

Abstract
Chikungunya virus (CHIKV), a positive-stranded alphavirus, causes epidemic febrile infections characterized by severe and prolonged arthralgia. In the present study, six CHIKV isolates (2006 RGCB03, RGCB05; 2007 RGCB80, RGCB120; 2008 RGCB355, RGCB356) from three consecutive Chikungunya outbreaks in Kerala, South India, were analyzed for genetic variations by sequencing the 11798 bp whole genome of the virus. A total of 37 novel mutations were identified and they were predominant in the 2007 and 2008 isolates among the six isolates studied. The previously identified E1 A226V critical mutation, which enhances mosquito adaptability, was present in the 2007 and 2008 samples. An important observation was the presence of two coding region substitutions, leading to nsP2 L539S and E2 K252Q change. These were identified in three isolates (2007 RGCB80 and RGCB120; 2008 RGCB355) by full-genome analysis, and also in 13 of the 31 additional samples (42%), obtained from various parts of the state, by sequencing the corresponding genomic regions. These mutations showed 100% co-occurrence in all these samples. In phylogenetic analysis, formation of a new genetic clade by these isolates within the East, Central and South African (ECSA) genotypes was observed. Homology modeling followed by mapping revealed that at least 20 of the identified mutations fall into functionally significant domains of the viral proteins and are predicted to affect protein structure. Eighteen of the identified mutations in structural proteins, including the E2 K252Q change, are predicted to disrupt T-cell epitope immunogenicity. Our study reveals that CHIK virus with novel genetic changes were present in the severe Chikungunya outbreaks in 2007 and 2008 in South India.
Electronic supplementary material
The online version of this article (doi:10.1007/s11262-009-0411-9) contains supplementary material, which is available to authorized users.
Keywords: Alphavirus, Positive-stranded RNA, Lineage, Virulence, Mutations, Epitope
Introduction
Increase in the virulence of emerging pathogens, such as Chikungunya virus (CHIKV), Dengue virus, and other arboviruses, is of grave concern [1–3]. Chikungunya (CHIK), a mosquito-transmitted zoonotic viral infection, is characterized by high fever, headache, myalgia, severe and prolonged arthralgia, and erythematous skin rashes. The main mosquito vectors are the members of Aedes family, Ae. aegypti and Ae. albopictus. Recent outbreaks of CHIK have exhibited unusual severity and suspected mortality among the affected people in several parts of the world [4–6].
CHIK was reported from India as early as in 1963 [7] and later it was thought to have disappeared from the sub-continent [8]. Among the three genotypes of Chikungunya virus [Asian; West African; and East, Central and South African (ECSA)], earlier outbreaks in India were caused by the Asian genotype. With the identification of the ECSA genotype of the virus from a field-caught mosquito in 2000 [9], the disease gained attention as a re-emerging infection. A number of CHIK outbreaks have been reported from several parts of the country during the period 2005–2008 [10, 11]. One of the worst affected states was Kerala in South India. As per the statistics available with the Department of Health and Family Welfare, Government of Kerala, the number of CHIKV suspected cases were 70,780 in 2006; 24,052 in 2007; and 24,667 in 2008. The disease in 2007 and 2008 was unusual with a rapid spread, lymphadenitis, hemorrhages, hepatic involvement, severe, prolonged arthralgia, and even suspected incidences of mortality [6, 12, 13]. Genetic changes in the virus were speculated as the plausible reason for this [12, 13].
CHIKV is a positive-stranded RNA virus belonging to the alphavirus of the Togaviridae family [1]. The 11.8 kb viral genome of the CHIKV codes for four non-structural proteins (nsP1, nsP2, nsP3, and nsP4), a core protein, and three envelope proteins (E1, E2, E3; E1 and E2 connected by a short 6K region). Previous studies show that in alphaviruses, the infectivity and virulence-associated gene mutations are distributed both in the structural and non-structural protein coding regions of the viral genome [14–16]. Several mutations in the structural and non-structural protein coding regions were detected in the CHIKV isolates from the Re’Union outbreak [4]. A point mutation in the E1 protein (A226V) has been proven to be the main reason for altered vector specificity and epidemic potential of the re-emerging CHIKV [17, 18]. This mutation has been documented in the recent viral isolates (2007) from the Indian subcontinent, including those from Kerala [6, 12].
Only few studies have looked into the variations in the genome regions other than the envelope protein coding region of the CHIKV isolates from India. Mutations in the nsP1 region (T128K and T376M) and capsid region (P23S) of 2006 isolates [11], and in the E1 region (V14A and A226V) and in the nsP1 region (M184T) of a 2007 isolate [6] have been previously reported. The present study was carried out to understand the gene mutations in the virus that caused an explosive febrile infection in three consecutive CHIK outbreaks in Kerala (2006–2008). It revealed novel mutations, phylogenetic evolution of a new viral genetic clade, and in silico predicted amino acid substitutions affecting several potential T-cell epitopes.
Materials and methods
Virus
CHIKV was isolated from serum samples of patients suffering from the classical symptoms such as fever, polyarthralgia, and skin rashes. Samples were collected during the July–December 2006, May–October, 2007, and May–September, 2008 outbreaks in Kerala. The presence of virus was detected by RT-PCR as previously reported [19]. The viral RNA was isolated using QiaAmp Viral RNA isolation kit (Qiagen, GmBH, Germany) and the reverse-transcription polymerase chain reaction (RT-PCR) was carried out using Genei One-step AMV RT-PCR kit (Bangalore Genei, Bangalore, India). Virus isolation was carried out using confluent monolayer culture of Vero cells. CHIKV in 200-μl patient serum was used for infection as per standard protocols [4, 6]. Infected cells were incubated till the cytopathic effects were visible.
Full-genome sequencing
Two isolates from the same locality for each of the 2006, 2007, and 2008 outbreaks were subjected to sequencing. The details of the samples, place, and date of collection are given in Fig. 1 and the Online Resource Table 1. Seven overlapping genomic regions were RT-PCR amplified from the isolated viral RNA. The details of the primer sequences and cloning strategy are given in Table 1 and Fig. 2. RT-PCR was carried out using Fidelitaq RT-PCR kit (USB, Cleveland, Ohio). The conditions were a reverse transcription step at 50°C for 45 min and 35 cycles of thermal cycling, which included denaturation at 95°C for 1 min, annealing at 55°C for 1 min, and an extension at 68°C for 4 min. The fragments were subsequently cloned into pGEM-T easy vector (Promega) and were sequenced using specific primers, using the Big-dye Terminator Cycle sequencing kit in an ABI 3730 Genetic Analyzer automated DNA sequencer (PE Applied Biosystems, Foster City, CA). The sequence contigs were assembled using CAP contig assembly program in Bio-Edit software [20].
Fig. 1.
