

Abstract
To establish a rapid and economical method for the expression of viral proteins in high yield and purity by Pichia pastoris, the S protein of the SARS-CoV was selected in this study. Six S glycoprotein fragments were expressed in Escherichia coli BL21 and yeast KM71H strains. After purification by affinity chromatography, the protein identities were confirmed by western blot analysis, N-terminal sequencing and mass spectrometry. The proteins expressed in E. coli were low in solubility and bound by GroEL. They still formed soluble aggregates even when the GroEL was removed by urea. The proteins expressed in P. pastoris were relatively soluble. The maximal yield of the RBD reached 46 mg/l with purity greater than 95%. Pull-down assay revealed that ACE2 was specifically captured from cell lysate, indicating that the RBD was biologically active. The glycosylated and deglycosylated RBD was then subjected to SEC and results showed that deglycosylated RBD formed soluble aggregates again. Taken together, pure and biological active RBD of the S protein could be expressed in P. pastoris, and the P. pastoris expression platform will be a good alternative for the expression of viral proteins, in particular, the highly glycosylated surface proteins that mediate the tissue tropism and viral entry.
Keywords: SARS-CoV, Spike, RBD, Pichia pastoris, ACE2
Introduction
In 2003, there were totally 8422 people worldwide, who suffered from severe acute respiratory syndrome (SARS), with 916 deaths reported from 32 countries. The SARS-CoV (SARS-coronavirus) is a single-stranded plus-sense RNA virus, approximately 30 kb in length with genomic sequence not closely resembling any of the previously characterized coronaviruses. The genome of SARS-CoV contains 15 putative open reading frames that encode four structural proteins, including the transmembrane spike (S) glycoprotein. The S protein is important for viral entry and defines host range and tissue tropism. Moreover, this protein can be divided into S1 and S2 domains, which are involved in cellular receptor interaction and membrane fusion respectively [1]. Amino acid residues 318–510 in the S1 domain was defined as the receptor binding domain (RBD) interacting with angiotensin converting enzyme 2 (ACE2), which is the SARS-CoV functional receptor [2, 3].
Expression of functional SARS-CoV proteins in high level and high purity is crucial for research to combat the possible future outbreak. Currently, most laboratories use the simple and economical bacterial expression system to produce viral proteins for structural and functional analysis. However, the lack of post-translation modifications, limited disulfide-bond formation and the absence of various chaperones often hinder the generation of properly folded and fully functional viral proteins, especially when the hosts of viruses are mammalian species.
To produce functionally active surface antigens for combating the future epidemics, one of the attractive possibilities is the yeast expression system. The methylotrophic yeast Pichia pastoris (P. pastoris) gives high yields of recombinant proteins, can be grown to high cell densities using defined minimal media and offers a cost-effective method for 13C-labelled protein production for NMR-based structural analyses [4]. Moreover, presence of α-signal sequence at the N-terminal end of the recombinant protein induces secretion to culture medium, leading to minimization of purification steps, improvement of recovery yield as well as reduction of the chance of protein degradation by endogenous proteases.
In order to compare the effectiveness of the Escherichia coli (E. coli) and P. pastoris systems in the expression of viral antigens, the S protein of the SARS-CoV was selected as the protein target. Six regions of the S protein were expressed in both E. coli and P. pastoris. Afterwards, the yields, purity and function of the recombinant proteins were compared. We found that the glycosylated proteins expressed in P. pastoris have much higher solubility and purity. Moreover, the RBD of the S protein can be produced in high yield and is functionally active.
Materials and methods
Cloning of DNA fragments into pGEX-6P-1 and pPICZα-A
Hydrophobicity and secondary structure of the S protein were predicted by Jpred (http://www.compbio.dundee.ac.uk/~www-jpred/). DNA encoding amino acid residues 13–672, 680–1192, 15–317, 318–510, 587–826 and 903–1187 of the S protein were amplified using the cDNA of SARS-CoV strain CUHK-Su10 as DNA template [5]. PCR amplification was performed as follows: initial denaturation at 94°C for 3 min, followed by 35 cycles (each at 94°C for 36 s, at 55°C for 45 s, at 72°C for 2–6 min) and a final extension at 72°C for 10 min. The PCR products were ligated into pGEX-6P-1 and pPICZα-A, followed by transforming into E. coli strain DH5α. Colony PCR, and sequencing using vector primers were performed to determine whether the cDNA was ligated into the correct sites.
