

Abstract
Alzheimer’s disease (AD) therapies predominantly focus on amyloid-β (Aβ), but Aβ effects may be maximal before clinical symptoms appear. Downstream of Aβ, dendritic spine loss correlates most strongly with cognitive decline in AD. Rho-associated kinases (ROCK1 and ROCK2) regulate the actin cytoskeleton, and ROCK1 and ROCK2 protein abundances are increased in early AD. Here, we found that the increased abundance of ROCK1 in cultured primary rat hippocampal neurons reduced dendritic spine length through a myosin-based pathway, whereas the increased abundance of ROCK2 induced spine loss through the serine and threonine kinase LIMK1. Aβ42 oligomers can activate ROCKs. Here, using static imaging studies combined with multi-electrode array analyses, we found that the ROCK2-LIMK1 pathway mediated Aβ42-induced spine degeneration and neuronal hyperexcitability. Live-cell microscopy revealed that pharmacologic inhibition of LIMK1 rendered dendritic spines resilient to Aβ42 oligomers. Treatment of hAPP mice with a LIMK1 inhibitor rescued Aβ-induced hippocampal spine loss and morphologic aberrations. Our data suggest that therapeutically targeting LIMK1 may provide dendritic spine resilience to Aβ, and therefore may benefit cognitively normal patients that are at high risk for developing dementia.
Introduction
Cognitive decline in Alzheimer’s disease (AD) is the result of synapse loss in brain regions that are critical for memory processes. Synapse or dendritic spine loss correlates more strongly with cognitive impairment in AD than amyloid-β (Aβ) or neurofibrillary tangle pathology, yet few therapeutic strategies target spines or synapses (1–8). Synaptic strength and activity are inseparably linked to spine morphology (9). Several discoveries indicate that spine structure remodeling is a plausible mechanism to maintain synapses and provide cognitive resilience in patients with an apolipoprotein E (APOE) ε4 allele and/or AD pathology (7, 8). These findings emphasize dendritic spines as therapeutic substrates with potential to protect cognitively normal patients at high risk for dementia.
Aβ induces dendritic degeneration of neurons, and these detrimental effects cause neuronal hyperexcitability by rendering neurons more electrically compact (10). This leads to aberrant circuit synchronization and ultimately cognitive impairment in AD patients and hAPP mice (11–16). Aβ likely wreaks havoc on the dendritic cytoskeleton by activating the RhoA guanosine triphosphatase (GTPase) and its primary downstream effectors: the Rho-associated protein kinase (ROCK) isoforms, ROCK1 and ROCK2 (17–19). ROCKs regulate actin-myosin–mediated cytoskeleton contractility (20–24), and increased activity of ROCKs could have detrimental consequences on dendritic spine remodeling (25). Furthermore, ROCK1 and ROCK2 protein abundances are increased among mild cognitive impairment (MCI) and AD patients, implying that ROCKs may contribute to synaptic loss in early disease stages (17, 26). Pharmacologic studies with Fasudil and Y-27632, the most widely characterized pan-ROCK inhibitors, suggest beneficial effects of ROCK inhibitors in AD models (27, 28). However, these and other ROCK inhibitors are not isoform-specific and can inhibit other AGC family kinases, including protein kinase A (PKA) and protein kinase C (PKC) (29). Moreover, critical questions remain regarding the role of ROCKs in AD and the contribution of ROCK1 or ROCK2 to the observed beneficial effects of pan-ROCK inhibitors. Collectively, these barriers have stalled ROCK inhibitors from entering clinical trials for AD. Here, we elucidated distinct isoform-specific mechanisms by which ROCKs may drive dendritic spine degeneration in MCI and AD and identified the ROCK2-LIM domain kinase isoform 1 (LIMK1) pathway as a key therapeutic avenue to provide dendritic spine resilience against Aβ.
Results
ROCK1 and ROCK2 regulate dendritic spine length and density through isoform-specific mechanisms
Past studies showed that ROCK1 and ROCK2 protein abundances were increased in MCI and AD brains compared to age-matched pathology-free controls, and that increased ROCKs were not the result of microglia or astrocyte accumulation in disease cases (17, 26). These results suggest that activity of ROCKs is increased early and remains increased in neurons throughout AD progression, possibly contributing to synapse loss. When ROCKs are active, neurite structural plasticity is repressed (30–33). Therefore, we hypothesized that increased protein abundance of ROCK1 or ROCK2 in neurons would induce detrimental AD-like structural effects on dendritic spines. To test this, rat hippocampal neurons were isolated at embryonic day 18 (E18) and cultured at high-density on glass coverslips, as previously described (25). At 14 days in vitro (DIV 14), neurons were transiently co-transfected with plasmids encoding Lifeact-GFP, a fluorescently-tagged small actin binding peptide (34), and ROCK1, ROCK2, or empty vector constructs (fig. S1A). Forty-eight hours after transfection, neurons were fixed and imaged using widefield microscopy. Z-series images were subjected to deconvolution followed by three-dimensional (3D) morphometry analysis (Fig. 1A). Dendritic spine length was reduced significantly in neurons expressing human ROCK1 compared to vector or Lifeact-GFP controls, whereas spine head diameter and density were similar among these conditions (Fig. 1B; fig. S1, B and C). Human ROCK2 expression reduced spine density significantly compared to vector or Lifeact-GFP; however, spine head diameter and length were not affected by ROCK2 (Fig. 1C; fig. S1, D and E). Notably, Lifeact-GFP alone or vector plus Lifeact-GFP were comparable on all spine readouts (Fig. 1, B and C; fig. S1, B to E). To test whether ROCK1 or ROCK2 kinase activity was required for their effects on spines, site-directed mutagenesis was employed to substitute leucine 105 or 121 for glycine in the ROCK1 or ROCK2 kinase domain ATP-binding pocket, respectively, rendering the enzymes inactive (35). Spine density and morphology in neurons expressing ROCK1-L105G or ROCK2-L121G were comparable to vector controls, indicating kinase activity of ROCKs is required for their effects on spines (Fig. 1, B and C; fig. S1, B to E). Expression of ROCK1 and ROCK1-L105G or ROCK2 and ROCK2-L121G appeared similar in neuroblastoma cells, suggesting that mutation of Leu105 or Leu121 to glycine does not perceptibly alter ROCK1 or ROCK2 protein stability, respectively (fig. S1A).
Figure 1. ROCK1 and ROCK2 regulate dendritic spine length and density through isoform-specific mechanisms.
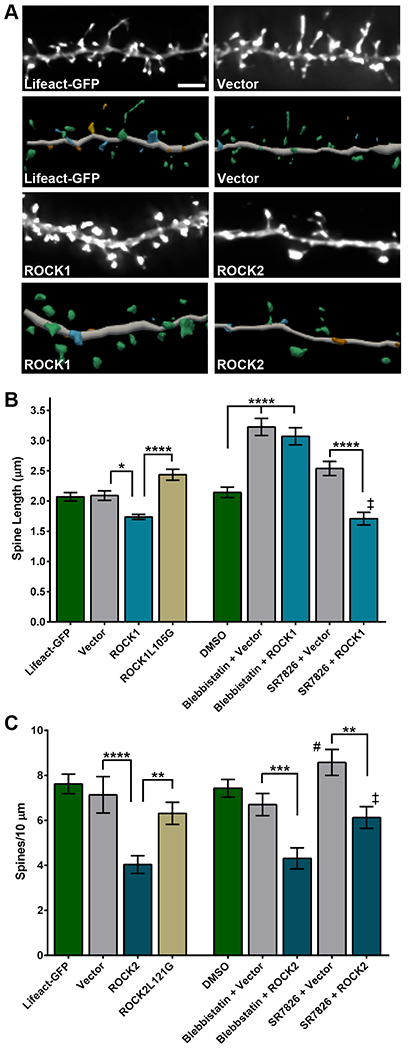
(A) Representative maximum-intensity widefield fluorescent images, after deconvolution, of hippocampal neurons expressing vector, ROCK1 or ROCK2 compared with the Lifeact-GFP control (top). Scale bar, 5 μm. Three-dimensional digital reconstructions of dendrites (bottom). Reconstructions were generated in Neurolucida 360. N=10–17 neurons (one dendrite per neuron) were analyzed per experimental condition in 3 independent cultures. (B) Dendritic spine length in hippocampal neurons expressing vector, wild-type human ROCK1, or ROCK1-L105G, and treated with blebbistatin or SR7826. Controls were transfected with Lifeact-GFP and treated with DMSO. Data are means ± SEM of 3 experiments. ****P<0.0001 and *P<0.05 (vs vector, actual P=0.0230; ‡ vs DMSO, P=0.0285) by one-way ANOVA with Sidak’s test. (C) Dendritic spine density in hippocampal neurons expressing vector, wild-type human ROCK2, or ROCK2-L121G and treated with blebbistatin or SR7826. Data are means ± SEM of 3 experiments. ****P<0.0001, ***P<0.001, **P<0.01 (vs DMSO, # actual P=0.0083), and *P<0.05 (vs SR7826, ‡ actual P=0.0207) by one-way ANOVA with Sidak’s test. Related data and analyses are shown in fig. S1.
ROCKs share protein substrates related to actin regulation, including myosin light chain (MLC), myosin light chain phosphatase, and LIMK1 (36–38). We hypothesized that the distinct ROCK1- or ROCK2-effects on spine length or density, respectively, may be governed by different mechanisms. To test this, neurons expressing ROCK1 or ROCK2 were treated with blebbistatin, which inhibits myosin ATPase and thus relaxes actin-myosin contractility, or SR7826, a small-molecule inhibitor of LIMK1 that impedes its activity on cofilin (fig. S1F) (39, 40). Blebbistatin, but not SR7826, rescued ROCK1-mediated reduction of spine length, whereas SR7826, but not blebbistatin, prevented ROCK2-mediated reduction of spine density (Fig. 1, B and C). Notably, blebbistatin and SR7826 significantly increased spine length and spine density, respectively, compared to DMSO controls (Fig. 1, B and C). These results suggest that ROCK1 kinase activity regulates spine length though myosin-actin pathways, while ROCK2 kinase activity controls spine density through LIMK1-cofilin-actin signaling. Moreover, our findings hint that increased protein abundance of ROCK1 and ROCK2 in MCI and AD may contribute to decreased spine structural plasticity and density that is observed in disease cases (8).
