

Abstract
To identify the potential B-cell antigenic epitopes within the N-terminus of SARS-CoV (SARS-associated coronavirus, SARS-CoV) M protein and characterize monoclonal antibody (MAb) against the protein as well as its recognizing region, we expressed and purified a portion of SARS-CoV M protein (amino acid 1–43) in Escherichia coli (E. coli). By using Western blot and enzyme-linked immunosorbent assay (ELISA), we showed that the purified recombinant M protein could be recognized by four SARS-CoV-positive human sera even when those sera were 12,800-fold diluted. Furthermore, we characterized one representative IgG2 MAb, 3H9, which exhibited a strong immunoreaction to both recombinant M protein and native viral protein of SARS-CoV. We found a B-cell antigenic epitope located between amino acid 1–15 and defined the MAb recognizing region within amino acid 16–28 of M. These findings not only suggest that both recombinant M protein and its specific MAbs may be used as the diagnostic reagents for SARS, but also provide a potential target site for the design of an epitope-based vaccine against SARS.
Keywords: SARS-associated coronavirus, Recombinant M protein, B-cell antigenic epitope, Monoclonal antibody, Western blot, ELISA
Introduction
Severe acute respiratory syndrome (SARS) is an emerging infectious disease and its etiological agent is known as SARS-associated coronavirus (SARS-CoV) [1, 2]. SARS-CoV, a novel human CoV (order Nidovirales, family Coronaviridae, genus Coronavirus), is a large, enveloped, positive stranded RNA virus, and structurally similar to other coronaviruses. The human coronaviruses, HCoV-229E and HCoV-OC43, were commonly found in upper respiratory tract infection. So far, little has been done about their immunogenicities and vaccines. The structural proteins of SARS-CoV include membrane glycoproteins S, M, E and nucleocapsid protein (N). Recently, some researches reported that the S and N proteins of SARS-CoV possessed neutralizing epitopes [3–6]. However, other reports showed that SARS-CoV M protein might elicit the specific antibody [7, 8] and M protein might be involved in virus assembly [9]. The exact function of M protein remains unknown.
Currently, the laboratory diagnostic methods of SARS mainly include reverse transcriptase-polymerase chain reaction (RT-PCR), real-time quantitative PCR (both detect SARS-CoV gene), and antibody detection methods such as immunofluorescence assay (IFA) and ELISA [10–16]. Many of these methods involve live viruses with high infectivity and mortality. Special facilities like the bio-safety level 3 laboratory are required. On the other hand, preparation of large amounts of recombinant SARS-CoV proteins with good immunogenicity and antigenicity as well as their specific monoclonal antibodies (MAbs) is very important for the usage of these diagnostic reagents and for evaluation of the safety effectiveness of vaccine. It has been established that M protein plays an important role in other coronaviruses’ life cycle and inducing immunity [17–23]. Therefore, we hypothesize that in SARS-CoV, M protein may also have such functions and may be an attractive target for anti-SARS drug research, vaccine development and the establishment of a serological detection assay. To testify this hypothesis, in the present study, we identified a B-cell antigenic epitope at the N-terminus of SARS-CoV M protein, characterized a monoclonal antibody against the protein, and defined the MAb recognizing region.
Materials and methods
Expression plasmid constructs
Gene sequences encoding the N-terminal amino acid (aa) residues (1–43) of M protein of SARS-CoV strain BJ01 (GenBank accession no. AY278488) were synthesized by TaKaRa Biotechnology Co. Ltd (Dalian, China) which includes two restriction enzyme sites of BamHI and XhoI on the 5′ and 3′ end to facilitate directional cloning. The synthesized M gene fragment was digested by BamHI and XhoI and subcloned into pGEX-6P-l (Amersham) to create a GST fusion protein-expressing plasmid pSARS-M. E. coli DH5α competent cells (Amersham) was transformed with the recombinant plasmid and clones were identified by restriction digestion and sequencing. The sequence was further analyzed by DNAMAN biological software. To generate two recombinants containing truncated M protein coding gene, PCR was performed using plasmid pSARS-M as a template with two sets of PCR primers. The sequences of the first set primer were: Forward 5′-TCC AGG GGC CCC TGG GAT CCA TGG CAG-3′, Reverse 5′-CTG ACT CTC GAG TCA TAG GAA TAG GAA ACC-3′, which were used to amplify the N-terminal 1–28 aa coding gene of M protein. The sequences of the second set primer were: Forward 5′-GGG CCC CTG GGA TCC CTG GAA CAA TGG-3′, Reverse 5′-CTG ACT CTC GAG TCA CCT GTT CCG ATT AGA-3′, which were employed to amplify the N-terminal 16–43 aa coding gene of M protein. BamHI and XhoI sites are underlined and non-specific nucleotides were positioned 5′ of restriction sites to facilitate enzyme digestion. The two DNA fragments had a 39-base pair (bp)-overlapping region, which encoded 13 aa. Procedures for cloning of the PCR products and identification of the recombinant plasmids were performed as described above. The recombinant plasmids were designated as pSARS-M1 and pSARS-M2, respectively.