Transformation of P. pastoris by electroporation
The competent cell preparation and the transformation of P. pastoris strain KM71H were performed as previously described [6]. In brief, 10 μg of each linearized plasmid (1 μg/l) was mixed with 80 μl of competent cells in an ice-cold 0.2 cm electroporation cuvette. After staying on ice for 5 min, electroporation was performed using Eppendorf electroporator 2510 with the following parameters: 1.25 kV/cm, 10 μF, 600 Ω and 5 ms. One milliliter of ice-cold 1 M sorbitol was immediately added to the competent cells, followed by incubation at 28°C for 90 min. The transformants were spread on Yeast Peptone Dextrose-agar plates with 100 mg/l zeocin and incubated at 28°C for three to ten days. Colonies with inserts were further streaked on Yeast Peptone Dextrose-agar plates with 500 mg/l, 800 mg/l and 1 g/l zeocin and incubated at 28°C for three to ten days.
Expression and purification of GST tagged S protein domains in E. coli
A single BL21 transformant was inoculated in LB medium and shaken at 37°C overnight until A600 reached 0.4–0.6. IPTG was added to a final concentration of 0.1 mM, followed by shaking overnight at 16°C. Harvested cell pellet was resuspended in phosphate buffer saline (PBS) and disrupted by using the Sonoplus ultrasonic homogenizer system (Bandeln). After centrifugating at 48,000g for 30 min, the supernatant was mixed with glutathione sepharose and gently shaken for 1 h. The resin was washed by PBS and then eluted by PBS with 10 mM reduced glutathione sepharose. To remove the GroEL bound on the S protein domains, PBS with 0.25–2.5 M of urea was used for washing.
Expression and purification of S protein domains in P. pastoris
A single KM71H transformant with each plasmid was inoculated in 10 ml of Buffered Glycerol-complex Medium (BMGY) and incubated at 28°C overnight until A600 reached 2–6. The inoculum was transferred to 1 l of BMGY and further shaken at 28°C overnight until A600 reached 2–6. The yeast was harvested and then resuspended in 100 ml of Buffered Methanol-complex Medium (BMMY). Methanol was added to a final concentration of 5 ml/l to induce the protein expression. The culture was incubated at 28°C and methanol was added every 24 h to compensate for evaporation. To harvest yeast cells from the culture, the culture was centrifuged at 48,000g for 30 min. After incubating the supernatant with the His-bind resin, the resin was washed and then eluted by PBS with 50 and 500 mM imidazole respectively. The Bradford assay was performed to determine the protein concentration in the eluate.
SDS-PAGE and immunoblot
The protein in the SDS-PAGE gel was transferred to a PVDF membrane using the Semi-dry transfer Units. The membrane was probed by 1:2000 diluted mouse anti-c-myc antibody (Santa Cruz), 1:2000 diluted mouse anti-His antibody (Santa Cruz), 1:2000 diluted mouse anti-glutathione S-transferase (GST) antibody (Santa Cruz) or 1:500 diluted mouse anti-hACE2 primary antibody (Roger), followed by 1:2000 diluted sheep anti-mouse antibody (DAKO). After immersing the membrane in Western Lightning Chemiluminescence Reagents, signal was captured by an X-ray film.
N-terminal sequencing
The PVDF membrane was immersed in Coomassie brilliant blue staining solution for 30 s, followed by destaining solution until bands were observed. The membrane containing approximately 100 pM of the target protein was excised and the extracted protein was sequenced by Precise Peptide Sequencing System 492.