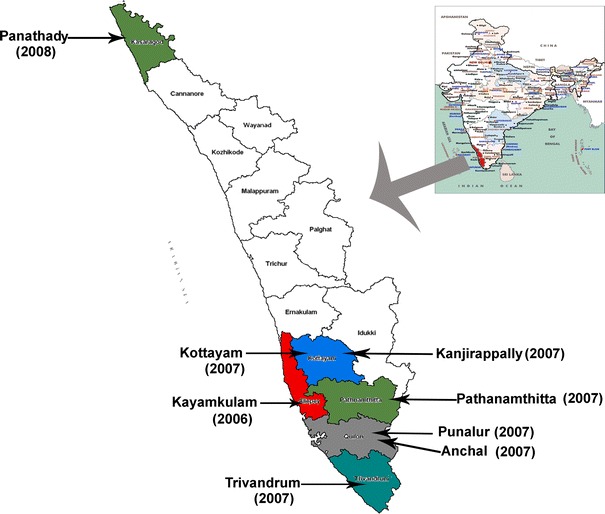
Locations in Kerala from where samples were collected for analysis in the study during CHIKV outbreaks in 2006, 2007, and 2008
Table 1.
Primers used for whole genome sequencing
| Name | Forward primer sequence (5′ → 3′) | Position (with respect to S27) | Name | Reverse primer sequence (5′ → 3′) in the complementary strand |
|---|---|---|---|---|
| chf1 | atggctgcgtgagacacac | 1–19 | chr1 | Nil |
| chf2 | cgtctatgctgtacacgca | 532–550 | chr2 | tgcgtgtacagcatagacg |
| chf3 | gacggattcctgatgtgc | 1004–1021 | chr3 | gcacatcaggaatccgtc |
| chf4 | gacagctttgtggtaccga | 1418–1436 | chr4 | tcggtaccacaaagctgtc |
| chf5 | cggtcgaagcgtacgacg | 1863–1880 | chr5 | cgtcgtacgcttcgaccg |
| chf6 | ctgcacgtacggttgactcgc | 2370–2390 | chr6 | gcgagtcaaccgtacgtgcag |
| chf7 | cgtgttaacgtgcttcagag | 2746–2765 | chr7 | ctctgaagcacgttaacacg |
| chf8 | cctatcctcgaaacagcg | 3134–3151 | chr8 | cgctgtttcgaggatagg |
| chf9 | gcctactaagagagtcac | 3619–3636 | chr9 | gtgactctcttagtaggc |
| chf10 | ggatgtgcaccgtcgtac | 4070–4087 | chr10 | gtacgacggtgcacatcc |
| chf11 | gacccagtcactgaacca | 4438–4455 | chr11 | tggttcagtgactgggtc |
| chf12 | gaaatgcccggtggatga | 4807–4823 | chr12 | tcatccaccgggcatttc |
| chf13 | ggccgtgtctgactgggt | 5236–5253 | chr13 | acccagtcagacacggcc |
| chf14 | gccggtgaacaccctggag | 5731–5749 | chr14 | ctccagggtgttcaccggc |
| chf15 | gagcgacattcaatccgtc | 6147–6165 | chr15 | gacggattgaatgtcgctc |
| chf16 | atacaggcggctgaaccct | 6599–6617 | chr16 | agggttcagccgcctgtat |
| chf17 | cgtctgacaaaatccgcgt | 7025–7043 | chr17 | acgcggattttgtcagacg |
| chf18 | ggtacgcactacagctacc | 7501–7519 | chr18 | ggtagctgtagtgcgtacc |
| chf19 | cgaagtcaagcacgaaggt | 7911–7929 | chr19 | accttcgtgcttgacttcg |
| chf20 | agagtggagtcttgccatc | 8343–8361 | chr20 | gatggcaagactccactct |
| chf21 | gggacacttcatcctggc | 8832–8849 | chr21 | gccaggatgaagtgtccc |
| chf22 | gtttccgctggcaaatgt | 9315–9332 | chr22 | acatttgccagcggaaac |
| chf23 | ccaggagctaccgtccctt | 9751–9769 | chr23 | aagggacggtagctcctgg |
| chf24 | acagctgtaaggtcttcaccgg | 10220–10241 | chr24 | ccggtgaagaccttacagctgt |
| chf25 | gtacggtacacgtgccatact | 10673–10693 | chr25 | agtatggcacgtgtaccgtac |
| chf26 | aggaccacatagtcaactacc | 11144–11164 | chr26 | ggtagttgactatgtggtcct |
| chf27 | gtcccctaagagacacattg | 11486–11505 | chr27 | caatgtgtctcttaggggac |
| Chf28 | Nil | 11780–11798 | Chr28 | tacgtccctgtgggttcggagaat |
All the primers are newly designed in the study. From each location, one forward and one reverse primer were designed based on the S-27 sequence
Primers used for generating the clones are highlighted in bold in the table
Fig. 2.
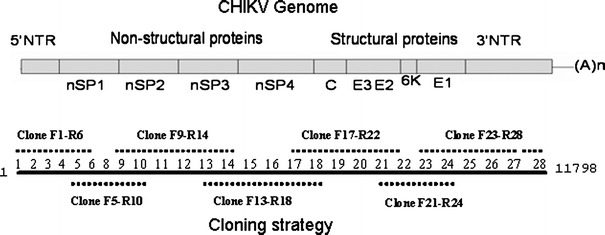
Cloning strategy adopted for full-genome sequencing
RT-PCR amplification and sequencing of partial nsP2, E1, and E2 sequences
Partial nsP2, E1, and E2 sequences were amplified from 31 samples (Online Resource Table 1) by RT-PCR. The primers used for amplification were ch8F and ch9R for nsP2 region; ch24F and ch25R for the E1 region; and ch21F and ch22R for the E2 region (Table 1). The RT-PCR was done as described above. The amplified products were purified using GFX Gel band purification system (GE Healthcare) and were subjected to automated DNA sequencing.
Phylogenetic analysis
CHIKV full-length coding sequences (11237 bp) were aligned with corresponding sequences obtained from NCBI GenBank using Clustal W program of MEGA3.1 [21] software, with Kimura-2 distance correction. The phylogeny was reconstructed by Neighbor-Joining method with 10000 bootstrap replications.
Mapping of mutations
Homology models for the CHIKV proteins were made from structural templates that were identified by BLAST search against the PDB chain database. Details of the selected templates, along with the regions showing reliable alignments, are given in Table 2. To avoid problems with automated alignments, the target sequences were manually threaded into the template structures using Deepview (Swiss-pdb Viewer) [22]. The resulting project files were sent to the SWISSMODEL automated homology model-building server for model calculation. Stereochemical quality checks were performed on the returned models using VADAR server [23]. A few problematic side chain conformations were identified and rectified. The resulting structures were energy minimized using DeepView iteratively using 200 cycles of steepest descent. Structural representations of the models were generated with PyMOL (http://www.pymol.org/) and the mutations identified in the study were mapped.
Table 2.