Expression and purification of M fusion protein
E. coli BL21 competent cells (Invitrogen) were transformed with the recombinant plasmid pSARS-M. The transformed BL21 strains were cultured in Luria–Bertani medium (LB) with continuous shaking at 37°C until the optical density (OD) at 600 nm reached 0.7, followed by induction with 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG; Sigma) at 30°C for additional 4-h culture. Control cultures containing the empty pGEX-6p-1 plasmid were processed in parallel. The cultured cells were centrifuged and harvested. Cells from a liter of culture were resuspended in 50 ml of PBS (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4, pH 7.3). The expression of fusion proteins were determined by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE). For purification, the crude materia of culture from pSARS-M plasmid-transformed BL21 were subsequently subjected to centrifugation at 4,000 × g, 4°C for 10 mm, and the precipitation was resuspended in PBS (pH 7.3) at 4°C. After sonication, the insoluble material was removed by centrifugation at 10,000 × g. The supernatants were filtered through a 0.45 μm nitrocellulose membrane and were then loaded onto Glutathione Sepharose 4B affinity chromotography column (Amersham). Purification of fusion protein was performed according to manufacturer’s instructions. The purified proteins were detected by SDS-PAGE.
Serologic samples and viruses
The sources and properties of human serum samples used in this study were listed in Table 1. All four SARS patients (age 30–45) had history of contact with other patients who were infected with SARS-CoV and exhibited symptoms including persistent fever (>38.5°C), cough and shortness of breath for several days before they were hospitalized. Patients were subsequently confirmed to be infected with SARS-CoV by clinical diagnosis combined with laboratory diagnostic methods according to the WHO criteria. After deactivation at 56°C for 45 min, the serologic samples were stored at −20°C until use. Both animal coronaviruses including infectious bronchitis virus (IBV) and transmissible gastroenteritis virus (TGEV) and sera from IBV-infected chicken and TGEV-infected swine were kindly provided by Dr. Y. Li from Jiangsu Academy of Agricultural Sciences, China, which were developed according to the methods as described elsewhere [24].
Table 1.
The properties and source of sera used in the Western blot and ELISA
| Serum group | Sample No. | Origin of serum samples (n) |
|---|---|---|
| Confirmed SARS patients’ | Sample 1 and 2 | The 2nd Hospital of Nanjing, China (2) |
| sera (collected 20–60 days after onset of fever) | Sample 3 and 4 | Beijing HuaDajiBiAi Bio-Technology Co., China (2) |
| Healthy human sera | Sample 5 and 6 | The 1st Affiliated Hospital of Nanjing Medical University, China (2) |
| Sample 7 and 8 | The 2nd Hospital of Nanjing, China (2) |
Western blot
Purified recombinant M protein was separated by SDS-PAGE and transferred to a polyvinylidene difluoride membrane (PVDF) by the Trans Blot system (Bio-Rad). The membrane was blocked with PBS-T (14 mM NaCl, 2.7 mM KCl, 10 mM NaHPO4, 1.8 mM KH2PO4 and 0.02% Tween-20) containing 5% non-fat milk powdered for 2 h at 4°C. The membrane was probed with SARS-CoV-positive human serum (1:1,000 dilution) for 3 h at room temperature. After washing three times in an appropriate volume of PBS-T, the membrane was incubated for 1 h with horseradish peroxidase (HRP)-conjugated goat anti-human IgG (Santa Cruz) at 1:2,000 concentration. The membrane was washed three times with PBS-T and once with distilled water for 10 min each time. Finally, an appropriate volume of enhanced chemiluminescence (ECL) solution (Amersham) was added to the washed membrane at room temperature for 1 min. Signal was detected by exposing to Kodak film for 1–3 min.
ELISA
The purified M protein was diluted with 50 mM carbonate buffer (35 mM NaHCO3, 15 mM Na2CO3, [pH 9.6]), and then used to coat 96-well plates (Costar Inc.) (0.4 μg/well) overnight at 4°C. After washing with PBS-T (PBS containing 0.5% Tween-20, pH 7.4) for three times, each well of the plate was incubated with blocking solution (0.1 M Tris–HCl, 0.5% BSA, 1% sucrose) at 37°C for 2 h. SARS-CoV-positive human serum diluted in a twofold series from 1:100 to 1:12,800 was added into each well in duplicate (100 μl/well) and incubated at 37°C for 1 h, followed by the addition of goat anti-human IgG antibody labeled with HRP (1:50,000; Sigma) at 37°C for 30 min. 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (100 μl/well) was added after six times washing with PBS-T and the wells were incubated for 10 min at 37°C. Fifty microlitres of stop buffer (2 M H2SO4) was added to each well and the OD value was read at 450 nm in CliniBio 128C reader. The cut-off value was defined as the mean OD plus 2.1 standard deviations calculated from the four negative samples used as controls.