Mass spectrometry
The SDS-PAGE gel containing the target protein was excised and sliced into small pieces. The sample was shaken in destaining solution until decolourization was complete, followed by incubating in 100 μl of 200 mM (NH4)2CO3 and 100 μl of acetonitrile for 10 and 5 min respectively. After drying by vacuum, the sample was transferred to 5 μl of 50 mM (NH4)2CO3 containing 100 ng of trypsin (Promega) and kept on ice for 30 min, followed by incubating at 30°C overnight for digestion. Three microlitres of supernatant was crystallized by 0.5 μl of saturated cinnamic acid and then analysed by the 4700 Proteomics Discovery System (Applied Biosystems).
Size exclusion chromatography
The protein was applied to the equilibrated HiLoad 16/60 Superdex 200 preparative grade column (GE Healthcare) with a flow rate of 3 ml/min. Volume of each fraction was 2 ml. The native size of the target protein was determined by comparing with the elution volumes of standard proteins including Blue dextran (Void volume), Ferritin (440 kDa), Ovalbumin (43 kDa) and Ribonuclease A (13.7 kDa) (GE Healthcare).
Cell culture and pull-down assay
Vero E6 cell was cultivated in Dulbecco’s Modified Eagle Medium (pH 7.4) with 100 ml/l fetal bovine serum and 10 ml/l penicillin–streptomycin at 37°C. To harvest the cells, cells with 90% confluence were detached by trypsin digestion. After washing with PBS, the cells were stored at −80°C for future use. To perform the pull-down assay, 5 × 106 Vero E6 cells were lysed by 5 ml of lysis buffer. The lysate was centrifuged at 12,000g for 10 min at 4°C. The supernatant was mixed with His-bind resin binding to 100 μg of S1c and shaken gently for 2 h at 4°C. The eluates were then examined by western blot analysis.
Deglycosylation
The S1c protein domain expressed in P. pastoris was deglycosylated by peptide-N-glycanase F (PNGase F) (New England BioLabs), which removes carbohydrate residues from proteins by hydrolysis of the bonding between N-glycan and asparagine residues. Two hundred nanolitres of PNGase F was added to 10 μg of protein, and the mixture was then incubated at 37°C for 3 h.
Results
Low solubility and aggregation of S protein fragments expressed in E. coli
Based on the bioinformatics analysis of the hydrophobicity and secondary structure of the S protein, the protein was divided into six overlapping regions as shown in Fig. 1. Complementary DNA corresponding to these six regions (S1a, S1b, S1c, S2a, S2b and S2c) were independently cloned into E. coli and P. pastoris expression vectors. Expression of the six GST-tagged S protein fragments in E. coli was induced by 0.1 mM IPTG. Soluble fractions were isolated and then purified by affinity chromatography. SDS-PAGE results revealed that S1b, S1c, S2b and S2c were successfully expressed but their solubility was moderately low. After purification, the protein amounts were between 20 μg/l and 100 μg/l. The expression levels and solubility of both S1a and S2a were very low (Fig. 2). Optimizing the amounts of IPTG, expression temperature and expression time could not improve the expression levels and the solubility of the proteins (data not shown).
Fig. 1.
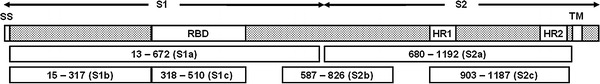
Schematic Diagram of SARS-CoV S glycoprotein. S glycoprotein can be divided into S1 and S2 domains that contain signal sequence (SS), receptor binding domain (RBD), heptad repeat 1 (HR1), heptad repeat 2 (HR2) and transmembrane region (TM). According to the locations of functional domains and the secondary structure prediction, six protein regions containing amino acid residues 13–672 (S1a), 15–317 (S1b), 318–510 (S1c), 680–1192 (S2a), 587–826 (S2b) and 903–1187 (S2c) were chosen for cloning and expression
Fig. 2.
