to dysfunction of several proteins [29]. It was demonstrated that disruption of lysosomal function impairs maturation of viruses through inhibiting the low-pH dependent proteases in trans-Golgi vesicles in HIV and the SARS coronavirus infection in vitro [15, 29]. However, little is understood about the mechanism of its antiviral effect. In previous reports, various drugs which possess inhibitory effects on the replication of HCV and have a synergistic action with IFNα have been proposed as new therapeutic agents to treat HCV. Some of them have proved to exhibit their anti-HCV effects through augmentation of IFN-induced antiviral gene responses [30, 31]. However, the anti-HCV effect of chloroquine was not associated with activation of one of IFN receptors signaling molecule PKR (Fig. 5a). Our results showed chloroquine induced the accumulation of ineffective autophagosomes in cytoplasm of Huh7/Rep-Feo cells (Fig. 5b) and inhibited the degradation of long-lived protein leucine (Fig. 6). These findings imply that chloroquine effectively impairs the function of autophagy in our experiment. These results indicated that chloroquine is a new anti-HCV agent that targets the autophagic proteolysis.
Previous reports have shown that chloroquine possesses anti-viral effects on various RNA viruses. Its best-studied effects are those against HIV replication, which are being tested in clinical trials [17, 18]. HCV co-infection is common in HIV-positive patients in USA and Europe [32, 33]. Since HIV infection accelerates the progression of HCV-related liver disease, treatment of HCV is generally recommended. However, co-infected patients have a greater risk of antiretroviral therapy-associated hepatotoxicity than patients with HIV only [34]. Moreover, treatment with ribavirin is believed to increase the risk of anemia in patients taking the HIV drug zidovudine [35]. A clinical study designed for HIV patients showed the safety and efficacy of chloroquine used for long terms up to 48 weeks [36]. Therefore, the combination therapy of interferon and chloroquine is, possibly, a hopeful therapy for HCV–HIV co-infected patients. Since chloroquine is known as one of the inexpensive drugs, therefore, chloroquine might provide a new effective, safe and economical therapeutic option for patients with HCV. In conclusion, autophagic proteolysis might be a new therapeutic target on the replication of HCV.
Contributor Information
Tomokazu Mizui, Phone: +81-3-38133111, FAX: +81-3-38138862, don-chip@aqua.email.ne.jp.
Shunhei Yamashina, Phone: +81-3-38133111, FAX: +81-3-38138862, Email: ryou0607jp@ybb.ne.jp.
Isei Tanida, Phone: +81-3-52851111, FAX: +81-3-52851150, Email: tanida@nih.go.jp.
Yoshiyuki Takei, Phone: +81-59-2315017, FAX: +81-59-2315223, Email: ytakei@clin.medic.mie-u.ac.jp.
Takashi Ueno, Phone: +81-3-38133111, FAX: +81-3-58025889, Email: upfield@juntendo.ac.jp.
Naoya Sakamoto, Phone: +81-3-38035877, FAX: +81-3-58030268, Email: nsakamoto.gast@tmd.ac.jp.
Kenichi Ikejima, Phone: +81-3-38133111, FAX: +81-3-38138862, Email: ikejima@juntendo.ac.jp.
Tsuneo Kitamura, Phone: +81-3-38133111, FAX: +81-3-38138862, Email: kitamura@juntendo.ac.jp.
Nobuyuki Enomoto, Phone: +81-55-2739581, FAX: +81-55-2736748, Email: enomoto@yamanashi.ac.jp.
Tatsuo Sakai, Phone: +81-3-38133111, FAX: +81-3-56896923, Email: tatsuo@juntendo.ac.jp.
Eiki Kominami, Phone: +81-3-38133111, FAX: +81-3-58025889, Email: kominami@juntendo.ac.jp.
Sumio Watanabe, Phone: +81-3-38133111, FAX: +81-3-38138862, Email: sumio@juntendo.ac.jp.