Establishment of MAb
Five 4- to 6-week-old Balb/C mice (females; the Center of Experimental Animal, Chinese Academy of Science Shanghai Branch) were injected subcutaneously with 25 μg of purified recombinant M protein emulsified with an equal volume of Freund’s Complete Adjuvant (Sigma) for three times with a 2-week interval. Mice received a final booster injection with 25 μg of antigen in 200 μl PBS to the intraperitoneal cavity 2 days prior to hybridoma fusion. Mice were sacrificed and their spleen cells were harvested. Mice S/P2.0 myeloma cells were in log-phase growth prior to fusion with spleen cells [25]. Hybridoma culture supernatants were simultaneously screened for antibody production by using indirect ELISA assay with both purified M protein and glutathione S-transferase (GST) protein as coating antigens, respectively. The procedures of ELISA assay were same as described above, except that the second antibody was a 1:2,000 dilution of HRP labeled rabbit anti-mouse immunoglobulin (Dako, Denmark). Only the hybridoma, whose cultural supernatants could react with the M fusion protein, but not with GST, was regarded as positive clone. Positive clones were continually selected and subcloned by limiting dilution for at least three times until the positive rate reached 100%. When the desired clones were identified, they were expanded in 75 cm2 flasks. One week later, the hybridoma suspension was harvested and cell debris were removed by centrifugation at 400 × g for 10 min. To prepare the ascites fluid of MAb, the Balb/C mouse, which was injected with liquid paraffin days before, was inoculated with 2 × 106 of positive hybridoina cells and continued to raise for 7–10 days until abdominal cavity appeared to be tympanites. Then ascites fluid was collected. The collected supernatants and ascites fluid were added with 1/100 volume of 1 M Tris–HCl (pH 7.4) and 1/500 volume of 10% NaN3, directly loaded on the Protein G-Sepharose 6B column (Amersham Biosciences). The column was washed with PBS and eluted with Glycine/HCl (pH 2.8). After measuring OD280 of the fractions, protein containing fractions were pooled and added with the equal volume of saturated (NH4)2SO4. Precipitated proteins were dissolved in PBS, dialyzed against PBS and stored at −80°C.
Characterizations of MAb
The identification of subclass and subtype of MAb was performed by Mouse Monoclonal Antibody Isotyping Kit as recommended by the supplier (Roche). To examine the specificity of MAb to SARS-CoV, Western blot analysis was performed using the purified M fusion protein; or diagnostic kit for antibody to SARS virus from Beijing HuaDaJiBiAi Bio-Technology Co. was used in indirect ELISA analysis. A total of 96-well plates from the kit, which were coated with the UV-irradiated and extracted viral protein of SARS-CoV, were used to determine the titers of MAb in the supernatants and ascites fluid. Antigens from viral proteins of both IBV and TGEV were also applied to 96-well plates as controls. All ELISA assays were performed as described above.
Results
Expression and purification of M fusion protein
Recombinant plasmids pSARS-M, pSARS-M1 and pSARS-M2 were digested with BamHI and XhoI, respectively, and the products were subjected to a 1% agarose gel and visualized by ethidium bromide staining. As expected, a DNA band corresponding to the molecular mass of 130, 115, and 115 bp was observed, respectively (data not shown). The nucleotide sequence alignment performed by DNAMAN software indicated that the inserted sequences were 100% identical with that of M gene fragment of SARS-CoV (BJ01) in GenBank. Moreover, expression of M fusion protein was induced in E. coli BL21 by IPTG and demonstrated by SDS-AGE analysis. We found an apparent protein band with the relative molecular mass of 30 KDa in BL21 transformed with pSARS-M plasmid, but not in BL21 and BL21 transformed with the empty plasmid pGEX-6P-1 (Fig. 1a, b). Furthermore, the recombinant M protein was purified by gel filtration of Glutathione Sepharose 4B affinity chromatography and the purity was confirmed by SDS-PAGE with a single band (Fig. 1b). These results suggest that GST/SARS-M fusion protein was successfully expressed in E. coli and effectively purified.
Fig. 1.
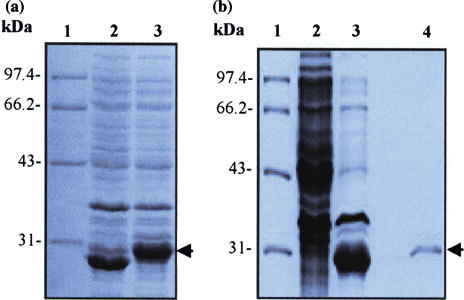
SDS-PAGE analysis on expression and purity of M protein in E. coli BL21. (a) SDS-PAGE analysis of expression of M protein in E. coli BL21. E. coli BL21 was transformed with the recombinant plasmid pSARS-M, followed by induction with IPTG. The bacterial cultural crude materials were subjected to the 10% SDS-PAGE. Lane 1: molecular weight marker; Lane 2: cellular extracts of TPTG-induced BL21 transformed with the empty plasmid pGEX-6P-1 (control); Lane 3: cellular extracts of IPTG-induced BL2 1 transformed with the recombinant pSARS-M construct. (b) SDS-PAGE analysis of purity of recombinant M protein after purified by affinity chromatography. The crude material of BL21 transformed with pSARS-M plasmid was subjected to sonication. The supernatants were filtered through a 0.45 μm nitrocellulose membrane and loaded onto Glutathione Sepharose 4B affinity chromotography column. The purified proteins were detected by SDS-PAGE. Lane 1: molecular weight marker; Lane 2: total cellular extracts of IPTG-induced BL21 (control); Lane 3: total cellular extracts of IPTG-induced BL21 transformed with the recombinant pSARS-M construct (control); Lane 4: eluted protein of IPTG-induced BL21 transformed with the recombinant pSARS-M construct from affinity chromatography purification. Arrows indicate M fusion protein expressed in E coli BL21 (left) and purified M fusion protein (right). The gel was stained by Coomassie blue. The molecular mass size markers (in KDa) are reported on the left of each Figure