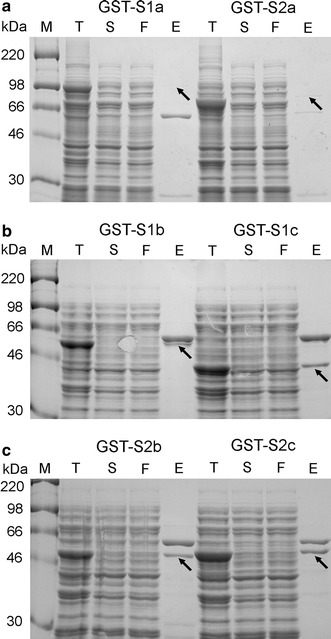
Expression of S protein domains in E. coli. SDS-PAGE analysis of expression of six GST-tagged S protein domains (GST-S1a, GST-S1b, GST-S1c, GST-S2a, GST-S2b and GST-S2c) in E. coli. Lane M: prestained protein markers; lane T: total lysate of the induced culture of transformed E. coli; lane S: supernatant of the induced culture of transformed E. coli; lane F: flow through after purification by the glutathione sepharose; lane E: proteins eluted from the glutathione sepharose. Arrows indicate the location of recombinant S protein fragments
Moreover, a protein of about 60 kDa was detected in all eluates of different S protein domains. Mass spectrometry analysis indicated that it is a bacterial chaperone called GroEL. After screening a number of chemicals and detergents, we found that urea at a concentration higher than 1.75 M can be used to remove the GroEL from the S1c (Fig. 3). However, the size exclusion chromatography (SEC) results revealed that the purified S1c protein domain formed aggregates in the absence of GroEL (data not shown). Taken together, functional S protein domains could not be successfully produced in the E. coli expression system.
Fig. 3.
Removal of GroEL by urea. SDS-PAGE analysis of GroEL-bound S1c protein domain treated by urea. The results suggest that GroEL was removed when GST-S1c was washed by urea at a concentration higher than 1.75 M. Lane M: prestained protein markers; lanes 1–9: GroEL-bound S1c protein domain treated with different concentrations of urea; lane 1: 0 M; lane 2: 0.5 M; lane 3: 0.75 M; lane 4: 1 M; lane 4: 1.25 M; lane 6: 1.5 M; lane 7: 1.75 M; lane 8: 2 M; lane 9: 2.5 M
Expression of S protein domains in P. pastoris
After transformation and screening by various zeocin concentrations, no colony could be found on the plate with 1 g/l zeocin whereas a few colonies survived on the plate with 800 mg/l zeocin. To express the S protein fragments in P. pastoris, a single positive transformant, which could survive on the plate with 800 mg/l zeocin, of each plasmid was inoculated until A600 reached 2–6. The expression was induced by adding methanol to a final concentration of 5 ml/l every 24 h. To monitor the expression level at different time points, 1 ml of the medium after 0, 24, 48, 72, 96, 120, 144 and 240 h of induction were purified by His-binding resin. The comparison the yield and properties of different protein domains expressed in E. coli and P. pastoris is summarized in Table 1. Among all S protein domains, the S1c has the highest solubility. A major band of about 35 kDa with a smear of higher molecular weight was detected by SDS-PAGE (Fig. 4a). The protein yield of S1c reached a maximum after 144 h of induction and the maximal yield was 46 mg/l. The protein identity was confirmed by western blot analysis using anti-c-myc and anti-His antibodies, N-terminal sequencing as well as mass spectrometry (data not shown). Based on the results from SEC, the purity of the S1c was higher than 95%.
Table 1.
The comparison of the expression of S protein domains in E. coli and P. pastoris
| Name | a. a. | Expressed in E. coli | Expressed in P. pastoris |
|---|---|---|---|
| S1a | 13–672 | Very low solubility, bound by GroEL | Fail to express |
| S1b | 15–317 | Low solubility, bound by GroEL | Low expression level, soluble, secretory |
| S1c | 318–510 | Low solubility, bound by GroEL | High expression level, soluble, secretory |
| S2a | 680–1192 | Very low solubility, bound by GroEL | Low expression level, insoluble |
| S2b | 587–826 | Low solubility, bound by GroEL | Fail to express |
| S2c | 903–1187 | Low solubility, bound by GroEL | Low expression level, soluble, non-secretory |
The yield and properties of different S protein domains expressed in E. coli and P. pastoris is summarized
Fig. 4.