Aβ-induced dendritic spine degeneration is mediated by the ROCK2-LIMK1 pathway
Aβ oligomers can wreak havoc on dendritic structure and degenerate spines in cellular and animal models of AD (10, 41, 42). Recent studies indicate that Aβ oligomers have detrimental effects on actin cytoskeleton rearrangement in neurons and that Rho-GTPase pathways may be involved (27, 28). Aβ42 oligomers can activate ROCKs, leading to increased phosphorylation of LIMK1 (17). To test whether ROCK1 or ROCK2 is necessary for Aβ-induced dendritic spine degeneration, rat hippocampal neurons were transduced with lentivirus expressing ROCK1- or ROCK2-targeted or scramble short hairpin RNA (shRNA) (fig. S2, A and B). Ninety-six hours later, cultures were treated with Aβ42 oligomers or DMSO for 6 hours then fixed, imaged, and processed for 3D morphometry analyses. Neurons that were untransduced or those transduced with scramble shRNA or ROCK1 shRNA displayed similar and significant reductions in spine density after exposure to Aβ42 compared to DMSO-treated counterparts. However, shRNA-mediated depletion of ROCK2 significantly curbed Aβ42-induced spine loss compared to scramble shRNA-transduced neurons treated with Aβ42 (Fig. 2, A and B). Notably, reduction of ROCK1 or ROCK2 did not significantly alter spine density, length, or head diameter in comparison to scramble controls (Fig. 2B; fig. S2, C and D). These results suggest that Aβ42-induced spine degeneration is predominantly mediated by ROCK2, rather than ROCK1. Based on this data, and those above demonstrating that LIMK1 inhibition blocked ROCK2-mediated spine loss (Fig. 1C), we hypothesized that suppressing LIMK1 activity would modulate Aβ42-induced spine degeneration. To test this, rat hippocampal neurons were treated simultaneously with SR7826 and Aβ42 oligomers for 6 hours. Aβ42 had no effect on spine density or morphology among neurons exposed to SR7826, indicating that LIMK1 inhibition prevented Aβ42-induced spine degeneration (Fig. 2C; fig. S2, E and F). Fasudil has been shown to block the negative effects of Aβ oligomers on dendritic spines (27, 28); similarly, our data here revealed that simultaneous exposure to fasudil and Aβ42 oligomers for 6 hours had no effect on spine density or morphology (fig. S2G).
Figure 2. Aβ-induced dendritic spine degeneration is mediated by the ROCK2-LIMK1 pathway.
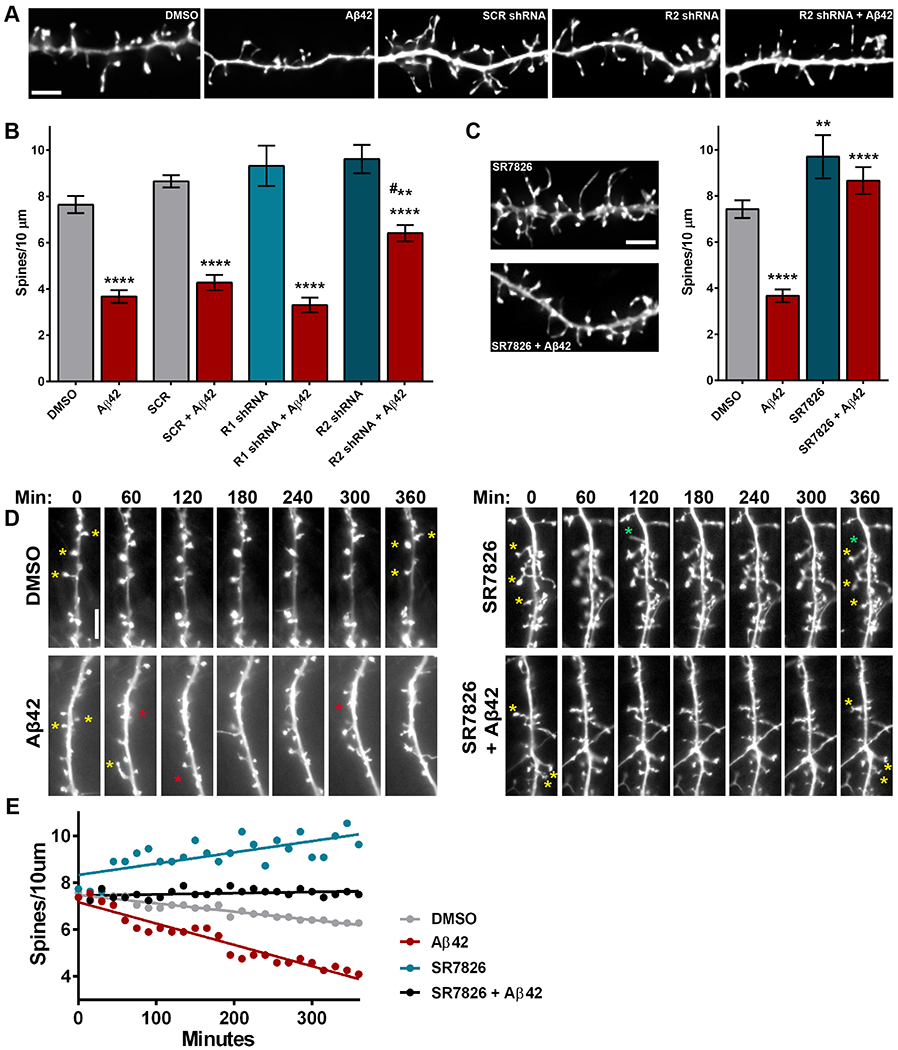
(A) Representative maximum-intensity widefield fluorescent images of hippocampal neurons after deconvolution. Scale bar, 5 μm. N=9-17 neurons (one dendrite per neuron) were analyzed per experimental condition in 3 independent cultures. (B) Dendritic spine density in hippocampal neurons transduced with lentivirus expressing scramble (SCR) or ROCK1- (R1), or ROCK2 (R2)-targeted shRNA and exposed to DMSO or oligomeric Aβ42 (500 nM). Data are means ± SEM of 3 experiments. ****P<0.0001 (Aβ42 vs DMSO controls) and **P<0.001 (vs SCR+Aβ42, # actual P=0.0069) by one-way ANOVA with Sidak’s test. (C) Representative maximum-intensity widefield fluorescent images (after deconvolution) of hippocampal neurons exposed to SR7826 (10 μM) with or without Aβ42 (500 nM). Scale bar, 5 μm. Data (right) are means ± SEM of 3 experiments; N=6–17 neurons (one dendrite per neuron) were analyzed per experimental condition in 3 independent cultures. ****P<0.0001 and **P<0.01 (actual P=0.0072) by one-way ANOVA with Sidak’s test. (D) Representative widefield live-cell fluorescent images of hippocampal neurons over time, exposed to DMSO, Aβ42, SR7826, or SR7826 + Aβ42. Stars highlight loss (red), maintenance (yellow), or formation (green) of dendritic spines. Scale bar, 5 μm. (E) Representative spine density counts in hippocampal neurons over 6 hours with the indicated treatments. Dots represent the spine density (spines per 10 μm) for a single dendrite at 15 minute intervals over 6 hours. N=3–5 neurons (one dendrite per neuron) were analyzed per experimental condition in 3 independent cultures Related data are shown in fig. S2.
Maintenance and retention of dendritic spines is hypothesized to facilitate memory and information processing in patients who harbor substantial Aβ pathology but are cognitively normal (7, 8). Therefore, therapeutics that protect spines from Aβ could be useful to prevent dementia onset. To this end, we tested whether SR7826 protected spines from Aβ42 oligomers or SR7826 generated new spines to compensate for Aβ42-induced spine loss. Treatment of hippocampal neurons with SR7826 and/or Aβ42 oligomers was performed for 6 hours on neurons transfected with plasmid expressing Lifeact-GFP. Live-cell imaging indicated that over 6 hours, spine loss occurred more rapidly among neurons exposed to Aβ42 compared to DMSO controls (Fig. 2, D and E). Spine density increased over time in cultures treated with SR7826, while spine density remained static in neurons simultaneously exposed to Aβ42 and SR7826 (Fig. 2, D and E). These findings suggest that pharmacologic inhibition of LIMK1 can generate spines under normal conditions but protects spines in the presence of Aβ42 oligomers.
LIMK1 inhibition protects against Aβ-induced neuronal hyperexcitability
Dendritic degeneration in hAPP mice causes neuronal hyperexcitability by rendering neurons more electrically compact (10). These detrimental effects ultimately drive aberrant circuit synchronization and likely contribute to cognitive impairment in AD patients. To evaluate the electrophysiological consequences of Aβ42-induced spine loss in hippocampal neurons, we seeded cells directly on multi-electrode arrays (MEAs) in individual cell culture plate wells and performed a baseline recording at DIV 14 (Fig. 3A). Immediately after the baseline recording, neurons were exposed to Aβ42 oligomers or DMSO for 6 hours, and at the end of 6 hours, a second recording was performed. Treatment with Aβ42 increased action potential frequency as well as the frequency of action potential bursts significantly compared to DMSO controls (Fig. 3, B to D). These results mirrored findings on hippocampal neuron hyperexcitability at the cellular and network level in hAPP mice (10). Neuronal hyperexcitability was not observed following simultaneous exposure to Aβ42 oligomers and SR7826 (Fig. 3, B to D). Notably, Fasudil blocked Aβ42-induced hyperexcitability of neurons (fig. S3). This suggests that the dendritic spine resilience provided by LIMK1 inhibition is protective against the toxic hyperexcitability induced by Aβ42 oligomers.