Antigenicity analysis of expressed protein
To examine the antigenicity of the expressed GST/SARS-M fusion protein, Western blot analysis was performed using SARS-CoV-positive human serum. As shown in Fig. 2a, the human serum specifically recognized a 30 KDa protein in the purified supernatants from IPTG-induced BL21 transformed with the plasmid pSARS-M, whereas no immunoreactive protein band was seen in the purified supernatants from IPTG-induced BL21 transformed with the empty plasmid pGEX-6P-l (Fig. 2b). These results indicated that no antibodies against GST protein were present in SARS-CoV-positive human serum. To determine whether the cross antigens are present in M proteins between SARS-CoV and other animal coronaviruses, Western blot was also performed using the sera from IBV-infected chicken and TGEV-infected swine, respectively. The results demonstrated that both IBV and TGEV sera failed to react with this 30 KDa of protein (data not shown). Indirect ELISA assay was performed to examine whether the purified GST/SARS-M fusion protein could be used as an antigen to detect the SARS-CoV-positive human sera. The results indicated that the purified M fusion protein still reacted strongly with all four SARS-CoV-positive human sera even when the sera were diluted to 1:12,800 (Fig. 2b). Our data also showed that purified M fusion protein had no cross-reaction with the sera of healthy donors (Fig. 2b). In addition, to confirm that no antibodies against GST proteins were present in SARS-CoV-positive human sera, ELISA assays were performed using purified GST proteins as antigen to coat 96-well plates. It was showed that no statistical significances were found in OD values between SARS-CoV-positive human sera and healthy human sera (data not shown). Taken together, these data suggest that the expressed GST/SARS-M fusion protein not only shows a good antigenic specificity and reactiongenicity, but also can serve as an ELISA antigen suitable for detection of SARS antibody.
Fig. 2.
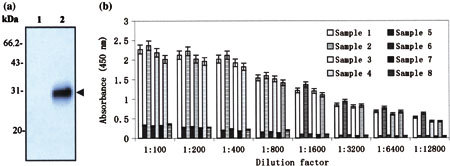
Analysis of antigenicity of expressed M protein. (a) Western blot analysis on the purified M fusion protein with the sera of SARS-CoV-positive patients. M protein was purified from lysates of BL21 cells transformed with pSARS-M plasmid through Glutathione Sepharose 4B affinity chromotography column. Purified proteins were subjected to SDS-PAGE and subsequently transferred to PVDF membrane for Western blot with the sera of SARS-CoV-positive patients. Lane 1: eluted protein of IPTG-induced BL21 transformed with the empty plasmid pGEX-6P-1 (control); Lane 2: eluted protein of IPTG-induced BL21 transformed with the recombinant pSARS-M construct. Results are Western blot probed with the serum from sample 1. Additional three experiments with the sera from sample 2, 3 and 4, respectively, were also performed and gave similar results. The properties of sera from four SARS-CoV-positive patients were listed in Table 1. Arrow indicates purified M fusion protein immunoblotting with the serum of SARS-CoV-positive patient. (b) Detection of eight human sera by indirect ELISA by using purified M fusion protein as antigen. The purified recombinant M protein was diluted and used to coat 96-well plates. SARS-CoV-positive human serum diluted in a twofold series from 1:100 to 1:12,800 was added into each well in duplicate. The OD value was read at 450 nm in CliniBio 128C reader and the cut-off value was defined as the mean OD plus 2.1 standard deviations calculated from the four negative samples used as controls. Sample 1–4 were sera from four confirmed SARS patients and 5–8 from four healthy people (controls), whose source and properties were listed in Table 1
Localization of B-cell antigenic epitope in M protein
Since the purified M fusion protein could react well with all four SARS-CoV-positive human sera (Fig. 2), theoretically, there should be B-cell antigenic epitope(s) within this M protein fragment. To search and localize the potential B-cell antigenic epitope(s), two constructs designed as pSARS-Ml and pSARS-M2 that expressed recombinant truncated M protein-coding proteins were generated. SDS-PAGE analysis demonstrated that both pSARS-Ml and pSARS-M2 constructs produced a prominent protein band with the relative molecular mass of 29 KDa (Fig. 3a). Furthermore, Western blot analysis was performed with SARS-CoV-positive human serum. As shown in Fig. 3b, the human serum only recognized protein that was expressed by pSARS-Ml construct in IPTG-induced BL21 but not by pSARS-M2 construct. These data suggest that, at least, one B-cell antigenic epitope was located in the N-terminal 1–15 amino acids of M protein.
Fig. 3.