References
- 1.Yotsuyanagi H, Koike K. Drug resistance in antiviral treatment for infections with hepatitis B and C viruses. J Gastroenterol. 2007;42:329–335. doi: 10.1007/s00535-007-2034-z. [DOI] [PubMed] [Google Scholar]
- 2.Bruno S, Stroffolini T, Colombo M, Bollani S, Benvegnù L, Mazzella G, et al. Sustained virological response to interferon-alpha is associated with improved outcome in HCV-related cirrhosis: a retrospective study. Hepatology. 2007;45:579–587. doi: 10.1002/hep.21492. [DOI] [PubMed] [Google Scholar]
- 3.Shiffman ML, Cooksley WG, Dusheiko GM, Lee SS, Balart L, Reindollar R, et al. Peginterferon alfa-2a in patients with chronic hepatitis C and cirrhosis. N Engl J Med. 2000;343:1673–1680. doi: 10.1056/NEJM200012073432302. [DOI] [PubMed] [Google Scholar]
- 4.Sezaki H, Suzuki F, Kawamura Y, Yatsuji H, Hosaka T, Akuta N, et al. Poor response to pegylated interferon and ribavirin in older women infected with hepatitis C virus of genotype 1b in high viral load. Dig Dis Sci. 2009;54:1317–1324. doi: 10.1007/s10620-008-0500-y. [DOI] [PubMed] [Google Scholar]
- 5.Honda T, Katano Y, Urano F, Murayama M, Hayashi K, Ishigami M, et al. Efficacy of ribavirin plus interferon-α in patients aged 60 years with chronic hepatitis C. J Gastroenterol Hepatol. 2007;22:989–995. doi: 10.1111/j.1440-1746.2006.04773.x. [DOI] [PubMed] [Google Scholar]
- 6.Wang CW, Klionsky DJ. The molecular mechanism of autophagy. Mol Med. 2003;9:65–76. [PMC free article] [PubMed] [Google Scholar]
- 7.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reggiori F, Klionsky DJ. Autophagy in the eukaryotic cell. Eukaryot Cell. 2002;1:11–21. doi: 10.1128/EC.01.1.11-21.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem. 2004;279:10136–10141. doi: 10.1074/jbc.M306124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Randolph VB, Winkler G, Stollar V. Acidotropic amines inhibit proteolytic processing of flavivirus prM protein. Virology. 1990;174:450–458. doi: 10.1016/0042-6822(90)90099-D. [DOI] [PubMed] [Google Scholar]
- 11.Posthuma CC, Pedersen KW, Lu Z, Joosten RG, Roos N, Zevenhoven-Dobbe JC, et al. Formation of the arterivirus replication/transcription complex: a key role for nonstructural protein 3 in the remodeling of intracellular membranes. J Virol. 2008;82:4480–4491. doi: 10.1128/JVI.02756-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suhy DA, Giddings TH, Jr, Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J Virol. 2000;74:8953–8965. doi: 10.1128/JVI.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology. 2008;48:1054–1061. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poole B, Ohkuma S. Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J Cell Biol. 1981;90:665–669. doi: 10.1083/jcb.90.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J. 2005;2:69. doi: 10.1186/1743-422X-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savarino A, Gennero L, Chen HC, Serrano D, Malavasi F, Boelaert JR, et al. Anti-HIV effects of chloroquine: mechanisms of inhibition and spectrum of activity. AIDS. 2001;15:2221–2229. doi: 10.1097/00002030-200111230-00002. [DOI] [PubMed] [Google Scholar]
- 17.Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R. Effects of chloroquine on viral infections: an old drug against today’s diseases? Lancet Infect Dis. 2003;3:722–727. doi: 10.1016/S1473-3099(03)00806-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sperber K, Chiang G, Chen H, Ross W, Chusid E, Gonchar M, et al. Comparison of hydroxychloroquine with zidovudine in asymptomatic patients infected with human immunodeficiency virus type 1. Clin Ther. 1997;19:913–923. doi: 10.1016/S0149-2918(97)80045-8. [DOI] [PubMed] [Google Scholar]
- 19.Paton NI, Aboulhab J. Hydroxychloroquine, hydroxyurea and didanosine as initial therapy for HIV-infected patients with low viral load: safety, efficacy and resistance profile after 144 weeks. HIV Med. 2005;6:13–20. doi: 10.1111/j.1468-1293.2005.00259.x. [DOI] [PubMed] [Google Scholar]