Figure 3. LIMK1 inhibition protects against Aβ-induced neuronal hyperexcitability.
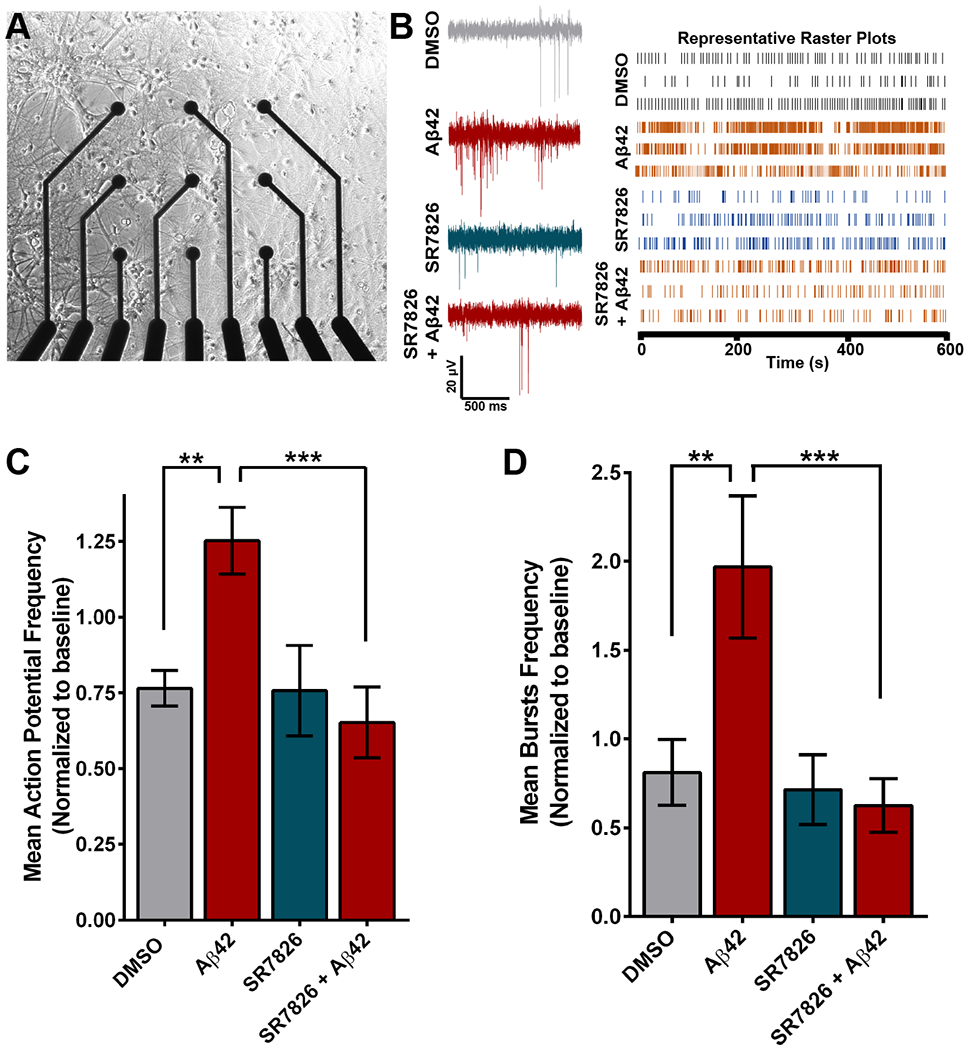
(A) Representative brightfield image of primary hippocampal neuron cultures grown on a multi-electrode array (MEA) plate. (B) Representative traces (left) and raster plots from 3 units (right) after exposure to DMSO, Aβ42, SR7826, or SR7826 + Aβ42. N=17–24 wells per condition which includes 4–6 neurons per well from 3 independent cultures. (C and D) Mean action potential frequency (C) and mean bursts frequency (D) over baseline in hippocampal neurons treated with DMSO, Aβ42, or SR7826 with or without Aβ42. Data are means ± SEM of 3 experiments. ***P<0.001 [actual P= 0.0009 (C), 0.0003 (D)] and **P<0.01 [actual P= 0.0062(C), 0.0016 (D)] by one-way ANOVA with Sidak’s test.
LIMK1 inhibition rescues hippocampal thin spine loss in hAPP mice
Past studies indicated that Aβ can activate RhoA in brain, but whether hAPP leads to downstream activation of ROCKs remained to be determined (19). To address this, we evaluated hippocampal tissue homogenates from 6-month-old hAPPJ20 mice and age-matched non-transgenic (NTG) littermate controls by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and subsequent immunoblot (Fig. 4A). Densitometry analysis indicated that ROCK2, but not ROCK1, protein levels were elevated significantly in hippocampal homogenates from hAPPJ20 brains compared to NTG controls. Moreover, phosphorylation of LIMK1 at Thr508 (pLIMK1) was increased significantly in hAPPJ20 mice compared to NTG littermates, indicating heightened activity of ROCKs in the hippocampus of hAPP mice (Fig. 4B). Notably, these results are consistent with increased amounts of ROCK2 protein and pLIMK1 in AD brains (18, 26).
Figure 4. LIMK1 inhibition rescues hippocampal thin spine loss in hAPP mice.
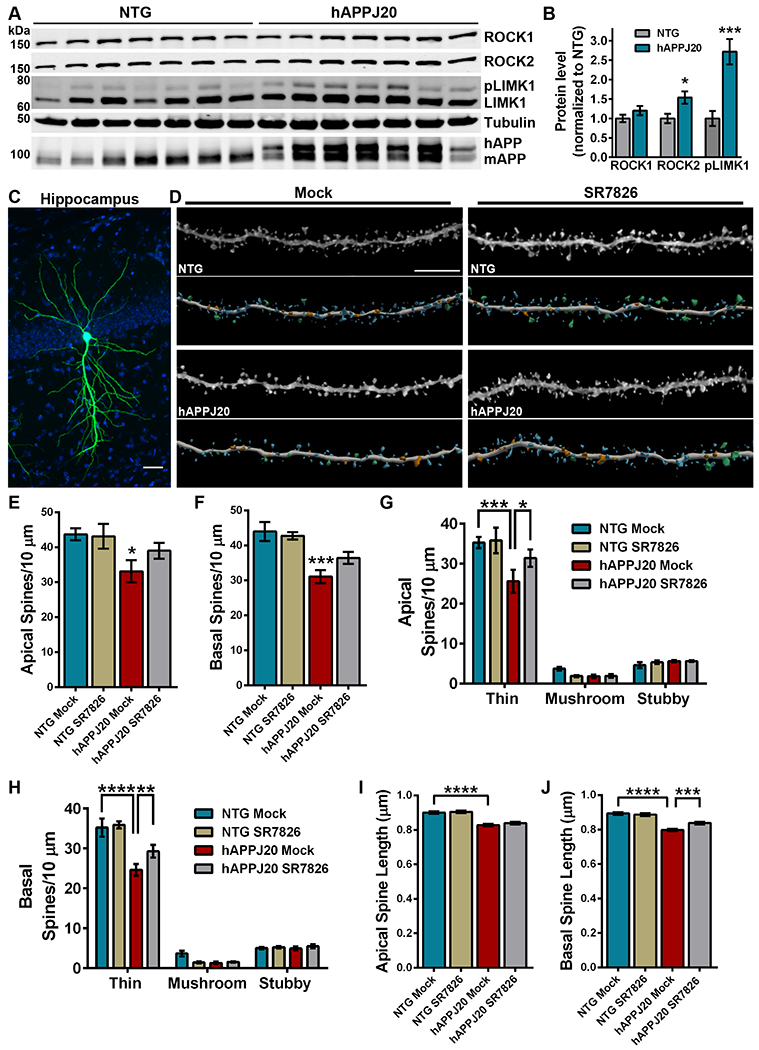
(A and B) Representative immunoblots (A) and densitometry analysis (B) of ROCK2 and phosphorylated LIMK1 protein abundance in the hippocampus of hAPPJ20 mice relative to each in non-transgenic (NTG) littermates. For densitometry, pLIMK1 was normalized to levels of LIMK1. Immunoblot for amyloid precursor protein (APP) identifies human APP in hAPPJ20 mice. Data are means ± SEM of 7 mice (3 males and 4 females per genotype). *P<0.05 (actual P=0.0191) and ***P<0.001 (actual P=0.0007) by an unpaired t-test. (C) Representative maximum-intensity image of a CA1 pyramidal neuron in the hippocampus iontophoretically-filled with Lucifer yellow. Blue signal is DAPI. Scale bar, 50 μm. (D) Representative maximum-intensity high-resolution confocal microscope images of dye-filled dendrites, from mock- or SR7826-treated hAPPJ20 and NTG mice, after deconvolution and corresponding three-dimensional digital reconstruction models of dendrites. Scale bar, 5 μm. Colors in digital reconstructions correspond to dendritic protrusion classes: blue represents thin spines; orange, stubby spines; green, mushroom spines; and yellow, dendritic filopodia. (E and F) Mean number of apical (E) and basal (F) spines per 10 μm in dendrites from mock- or SR7826-treated hAPPJ20 and NTG mice. Data are means ± SEM. The apical dendrite conditions are as follows: N=26 dendrites from 5 NTG mock mice (3 females, 2 males), N=26 dendrites from 5 NTG SR7826 mice (1 female, 4 males), N=33 dendrites from 5 hAPPJ20 mock mice (2 females, 3 males), and N=30 dendrites from 5 hAPPJ20 SR7826 mice (2 females, 3 males) for a total of 3,546 μm analyzed. The basal dendrite conditions are as follows: N=22 dendrites from 5 NTG mock mice (3 females, 2 males), N=23 dendrites from 5 NTG SR7826 mice (1 female, 4 males), N=26 dendrites from 5 hAPPJ20 mock mice (2 females, 3 males), and N=26 dendrites from 5 hAPPJ20 SR7826 mice (2 females, 3 males) for a total of 3,126 μm analyzed. *P<0.05 (actual P=0.0313) and ***P<0.001 (actual P=0.0005) by one-way ANOVA with Sidak’s test. (G and H) Mean number of thin, stubby, or mushroom spines per 10 μm of apical (G) or basal (H) dendrites from mock- or SR7826-treated hAPPJ20 and NTG mice. Data are means ± SEM; N as given in (E and F). *P<0.05 (actual P=0.0398), **P<0.01 (actual P=0.0098), ***P<0.001 (actual P=0.0002), and ****P<0.0001 by two-way ANOVA with Tukey’s test. (I and J) Mean spine length of apical (I) and basal (J) spines among CA1 pyramidal neurons in the hippocampus from mock- or SR7826-treated hAPPJ20 and NTG mice. Data are means ± SEM; N as given in (E and F). ***P<0.001 (actual P=0.0003) and ****P<0.0001 by one-way ANOVA with Sidak’s test.