CHIKV proteins and their regions used in molecular modeling
| Sl. no. | CHIKV protein/domains modeled (target) | Template | PDB id | % identity with template | Region of target modeled (amino acids) |
|---|---|---|---|---|---|
| 1 | Helicase (nsP2) | Hydrolase ( Deinococcus radiodurans recd2) | 3E1s chain A | 26.3 | 1–102 |
| 2 | Peptidase C9 (nsP2) | Helicase nsp2. Chain: a. Venezuelan equine encephalitis virus | 2hwk chain A | 44.4 | 41–202 |
| 3 | Macrodomain (nsP3) | Non-structural protein 3. Chain: a Human coronavirus 229e | 3ewr chain A | 23 | 6–139 |
| 4 | S3Peptidase (Capsid) | Semliki forest virus capsid protein | 1Vcp chain C | 93.2 | 8–156 |
| 5 | E1 (Structural protein) | Glycoprotein e1 from Semliki forest virus. | 1rer chain A | 63.5 | 67–456 |
T-cell epitope prediction
T-cell epitopes of the CHIKV structural proteins were predicted using the EpiJen online server [24]. The entire structural polyprotein sequence of CHIKV was used as input to EpiJen and the server was run in the default mode. The appropriate proteasomal and Tap cut-off values were selected while getting epitope predictions for the 18 different HLA alleles that are currently being offered at EpiJen.
Results
RT-PCR and virus isolation
In RT-PCR of the clinical samples, 9 out of 11 (81.18%) samples from 2006, 33 out of 36 (91.6%) samples from 2007, and 11 out of 19 (57.8%) samples from 2008 were positive for the presence of viral RNA. During virus isolation in Vero cells from RT-PCR-positive samples, cytopathic effect (CPE) development was observed in 48–96 h. The CPE was characterized by the roundening of cells, increased granularity and vacuolation, followed by cell death and disruption of the monolayer by detachment of the dead cells. RNA isolation and RT-PCR amplification from the culture supernatant further confirmed the presence of the virus.
Full-genome sequencing of 2006, 2007, and 2008 isolates
Single passage virus and high-fidelity PCR enzymes were used to minimize experimentally induced genetic alterations in whole genome analysis. Six isolates—RGCB03, RGCB05, RGCB80, RGCB120, RGCB355, and RGCB356—were subjected to full genome sequencing. The year of collection of these isolates, GenBank accession numbers of the sequences and the number of nucleotide changes observed with respect to the reference sequence of S-27 African strain (GenBank accession No. AF369024) are given in Table 3. The full-length assembled CHIKV genome consisted of 11,798 base pairs and corresponds to the positions 1–11,798 of the reference sequence. The Kerala isolates had an average whole genome nucleotide identity of 96.95% with the S-27 sequence, 98.07% with the 2000 Indian (Yawat) isolate, and 99.68-99.8% with the 2006 Indian and Re’Union isolates. The sequences had 99.7% identity with the 2007 ITA07-RA strain, which was isolated in Italy. The comparative analysis of the predicted amino acid substitutions observed in the six Kerala isolates with that of other Indian isolates, Re’Union and Italian ECSA isolates are given in Table 4.
Table 3.
Nucleotide changes in RGCB isolates with respect to S-27 reference strain
| Sl. no. | Isolate | Year of collection | GenBank accession no. | Number of nucleotide changes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Coding region (synonymous/non-synonymous) | 5′NTR | 3′NTR | ||||||||||||
| Non-structural protein | Structural protein | |||||||||||||
| nsP1 | nsP2 | nsP3 | nsP4 | C | E3 | E2 | 6K | E1 | ||||||
| 1 | RGCB03/KL06 | 2006 | GQ428210 | 31/9 | 51/5 | 47/12 | 44/7 | 16/3 | 3/1 | 29/15 | 2/2 | 28/6 | 1 | 16 |
| 2 | RGCB05/KL06 | 2006 | GQ428211 | 32/9 | 50/5 | 48/12 | 42/6 | 15/4 | 3/1 | 31/14 | 2/2 | 28/4 | 1 | 17 |
| 3 | RGCB80/KL07 | 2007 | GQ428212 | 33/10 | 55/9 | 47/12 | 42/7 | 16/3 | 3/1 | 31/15 | 2/2 | 29/5 | 1 | 19 |
| 4 | RGCB120/KL07 | 2007 | GQ428213 | 33/9 | 49/8 | 48/11 | 44/6 | 16/3 | 3/1 | 32/17 | 2/3 | 29/5 | 1 | 19 |
| 5 | RGCB355/KL08 | 2008 | GQ428214 | 33/9 | 51/7 | 50/13 | 45/9 | 16/3 | 3/1 | 31/16 | 2/2 | 29/6 | 1 | 19 |
| 6 | RGCB356/KL08 | 2008 | GQ428215 | 35/10 | 52/6 | 47/12 | 42/6 | 16/2 | 3/2 | 29/17 | 2/2 | 28/6 | 1 | 17 |
Table 4.
Comparison of amino acid substitutions identified in Kerala isolates with that of S-27 and other closely related ECSA genotypes of CHIKV
| Protein | Position | Isolates | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Identified protein domains in the corresponding region | Polypeptide | Protein | S-27 | IND-00-MH-4 (Yawat) | LR2006-OPY1 | DRDE-06 | IND-06-RJ1 | IND-06-MH2 | IND-06-AP3 | IND-06-KA15 | IND-06-TN1 | DRDE-07 | RGCB03-06 | RGCB 05-06 | RGCB 80-07 | RGCB 120-07 | RGCB 355-08 | RGCB 356-08 | ITA07-RA1 | |
| nsP1 | Viral methyl transferase | 105 | 105 | G | . | . | . | . | . | . | . | . | . | . | . | . | . | . | R | . |
| 128 | 128 | T | . | . | K | K | K | K | K | K | K | K | K | K | K | K | K | K | ||
| 258 | 258 | W | . | . | . | . | . | . | . | . | . | . | . | . | . | . | R | . | ||
| 314 | 314 | M | . | . | L | . | . | . | . | . | L | . | . | . | . | . | . | . | ||
| 376 | 376 | T | . | . | M | M | M | M | M | M | M | M | M | M | M | M | M | M | ||
| 488 | 488 | Q | . | R | R | R | R | R | R | R | R | R | R | R | R | R | R | R | ||
| 536 | 536 | D | . | . | . | . | . | . | . | . | . | . | . | G | . | . | . | . | ||
| nsP2 | 583 | 48 | V | . | . | . | . | . | . | . | . | . | . | . | A | . | . | . | . | |
| 589 | 54 | S | . | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | ||
| Viral helicase | 716 | 181 | V | . | . | . | . | . | . | . | . | . | . | . | A | . | . | . | . | |
| 772 | 237 | L | . | . | . | . | . | . | . | . | . | . | . | . | R | . | . | . | ||
| 773 | 238 | L | . | . | . | . | . | . | . | . | . | . | . | . | P | . | . | . | ||