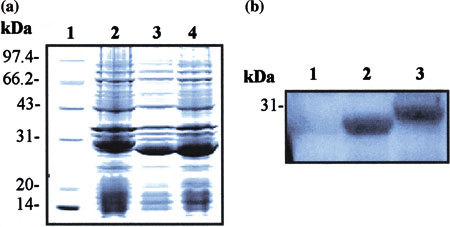
Localization of B-cell antigenic epitope in M protein. (a) SDS-PAGE analysis of truncated M protein expressed in E. coli BL21. BL21 was transformed with two truncated recombinant M protein-coding genes constructs, followed by induction with IPTG. The bacterial cultural crude materials were detected by the 10% SDS-PAGE and then stained with Coomassie blue. Lane 1: molecular weight marker; Lane 2: cellular extracts of IPTG-induced BL21 transformed with the recombinant pSARS-M construct (control); Lane 3: cellular extracts of IPTG-induced BL21 transformed with the recombinant pSARS-Ml construct; Lane 4: cellular extracts of IPTG-induced BL21 transformed with the recombinant pSARS-M2 construct. (b) Localization of B-cell antigenic epitope in M protein by Western blot with the sera of SARS-CoV-positive patients. The crude materials of inductive culture of E. coli BL21 were subjected to SDS-PAGE and subsequently transferred to PVDF membrane for Western blot analysis with the SARS-CoV-positive human serum. Lane 1: total cellular extracts of IPTG-induced BL21 transformed with pSARS-M2 construct; Lane 2: total cellular extracts of IPTG-induced BL21 transformed with pSARS M1 construct; Lane 3: total cellular extracts of IPTG-induced BL21 transformed with pSARS-M construct (control). A representative experiment is shown with the serum from sample 1. Additional three experiments with the sera from sample 2, 3 and 4, respectively, were also performed and gave similar results. The properties of sera from four-SARS-CoV-positive patients were listed in Table 1
Production and characterization of MAb against M protein
Fusion of spleen cells from immunized Balb/C mice with S/P2.0 myeloma cells produced several hybridoma clones secreting MAbs against recombinant M protein fragment (data not shown). Positive clones were determined through indirect ELISA, which gave an absorbance threefold higher than that of the background. The positive clones showed great variation in their abilities to secret MAbs. The hybridoma cell lines yielding the highest antibody titer, 3H9, were selected to produce MAb for further analyses and experiments.
The results from MAb isotyping test indicated that the MAb, 3H9, was IgG2a with the Kappa light chain. To determine the specificity of MAb, we further performed Western blot. As shown in Fig. 4a, the MAb bound specifically to recombinant M fusion protein and gave a single band with the M fusion protein at the exact location (about 30 KDa in relative molecular mass). However, the MAb could not react with cellular extracts of IPTG-induced BL21 and BL21 transformed with plasmid pGEX-6P-1 (Fig. 4a).
Fig. 4.
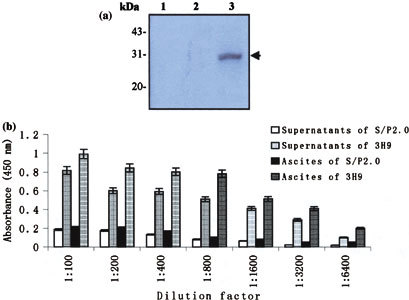
Identification of the MAb specificity. (a) Western blot analysis of the expressed M fusion protein with the MAb, 3H9. The crude materials of inductive culture of E. coli BL21 were subjected to SDS-PAGE and subsequently transferred to PVDF membrane for Western blot with the MAb, 3H9. Lane 1: total cellular extracts of IPTG-induced BL21 (control); Lane 2: total cellular extracts of IPTG-induced BL21 transformed with pGEX-6p-l (control); Lane 3: total cellular extracts of IPTG-induced BL21 transformed with the recombinant pSARS-M construct. Arrow indicates expressed M fusion protein immunoblotting with the MAb. (b) ELISA reactivity of the MAb, 3H9, to viral protein of SARS-CoV. A total of 96-well plates from the diagnostic kit detecting antibody specific to SARS virus, which were coated with the UV-irradiated and extracted viral protein of SARS-CoV, were used to determine the titers of MAb in the supernatants and ascites fluid by indirect ELISA. Supernatants and ascites fluid from 3H9 and S/P2.0 (controls) diluted in a two-fold series from 1:100 to 1:6,400 were added into each well in duplicate, respectively. The OD value was read at 450 nm in CliniBio 128C reader and the cut-off value was defined as the mean OD plus 2.1 standard deviations calculated from the four negative samples used as controls
To examine whether the MAb can be used as an ELISA reagent for detection of SARS-CoV viral protein, indirect ELISA was performed by using a commercially available SARS virus diagnostic kit detecting antibody specific to SARS-CoV. The results revealed that both supernatants and ascites fluid produced by 3H9 with the titer up to 1:6,400 could react specially with the viral protein of SARS-CoV coated on the 96-well plates (Fig. 4b). Our data also showed that the MAb from 3H9 has no cross-reaction with IBV and TGEV viral proteins (data not shown). Together, these data suggest that the MAb of 3H9 is specific to SARS-CoV and can be served as an ELISA antibody to detect the viral protein of SARS-CoV.
To localize the MAb recognizing region in recombinant M protein, we further perform Western blot using this antibody to probe the membrane on which both full length and the truncated M proteins were transferred. The results demonstrated that the MAb could react with all of the three proteins expressed by pSARS-M, pSARS-Ml, and pSARS-M2 construct, respectively, exhibiting a prominent immunoreactive protein band (Fig. 5). These data suggest that the MAb recognizing region was located in overlapping region (N-terminal 16–28 aa) between the expressed proteins by pSARS-Ml and pSARS-M2 construct.
Fig. 5.