Past studies demonstrated that hippocampal spine loss occurs in hAPPJ20 mice at 9 months of age when amyloid plaque load is still minimal (43). Based on our results above (Figure 1C), increased amounts of ROCK2 and pLIMK1 at 6 months old would likely reduce spine density in the hippocampus of hAPPJ20 mice. Moreover, LIMK1 inhibition prevented Aβ42-induced spine loss in hippocampal neurons when SR7826 and Aβ42 oligomers were applied simultaneously (Figure 2, C to E). Therefore, we wondered whether treatment with SR7826 would benefit hippocampal spines undergoing degeneration in hAPPJ20 mice. Initially, we dosed 6 month old hAPPJ20 mice and age-matched NTG controls with 10 mg/kg SR7826 or equivalent volume DMSO (mock) by oral gavage and harvested brains 6 hours later to verify SR7826 target engagement. SDS-PAGE and immunoblot of synaptosome fractions from hippocampal tissue homogenates revealed that phosphorylation of cofilin at Ser3 (pCofilin), a LIMK1 substrate (44, 45), was reduced significantly in mice treated with SR7826 (Figure S4, A and B). To test the therapeutic potential of LIMK1 inhibition, we dosed 6-month-old hAPPJ20 mice and age-matched NTG controls with 10 mg/kg SR7826 or mock once a day by oral gavage for 11 days. At the end of 11 days, all mice were weighed, transcardially perfused, and organs were collected for analysis. Treatment with SR7826 did not alter weight in hAPPJ20 or NTG mice, and histological examination of liver samples did not indicate SR7826-induced toxicity (fig. S4, C and D).
To evaluate spines, individual CA1 pyramidal neurons in the hippocampus were targeted for iontophoretic microinjection of the fluorescent dye Lucifer yellow, followed by high-resolution confocal laser scanning microscopy and dendritic 3D reconstructions for morphometry analysis (Fig. 4, C and D). Comparison of apical and basal dendrites revealed significant reductions in spine density among hAPPJ20 mock animals compared to NTG mock, supporting our hypothesis that Aβ-induced activity of the ROCK2-LIMK1 pathway causes robust spine loss. Global apical and basal spine densities in SR7826-treated hAPPJ20 mice were increased, but not significantly, compared to mock hAPPJ20 (Fig. 4, E and F). Spine morphology influences excitatory neurotransmission and synaptic plasticity, and spines can be classified on the basis of their 3D structure as stubby, mushroom, or thin (9, 46, 47). Examination of thin, mushroom, and stubby spine populations on apical and basal dendrites among each experimental condition revealed robust loss of thin spines in hAPPJ20 mock animals compared to NTG mock. This indicates that the reduction in thin spine density drove the global decrease in spine density among hAPPJ20 mock samples. Treatment with SR7826 increased both apical and basal thin spine density significantly in hAPPJ20 mice over mock-treated animals (Fig. 4, G and H). However, no significant changes in density were observed in mushroom or stubby spine populations among the experimental conditions.
To further analyze spine structure, mean apical and basal spine length and head diameters were plotted for each experimental condition. Significant reductions in both apical and basal spine length were identified in hAPPJ20 mock animals compared to NTG mock, however treatment with SR7826 increased basal spine length significantly in hAPPJ20 mice (Fig. 4, I and J). Mean apical and basal spine head diameters were similar among hAPPJ20 mock and NTG mock animals, while SR7826 reduced apical spine head diameter in NTG mice but increased basal spine head diameter in hAPPJ20 mice (fig. S5, A and B). Notably, we did not observe changes in soluble or insoluble Aβ42 or thioflavin S staining among hAPPJ20 mice treated with SR7826, suggesting that the beneficial effects of SR7826 on spines were not due to reductions in Aβ (fig. S6, A to C). Collectively, our findings link experimental models with human disease by demonstrating that Aβ-induced changes in ROCK2-LIMK1 signaling likely contribute to dendritic spine degeneration in AD. Moreover, pharmacologic inhibition of LIMK1 is identified as a rational therapeutic approach to protect spines from Aβ.
Discussion
As the human population ages, the ability to maintain cognitive function with a brain that is accumulating AD pathology is likely linked to the preservation and maintenance of synapses and dendritic spines. Therefore, protecting dendritic spines from the degenerating forces of AD is critical for preventative therapeutics. In this report, we reveal that ROCKs govern dendritic spine density and morphology through isoform-specific cell biological mechanisms, and we discuss how this may impact spine structure in AD. Our findings support the hypothesis that Aβ42 oligomers induce hippocampal neuron spine degeneration and hyperexcitability through the ROCK2-LIMK1 pathway, and we assess the therapeutic potential of LIMK1 inhibition in cellular and animal models of AD.
The amount and activity of ROCK1 and ROCK2 proteins were increased in AD brains assessed in our study, and these changes are likely due to accumulation of Aβ (17, 18, 26). Here, we found that enhanced activity of ROCK1-myosin-actin or ROCK2-LIMK1-cofilin-actin signaling decreased spine length or density, respectively, in neurons. These findings implicate the ROCK2-LIMK1 pathway as a potential culprit of reduced spine density in AD, however spine length was reported similar among age-matched pathology-free controls and AD cases (8). Increased spine length was observed exclusively in cognitively normal patients with AD pathology (CAD), suggesting that (i) enhancing spine length in MCI patients may be beneficial, or (ii) that increased spine length is a resilience mechanism prior to cognitive decline (7, 8). Whether ROCK1 amounts are decreased in CAD cases is unclear, and although these studies do not eliminate the rationale for pharmacologic inhibition of ROCK1, other caveats may and are discussed below.
Epileptiform activity is an indicator of network hyperexcitability in hAPP mice, and high rates of subclinical epileptiform activity are detected in AD patients (11, 14, 48). Network hyperexcitability in hAPP mice is driven by degeneration of hippocampal pyramidal neurons’ dendrites and dendritic spines (10). Loss of dendrites and spines cause neuronal hyperexcitability by reducing the total surface area of the cell and thus rendering neurons more electrically compact. In a more compact neuron, synapse currents would be translated more efficiently into post-synaptic and axon-somatic depolarization, leading to increased action potential output (49). Consequences of this mechanism may include hyperexcitability of the neuron as well as aberrant circuit synchronization. Our culture work here directly supports these findings by linking Aβ-induced dendritic spine loss to increased neuronal firing rates. Including our data herein, we now know that these Aβ-induced structural and electrophysiological phenotypes are shared across humans, rodents and cellular models of AD. Past studies provide evidence that Aβ oligomers can directly interact with the cellular prion protein (PrPC) (50), which may induce PrPC-mediated signaling of RhoA (51). These pathways would link extracellular Aβ to intracellular ROCK1 and/or ROCK2 signaling. Our data from this study showed that while spine morphology was similar between Aβ42-treated samples and controls in cultured hippocampal neurons, hAPPJ20 mice displayed substantial reductions in both spine density and length among hippocampal neurons, suggesting that activity of both ROCK1 and ROCK2 signaling pathways contributed to these effects. Moreover, thioflavin S staining was minimal in the 6-month-old hAPPJ20 mice, supporting the hypothesis that structural deficits and spine loss are most likely linked to Aβ oligomers rather than accumulation of plaques.
Two pan-ROCK inhibitors, Fasudil and Ripasudil, have been used to treat human disease (52). Prior studies have explored the potential to repurpose ROCK inhibitors for neurodegenerative disorders, including AD, frontotemporal dementia, Parkinson’s disease, and amyotrophic lateral sclerosis (26, 28, 53–56). Despite the translational potential these compounds exhibit, target-selectivity caveats and ambiguity over ROCK1- or ROCK2-specific functions in neurons have stalled ROCK-based therapeutics for cognitive treatment (52, 57). Blood pressure reduction is an effect of pan-ROCK inhibitors and is predominantly due to ROCK1 inhibition, therefore ROCK2-selective pathways and drugs may provide a better safety profile (58). Fasudil was shown to block negative effects of Aβ oligomers on dendritic spines (27, 28). Our data here revealed that simultaneous exposure to Fasudil and Aβ42 oligomers for 6 hours had no effect on spine density or morphology, supporting the published findings, and that Fasudil blocked Aβ42-induced hyperexcitability of neurons. Our shRNA results suggest that the beneficial effects of Fasudil on Aβ-induced spine toxicity are predominantly modulated through inhibition of ROCK2 signaling. However, Fasudil is not specific to ROCKs (29), therefore moving down the ROCK2 pathway to LIMK1 inhibitors may be a safer, more efficient therapeutic strategy with fewer off-target effects. While LIMK1 is predominantly expressed in brain, both LIMK1 and LIMK2 can phosphorylate the actin-severing protein cofilin at Ser3 (45, 59). However, the identification of additional molecular substrates of LIMKs has been extremely limited (60). In the 6 years prior to this study, several LIMK inhibitors have been discovered, some of which are highly selective, including SR7826, and are now undergoing further development and optimization (40, 60, 61). Future use of these compounds will increase our understanding of the LIMK isoforms’ functions as well as fuel new therapeutic avenues.