| 864 | 329 | K | . | . | . | . | . | . | . | . | . | . | . | E | . | . | . | . | ||
| 946 | 411 | N | . | . | . | . | . | . | . | . | . | . | . | . | . | . | D | . | ||
| Peptidase C9 | 1074 | 539 | L | . | . | . | . | . | . | . | . | . | . | . | S | S | S | . | . | |
| 1243 | 708 | L | . | . | . | . | . | . | . | . | . | . | . | . | . | P | . | . | ||
| 1328 | 793 | A | . | V | V | V | V | V | V | V | V | V | V | V | V | V | V | V | ||
| nsP3 | Macrodomain | 1359 | 26 | P | . | . | . | . | . | . | . | . | . | . | S | . | . | . | . | . |
| 1427 | 94 | K | . | . | . | . | . | . | . | . | . | . | . | Q | . | . | . | . | ||
| 1534 | 201 | L | . | . | . | . | . | . | . | . | . | . | . | . | . | R | . | . | ||
| 1583 | 250 | D | . | . | . | . | . | . | . | . | . | E | . | . | . | . | . | . | ||
| 1709 | 376 | I | T | T | T | T | T | T | T | T | T | T | T | T | T | T | T | . | ||
| 1773 | 440 | E | . | . | . | . | . | . | . | . | . | . | . | . | . | G | . | . | ||
| 1794 | 461 | L | . | P | P | P | P | P | P | P | P | P | P | P | P | P | P | P | ||
| 1804 | 471 | P | . | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | ||
| nsP4 | 1938 | 75 | T | . | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | |
| 1979 | 116 | S | . | . | . | . | . | . | . | . | . | . | . | . | . | P | . | . | ||
| RNA dependent RNA polymerase (RdRP) | 2115 | 252 | N | . | . | . | . | . | . | . | . | . | . | . | . | . | D | . | . | |
| 2117 | 254 | T | . | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| 2152 | 289 | V | . | . | . | . | . | . | . | . | . | . | . | A | . | . | . | . | ||
| 2342 | 479 | M | . | . | . | . | . | . | . | . | . | . | . | . | . | V | . | . | ||
| 2352 | 489 | M | . | . | . | . | . | . | . | . | K | . | . | . | . | . | . | |||
| Capsid | 23 | 23 | P | . | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | |
| 27 | 27 | V | . | . | I | I | I | . | I | I | . | I | I | I | I | I | . | I | ||
| 28 | 28 | I | . | . | . | . | . | . | . | . | . | . | . | . | . | . | T | . | ||
| S3 Peptidase | 199 | 199 | G | . | . | . | . | . | . | . | . | . | . | D | . | . | . | . | . | |
| E3 | 305 | 44 | R | . | . | . | . | . | . | . | . | . | . | . | . | . | . | G | . | |
| E2 | 397 | 72 | N | . | . | . | . | . | . | . | . | . | . | . | . | . | S | . | . | |
| 554 | 229 | V | . | . | . | . | . | . | . | . | . | . | . | . | . | . | I | . | ||
| 577 | 252 | K | . | . | . | . | . | . | . | . | . | . | . | Q | Q | Q | . | . | ||
| 641 | 316 | E | . | . | . | . | . | . | . | . | . | K | . | . | . | . | . | . | ||
| 675 | 350 | G | . | . | . | . | . | . | . | . | . | . | . | . | . | . | S | . | ||
| 686 | 361 | E | . | . | . | . | . | . | . | . | . | . | . | . | G | . | . | . | ||
| 700 | 375 | S | . | T | T | T | T | T | T | T | T | T | T | T | T | T | T | T | ||
| 711 | 386 | V | . | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| 741 | 416 | C | . | . | . | . | . | . | . | . | . | . | . | . | G | . | . | . | ||
| 6K | 756 | 8 | V | . | I | I | I | I | I | I | I | I | I | I | I | I | I | I | I | |
| 808 | 60 | S | . | . | . | . | . | . | . | . | . | . | . | . | G | . | . | . | ||
| E1 | 813 | 04 | V | . | . | . | . | . | . | . | . | A | . | . | . | . | . | A | . | |
| 926 | 127 | E | . | . | . | . | . | . | . | . | . | G | . | . | . | . | . | . | ||
| 983 | 184 | D | . | . | . | . | . | . | . | . | . | . | . | . | G | . | . | . | ||
| 994 | 195 | Y | . | . | . | . | . | . | . | . | . | H | . | . | . | . | . | . | ||
| 996 | 197 | M | . | . | . | . | . | . | . | . | . | . | V | . | . | . | . | . | ||
| 1035 | 226 | A | . | V | . | . | . | . | . | . | V | . | . | V | V | V | V | V | ||
| 1057 | 248 | G | . | . | . | . | . | . | . | . | . | . | . | . | . | E | . | . | ||
| 1093 | 284 | D | . | E | E | E | E | E | E | E | E | E | E | E | E | E | E | E | ||
| 1113 | 304 | P | . | . | . | . | . | . | . | . | . | . | . | . | . | . | L | . | ||
| 1205 | 396 | T | . | . | . | . | . | . | . | . | . | A | . | . | . | . | . | . | ||
| 1220 | 411 | K | . | . | . | . | . | . | . | . | . | . | . | . | . | R | . | . | ||
| 1242 | 433 | C | . | . | . | . | . | . | . | . | . | . | . | R | . | . | . | . | ||
Identical amino acids with S-27 strain are indicated by “.”
Isolates of the present study are indicated in italics. Unique mutations are identified in bold
Sequence analysis of the non-structural protein coding region
Several nucleotide changes were found in the non-structural protein coding region in comparison with the S-27 prototype strain. Some of these nucleotide substitutions were non-synonymous and resulted in amino acid change (Tables 3 and 4). Most of these changes were also found in other CHIKV isolates from 2006 and 2007 outbreaks in India and other parts of the world. However, some nucleotide changes were unique to the Kerala isolates studied (Online Resource Table 2). Six nucleotide changes (T1381G; T3297C; T3397C; C5014T; A6076G; C7450T) were common to the 2007 isolates (RGCB80, RGCB120) and to one of the 2008 isolates (RGCB355).
Sequence analysis of structural protein coding region
Structural protein coding region also showed several nucleotide changes (Table 3). As in the non-structural protein coding region, some of the changes were present only in RGCB isolates (Online Resource Table 2). Among this, five unique nucleotide changes (C7983T; A9295C; G9654A; G9681A; A10356G) were uniformly found in RGCB80, RGCB120, and RGCB355. The A9295C change caused a K252Q mutation in the E2 coding region of the three isolates. The nucleotide substitution C10670T causing the A226 V mutation in the E1 protein was present only in 2007 and 2008 RGCB isolates (viz. RGCB80, RGCB120, RGCB355, and RGCB356). Predicted amino acid changes observed in the structural protein region are shown in Table 4.