Expression of S1c domain in P. pastoris. SDS-PAGE analysis of expression of (a) S1c and (b) S1b, as well as western blotting analysis of expression of (c) S2a and S2c of protein domains in P. pastoris. Lane M: prestained protein markers; Numbers of subsequent lanes: the number of hours after induction; lane T: total lysate; lane S: soluble fraction; lane E: eluent
At least three more protein domains spanning the amino acid residues 15–317, and 680–1187 and 903–1187 were successfully expressed. As S1c, the protein yield of S1b reached a maximum after 144 h of induction but the expression level was low (Fig. 4b). S2a and S2c were only to be found in total lysate and soluble fraction of total lysate respectively, implying that S2a was insoluble while S2c was soluble but unable to be secreted (Fig. 4c). Because of the high expression level of S1c, this protein domain was chosen for subsequent functional analysis.
Pull-down of ACE2 by the S1c protein domain expressed in P. pastoris
ACE2 is the functional receptor of SARS-CoV, which can interact with RBD of the S-protein. The amino acid sequence of the RBD is the same as S1c. In order to determine whether S1c was properly folded and biologically active, pull-down assay using the S1c as bait was undergone, followed by western blot analysis using anti-human ACE2 antibody to detect the presence of ACE2. As shown in Fig. 5, the ACE2 of 120 kDa in size was detected only in the pull-down fraction, revealing that the S1c expressed in P. pastoris could specifically interact with ACE2.
Fig. 5.
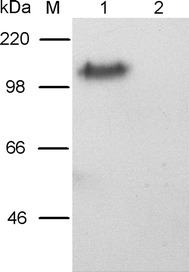
Interaction between S1c and ACE2. Pull-down assay of S1c protein domain expressed by in P. pastoris. Western blot analysis using anti-hACE2 antibody was used to detect ACE2. Lane M: prestained protein markers; lane 1: purified S1c protein domain bound to His-bind resin was used as bait to incubate with protein lysate of VeroE6 cells; lane 2: His-bind resin in the absence of S1c was used as bait to incubate with protein lysate of VeroE6 cells
The deglycosylated S1c with a higher molecular weight
The apparent and calculated molecular weights of the S1c were 35 kDa and 24 kDa, respectively. Moreover, the purified S1c was in the form of a smear rather than a sharp band. This might be caused by the presence of post-translational modifications, including glycosylation with side chains in various lengths. To confirm this, S1c was treated by PNGase F. SDS-PAGE analysis showed that both the major band and the smear were shifted to a band of 29 kDa in size (Fig. 6), which was still higher than the calculated molecular weight based on the amino acid sequence. The possibility of incomplete removal of α-signal peptide was ruled out since the N-terminal sequencing result showed that S1c was successfully cleaved by Kex2 (data not shown). Moreover, in the peptide mass fingerprint, the peak of C-terminal peptide was detected.
Fig. 6.
De-N-glycosylation of S1c by PNGase F. SDS-PAGE analysis of glycosylated and de-N-glycosylated S1c protein domain expressed in P. pastoris. Lane M: prestained protein marker; lane 1: S1c protein domain expressed before de-N-glycosylation; lane 2: S1c protein domain after treating with PNGase F
Aggregation of the deglycosylated S1c
When the native sizes of the glycosylated and deglycosylated S1c were determined by SEC, the glycosylated S1c was eluted in a board peak (Fig. 7a). By comparing with the standards, the native size of the major S1c was 60 kDa, suggesting that the glycosylated S1c was monomeric (Fig. 7b). After deglycosylation by PNGase F, S1c was eluted in the void volume, indicating that S1c formed aggregates again (Fig. 7c). This suggests that the glycosylation of S1c is very crucial for the solubility, and very probably for the proper folding, too, of the protein.
Fig. 7.