Thin spine loss is a shared phenotype among hAPPJ20 mice (observed here) and AD patients (8). Spine structure is inseparably linked to spine function, and spines can be classified on the basis of their 3D morphology as stubby, mushroom, or thin (46, 47, 62, 63). Stubby spines are hypothesized as transitional structures that may enlarge, possibly to mushroom spines, which are more stable entities with a wide head and thin neck. Thin spines are more dynamic, and lack the wide head of mushroom spines. Spine morphology can robustly affect molecular diffusion. For instance, length and width of spine necks is a predominant mediator of compartmentalization, facilitating efficient regulation of synaptic biochemical and electrical components (64). Our study revealed that thin spine loss and mean reduction of spine length were observed among both apical and basal dendrites in the CA1 region of the hippocampus of hAPPJ20s. While LIMK1 inhibition increased apical and basal thin spine density, spine length was increased more robustly on basal dendrites. Moreover, SR7826 reduced mean spine head diameter on apical, but not basal, dendrites in NTG mice, whereas SR7826 increased mean spine head diameter significantly on basal dendrites in hAPPJ20 mice. These results are challenging to interpret but may suggest that LIMK1 regulates different aspects of spine morphology depending on the geographical location of the spine, and Aβ accumulation seemingly layers an additional level of complexity. Likely, consequences on dendritic spine structure following LIMK1 inhibition is steered by the electrophysiological activity of the brain region and neuronal network during the time of drug dosing (64–68). Therefore, strong influences on brain environment such as age and disease state will need to be considered for therapeutic strategies targeting dendritic structure in AD (7, 69).
Materials and Methods
Primary rat hippocampal neuron and continuous cell culture
Rat hippocampal neurons were isolated from E18 Sprague-Dawley rat embryos and cultured at a density of 2 x 105 cells per coverslip on poly-L-lysine-coated 18mm glass coverslips as previously described with minor modifications (25). Briefly, neurons were cultured in Neurobasal medium (Invitrogen) supplemented with B27 that was conditioned by separate cultures of primary rat astrocytes and glia. Neurons were treated at days in vitro (DIV) 4 with 5μM Cytosine β-D-arabinofuranoside hydrochloride (Sigma Aldrich) to eliminate the presence of native astrocytes and glia on the glass coverslips. Media was changed every 3-4 days with new glia-conditioned Neurobasal medium for proper culture maintenance. At DIV 12 neurons were co-transfected with plasmids using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. Neuro-2A (N2a) mouse neuroblastoma cells were maintained in DMEM with 10% fetal bovine serum, and 1% penicillin/streptomycin.
DNA constructs, lentivirus, and shRNA
Lentivirus vectors for shRNA expression were constructed and generated as previously described (53, 70). For ROCK1 shRNA: 5’-GCCAATGACTTACTTAGGA; ROCK2 shRNA: 5’-ATCAGACAGCATCCTTTCT; and scramble: 5’-GGACTACTCTAGACGTATA. To generate ROCK1-L121G, cDNA encoding human ROCK1 was used as a template and QuikChange XL Site-directed Mutagenesis kit (Stratagene) was employed. Sense primer 5’- CCACCAGGAAGGTATATGCTATGGGGCTTCTCAGCAAATTTGAAA; antisense 5’- TTTCAAATTTGCTGAGAAGCCCCATAGCATATACCTTCCTGGTGG. To generate ROCK2-L121G, cDNA encoding human ROCK2 was used as a template. Sense primer 5’-GGCATCGCAGAAGGTTTATGCTATGGGGCTTCTTAGTAAGTTTGA; antisense 5’-TCAAACTTACTAAGAAGCCCCATAGCATAAACCTTCTGCGATGCC. Constructs were verified by sequencing. Plasmid encoding Lifeact-GFP was a generous gift from Dr. Gary Bassell, Emory University School of Medicine, Atlanta, GA, USA. Previous studies have demonstrated that Lifeact-expressing neurons display normal, physiological actin dynamics and dendritic spine morphology (34, 71).
Chemicals
Aβ42 (Bachem) oligomers were prepared as previously described (17). Aβ was re-suspended in 1X HBSS and Dimethyl sulfoxide (DMSO) then placed in 4°C overnight. At DIV14, primary hippocampal neurons were treated with 500 nM Aβ42 for 6 hrs. Fasudil (Selleckchem #S1573) and SR7826 (Tocris #562610) were reconstituted to a 10 mM stock in either water or DMSO, respectively. At DIV14, primary hippocampal neurons were dosed with either 10 μM SR7826, 30 μM Fasudil, or a combination of drug plus Aβ42 for 6 hrs. Six hours was chosen based on past studies demonstrating that Aβ42-induced spine loss in cultured neurons plateaus at approximately 6 hours post exposure (41), and pan-ROCK inhibitors induce robust changes in spine morphology on cultured hippocampal neurons following 6 hours of exposure (25). Blebbistatin (Tocris #1852) was reconstituted to a 10 mM stock in DMSO. At DIV14, primary hippocampal neurons were treated with 5 μM blebbistatin for 1 hour. The one hour incubation time was selected based on prior studies (72, 73).
Cell lysate preparation, immunoblots, and antibodies
Cells were lysed in phosphate-buffered saline (PBS) plus protease inhibitor cocktail (PIC; Roche Diagnostics, Risch-Rotkreuz, Switzerland), Halt phosphatase inhibitor cocktail (Pierce, Rockford, IL, USA), and lysis buffer containing 0.5% Nonidet P-40, 0.5% deoxycholate, 150 mM sodium chloride, and 50 mM Tris, pH 7.4. All lysates were subjected to a 15,871 × g spin for 5 minutes to remove nuclei and debris. Protein concentration was determined by bicinchoninic acid method (Pierce). Immunoblots were performed using standard procedures as described previously (74). A quantity of 50 μg protein per sample was loaded per lane. Tubulin was used as a loading control. Images were captured using an Odyssey Image Station (LiCor), and band intensities were quantified using Odyssey Application Software Version 3.0 (LiCor). Primary antibodies were incubated overnight at 4°C. Primary antibodies include: ROCK1 (Abcam 45171), ROCK2 (Santa Cruz 5561), LIMK (Cell Signaling 3842S), Phospho-LIMK (Cell Signaling 3841), Phospho-Cofilin (Cell Signaling 3313), Cofilin (Cell Signaling 3318), and Tubulin (Iowa Hybridoma Bank). Secondary antibodies include: AlexaFluor 680 goat anti-rabbit (Life Technologies A21109) and goat anti-mouse (Li-Cor 926-32210).
Oral gavage
All experimental procedures were performed under a protocol approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham. Six month old non-transgenic (NTG) and hAPPJ20 mice (B6.Cg-Zbtb20Tg(PDGFB-APPSwInd)20Lms/2Mmjax) (The Jackson Laboratory MMRRC Stock No: 34836-JAX J20) were treated once daily with mock (90% H20, 10% DMSO) or SR7826 (dissolved in (90% H20, 10% DMSO) for 11 days via oral gavage using plastic gavage tips (Instech Catalog #FTP-20-38). Treatment was given at 2:00 pm daily throughout the entirety of the treatment regimen. SR7826 was dissolved fresh each day at a concentration of 10 mg/kg (200 μL total volume per animal per day). Mice were sacrificed at the end of the treatment period for postmortem analyses. For all experiments, age-matched and sex-matched animals were used. When necessary, additional details on mouse sex are provided in figure legends.
Perfusions and brain tissue processing
Animals were anesthetized with Fatal Plus (Vortech Pharmaceuticals, Catalog #0298-9373-68). Mice were transcardially perfused with cold 1% paraformaldehyde (PFA; Sigma Aldrich, Catalog #P6148) for 1 min, followed by cold 4% PFA with 0.125% glutaraldehyde (Fisher Scientific, Catalog #BP2547) for 10 min. A peristaltic pump (Cole Parmer) was used for consistent administration of PFA. Immediately following perfusion, mice were decapitated, and the whole brain was removed and drop-fixed in 4% PFA containing glutaraldehyde for 8-12 hours at 4°C. After post-fixation, the brains were sliced in 250 μm coronal sections using a Leica vibratome (VT1000S) with a speed of 70, and frequency of 7. The platform was filled with cold 0.1 M PB buffer, and the brain was glued (Loctite) perpendicular to the stage, cerebellum side down. All slices were stored one slice per well in a 48-well plate containing 0.1% sodium azide (Fisher, Catalog#BP922I) in 0.1M PB at 4°C. Notably, these procedures were performed according to (75). For PBS perfusions, animals were anesthetized with Fatal Plus. Mice were transcardially perfused with cold 1X PBS for 2 minutes. Immediately following perfusion, the brain was extracted and dissected into two hemispheres. Each hemisphere was immediately flash frozen in 2-methylbutane (Sigma, Catalog#320404), placed on dry ice, and stored at −80°C.
Synaptosome preparations
Hemibrains were bathed in a petri dish of ice-cold PBS with protease (Sigma S8820) and phosphatase inhibitors (Thermo Scientific 1861277). The hippocampus was isolated from each hemibrain and synaptosomes were prepared using the following biochemical fractionation protocol, as previously described (76, 77). Sub-dissected tissue samples were bathed and homogenized for 30 s in TEVP buffer (10 mM Tris base, 5 mM NaF, 1 mM Na3VO4, 1 mM EDTA, 1 mM EDTA) with 320 mM sucrose and protease and phosphatase inhibitors. A small volume was saved as whole homogenate (WH). Remaining sample was centrifuged at 800 × g for 10 min at 4°C. The supernatant (S1) was removed, and the pellet (P1) was stored in TEVP + inhibitors. S1 was centrifuged at 9200 × g for 10 min at 4 °C. The supernatant (S2) was removed and stored. The pellet (P2) was resuspended in TEVP + 32 mM sucrose + inhibitors and centrifuged at 25,000 × g for 20 min at 4°C. The supernatant (LS1) was removed and stored. The pellet (synaptosome fraction) was resuspended in TEVP + inhibitors and stored at −80°C.