5′ and 3′ non-translated regions (NTRs)
5′NTR region in the Kerala isolates did not reveal any nucleotide change compared to the S-27 strain, except for G68T substitution, which was observed also in all the recent Indian and Re’ Union viral isolates. There were no deletions or insertions. However, the 3′NTR showed nucleotide substitutions (Table 3; Online Resource Table 2). The internal polyadenylation stretch at nucleotides 11445–11458, observed within the 3′NTR in the S-27 strain, was absent in the recent isolates of CHIKV ECSA strains [6]. This stretch of 14 adenine nucleotides was absent in the six isolates sequenced in this study also. Most of the nucleotide changes observed in the 3′NTR were common to the RGCB isolates and recent CHIKV isolates from India and other parts of the world. The C11614T substitution was observed only in three RGCB isolates (RGCB80, RGCB120, RGCB355), ITA07-RA1 and in IND-06-RJ1 strains. Another common nucleotide variation observed in Indian isolates (DRDE06, ITA-07-RA, IND-06-RJ1, IND-06-KA15, IND-06-TN1) and in RGCB isolates (RGCB05, RGCB80, RGCB120, and RGCB355) was the A11655G substitution. 3′NTR did not reveal any deletions or insertions.
Phylogenetic analysis based on full-length coding region
Phylogenetic analysis using 11237 bp full-length coding region nucleotide sequences of the six RGCB isolates, eight other Indian isolates available in GenBank, one isolate from Italy, one from Re’Union Island, and three from Mauritius indicated clear clustering of the 2006, 2007, and 2008 isolates (Fig. 3). The isolates Yawat-2000 and S-27 African prototype formed separate branches. Within the recent ECSA genotypes, the three Kerala isolates (RGCB80, RGCB120, and RGCB355) and the ITA-07-RA1 Italian isolate showed very close clustering. The three RGCB isolates formed a separate clade, with 100% boot-strap support. It was found that the presence of five unique nucleotide variations in the genome of these RGCB isolates (T3297C, C5014T, A6076G, G9654A, and A10356G) contributed to the formation of this clade.
Fig. 3.

Phylogenetic analysis based on full-length coding region nucleotide sequences (11237 bp) of selected ECSA strains with Kerala strains (filled triangle). GenBank Accession numbers and strain name are given. GenBank accession nos. of RGCB isolates are given in Table 3. Boot-strap values (>50%) are indicated at the nodes. Scale bar represent the number of substitutions per site
Sequence analysis of partial nsP2, E1, and E2 coding region
In 31 additional samples collected from different parts of the state from 2007 and 2008 outbreaks, nucleotide changes causing the nsP2 L539S and E2 K252Q were present in 13 samples (42%); and the nucleotide change causing the E1 A226V mutation was present in 28 of the samples (Online Resource, Table 1). Interestingly, the L539S change in the non-structural protein coding region and K252Q change in the structural protein coding region showed 100% co-occurrence. Since we resorted to direct sequencing of the PCR product in all these cases, the possibility of these viral strains being quasispecies circulating in the patients had to be excluded. In careful analysis of the sequencing chromatogram, all samples showed only one clear peak, which corresponded to the changed nucleotide. This confirmed the presence of a single species of virus in such samples.
Molecular modeling
Homology-based models of five CHIKV proteins are given in Fig. 4 with the mutations that have been mapped onto the three dimensional structure. Only five of the CHIKV proteins had reliable templates for modeling (Table 2). The models represent energy minimized structures and were not subjected to dynamic perturbation. Target-template identities ranged from 23% for the CHIKV macrodomain to about 93% for the peptidase S3. As shown, many of the mutations were mapped to areas with major secondary structures, and could affect the local protein structure.
Fig. 4.
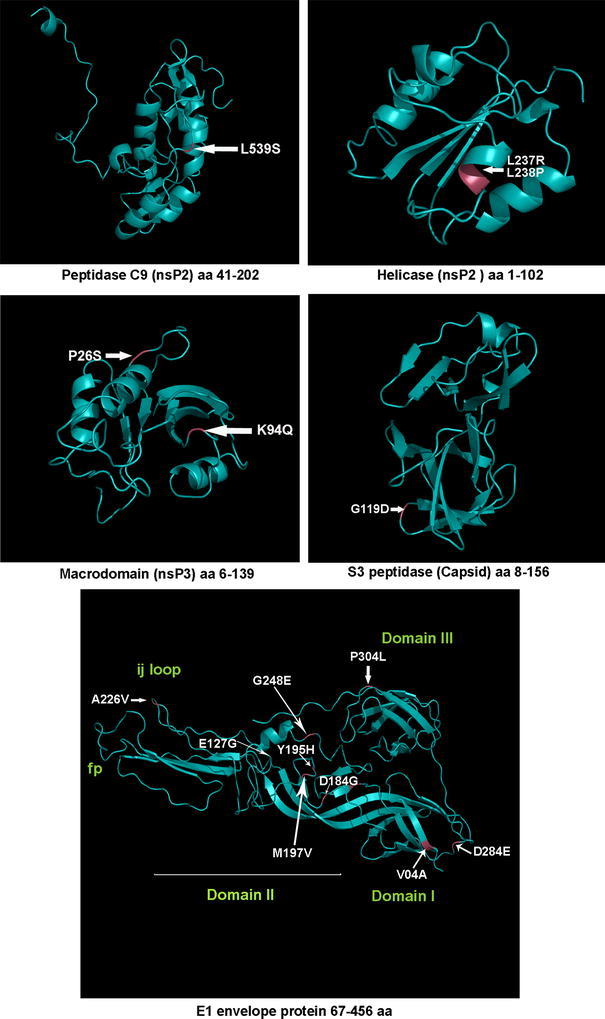
Mapping the positions of selected major mutations identified in CHIKV Kerala isolates on to the structural regions of the corresponding protein. The sequence of the reference strain S-27 was modeled using suitable templates as described in “Materials and Methods”
T-cell epitope prediction
Results from the T-cell epitope prediction server, EpiJen are shown in Table 5 and the actual peptide-epitope sequences are shown in Online Resource Tables 3 and 4. While ranking the epitopes for mutation mapping, epitopes with calculated IC50 values above 100 were not included since such peptides are unlikely to yield significant immunogenicity. It is interesting to note that majority of the identified mutations fall within predicted T-cell epitopes, and are therefore predicted to influence immunogenicity. The unique mutation K577Q in the E2 structural protein was linked to epitopes presented by HLA-A and HLA-B alleles. The E1 A226V mutation, implicated in the increased virulence of Re’Union isolates, also could be mapped to epitopes presented by a number of HLA alleles.
Table 5.