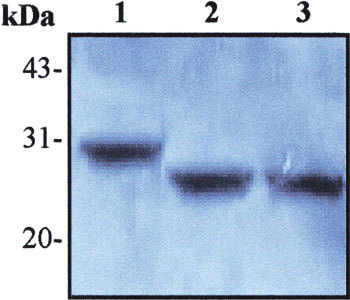
Localization of the recognizing region of the MAb, 3H9 in recombinant M protein by Western blot. The crude materials of E. coli BL21 culture were subjected to SDS-PAGE and subsequently transferred to PVDF membrane for Western blot with the MAb, 3H9. Lane 1: total cellular extracts from IPTG-induced BL21 transformed with recombinant pSARS-M construct (control); Lane 2: total cellular extracts from IPTG-induced BL21 transformed with recombinant pSARS-Ml construct; Lane 3: total cellular extracts from IPTG-induced BL21 transformed with recombinant pSARS-M2 construct
Discussion
The antigenic properties of SARS-CoV show both similarities to and differences from those of other coronaviruses [26]. SARS-CoV N protein was found to be highly antigenic and contain many linear and conformational epitopes, which are consistent with results from investigations of other coronaviruses [8, 27, 28]. Moreover, in other coronavirus, the S1 domain of the major envelope spike protein was the most antigenic. For instance, several antigenic sites were found in the N-terminal S1 domain of TGV [29]. However, in SARS-CoV, the S2 region of the envelope spike protein was the B-cell epitope-rich region, indicating that it is an immunodominant site on the viral envelope comprising the spike, matrix, and small envelope glycoprotein [6]. With respect to the M protein in other animal coronaviruses, the M protein is abundant among the structural proteins and can induce antibody-dependent complement-mediated virus neutralization. It is also involved in the assembly and budding of virions together with the E protein [17–23]. These known key roles of the M protein make it an attractive target for the development of vaccines and drugs as well as for effective diagnostic reagents. However, SARS-CoV differs from other coronaviruses in nucleotide sequence, and the exact function of its M protein is still unknown.
In our current study, we analyzed the antigenicity of recombinant SARS-CoV M protein and identified the specificity of its monoclonal antibody. Our results not only provide the potential materials, which could serve as ELISA reagents suitable for diagnosis of SARS, but also reveal several points in understanding B-cell epitope of SARS-CoV M protein and its MAb recognizing sequence.
First, our results provide direct experimental evidence that purified M fusion protein could serve as an ELISA reagent for SARS diagnosis. M protein has been previously shown to be high antigenicities in animal coronavirus. For instance, Elia et al. developed recombinant M protein-based ELISA test for detection of antibodies to canine coronavirus, which exhibited an excellent correlation with those of whole virus ELISA and Western blot [30]. Recently, although Han et al. have used the purified recombinant M protein (expressed in Pichia pastoris) of SARS-CoV as an ELISA antigen to detect four undiluted SARS-CoV-positive sera, its sensitivity remains to be evaluated [31]. Here we demonstrated that recombinant SARS-CoV M fusion protein (1–43 aa) expressed in E.coli was highly antigenic and specifically reactive with the sera from SARS-CoV-positive patients even when the sera were diluted to 1:12,800. Therefore, it suggests that our purified M fusion protein may be potentially useful as an ELISA antigen for diagnosis of SARS.
Second, we provide an experimental evidence to suggest that the MAb against M fusion protein fragment has a potential to detect viral antigen of SARS-CoV. Recently, monoclonal antibody-based ELISAs for SARS-CoV infection have been developed [32]. However, most of them were involved in using MAbs against S and N proteins. More recent report by Tripp et al. showed that two MAbs (292 and 283) against SARS-CoV M protein were developed and showed their reactivity with SARS-CoV in IFA and immunoprecipitation test. However, even when two MAbs were tested at a 1:80 dilutions for reactivity against SARS-CoV by ELISA, the positive-to-negative (P/N) ratios were no more than 3.7, implying their limiting usage as ELISA diagnostic reagent [33]. In the present study, we provided direct evidence that the MAb against M protein could be specifically reactive with the viral protein of SARS-CoV in ELISA even when it was diluted to 1:6,400. Whether this MAb-based antigen capture ELISA for detection of SARS-CoV virus can be developed needs further investigation.
Third, our experiment showed that one potential linear B-cell antigenic epitope located between 1 and 15 aa and the MAb recognizing region was within amino acid 16–28 of M protein. Since outbreak of SARS in 2003, B-cell epitopes have been of particular interest to investigators who study the humoral response and design epitope-based vaccine to prevent potential antibody-mediated immunopathy [3, 4, 6]. Many of these studies focused on SARS-CoV S and N proteins. For example, by using phage-display library technique, Duan et al. has characterized a human single chain Fv (scFv), B1, which could recognize an epitope within amino acids 1023–1189 of S2 protein and has the potent neutralizing activities against infection by pseudovirus expressing SARS-CoV S protein in vitro [34]. More recently, Hu et al. synthesized a 10-mer overlapping peptide library, which spanned the major structural proteins of SARS-CoV for screening and identification of linear B-cell epitopes of SARS-CoV. They demonstrated that five of assembled 22 longer peptides showed high cross-immunoreactivities to 42 SARS convalescent patients’ sera from the severest epidemic regions of the China mainland. Among them, S471–503, a peptide located at the receptor binding domain (RBD) of SARS-CoV, could specifically block the binding between the RBD and angiotensin-converting enzyme 2, resulting in the inhibition of SARS-CoV entrance into host cells in vitro [35]. In current study, we demonstrated a linear B-cell antigenic epitope located in the M protein between 1 and 15 aa, which was consistent with the previous reports [7, 8]. Interestingly, when we searched for potential B-cell antigenic epitopes within the full-length M protein using Bioinformatics tools, we found that there were seven predicted antigenic epitopes within M protein (C. Lu, personal communication). Among them, the first epitope was just located at 8–36 aa of the M protein. Of course, our results did not eliminate the possibility that other potential B-cell antigenic epitope(s) may still locate in this region. While Hu et al. has identified the B-cell antigenic epitope on S2 protein, they also found that two synthesized overlapping peptides, both of which were located within amino acids 29–48 of M protein, could react to mixture of 42 SARS convalescent patients’ sera, indicating that other potential B-cell antigenic epitope(s) may still locate in this region [35]. Meanwhile, our experiment showed that the recognizing region of the MAb against M fragment was in the M protein between amino acids 16 and 28. These data supported our primary hypothesis that the N-terminus of SARS-CoV M protein may play a critical role in induction of immune response, particularly in humoral immunity, in human and mouse.