Amyloid measurements
Soluble and insoluble Aβ42 were extracted according to the Human Brain Aβ42 ELISA (Millipore) manufacturer’s instructions,. Plates were read at 450 nm on a Spectra Max Plus plate reader (Molecular Devices). For Thioflavin S staining, perfused mouse brains were sectioned to 50 μm slices using a vibratome (Leica VT1000S). Slices were then subjected to the following washes: 70% EtOH for 1 minute, 80% EtOH for 1 minute, Thioflavin S in 80% EtOH for 15 minutes, 80% EtOH for 1 minute, 70% EtOH for 1 minute, and then two washes in DI H2O. Coverslips were then mounted on glass slides with Vectashield aqueous mounting media (Vector Labs, Catalog #H1000). Images were captured on a Nikon (Tokyo, Japan) Eclipse Ni upright microscope, using a Nikon Intensilight and Photometrics Coolsnap HQ2 camera to image Thioflavin S. Images were captured with Nikon Elements 4.20.02 image capture software using 4X objective (Nikon Plan Fluor 0.13 N.A. objective).
Multi-electrode array recording and analysis
Single neuron electrophysiological activity was recorded using a MEA2100 Lite recording system (Multi Channel Systems). E18 rat primary hippocampal neurons were harvested as described above, and plated in 6-well multi-electrode arrays (MEA) at a density of 125,000 cells/well. Each MEA well contained 9 extracellular recording electrodes and a ground electrode. At DIV 14, a 30 minute MEA prerecording was performed followed by application of Aβ42 or pharmacological inhibitors for 6 hours. After 6 hours, a follow-up 30minute MEA recording was performed to determine effects on neuronal firing properties. All recordings were performed while connected to a temperature-controlled headstage (37°C) with 5% CO2 and containing a 60-bit amplifier. Electrical activity was measured by an interface board at 30 kHz, digitized, and transmitted to an external PC for data acquisition and analysis in MC_Rack software (Multi Channel Systems). All data were filtered using dual 10 Hz (high pass) and 10,000 Hz (low-pass) Butterworth filters. Action potential thresholds were set manually for each electrode (typically > 4 standard deviations from the mean signal). Neuronal waveforms collected in MC_Rack were exported to Offline Sorter (Plexon) for sorting of distinct waveforms corresponding to multiple units on one electrode channel, and confirmation of waveform isolation using principal component analysis, inter-spike intervals, and auto- or cross-correlograms. Further analysis of burst activity and firing rate was performed in NeuroExplorer. Mean firing frequency and bursting were calculated by creating a ratio of firing or bursting at a 6 hour time point/baseline. Specifically, either the firing frequency or burst activity were averaged on a per well basis post-6 hr treatment, and then divided by the average on a per well basis at the baseline.
Static widefield microscopy
On DIV 14, neurons were fixed with room temperature 2% PFA in 0.1 M PBS, washed 2 times with 1X PBS, and coverslips were mounted on microscope slides (Fisher, Catalog #12-550-15) using Vectashield mounting media (Vector Labs, Catalog #H1000). A blinded experimenter performed all imaging. Images were captured on a Nikon (Tokyo, Japan) Eclipse Ni upright microscope, using a Nikon Intensilight and Photometrics Coolsnap HQ2 camera to image Lifeact-GFP. Images were captured with Nikon Elements 4.20.02 image capture software using 60X oil-immersion objective (Nikon Plan Apo, N.A. 1.40). Z-series images were acquired at 0.15μm increments through the entire visible dendrite. Dendrites were selected for imaging by using the following criteria: 1) minimum of 25 μm from the soma; 2) no overlap with other branches; 3) must be a secondary dendritic branch. Prior to analysis, captured images were deconvolved using Huygens Deconvolution System (16.05, Scientific Volume Imaging, the Netherlands) with the following settings: CMLE; maximum iterations: 50; signal to noise ratio: 40; quality: 0.01. Deconvolved images were saved in .tif format.
Live-cell widefield microscopy
Primary rat hippocampal E18 neurons were plated on 25mm round glass coverslips (Warner Instruments) at a density of 4x105 cells per coverslip. Cells were maintained as described above. Neurons were transfected at DIV14 with Lifeact-GFP using Lipofectamine 2000 (Invitrogen) according to manufacturer instructions. Neurons were imaged with a 60X oil immersion objective (Nikon Plan Apo, N.A. 1.40) on a Nikon (Tokyo, Japan) Ti2-E inverted microscope with a SOLA light source. Their environment was maintained with a Tokai Hit stage top incubation system, with settings as follows: Top heater 42.3°C; Stage Heater 38.3°C; Bath Heater 41°C; Lens Heater 41°C; CO2 concentration 5%. Neurons were imaged with the following parameters: SOLA light source, 10%; exposure, 200 ms; image size, 1028 × 1028 pixels. Images were captured with an ORCA-Flash 4.0 V3 CMOS camera (Hamamatsu, Hamamatsu City, Japan). An image was captured every 15 minutes for a total of 6 hours. DMSO, 500 nM Aβ42 and/or 10 μm SR7826 were added after the first two images were acquired. For spine density analysis, spines from a representative secondary dendrite at least 25 μm from the soma were counted at each time point and plotted over time.
Iontophoretic microinjection of fluorescent dye
Microinjections were executed using previously described methods (75, 78). A Nikon Eclipse FN1 upright microscope with a 10X objective and a 40X water objective was placed on an air table. The tissue chamber used was assembled in the lab, and consisted of a 50x75 mm plastic base with a 60x10 mm petri dish epoxied to the base. A platinum wire was attached so that the ground wire could be connected to the bath by an alligator clip. The negative terminal of the electric current source was connected to a glass micropipette filled with 2 μl of 8% Lucifer yellow dye (ThermoFisher, Catalog#L453). Micropipettes (A-M Systems, Catalog #603500) with highly tapered tips were pulled fresh the day of use. A manual micromanipulator was secured on the air table with magnets that provided a 45° angle for injection. Brain slices were placed into a small petri dish containing 1X PBS and 4′,6-diamidino-2-phenylindole (DAPI) for 5 min at room temperature. After incubation in DAPI, slices were placed on dental wax, and then a piece of filter paper was used to adhere the tissue. The filter paper was then transferred to the tissue chamber filled with 1X PBS, and weighted down for stability. The 10X objective was used to visualize advancement of the tip of the micropipette in XY and Z until the tip was just a few micrometers above the tissue. The 40X objective was then used while advancing the tip into the CA1 region of the hippocampus. Once the microelectrode contacted a neuron, 2 nA of negative current was used for 5 min to fill the neuron with Lucifer yellow. After 5 min, the current was turned off and the micropipette was removed from the neuron. Neuron impalement within the CA1 occurs randomly in a blind manner. If the entire neuron does not fill with dye after penetration, the electrode is removed and the neuron is not used for analysis. Multiple neurons were injected in each hemisphere of the hippocampus of each animal. After injection, the filter paper containing the tissue was moved back into the chamber containing 1X PBS. The tissue was carefully lifted off the paper and placed on a glass slide with two 125 μm spacers (Electron Microscopy Sciences, Catalog #70327-20S). Excess PBS was carefully removed with a Kimwipe, and the tissue air-dried for 1 min. One drop of Vectashield (Vector Labs, Catalog #H1000) was added directly to the slice; the coverslip (Warner, Catalog #64-0716) was added and sealed with nail polish. Injected tissue was stored at 4°C in the dark.
Confocal microscopy
Confocal microscopy was used to capture images of dendrites from the CA1 region of the hippocampus, based on previously described methods (75, 78). A blinded experimenter performed all imaging. Images were captured with a Nikon (Tokyo, Japan) Ti2 C2 confocal microscope. The experimenter identified secondary dendrites from dye-impregnated neurons and captured three-dimensional z-stacks of those meeting the following criteria: (1) within 80 μm working distance of microscope; (2) relatively parallel with the surface of the coronal section; (3) no overlap with other branches; (4) minimum of 50 μm from the soma; (5) maximum of 110 μm from the soma. For each dendrite, z-stacks were captured with a 60X oil-immersion objective (Nikon Plan Apo, N.A. 1.40) using the following parameters: z-step: 0.1 μm; image size: 1024 × 512 px (0.04 μm x 0.04 μm x 0.1 μm); zoom: 4.8x; line averaging: 4; acquisition rate: 1 frame/sec. Captured images were deconvolved using Huygens Deconvolution System (16.05, Scientific Volume Imaging, the Netherlands) and the following settings: GMLE; maximum iterations: 10; signal to noise ratio: 15; quality: 0.003. Deconvolved images were saved in .tif format.
Dendritic spine morphometry analysis
Automated image analysis was performed with Neurolucida 360 (2.70.1, MBF Biosciences, Williston, Vermont) based on previously described methods (79). Deconvolved image stacks were imported into Neurolucida 360, and the full dendrite length was traced with semi-automatic directional kernel algorithm. The experimenter manually confirmed that all assigned points matched dendrite diameter and position in X, Y, and Z planes and adjusted each reconstruction if necessary. For widefield microscopy, dendritic spine reconstruction was performed automatically using a voxel-clustering algorithm and the following parameters: outer range: 10.0 μm; minimum height: 0.5 μm; detector sensitivity 100%; minimum count: 8 voxels. For confocal microscopy, dendritic spine reconstruction was performed automatically using a voxel-clustering algorithm and the following parameters: outer range: 5.0 μm; minimum height: 0.3 μm; detector sensitivity 80%; minimum count: 8 voxels. Next, the experimenter manually verified that the classifier correctly identified all protrusions. When necessary, the experimenter added any protrusions semi-automatically by increasing detector sensitivity. The morphology and backbone points of each spine were verified to ensure a representative spine shape, and merge and slice tools were used to correct inconsistencies. Each dendritic protrusion was automatically classified as a dendritic filopodium, thin spine, stubby spine, or mushroom spine based on previously described morphological measurements (78). Reconstructions were collected in Neurolucida Explorer (2.70.1, MBF Biosciences, Williston, Vermont) for branched structure analysis, and then exported to Microsoft Excel (Redmond, WA). Spine density was calculated as the number of spines per 10 μm of dendrite length.