Association of unique structural protein mutations identified in RGCB isolates with potential T-cell epitopes
| Sl. no. | Viral protein | Position in the polypeptide | Mutation | Associated HLA allelesa | Isolates that show the presence of the mutation | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| RGCB03/2006 | RGCB05/2006 | RGCB80/2007 | RGCB120/2007 | RGCB355/2008 | RGCB356/2008 | |||||
| 1 | Capsid | 28 | I → T | HLA-B*27, HLA-B*51, HLA-B*53 | – | – | – | – | – | Y |
| 2 | 199 | G → D | HLA-A*0301, HLA-A*1101, HLA-A*24, HLA-A*6801, HLA-B*07, HLA-B*27 | – | Y | – | – | – | – | |
| 3 | E3 | 305 | R → G | HLA-A*0101, HLA-A*0201, HLA-A*0202, HLA-A*0203, HLA-B*27 | – | – | – | – | – | Y |
| 4 | E2 | 554 | V → I | HLA-A*0101, HLA-A*0206, HLA-A*0301, HLA-A*1101, HLA-A*6802 | – | – | – | – | – | Y |
| 5 | 577 | K → Q | HLA-A*0301, HLA-A*1101, HLA-B*40, HLA-B*44 | – | – | Y | Y | Y | – | |
| 6 | 641 | E → K | HLA-A*0201, HLA-A*0202, HLA-A*6802, HLA-B*40, HLA-B*44 | Y | – | – | – | – | – | |
| 7 | 675 | G → S | HLA-B*40, HLA-B*44, HLA-B*51 | – | – | – | – | – | Y | |
| 8 | 686 | E → G | HLA-A*0101, HLA-A*0201, HLA-A*0203, HLA-A*0301, HLA-A*24, HLA-B*40, HLA-B*44 | – | – | – | Y | – | – | |
| 9 | 741 | C → G | HLA-A*0201, HLA-A*0202, HLA-A*0203, HLA-A*0206, HLA-A*0301, HLA-A*24, HLA-A*3101, HLA-A*6802 | – | – | – | Y | – | – | |
| 10 | 6K | 808 | S → G | HLA-A*0101, HLA-A*0201, HLA- A*0202, HLA-A*0206, HLA-B*51 | – | – | – | Y | – | – |
| 11 | E1 | 813 | V → A | HLA-A*0201, HLA-A*0202, HLA-A*0206, HLA-A*24, HLA-A*6802, HLA-B*51 | – | – | – | – | – | Y |
| 12 | 926 | E → G | HLA-A*0301, HLA-A*1101, HLA-A*6801, HLA-B*40, HLA-B*44 | Y | – | – | – | – | – | |
| 13 | 983 | D → G | HLA-A*0202, HLA-A*0203, HLA-A*0206, HLA-A*0301, HLA-A*1101, HLA-A*24, HLA-A*6801, HLA-A*6802, HLA-B*51 | – | – | – | Y | – | – | |
| 14 | 994 | Y → H | HLA-A*0101, HLA-A*24, HLA-B*07 | Y | – | – | – | – | – | |
| 15 | 1035 | A → V | HLA-A*0101, HLA-A*0201, HLA-A*0202, HLA-A*0203, HLA-A*0206, HLA-A*0301, HLA-B*07, HLA-B*27, HLA-B*51 | – | – | Y | Y | Y | Y | |
| 16 | 1057 | G → E | HLA-A*0201, HLA-A*0202, HLA-A*0203, HLA-A*24 | – | – | – | – | Y | – | |
| 17 | 1205 | T → A | HLA-A*0201, HLA-A*0203, HLA-A*0206, HLA-A*24, HLA-B*51 | Y | – | – | – | – | – | |
| 18 | 1220 | K → R | HLA-A*0202, HLA-A*0206, HLA-A*0301, HLA-A*1101, HLA-A*6802, HLA-B*51 | – | – | – | – | Y | – | |
aPredicted epitopes are given in Supplementary Tables 2 and 3. Y Indicates presence of mutation
Discussion
Kerala was one of the South Indian states that witnessed wide-spread outbreak of CHIKV in the years 2006, 2007, and 2008. These outbreaks differed in the geographical region involved (coastal region in 2006 versus hilly areas with rubber plantations and abundance of Ae. albopictus mosquitoes in 2007 and 2008) and in disease severity (the 2007 and 2008 outbreaks more severe than the 2006 outbreak) [6, 12, 13]. In the outbreaks in 2007 and 2008, one of the unique complications observed was joint swelling (61.3% cases) along with painful arthralgia, which persisted for long periods [13, 25]. Another major difference in the clinical symptoms compared to the outbreaks in other parts of the world was the increased presence of skin rashes/itching. In one of the studies, these symptoms were observed in 80.8% patients in the Kerala outbreak in 2007–2008 [13]. Similar symptoms were seen only in 19.3% of the patients in the Re’Union outbreak [26]. Skin manifestations were predominant in pediatric patients also. 75% of the CHIKV affected infants exhibited peripheral cyanosis and dermatological manifestations like generalized erythema, maculopapular rash, vesiculobullous lesions or skin peeling, as characteristic symptoms [27]. A rapid spread of the disease was observed in 2007 and 2008, than in 2006, which has been linked to the outbreak of the disease in areas with abundance of Ae. albopictus. This vector permits rapid replication of the E1 A226V mutant virus [17, 18], that was circulating in Kerala during this period [12]. Though mortalities were observed, they were not conclusively attributed to CHIKV infection [25].
The increased severity of recent CHIK infections in other parts of the world has been linked to the genetic alterations in the virus [4, 17, 18]. In order to identify possible correlation of the clinical severity of the disease in Kerala with CHIKV genomic changes, we analyzed full-genome sequences of the viral isolates obtained from these outbreaks. Our study exposed greater genetic heterogeneity in the CHIKV circulating in Kerala, than that was observed in earlier studies. Both coding region changes and changes in the non-coding region, especially 3′NTR were observed in the study. Except for the 4 mutations—E1 (V04A) [6]; E2 (K252Q), and nsP2 (V48A, L539S) [28]—all the 18 mutations in the structural protein coding region and 19 mutations in the non-structural protein coding region identified in this study are new. Mutations were present throughout the genome. However, they were predominant in nsP2, E1, and E2 coding regions, which play major role in alphavirus infection and replication. Interestingly, they were more in the 2007 and 2008 isolates (Table 4). Along with the A226V mutation in the E1 protein, K252Q mutation in the E2 protein and L539S in the nsP2 protein were common features of most of the 2007–2008 Kerala isolates (Table 4). These changes and possibly other mutations in the viral proteins of the Kerala isolates might have contributed to the pathogenesis of CHIKV in these outbreaks.
The non-structural protein nsP2 plays a critical role in the cytopathogenicity of alphaviruses [16, 29, 30]. The Viral Helicase and Peptidase C9 cysteine-protease domains of this protein had predicted amino acid changes in the RGCB CHIKV isolates (Table 4). In protein model, the two observed mutations in the viral helicase domain, L237R and L238P in RGCB120 were mapped to an α-helix forming region (Fig. 4). These are non-conservative changes that could alter the conformation of this region. Mutation that was observed in the cysteine-protease domain was the L539S in the RGCB80, RGCB120, and RGCB355 (Fig. 4), which fell closer to the H548. The H548 residue forms the catalytic diad along with C478 in other alphaviruses [31]. This change from a hydrophobic amino acid to polar amino acid also has the potential to affect the secondary structure of this region.