Glycosylated S1c is monomeric while deglycosylated S1c forms aggregates. a Elution profile of glycosylated S1c showed that a board peak between 50 and 90 ml was detected while the peak height reached a maximum at 78 ml. b Fractions of glycosylated S1c separated by SEC were analyzed by SDS-PAGE. The major band of 35 kDa was eluted in 80–85 ml. Lane M: prestained protein markers; subsequent numbers: the numbers of fractions collected during the analysis of S1c by SEC. c When the deglycosylated S1c was analysed by SEC, the protein was eluted at void volume, suggesting that the deglycosylated protein formed soluble aggregates
Discussion
Since the emergence of molecular biology, the E. coli expression system has offered an excellent platform for the rapid and economical protein production with a high yield. However, the expression of highly glycosylated viral surface antigens in the bacterial expression system is always a difficult task. In the absence of hydrophilic glycosylation, the expressed proteins are either insoluble or aggregated. Despite the fact that the baculovirus expression system is widely used for the expression of glycosylated mammalian proteins, the system is hampered by three very slow and tedious procedures, namely, generation of high titer baculovirus stock, determination of the virus titer and discovery of the best conditions for protein expression [7]. The mammalian expression system is a newly emerging attractive option but it was generally regarded as being cumbersome, tedious and expensive [8].
In this study, we explore the possibility of using yeast expression system as an efficient and low-cost option to supplement the bacterial system. Previous reports have demonstrated the feasibility of using the P. pastoris as host to express the SARS-CoV proteins, including the membrane [9] and the nucleocapsid [10, 11] protein. Although Lu et al. [12] expressed a small fragment of the S1 domain (amino acid residues 251–561) in the P. pastoris, they had not demonstrated the ability of this fragment to specifically pull-down ACE2 from cell lysate. In this study, we report the expression of the S protein domains in E. coli and P. pastoris.
When the S protein domains were expressed in E. coli, the proteins formed aggregates or even could not be expressed. Moreover, all soluble GST-tagged S protein domains were bound by GroEL, which was irremovable by affinity chromatography. GroEL is an endogenous bacterial chaperone preventing irreversible protein aggregation as well as assisting protein folding into the native conformation [13]. Assistance of protein folding by GroEL is a common phenomenon because approximately 10% of newly synthesized polypeptides in E. coli were the substrates [14]. When the GroEL was removed from S1c by urea, the protein domain formed soluble aggregates. Our results suggest that properly folded and hence functional protein domain of S protein cannot be obtained in the E. coli expression system.
Regarding the expression in P. pastoris, at least five out of six domains have been successfully expressed. The yield of the most soluble S1c domain can attain 46 mg/l. A possible reason leading to the difference in expression levels of S protein domains in P. pastoris is the codon bias. It has been reported that the expression level of a protein encoded by rare codons can be increased by five-to-ten folds after codon optimization [15, 16]. When the nucleotide sequences encoding S protein domains were submitted to two databases for rare codon analysis, we found that the expression levels are negatively related to the number of rare codons (Table 2). S1c contains the lowest number of rare codons, followed by S2c and S1b. The S2b is the only exceptional case, in which the number of rare codons was similar to that of S1b but the expression level was much lower. However, when we consider only the codons with the lowest frequencies (2.0 or 2.2/1000) in the database of the “Rare codons’ Search”, S2b contains two of these codons whereas other fragments contain one or less. This might explain why S2b has such a low expression level in P. pastoris. Taken together, we believe that the expression levels of S protein domains could be improved after codon optimization.
Table 2.