Statistical analysis
All analyses were conducted with Prism 6.0 (GraphPad Software, La Jolla, CA). Data are presented as mean ± SEM, and all graph error bars represent SEM. All statistical tests were two-tailed with threshold for statistical significance set at 0.05. Statistical comparisons are indicated in figure legends and included unpaired t-test; two-way ANOVA with Tukey comparison’s test; and one-way ANOVA with Šidák post-hoc analysis. To compare aggregate spine densities or morphologies among experimental conditions, the mean spine density or morphologic measurement was calculated per experimental replicate (or N). These experiment means were then averaged per experimental condition and reported as a condition mean. See figure legends for details on N per experiment.
Supplementary Material
Figure S1. Expression of human ROCKs in hippocampal neurons.
Figure S2. Aβ42-induced spine loss is prevented by Fasudil.
Figure S3. Fasudil protects against Aβ-induced neuronal hyperexcitability.
Figure S4. SR7826 reduces the abundance of phosphorylated cofilin in the hippocampus.
Figure S5. SR7826 alters mean spine head diameter.
Figure S6. Aβ deposits are not substantially altered by SR7826.
Acknowledgments:
We thank Dr. Yangbo Feng at Reaction Biology and members of Dr. Erik Roberson’s laboratory at UAB for helpful discussions. H & E staining was provided by the UAB Pathology Core Research Lab.
Funding: This work was supported by the National Institutes of Health through NIA AG061800 to J.H.H., NIA AG054719 to J.H.H., NIA AG043552 to J.H.H., Emory Neuroscience NINDS Core Facilities grant P30NS055077, and B.W.H. was supported by T32 NS 061788. Additional support stemmed from a New Investigator Research Grant 2015-NIRG-339422 to J.H.H. from the Alzheimer’s Association.
Footnotes
Competing interests: The authors declare they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.DeKosky ST, and Scheff SW. 1990. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Annals of neurology 27: 457–464. [DOI] [PubMed] [Google Scholar]
- 2.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, and Katzman R. 1991. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Annals of neurology 30: 572–580. [DOI] [PubMed] [Google Scholar]
- 3.Scheff SW, Price DA, Schmitt FA, and Mufson EJ. 2006. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiology of aging 27: 1372–1384. [DOI] [PubMed] [Google Scholar]
- 4.Scheff SW, and Price DA. 2003. Synaptic pathology in Alzheimer’s disease: a review of ultrastructural studies. Neurobiology of aging 24: 1029–1046. [DOI] [PubMed] [Google Scholar]
- 5.Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, and Martin LJ. 1997. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. Journal of neuropathology and experimental neurology 56: 933–944. [DOI] [PubMed] [Google Scholar]
- 6.Braak H, and Braak E. 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: 239–259. [DOI] [PubMed] [Google Scholar]
- 7.Boros BD, Greathouse KM, Gearing M, and Herskowitz JH. 2019. Dendritic spine remodeling accompanies Alzheimer’s disease pathology and genetic susceptibility in cognitively normal aging. Neurobiology of aging 73: 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boros BD, Greathouse KM, Gentry EG, Curtis KA, Birchall EL, Gearing M, and Herskowitz JH. 2017. Dendritic spines provide cognitive resilience against Alzheimer’s disease. Annals of neurology 82: 602–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayashi Y, and Majewska AK. 2005. Dendritic spine geometry: functional implication and regulation. Neuron 46: 529–532. [DOI] [PubMed] [Google Scholar]
- 10.Siskova Z, Justus D, Kaneko H, Friedrichs D, Henneberg N, Beutel T, Pitsch J, Schoch S, Becker A, von der Kammer H, and Remy S. 2014. Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer’s disease. Neuron 84: 1023–1033. [DOI] [PubMed] [Google Scholar]
- 11.Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, and Mucke L. 2007. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55: 697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fulop L, Penke B, Zilberter Y, Harkany T, Pitkanen A, and Tanila H. 2009. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci 29: 3453–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, Cornes SB, Henry ML, Nelson AB, Seeley WW, Geschwind MD, Gorno-Tempini ML, Shih T, Kirsch HE, Garcia PA, Miller BL, and Mucke L. 2013. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA neurology 70: 1158–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu GQ, Palop JJ, and Mucke L. 2012. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proceedings of the National Academy of Sciences of the United States of America 109: E2895–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, and Garaschuk O. 2008. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321: 1686–1689. [DOI] [PubMed] [Google Scholar]
- 16.Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, and Konnerth A. 2012. Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America 109: 8740–8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henderson BW, Gentry EG, Rush T, Troncoso JC, Thambisetty M, Montine TJ, and Herskowitz JH. 2016. Rho-associated protein kinase 1 (ROCK1) is increased in Alzheimer’s disease and ROCK1 depletion reduces amyloid-beta levels in brain. Journal of neurochemistry 138: 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heredia L, Helguera P, de Olmos S, Kedikian G, Sola Vigo F, LaFerla F, Staufenbiel M, de Olmos J, Busciglio J, Caceres A, and Lorenzo A. 2006. Phosphorylation of actin-depolymerizing factor/cofilin by LIM-kinase mediates amyloid beta-induced degeneration: a potential mechanism of neuronal dystrophy in Alzheimer’s disease. J Neurosci 26: 6533–6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petratos S, Li QX, George AJ, Hou X, Kerr ML, Unabia SE, Hatzinisiriou I, Maksel D, Aguilar MI, and Small DH. 2008. The beta-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain : a journal of neurology 131: 90–108. [DOI] [PubMed] [Google Scholar]
- 20.Leung T, Chen XQ, Manser E, and Lim L. 1996. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Molecular and cellular biology 16: 5313–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leung T, Manser E, Tan L, and Lim L. 1995. A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. The Journal of biological chemistry 270: 29051–29054. [DOI] [PubMed] [Google Scholar]
- 22.Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, Nakano T, Okawa K, Iwamatsu A, and Kaibuchi K. 1996. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. The EMBO journal 15: 2208–2216. [PMC free article] [PubMed] [Google Scholar]
- 23.Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, Watanabe N, Saito Y, Kakizuka A, Morii N, and Narumiya S. 1996. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. The EMBO journal 15: 1885–1893. [PMC free article] [PubMed] [Google Scholar]
- 24.Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, and Narumiya S. 1996. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett 392: 189–193. [DOI] [PubMed] [Google Scholar]
- 25.Swanger SA, Mattheyses AL, Gentry EG, and Herskowitz JH. 2015. ROCK1 and ROCK2 inhibition alters dendritic spine morphology in hippocampal neurons. Cell Logist 5: e1133266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herskowitz JH, Feng Y, Mattheyses AL, Hales CM, Higginbotham LA, Duong DM, Montine TJ, Troncoso JC, Thambisetty M, Seyfried NT, Levey AI, and Lah JJ. 2013. Pharmacologic inhibition of ROCK2 suppresses amyloid-beta production in an Alzheimer’s disease mouse model. J Neurosci 33: 19086–19098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rush T, Martinez-Hernandez J, Dollmeyer M, Frandemiche ML, Borel E, Boisseau S, Jacquier-Sarlin M, and Buisson A. 2018. Synaptotoxicity in Alzheimer’s Disease Involved a Dysregulation of Actin Cytoskeleton Dynamics through Cofilin 1 Phosphorylation. J Neurosci 38: 10349–10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sellers KJ, Elliott C, Jackson J, Ghosh A, Ribe E, Rojo AI, Jarosz-Griffiths HH, Watson IA, Xia W, Semenov M, Morin P, Hooper NM, Porter R, Preston J, Al-Shawi R, Baillie G, Lovestone S, Cuadrado A, Harte M, Simons P, Srivastava DP, and Killick R. 2018. Amyloid beta synaptotoxicity is Wnt-PCP dependent and blocked by fasudil. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 14: 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davies SP, Reddy H, Caivano M, and Cohen P. 2000. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351: 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bito H, Furuyashiki T, Ishihara H, Shibasaki Y, Ohashi K, Mizuno K, Maekawa M, Ishizaki T, and Narumiya S. 2000. A critical role for a Rho-associated kinase, p160ROCK, in determining axon outgrowth in mammalian CNS neurons. Neuron 26: 431–441. [DOI] [PubMed] [Google Scholar]
- 31.Woo S, and Gomez TM. 2006. Rac1 and RhoA promote neurite outgrowth through formation and stabilization of growth cone point contacts. J Neurosci 26: 1418–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan XB, Jin M, Xu X, Song YQ, Wu CP, Poo MM, and Duan S. 2003. Signalling and crosstalk of Rho GTPases in mediating axon guidance. Nat Cell Biol 5: 38–45. [DOI] [PubMed] [Google Scholar]
- 33.Zhang XF, Schaefer AW, Burnette DT, Schoonderwoert VT, and Forscher P. 2003. Rho-dependent contractile responses in the neuronal growth cone are independent of classical peripheral retrograde actin flow. Neuron 40: 931–944. [DOI] [PubMed] [Google Scholar]
- 34.Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, and Wedlich-Soldner R. 2008. Lifeact: a versatile marker to visualize F-actin. Nature methods 5: 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amano M, Chihara K, Kimura K, Fukata Y, Nakamura N, Matsuura Y, and Kaibuchi K. 1997. Formation of actin stress fibers and focal adhesions enhanced by Rho-kinase. Science 275: 1308–1311. [DOI] [PubMed] [Google Scholar]
- 36.Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, and Kaibuchi K. 1996. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). The Journal of biological chemistry 271: 20246–20249. [DOI] [PubMed] [Google Scholar]