Among the structural proteins, E1 and E2 proteins play major role in the cell entry of alphavirus. Domains I and II of the E1 protein are involved in E1 trimerization during the viral fusion process at the time of infection. Domain II mediates the E1–E2 interaction during the virus maturation and budding from infected cells [32–34]. In RGCB CHIKV isolates, the observed mutations in the E1 protein coding region were spread across all these functional regions (Fig. 4). Most of the mutations were in the loops of the domain II, indicating that this region plays major role in the biology of CHIKV E1 protein. Some of the changes were non-conservative (E127G, D184G, G248E), which might affect the mobility and strength of E1–E1 and E1–E2 interactions. The folding back of domain III and its interactions with the E1 trimer during fusion are critical events in alphavirus entry [35]. Domain III in one of the RGCB CHIKV isolates (RGCB356) had a major mutation (P304L) in one of the loops that could destabilize the local structure.
In the E2 protein, most of the mutations fell in the region between amino acids (aa) 229 and 361. This stretch of the protein (aa 1–364) in alphaviruses forms a highly hydrophilic domain. In Hopps and Woods [36] hydrophilicity prediction, the CHIKV E2 peptide 249GDRKGKIH256 within this region was the most hydrophilic. Hence it could be a potential antigenic epitope. The K252Q basic to neutral amino acid change, observed consistently in many Kerala isolates, involved this epitope and was found to reduce the predicted hydrophilicity. This, in turn, indicates altered immunogenicity of the E2 protein of the isolates. The residues corresponding to the receptor binding region of E2 in alphaviruses (aa 170–220) [37] did not show mutation in the six isolates studied. The 391–423 aa stretch is identified as cytoplasmic domain of E2 protein in Sindbis virus [38], and the C416G mutation that was observed in RGCB120 falls within this region. This mutation changes one of the two conserved cysteine residues. Palmitoylation of these cysteine residues are thought to be important in anchoring of the E2 to the plasma membrane, and favor interaction with the capsid protein during Sindbis virus assembly [38, 39].
The non-coding region changes were mostly in the 3′NTR region, which play role in the viral genome replication during minus strand synthesis in alphaviruses [40]. The 3′NTR of alphaviruses contain repeat sequence elements (RSEs) (Pfeffer et al., 1998) and the CHIKV 3′NTR has three RSEs at positions 11382–11416, 11525–11559, and 11611–11646 [41]. Interestingly, one of the 3′NTR nucleotide changes (C11614T) observed in three RGCB isolates (RGCB80, RGCB120, and RGCB355) and in two other isolates (ITA07-RA and IND-06-RJ1) fell within the third RSE region. However, this did not change the predicted RNA secondary structure at the region.
Analysis of the changes possibly affecting the immunogenicity of these isolates indicated that several mutations in the structural protein region fell within the predicted T-cell epitopes (Table 5). Epitopes presented by major HLA alleles, such as HLA-A*02 and HLA-A*11 [42], were shown to be involved. The latter is one of the main HLA types present in populations world over and is highly prevalent in South East Asia [43]. Classical CHIKV infections are known to offer long-term protection [44]. However, the variations in these epitopes have the potential to affect the cell-mediated immune response to CHIKV antigens.
In phylogenetic analysis, the 2007 and 2008 isolates showed closer clustering, even though the 2008 isolates were from a geographically distant location. This indicates that the viral strains causing the latter outbreaks were similar to the strains that were circulating in 2007. The timelines of the outbreak with an inter-epidemic period of almost a year (May–October 2007 and May–September 2008) clearly points out that the viral strains were maintained in active form in the region during this period. Further investigations are essential to understand whether this was contributed by sporadic incidences of the disease, which was prevailing undetected in the population, or through natural reservoirs of the Chikungunya virus such as non-human primates or vector mosquitoes [1].
In conclusion, our study reveals circulation of CHIKV with novel genetic changes in Kerala, South India, in the 2007 and 2008 disease outbreaks. Evolvement of a new genetic clade among these viruses was also observed in phylogenetic analysis. The samples analyzed represent only a small cross section of the cases from massive Chikungunya outbreaks in the state. However, the study points out genome microevolution of the viral strains, which might have affected the disease profile. Functional studies on these mutant viruses would help to understand the correlation of these genetic changes to CHIKV virulence and pathogenesis.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The work was funded by Department of Biotechnology, Govt. of India (Grant No. BT/PR9101/MED/29/04/2007). The authors are thankful to Prof. C.C. Kartha, Professor of Eminence, Cardiovascular Disease Biology, RGCB for critical suggestions. Authors are grateful to the Indian Medical Association, Thiruvananthapuram, and the Department of Health, Govt. of Kerala, for the help rendered during the study.
Footnotes
Aneesh Issac and Sajith Nair contributed equally to this work.