Rare codons of S protein fragments expressed in P. pastoris
| No. of a. a. residues | Codon | a. a. residues encoded | Frequency (Database 1) | Frequency (Database 2) | a. a. residues of S protein | |
|---|---|---|---|---|---|---|
| 18 | cgg | Arg | 2 | 5 | S1a | S1b |
| 19 | ugc | Cys | 4.2 | – | ||
| 39 | ggg | Gly | – | 10 | ||
| 68 | ggg | Gly | – | 10 | ||
| 113 | ucg | Ser | – | 8 | ||
| 126 | cga | Arg | 4.4 | 10 | ||
| 159 | ugc | Cys | 4.2 | – | ||
| 169 | ucg | Ser | – | 8 | ||
| 183 | cga | Arg | 4.4 | 10 | ||
| 192 | ggg | Gly | – | 10 | ||
| 194 | cuc | Leu | – | 8 | ||
| 264 | cuc | Leu | – | 8 | ||
| 286 | cuc | Leu | – | 8 | ||
| 288 | ugc | Cys | 4.2 | – | ||
| 292 | agc | Ser | – | 9 | ||
| 355 | cuc | Leu | – | 8 | S1c | |
| 366 | ugc | Cys | 4.2 | – | ||
| 378 | ugc | Cys | 4.2 | – | ||
| 398 | gcg | Ala | 3.8 | 5 | ||
| 467 | ugc | Cys | 4.2 | – | ||
| 507 | ccg | Pro | 4.2 | 10 | ||
| 532 | cuc | Leu | – | 8 | ||
| 563 | cga | Arg | – | 10 | ||
| 563 | cga | Arg | 4.4 | – | ||
| 576 | ugc | Cys | 4.2 | 10 | ||
| 603 | ugc | Cys | 4.2 | – | S2b | |
| 615 | cuc | Leu | – | 8 | ||
| 620 | cgc | Arg | 2.2 | 6 | ||
| 648 | ugc | Cys | 4.2 | – | ||
| 670 | agc | Ser | – | 9 | ||
| 703 | agc | Ser | – | 9 | S2a | |
| 725 | ugc | Cys | 4.2 | – | ||
| 736 | cuc | Leu | – | 8 | ||
| 740 | agc | Ser | – | 9 | ||
| 742 | ugc | Cys | 4.2 | – | ||
| 749 | cuc | Leu | – | 8 | ||
| 758 | cgc | Arg | 2.2 | 6 | ||
| 804 | cuc | Leu | – | 8 | ||
| 810 | cuc | Leu | – | 8 | ||
| 822 | ugc | Cys | 4.2 | – | ||
| 831 | cuc | Leu | – | 8 | ||
| 834 | gcg | Ala | 3.8 | 5 | ||
| 847 | cuc | Leu | – | 8 | ||
| 898 | cuc | Leu | – | 8 | ||
| 902 | caa | Gln | – | 5 | ||
| 912 | gcg | Ala | 3.8 | – | S2c | |
| 949 | agc | Ser | – | 9 | ||
| 964 | ucg | Ser | – | 8 | ||
| 965 | cga | Arg | 4.4 | 10 | ||
| 971 | gcg | Ala | 3.8 | 5 | ||
| 985 | agc | Ser | – | 9 | ||
| 1039 | ccg | Pro | 4.2 | 10 | ||
| 1060 | gcg | Ala | 3.8 | 5 | ||
| 1167 | cgc | Arg | 2.2 | 6 | ||
| 1168 | cuc | Leu | – | 8 | ||
| 1179 | cuc | Leu | – | 8 | ||
The nucleotide sequencing of S protein was analysed by “Rare codons’ Search” (http://molbiol.ru/eng/scripts/01_11.html) and “Graphical Codon Usage Ana lyzer” (http://gcua.schoedl.de/) that are represented by database 1 and 2, respectively. The total frequency is 1000. Bold numbers in database 1 indicate the frequencies are 2 or 2.2
Results of SEC showed that the native size of S1c was between 27 and 478 kDa, while the peak height was in the location of 60 kDa. Thus, we can conclude that S1c expressed in P. pastoris appears as a monomer protein. The native size of the smear determined by SEC was much higher than that determined by SDS-PAGE. It might be caused by the extension of carbohydrate side chains, which could further increase the apparent size of a globular protein. When the S1c domain was deglycosylated, it formed soluble aggregates, indicating that the glycosylation is essential to prevent the protein aggregation. The size of de-N-glycosylated S1c was still 5–10 kDa higher than the calculated molecular weight. A probable reason is the presence of O-linked mannose residues linking to serine or threonine residues. Another possibility is the incomplete de-N-glycosylation by PNGase F. More N-linked glycans can be removed by using both PNGase F and other N-glycanases such as Endo H and Endo F [17]. To confirm that the protein domains expressed in P. pastoris are functionally active, we have performed the ACE2 pull-down from VeroE6 cell lysate using the glycosylated S1c domain. The results have unambiguously showed that the protein domain has the native fold that can enable its specific binding to the receptor.