In summary, we have expressed and purified SARS-CoV M fragment and characterized its MAb, both of which exhibited the potential for ELISA diagnostic reagents. And we also experimentally identified one B-cell antigenic epitope and defined the MAb recognizing region in the M fragment. Our findings may shed light on the establishment of other valid and effective detection methods and development of epitope-based vaccine for SARS-CoV infection. Further studies are needed to better understand whether this B-cell antigenic epitope may induce neutralizing antibody in human body and to better evaluate the sensitivity of both M protein- and its MAb-based diagnostic method for SARS infection.
Acknowledgements
We thank Dr. Zan Huang and Dr. Shengnan Li for critical reading of the manuscript. This work was supported by grants from the Ministry of Health, Jiangsu Province, P.R. China (H200308 to C.L.).
Footnotes
Chao Qian and Di Qin contributed equally to this work.
References
- 1.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 2.Peiris J.S., Lai S.T., Poon L.L.M., Guan Y., Yam L.Y.C., Lim W., Nicholls J., Yee W.K.S., Yan W.W., Cheung M.T., Cheng V.C.C., Chan K.H., Tsang D.N.C., Yung R.W.H., Ng T.K., Yuen K.Y. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu L., Manopo I., Leung B.P., Chng H.H., Ling A.E., Chee L.L., Ooi E.E., Chan S.W., Kwang J. J. Clin. Microbiol. 2004;42:1570–1576. doi: 10.1128/JCM.42.4.1570-1576.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang S., Chou T.W., Sakhatskyy P.V., Huang S., Lawrence J.M., Cao H., Huang X., Lu S. J. Virol. 2005;79:1906–1910. doi: 10.1128/JVI.79.3.1906-1910.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H., Wang G., Li J., Nie Y., Shi X., Lian G., Wang W., Yin X., Zhao Y., Qu X., Ding M., Deng H. J. Virol. 2004;78:6938–6945. doi: 10.1128/JVI.78.13.6938-6945.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhong X., Yang H., Guo Z.F., Sin W.Y., Chen W., Xu J., Fu L., Wu J., Mak C.K., Cheng C.S., Yang Y., Cao S., Wong T.Y., Lai S.T., Xie Y., Guo Z. J. Virol. 2005;79:3401–3408. doi: 10.1128/JVI.79.6.3401-3408.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shichijo S., Keicho N., Long H.T., Quy T., Phi N.C., Ha L.D., Ban V.V., Itoyama S., Hu C.-J., Komatsu N., Kirikae T., Kirikae F., Shirasawa S., Kaji M., Fukuda T., Sata M., Kuratsuji T., Itoh K., Sasazuki T. Tissue Antigens. 2004;64:600–607. doi: 10.1111/j.1399-0039.2004.00314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J., Wen J., Li J., Yin J., Zhu Q., Wang H., Yang Y., Qin E., You B., Li W., Li X., Huang S., Yang R., Zhang X., Yang L., Zhang T., Yin Y., Cui X., Tang X., Wang L., He B., Ma L., Lei T., Zeng C., Fang J., Yu J., Wang J., Yang H., West M.B., Bhatnagar A., Lu Y., Xu N., Liu S. Clin. Chem. 2003;49:1989–1996. doi: 10.1373/clinchem.2003.023184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang Y., Yang Z., Kong W., Nabel G.J. J. Virol. 2004;78:12557–12565. doi: 10.1128/JVI.78.22.12557-12565.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan P.K., Ng K.C., Chan R.C., Lam R.K., Chow V.C., Hui M., Wu A., Lee N., Yap F.H., Cheng F.W., Sung J.J., Tam J.S. Emerg. Infect. Dis. 2004;10:530–532. doi: 10.3201/eid1003.030493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan P.K., To W.K., Ng K.C., Lam R.K., Ng T.K., Chan R.C., Wu A., Yu W.C., Lee N., Hui D.S., Lai S.T., Hon E.K., Li C.K., Sung J.J., Tam J.S. Emerg. Infect. Dis. 2004;10:825–831. doi: 10.3201/eid1005.030682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guan M., Chen H.Y., Foo S.Y., Tan Y.J., Goh P.Y., Wee S.H. Clin. Diag. Lab. Immunol. 2004;11:287–291. doi: 10.1128/CDLI.11.2.287-291.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He Q., Chong K.H., Chng H.H., Leung B., Ling A.E., Wei T., Chan S.W., Ooi E., Kwang J. Clin. Diag. Lab. Immunol. 2004;11:417–422. doi: 10.1128/CDLI.11.2.417-422.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poon L.L., Chan K.H., Wong O.K., Yam W.C., Yuen K.Y., Guan Y., Lo Y.M., Peiris J.S. J. Clin. Virol. 