References
- 1.Powers AM, Logue CH. J. Gen. Virol. 2007;88:2363. doi: 10.1099/vir.0.82858-0. [DOI] [PubMed] [Google Scholar]
- 2.Vasilakis N, Weaver SC. Adv. Virus. Res. 2008;72:1. doi: 10.1016/S0065-3527(08)00401-6. [DOI] [PubMed] [Google Scholar]
- 3.Gould EA, Solomon T. Lancet. 2008;371:500. doi: 10.1016/S0140-6736(08)60238-X. [DOI] [PubMed] [Google Scholar]
- 4.Schuffenecker I, Iteman I, Michault A, Murri S, Frangeul L, Vaney MC, Lavenir R, Pardigon N, Reynes JM, Pettinelli F, Biscornet L, Diancourt L, Michel S, Duquerroy S, Guigon G, Frenkiel MP, Brehin AC, Cubito N, Despres P, Kunst F, Rey FA, Zeller H, Brisse S. PLoS Med. 2006;3:e263. doi: 10.1371/journal.pmed.0030263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rezza G, Nicoletti L, Angelini R, Romi R, Finarelli AC, Panning M, Cordioli P, Fortuna C, Boros S, Magurano F, Silvi G, Angelini P, Dottori M, Ciufolini MG, Majori GC, Cassone A. Lancet. 2007;370:1840. doi: 10.1016/S0140-6736(07)61779-6. [DOI] [PubMed] [Google Scholar]
- 6.Santhosh SR, Dash PK, Parida MM, Khan M, Tiwari M, Lakshmana Rao PV. Virus Res. 2008;135:36. doi: 10.1016/j.virusres.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Shah KV, Gibbs CJ, Jr, Banerjee G. Indian J. Med. Res. 1964;52:676. [PubMed] [Google Scholar]
- 8.Pavri K. Trans. R Soc. Trop. Med. Hyg. 1986;80:491. doi: 10.1016/0035-9203(86)90358-5. [DOI] [PubMed] [Google Scholar]
- 9.Mourya DT, Thakare JR, Gokhale MD, Powers AM, Hundekar SL, Jayakumar PC, Bondre VP, Shouche YS, Padbidri VS. Acta Virol. 2001;45:305. [PubMed] [Google Scholar]
- 10.Yergolkar PN, Tandale BV, Arankalle VA, Sathe PS, Sudeep AB, Gandhe SS, Gokhle MD, Jacob GP, Hundekar SL, Mishra AC. Emerg. Infect. Dis. 2006;12:1580. doi: 10.3201/eid1210.060529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arankalle VA, Shrivastava S, Cherian S, Gunjikar RS, Walimbe AM, Jadhav SM, Sudeep AB, Mishra AC. J. Gen. Virol. 2007;88:1967. doi: 10.1099/vir.0.82714-0. [DOI] [PubMed] [Google Scholar]
- 12.Kumar NP, Joseph R, Kamaraj T, Jambulingam P. J. Gen. Virol. 2008;89:1945. doi: 10.1099/vir.0.83628-0. [DOI] [PubMed] [Google Scholar]
- 13.Kannan M, Rajendran R, Sunish IP, Balasubramaniam R, Arunachalam N, Paramsivan R, Tewari SC, Samuel PP, Tyagi BK. Indian J. Med. Res. 2009;129:311. [PubMed] [Google Scholar]
- 14.Kielian M, Klimjack MR, Ghosh S, Duffus WA. J. Cell Biol. 1996;134:863. doi: 10.1083/jcb.134.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fazakerley JK, Boyd A, Mikkola ML, Kaariainen L. J. Virol. 2002;76:392. doi: 10.1128/JVI.76.1.392-396.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayuri, Geders TW, Smith JL, Kuhn RJ. J. Virol. 2008;82:7284. doi: 10.1128/JVI.00224-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vazeille M, Moutailler S, Coudrier D, Rousseaux C, Khun H, Huerre M, Thiria J, Dehecq JS, Fontenille D, Schuffenecker I, Despres P, Failloux AB. PLoS One. 2007;2:e1168. doi: 10.1371/journal.pone.0001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsetsarkin KA, Vanlandingham DL, McGee CE, Higgs S. PLoS Pathog. 2007;3:e201. doi: 10.1371/journal.ppat.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasebe F, Parquet MC, Pandey BD, Mathenge EG, Morita K, Balasubramaniam V, Saat Z, Yusop A, Sinniah M, Natkunam S, Igarashi A. J. Med. Virol. 2002;67:370. doi: 10.1002/jmv.10085. [DOI] [PubMed] [Google Scholar]
- 20.Hall TA. Nucleic Acids Symp. Ser. 1999;41:95. doi: 10.1093/nass/42.1.95. [DOI] [PubMed] [Google Scholar]
- 21.Kumar S, Tamura K, Nei M. Brief Bioinform. 2004;5:150. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- 22.Guex N, Peitsch MC. Electrophoresis. 1997;18:2714. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 23.Willard L, Ranjan A, Zhang H, Monzavi H, Boyko RF, Sykes BD, Wishart DS. Nucleic Acids Res. 2003;31:3316. doi: 10.1093/nar/gkg565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doytchinova IA, Guan P, Flower DR. BMC Bioinformatics. 2006;7:131. doi: 10.1186/1471-2105-7-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sudeep AB, Parashar D. J. Biosci. 2008;33:443. doi: 10.1007/s12038-008-0063-2. [DOI] [PubMed] [Google Scholar]
- 26.Talarmin F, Staïkowsky F, Schoenlaub P, Risbourg A, Nicolas X, Zagnoli A, Boyer P. Med. Trop. 2007;67:167. [PubMed] [Google Scholar]
- 27.Valamparmpil JJ, Chirakkarot S, Letha S, Jayakumar C, Gopinathan KM. Ind. J. Paediatr. 2009;76:151. doi: 10.1007/s12098-009-0045-x. [DOI] [PubMed] [Google Scholar]
- 28.Cherian SS, Walimbe AM, Jadhav SM, Gandhe SS, Hundekar SL, Mishra AC, Arankalle VA. Infect. Genet. Evol. 2009;9:16. doi: 10.1016/j.meegid.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Garmashova N, Gorchakov R, Frolova E, Frolov I. J. Virol. 2006;80:5686. doi: 10.1128/JVI.02739-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tamm K, Merits A, Sarand I. J. Gen. Virol. 2008;89:676. doi: 10.1099/vir.0.83320-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Golubtsov A, Kaariainen L, Caldentey J. FEBS Lett. 2006;580:1502. doi: 10.1016/j.febslet.2006.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang W, Mukhopadhyay S, Pletnev SV, Baker TS, Kuhn RJ, Rossmann MG. J. Virol. 2002;76:11645. doi: 10.1128/JVI.76.22.11645-11658.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mukhopadhyay S, Zhang W, Gabler S, Chipman PR, Strauss EG, Strauss JH, Baker TS, Kuhn RJ, Rossmann MG. Structure. 2006;14:63. doi: 10.1016/j.str.2005.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kielian M. Virology. 2006;344:38. doi: 10.1016/j.virol.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 35.Kielian M, Rey FA. Nat. Rev. Microbiol. 2006;4:67. doi: 10.1038/nrmicro1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hopp TP, Woods KR. Proc. Natl Acad. Sci. USA. 1981;78:3824. doi: 10.1073/pnas.78.6.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith TJ, Cheng RH, Olson NH, Peterson P, Chase E, Kuhn RJ, Baker TS. Proc. Natl Acad. Sci. USA. 1995;92:10648. doi: 10.1073/pnas.92.23.10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilkinson TA, Tellinghuisen TL, Kuhn RJ, Post CB. Biochemistry. 2005;44:2800. doi: 10.1021/bi0479961. [DOI] [PubMed] [Google Scholar]
- 39.West J, Hernandez R, Ferreira D, Brown DT. J. Virol. 2006;80:4458. doi: 10.1128/JVI.80.9.4458-4468.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strauss JH, Strauss EG. Microbiol. Rev. 1994;58:491. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khan AH, Morita K, Parquet MC, Hasebe F, Mathenge EGM, Igarashi A. J. Gen. Virol. 2002;83:3075. doi: 10.1099/0022-1317-83-12-3075. [DOI] [PubMed] [Google Scholar]
- 42.Middleton D, Williams F. Methods Mol. Biol. 2003;210:67. doi: 10.1385/1-59259-291-0:67. [DOI] [PubMed] [Google Scholar]
- 43.Sidney J, Grey HM, Southwood S, Celis E, Wentworth PA, del Guercio MF, Kubo RT, Chesnut RW, Sette A. Hum. Immunol. 1996;45:79. doi: 10.1016/0198-8859(95)00173-5. [DOI] [PubMed] [Google Scholar]
- 44.Pialoux G, Gauzere BA, Jaureguiberry S, Strobel M. Lancet Infect. Dis. 2007;7:319. doi: 10.1016/S1473-3099(07)70107-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.