Six SARS-CoV S protein domains were expressed in E. coli and P. pastoris. Although some of the protein domains could be expressed in E. coli, the proteins were associated with GroEL. After the removal of GroEL by 2 M urea in PBS, the proteins formed aggregates that cannot be used for any functional analysis. On the other hand, at least three out of the six protein domains could be expressed in P. pastoris. Notably, two of them were secreted to the culture medium. The S1c domain produced in P. pastoris is biologically active because it could selectively pull-down the functional receptor ACE2 from the VeroE6 protein lysate. Glycosylation is important for the folding and therefore the function of the protein because deglycosylated S1c domain formed misfolded protein aggregates. The results reported here suggest that the P. pastoris expression system is an excellent alternative to express active viral antigens with extensive posttranslational modifications. The antigens can be subsequently used for antibody and vaccine production, receptor identification and other functional characterization.
Acknowledgements
This project team is supported by the Research Fund for the Control of Infectious Diseases (RFCID) from the Health, Welfare and Food Bureau of the Hong Kong SAR Government.
References
- 1.Yeung KS, Yamanaka GA, Meanwell NA. Med. Res. Rev. 2006;26:414. doi: 10.1002/med.20055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. Nature. 2003;426:450. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wong SK, Li W, Moore MJ, Choe H, Farzan M. J. Biol. Chem. 2004;279:3197. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daly R, Hearn MT. J. Mol. Recognit. 2005;18:119. doi: 10.1002/jmr.687. [DOI] [PubMed] [Google Scholar]
- 5.Tsui SKW, Chim SS, Lo YM, Chinese University of Hong Kong Molecular SARS Research Group N. Engl. J. Med. 2003;349:187. doi: 10.1056/NEJM200307103490216. [DOI] [PubMed] [Google Scholar]
- 6.Tan W, Gou DM, Tai E, Zhao YZ, Chow LM. Arch. Biochem. Biophys. 2006;452:119. doi: 10.1016/j.abb.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 7.Philipps B, Rotmann D, Wicki M, Mayr LM, Forstner M. Protein Expr. Purif. 2005;42:211. doi: 10.1016/j.pep.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 8.Geisse S, Henke M. J. Struct. Funct. Genomics. 2005;6:165. doi: 10.1007/s10969-005-2826-4. [DOI] [PubMed] [Google Scholar]
- 9.Han X, Bartlam M, Jin YH, Liu X, He X, Cai X, Xie Q, Rao Z. J. Virol. Methods. 2004;122:105. doi: 10.1016/j.jviromet.2004.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao S, Wang H, Luhur A, Wong SM. J. Virol. Methods. 2005;130:83. doi: 10.1016/j.jviromet.2005.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu RS, Yang KY, Lin J, Lin YW, Zhang ZH, Zhang J, Xia NS. World J. Gastroenterol. 2004;10:3602. doi: 10.3748/wjg.v10.i24.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu H, Yang G, Fei X, Guo H, Tan Y, Chen H, Guo A. Virus Genes. 2006;33:329. doi: 10.1007/s11262-006-0072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ewalt KL, Hendrick JP, Houry WA, Hartl FU. Cell. 1997;90:491. doi: 10.1016/S0092-8674(00)80509-7. [DOI] [PubMed] [Google Scholar]
- 14.Lin Z, Rye HS. Crit. Rev. Biochem. Mol. Biol. 2006;41:211. doi: 10.1080/10409230600760382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu S, Li L, Qiao J, Guo Y, Cheng L, Liu J. Protein Expr. Purif. 2006;47:249. doi: 10.1016/j.pep.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 16.Xiong AS, Yao QH, Peng RH, Han PL, Cheng ZM, Li Y. J. Appl. Microbiol. 2005;98:418. doi: 10.1111/j.1365-2672.2004.02476.x. [DOI] [PubMed] [Google Scholar]
- 17.Molhoj M, Ulvskov P, Dal Degan F. Plant Physiol. 2001;127:674. doi: 10.1104/pp.010269. [DOI] [PMC free article] [PubMed] [Google Scholar]