- 37.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, and Kaibuchi K. 1996. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 273: 245–248. [DOI] [PubMed] [Google Scholar]
- 38.Sumi T, Matsumoto K, and Nakamura T. 2001. Specific activation of LIM kinase 2 via phosphorylation of threonine 505 by ROCK, a Rho-dependent protein kinase. The Journal of biological chemistry 276: 670–676. [DOI] [PubMed] [Google Scholar]
- 39.Kovacs M, Toth J, Hetenyi C, Malnasi-Csizmadia A, and Sellers JR. 2004. Mechanism of blebbistatin inhibition of myosin II. The Journal of biological chemistry 279: 35557–35563. [DOI] [PubMed] [Google Scholar]
- 40.Yin Y, Zheng K, Eid N, Howard S, Jeong JH, Yi F, Guo J, Park CM, Bibian M, Wu W, Hernandez P, Park H, Wu Y, Luo JL, LoGrasso PV, and Feng Y. 2015. Bis-aryl urea derivatives as potent and selective LIM kinase (Limk) inhibitors. Journal of medicinal chemistry 58: 1846–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, and Klein WL. 2007. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci 27: 796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, and Hyman BT. 2005. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci 25: 7278–7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pozueta J, Lefort R, Ribe EM, Troy CM, Arancio O, and Shelanski M. 2013. Caspase-2 is required for dendritic spine and behavioural alterations in J20 APP transgenic mice. Nature communications 4: 1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, and Caroni P. 1998. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393: 805–809. [DOI] [PubMed] [Google Scholar]
- 45.Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, and Mizuno K. 1998. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393: 809–812. [DOI] [PubMed] [Google Scholar]
- 46.Harris KM, Jensen FE, and Tsao B. 1992. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci 12: 2685–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hering H, and Sheng M. 2001. Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci 2: 880–888. [DOI] [PubMed] [Google Scholar]
- 48.Vossel KA, Ranasinghe KG, Beagle AJ, Mizuiri D, Honma SM, Dowling AF, Darwish SM, Van Berlo V, Barnes DE, Mantle M, Karydas AM, Coppola G, Roberson ED, Miller BL, Garcia PA, Kirsch HE, Mucke L, and Nagarajan SS. 2016. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Annals of neurology 80: 858–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnston D, Magee JC, Colbert CM, and Cristie BR. 1996. Active properties of neuronal dendrites. Annual review of neuroscience 19: 165–186. [DOI] [PubMed] [Google Scholar]
- 50.Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, and Strittmatter SM. 2009. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457: 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim HJ, Choi HS, Park JH, Kim MJ, Lee HG, Petersen RB, Kim YS, Park JB, and Choi EK. 2017. Regulation of RhoA activity by the cellular prion protein. Cell death & disease 8: e2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feng Y, LoGrasso PV, Defert O, and Li R. 2015. Rho Kinase (ROCK) Inhibitors and Their Therapeutic Potential. Journal of medicinal chemistry. [DOI] [PubMed] [Google Scholar]
- 53.Gentry EG, Henderson BW, Arrant AE, Gearing M, Feng Y, Riddle NC, and Herskowitz JH. 2016. Rho Kinase Inhibition as a Therapeutic for Progressive Supranuclear Palsy and Corticobasal Degeneration. J Neurosci 36: 1316–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gunther R, Balck A, Koch JC, Nientiedt T, Sereda M, Bahr M, Lingor P, and Tonges L. 2017. Rho Kinase Inhibition with Fasudil in the SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis-Symptomatic Treatment Potential after Disease Onset. Front Pharmacol 8: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koch JC, Tonges L, Barski E, Michel U, Bahr M, and Lingor P. 2014. ROCK2 is a major regulator of axonal degeneration, neuronal death and axonal regeneration in the CNS. Cell death & disease 5: e1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tatenhorst L, Eckermann K, Dambeck V, Fonseca-Ornelas L, Walle H, Lopes da Fonseca T, Koch JC, Becker S, Tonges L, Bahr M, Outeiro TF, Zweckstetter M, and Lingor P. 2016. Fasudil attenuates aggregation of alpha-synuclein in models of Parkinson’s disease. Acta neuropathologica communications 4: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Julian L, and Olson MF. 2014. Rho-associated coiled-coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases 5: e29846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feng Y, and LoGrasso PV. 2014. Rho kinase inhibitors: a patent review (2012 - 2013). Expert Opin Ther Pat 24: 295–307. [DOI] [PubMed] [Google Scholar]
- 59.Foletta VC, Moussi N, Sarmiere PD, Bamburg JR, and Bernard O. 2004. LIM kinase 1, a key regulator of actin dynamics, is widely expressed in embryonic and adult tissues. Experimental cell research 294: 392–405. [DOI] [PubMed] [Google Scholar]
- 60.Prunier C, Prudent R, Kapur R, Sadoul K, and Lafanechere L. 2017. LIM kinases: cofilin and beyond. Oncotarget 8: 41749–41763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manetti F 2012. LIM kinases are attractive targets with many macromolecular partners and only a few small molecule regulators. Medicinal research reviews 32: 968–998. [DOI] [PubMed] [Google Scholar]
- 62.Chang FL, and Greenough WT. 1984. Transient and enduring morphological correlates of synaptic activity and efficacy change in the rat hippocampal slice. Brain research 309: 35–46. [DOI] [PubMed] [Google Scholar]
- 63.Peters A, and Kaiserman-Abramof IR. 1970. The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. Am J Anat 127: 321–355. [DOI] [PubMed] [Google Scholar]
- 64.Tonnesen J, Katona G, Rozsa B, and Nagerl UV. 2014. Spine neck plasticity regulates compartmentalization of synapses. Nature neuroscience 17: 678–685. [DOI] [PubMed] [Google Scholar]
- 65.Lang C, Barco A, Zablow L, Kandel ER, Siegelbaum SA, and Zakharenko SS. 2004. Transient expansion of synaptically connected dendritic spines upon induction of hippocampal long-term potentiation. Proceedings of the National Academy of Sciences of the United States of America 101: 16665–16670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matsuzaki M, Honkura N, Ellis-Davies GC, and Kasai H. 2004. Structural basis of long-term potentiation in single dendritic spines. Nature 429: 761–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tanaka J, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GC, and Kasai H. 2008. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science 319: 1683–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Harvey CD, and Svoboda K. 2007. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature 450: 1195–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dickstein DL, Weaver CM, Luebke JI, and Hof PR. 2012. Dendritic spine changes associated with normal aging. Neuroscience 251: 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Herskowitz JH, Offe K, Deshpande A, Kahn RA, Levey AI, and Lah JJ. 2012. GGA1-mediated endocytic traffic of LR11/SorLA alters APP intracellular distribution and amyloid-beta production. Mol Biol Cell 23: 2645–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Riedl J, Flynn KC, Raducanu A, Gartner F, Beck G, Bosl M, Bradke F, Massberg S, Aszodi A, Sixt M, and Wedlich-Soldner R. 2010. Lifeact mice for studying F-actin dynamics. Nature methods 7: 168–169. [DOI] [PubMed] [Google Scholar]
- 72.Bzymek R, Horsthemke M, Isfort K, Mohr S, Tjaden K, Muller-Tidow C, Thomann M, Schwerdtle T, Bahler M, Schwab A, and Hanley PJ. 2016. Real-time two- and three-dimensional imaging of monocyte motility and navigation on planar surfaces and in collagen matrices: roles of Rho. Sci Rep 6: 25016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maitre JL, Turlier H, Illukkumbura R, Eismann B, Niwayama R, Nedelec F, and Hiiragi T. 2016. Asymmetric division of contractile domains couples cell positioning and fate specification. Nature 536: 344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Herskowitz JH, Seyfried NT, Gearing M, Kahn RA, Peng J, Levey AI, and Lah JJ. 2011. Rho kinase II phosphorylation of the lipoprotein receptor LR11/SORLA alters amyloid-beta production. The Journal of biological chemistry 286: 6117–6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dumitriu D, Rodriguez A, and Morrison JH. 2011. High-throughput, detailed, cell-specific neuroanatomy of dendritic spines using microinjection and confocal microscopy. Nat Protoc 6: 1391–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hallett PJ, Collins TL, Standaert DG, and Dunah AW. 2008. Biochemical fractionation of brain tissue for studies of receptor distribution and trafficking. Current protocols in neuroscience Chapter 1: Unit 1 16. [DOI] [PubMed] [Google Scholar]
- 77.Warmus BA, Sekar DR, McCutchen E, Schellenberg GD, Roberts RC, McMahon LL, and Roberson ED. 2014. Tau-mediated NMDA receptor impairment underlies dysfunction of a selectively vulnerable network in a mouse model of frontotemporal dementia. J Neurosci 34: 16482–16495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Greathouse KM, Boros BD, Deslauriers JF, Henderson BW, Curtis KA, Gentry EG, and Herskowitz JH. 2018. Distinct and complementary functions of rho kinase isoforms ROCK1 and ROCK2 in prefrontal cortex structural plasticity. Brain structure & function 223: 4227–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dickstein DL, Dickstein DR, Janssen WG, Hof PR, Glaser JR, Rodriguez A, O’Connor N, Angstman P, and Tappan SJ. 2016. Automatic Dendritic Spine Quantification from Confocal Data with Neurolucida 360. Current protocols in neuroscience 77: 1 27 21–21 27 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Expression of human ROCKs in hippocampal neurons.
Figure S2. Aβ42-induced spine loss is prevented by Fasudil.
Figure S3. Fasudil protects against Aβ-induced neuronal hyperexcitability.
Figure S4. SR7826 reduces the abundance of phosphorylated cofilin in the hippocampus.
Figure S5. SR7826 alters mean spine head diameter.
Figure S6. Aβ deposits are not substantially altered by SR7826.