- 20.Tanabe Y, Sakamoto N, Enomoto N, Kurosaki M, Ueada E, Maekawa S, et al. Synergistic inhibition of intracellular hepatitis C virus replication by combination of ribavirin and interferon-alpha. J Infect Dis. 2004;189:1129–1139. doi: 10.1086/382595. [DOI] [PubMed] [Google Scholar]
- 21.Yokota T, Sakamoto N, Enomoto N, Tanabe Y, Miyagishi M, Maekawa S, et al. Inhibition of intracellular hepatitis C virus replication by synthetic and vector-derived small interfering RNAs. EMBO Rep. 2003;4:602–608. doi: 10.1038/sj.embor.embor840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ueno T, Ishidoh K, Mineki R, Tanida I, Murayama K, Kadowaki M, et al. Autolysosomal membrane-associated betaine homocysteine methyltransferase. Limited degradation fragment of a sequestered cytosolic enzyme monitoring autophagy. J Biol Chem. 1999;274:15222–15229. doi: 10.1074/jbc.274.21.15222. [DOI] [PubMed] [Google Scholar]
- 24.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gronostajski RM, Pardee AB. Protein degradation in 3T3 cells and tumorigenic transformed 3T3 cells. J Cell Physiol. 1984;119:127–132. doi: 10.1002/jcp.1041190120. [DOI] [PubMed] [Google Scholar]
- 26.Clemens MJ. PKR–a protein kinase regulated by double-stranded RNA. Int J Biochem Cell Biol. 1997;29:945–949. doi: 10.1016/S1357-2725(96)00169-0. [DOI] [PubMed] [Google Scholar]
- 27.Gale MJ, Jr, Korth MJ, Tang NM, Tan SL, Hopkins DA, Dever TE, et al. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- 28.Glaumann H, Ahlberg J. Comparison of different autophagic vacuoles with regard to ultrastracture, enzymatic composition, and degradation capacity—formation of crinosomes. Exp Mol Pathol. 1987;47:346–362. doi: 10.1016/0014-4800(87)90018-9. [DOI] [PubMed] [Google Scholar]
- 29.Thorens B, Vassalli P. Chloroquine and ammonium chloride prevent terminal glycosylation of immunoglobulins in plasma cells without affecting secretion. Nature. 1986;321:618–620. doi: 10.1038/321618a0. [DOI] [PubMed] [Google Scholar]
- 30.Dev A, Patel K, McHutchison JG. New therapies for chronic hepatitis C virus infection. Curr Gastroenterol Rep. 2004;6:77–86. doi: 10.1007/s11894-004-0030-5. [DOI] [PubMed] [Google Scholar]
- 31.Lin K, Kwong AD, Lin C. Combination of a hepatitis C virus NS3-NS4A protease inhibitor and alpha interferon synergistically inhibits viral RNA replication and facilitates viral RNA clearance in replicon cells. Antimicrob Agents Chemother. 2004;48:4784–4792. doi: 10.1128/AAC.48.12.4784-4792.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Staples CT, Jr, Rimland D, Dudas D. Hepatitis C in the HIV (human immunodeficiency virus) Atlanta V.A. (Veterans Affairs Medical Center) Cohort Study (HAVACS): the effect of coinfection on survival. Clin Infect Dis. 1999;29:150–154. doi: 10.1086/520144. [DOI] [PubMed] [Google Scholar]
- 33.Denis F, Adjide CC, Rogez S, Delpeyroux C, Rogez JP, Weinbreck P. Seroprevalence of HBV, HCV and HDV hepatitis markers in 500 patients infected with the human immunodeficiency virus. Pathol Biol (Paris) 1997;45:701–708. [PubMed] [Google Scholar]
- 34.Sulkowski MS, Benhamou Y. Therapeutic issues in HIV/HCV-coinfected patients. J Viral Hepat. 2007;14:371–386. doi: 10.1111/j.1365-2893.2006.00816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alvarez D, Dieterich DT, Brau N, Moorehead L, Ball L, Sulkowski MS. Zidovudine use but not weight-based ribavirin dosing impacts anaemia during HCV treatment in HIV-infected persons. J Viral Hepat. 2006;13:683–689. doi: 10.1111/j.1365-2893.2006.00749.x. [DOI] [PubMed] [Google Scholar]
- 36.Paton NI, Aboulhab J, Karim F. Hydroxychloroquine, hydroxycarbamide, and didanosine as economic treatment for HIV-1. Lancet. 2002;359:1667–1668. doi: 10.1016/S0140-6736(02)08557-4. [DOI] [PubMed] [Google Scholar]