2003;28:233–238. doi: 10.1016/j.jcv.2003.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poon L.L., Chan K.H., Wong O.K., Cheung T.K., Ng I., Zheng B., Seto W.H., Yuen K.U., Guan Y., Peiris J.S. Clin. Chem. 2004;50:67–72. doi: 10.1373/clinchem.2003.023663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi Y., Yi Y., Li P., Kuang T., Li L., Dong M., Ma Q., Cao C. J. Clin. Microbiol. 2003;41:5781–5782. doi: 10.1128/JCM.41.12.5781-5782.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corse E., Machamaer C.E. J. Virol. 2000;74:4319–4326. doi: 10.1128/JVI.74.9.4319-4326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Haan C.A., Vennema H., Rottier P.J. J. Virol. 2000;74:4967–4978. doi: 10.1128/JVI.74.11.4967-4978.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molekamp R., Spaan W.J. Virology. 1997;239:78–86. doi: 10.1006/viro.1997.8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narayanan K., Makino S. J. Virol. 2001;75:9059–9067. doi: 10.1128/JVI.75.19.9059-9067.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.P.J.M. Rottier, in The Coronavirus Membrane Protein, ed. by S.G. Siddell (Plenum Press, NY, 1995), pp. 115–139
- 22.Vennema H., Godecke G.J., Rossen J.W., Voorhout W.F., Horzinek M.C., Opstelten D.J., Rottier P.J. EMBO J. 1996;15:2020–2028. doi: 10.1002/j.1460-2075.1996.tb00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woods R.D., Wesley R., Kapke P.A. Adv. Exp. Med. Biol. 1987;218:493–500. doi: 10.1007/978-1-4684-1280-2_64. [DOI] [PubMed] [Google Scholar]
- 24.Yu L., Liu W., Schnitzlein W.M., Tripathy D.N., Kwang J. Avian Dis. 2001;45:340–348. doi: 10.2307/1592973. [DOI] [PubMed] [Google Scholar]
- 25.W.M. Yokoyama, in Production of Monoclonal Antibodies, ed. by J.E. Coligan, A.M. Kruisbeek, D.H. Margulies, E.M. Shevach, W. Strober (John, Wiley and Sons, Inc., NY, 1999), Sec. 16.1.1–16.1.17
- 26.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K.A., Petrescu S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z., Pei D., Jiang L., Song Y., Wang J., Wang H., Zhou D., Zhai J., Du Z., Li B., Qiu M., Han Y., Guo Z., Yang R. Clin. Chem. 2004;50:988–995. doi: 10.1373/clinchem.2004.031096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X., Shi Y., Li P., Li L., Yi Y., Ma Q., Cao C. Clin. Diagn. Lab. Immunol. 2004;11:227–228. doi: 10.1128/CDLI.11.1.227-228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delmas B., Rasschaert D., Godet M., Gelfi J., Laude H. J. Gen. Virol. 1990;71:1313–1323. doi: 10.1099/0022-1317-71-6-1313. [DOI] [PubMed] [Google Scholar]
- 30.Elia G., Fiermonte G., Pratelli A., Martella V., Camero M., Cirone F., Buonavoglia C. J. Virol. Meth. 2003;109:139–142. doi: 10.1016/S0166-0934(03)00064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han X., Bartlam M., Jin Y.H., Liu X., He X., Cai X., Xie Q., Rao Z. J. Virol. Meth. 2004;122:105–111. doi: 10.1016/j.jviromet.2004.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He Q., Du Q., Lau S., Manopo I., Lu L., Chan S.W., Fenner B.J., Kwang J. J. Virol. Meth. 2005;127:46–53. doi: 10.1016/j.jviromet.2005.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tripp R.A., Haynes L.M., Moore D., Anderson B., Tamin A., Harcourt B.H., Jones L.P., Yilla M., Babcock G.J., Greenough T., Ambrosino D.M., Alvarez R., Callaway J., Cavitt S., Kamrud K., Alterson H., Smith J., Harcourt J.L., Miao C., Razdan R., Comer J.A., Rollin P.E., Ksiazek T.G., Sanchez A., Rota P.A., Bellini W.J., Anderson L.J. J. Virol. Meth. 2005;128:21–28. doi: 10.1016/j.jviromet.2005.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duan J., Yan X., Guo X., Cao W., Han W., Qi C., Feng J., Yang D., Gao G., Jin G. Biochem. Biopys. Res. Commun. 2005;333:186–193. doi: 10.1016/j.bbrc.2005.05.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu H., Li L., Kao R.Y., Kou B., Wang Z., Zhang L., Zhang H., Hao Z., Tsui W.H., Ni A., Cui L., Fan B., Guo F., Rao S., Jiang C., Li Q., Sun M., He W., Liu G. J. Comb. Chem. 2005;7:648–656. doi: 10.1021/cc0500607. [DOI] [PubMed] [Google Scholar]