

Abstract
Rabies, an acute progressive encephalomyelitis caused by viruses in the genus Lyssavirus, is one of the oldest known infectious diseases. Although dogs and other carnivores represent the greatest threat to public health as rabies reservoirs, it is commonly accepted that bats are the primary evolutionary hosts of lyssaviruses. Despite early historical documentation of rabies, molecular clock analyses indicate a quite young age of lyssaviruses, which is confusing. For example, the results obtained for partial and complete nucleoprotein gene sequences of rabies viruses (RABV), or for a limited number of glycoprotein gene sequences, indicated that the time of the most recent common ancestor (TMRCA) for current bat RABV diversity in the Americas lies in the seventeenth to eighteenth centuries and might be directly or indirectly associated with the European colonization. Conversely, several other reports demonstrated high genetic similarity between lyssavirus isolates, including RABV, obtained within a time interval of 25–50 years. In the present study, we attempted to re-estimate the age of several North American bat RABV lineages based on the largest set of complete and partial glycoprotein gene sequences compiled to date (n = 201) employing a codon substitution model. Although our results overlap with previous estimates in marginal areas of the 95 % high probability density (HPD), they suggest a longer evolutionary history of American bat RABV lineages (TMRCA at least 732 years, with a 95 % HPD 436–1107 years).
Electronic supplementary material
The online version of this article (doi:10.1007/s11262-013-0952-9) contains supplementary material, which is available to authorized users.
Keywords: Rabies, Virus, Glycoprotein, Viral evolution, Bat rabies, Phylogenetic analysis
Introduction
Rabies is characterized clinically by acute progressive encephalomyelitis and is one of the oldest diseases known to humankind. The dangers of dog bites were mentioned as early as the twenty-third century bc in the pre-Mosaic Eshnunna Code of Mesopotamia [1]. While different etiologies have been attributed to the development of rabies historically, it is now understood that all viruses classified in the genus Lyssavirus (family Rhabdoviridae) can cause the disease. Though all mammals are susceptible to rabies, the major natural reservoir hosts of lyssaviruses are carnivores and bats [2]. Rabies virus (RABV) is the type species of genus Lyssavirus [3]. RABV is distributed in carnivores worldwide, except Antarctica and several insular territories. However, its presence in bats is observed exclusively in the New World. Lyssaviruses of other species have more limited distribution in areas of the Old World. The most prominent diversity of lyssaviruses has been found in Africa. Moreover, either in Africa or other continents of the Old World, the majority of divergent lyssaviruses has been documented in bats. For this reason, it was proposed that Africa was the continent where this viral genus initially evolved [4], and bats were suggested as the primary evolutionary reservoir of lyssaviruses [5]. Yet, it is unclear why RABV is not present in the Old World bats, nor is it completely understood how the New World bats acquired this virus [6].
One problem is that evolutionary estimates based on molecular clock analysis indicate a relatively young age of lyssaviruses, including RABV [5, 7–9]. For example, based on a limited number of glycoprotein (G) sequences, the time of the proposed host switch of lyssaviruses from bats to carnivores was suggested to have occurred only approximately 888–1,459 years ago [5]. One molecular clock estimate performed on complete and partial nucleoprotein (N) gene sequences suggested that the time of the most recent common ancestor (TMRCA) for current bat RABV diversity in the Americas lies in the seventeenth century, although with broad 95 % high probability density (HPD) values, ranging between the years 1254 and 1782 [7]. Similarly, based on RABV N gene sequences, the age of genetic diversity of RABV was estimated as broadly similar in bats, with mean estimates of 118–233 years [8]. However, a discordant estimation of 739 years was obtained in the same study for phosphoprotein (P) gene sequences. This estimation included very broad HPD values (114–2291 years), likely due to greater genetic variability of the P in comparison to the N as well as limitations of the dataset [8]. In another study, the TMRCA for carnivore RABV variants was estimated at 761 years (95 % HPD 373–1222 years), whereas the TMRCA for bat RABV lineages was estimated at only 180 years (95 % HPD 69–342 years), based on limited numbers of N and G gene sequences of bat RABV samples [9]. Unfortunately these studies did not indicate whether they employed a simple substitution model or codon-based models. Superiority of the latter was demonstrated for ancient viruses [10].
In contrast, several studies reported a high genetic similarity between lyssaviruses of certain phylogenetic lineages, isolated within time intervals of 25–50 years [11–13]. As there was no sufficient dataset of older viral gene sequences from the same phylogenetic lineages for comparison, such records were not used for adjustments of age estimates for viral lineages. However, by inference, they suggest that studies performed on sequences of recent RABV isolates, or estimates based on limited numbers of sequences, may lack resolution.
In the present study, we re-estimated the age of several North American bat RABV lineages based on the largest set of complete and partial G gene sequences analyzed to date implementing a codon-based substitution model. After the P, the G is the most variable of the five structural lyssavirus proteins, likely because it is responsible for significant virus-host interactions and adaptation to new species during host shifts [14, 15]. Nevertheless, no significant or substantial positive selection was detected in the G and other RABV genes to date. The majority of codons are subjected to purifying selection. However, the G is neither as strongly subjected to purifying selection as the N, nor is such hypervariable as non-coding regions of RABV genome [5, 8, 9, 15]. Therefore it is a good candidate for evolutionary analysis, particularly in comparison with the results obtained for other RABV genes in previous studies.
Materials and methods
Brain tissue samples from rabid animals were obtained via routine surveillance activity of the CDC (Atlanta, GA, USA), as well as via national and international requests in the framework of activity of the World Health Organization Collaborating Center for Reference and Research on Rabies. On several occasions, where the quality of the original brain sample was poor (i.e., sample deterioration or a limited amount of tissue which would not allow for reliable amplification of viral G by RT-PCR), a single suckling mouse brain passage was performed.
Total RNA was extracted from infected brain tissues using TRIZol reagent (Invitrogen). Primers were designed according to the RABV gene sequences available from GenBank. The RT-PCR was performed as described elsewhere [13]. In brief, cDNA was obtained during reverse transcription with a sense primer (90 min at 42 °C) in the presence of dNTPs and RT AMV (Roche Diagnostics Corp) and subjected to 40 PCR cycles: 94 °C, 30 s; 37 °C, 30 s; 72 °C, 90 s supplemented by a final extension for 10 min at 70 °C in the presence of both sense and anti-sense primers and Taq polymerase (Roche Molecular Systems Inc.). The RT-PCR products were purified and subjected to direct sequencing with subsequent processing on an ABI 3730 DNA Sequencer (Applied Biosystems). The complete G sequences were assembled and aligned using the Bio Edit program [17]. The dataset was supplemented with dated complete and partial RABV G sequences available from GenBank.
Positive selection analyses were performed under the single likelihood ancestor counting (SLAC), fixed effect likelihood (FEL), mixed effects model of episodic selection (MEME), and fast unbiased Bayesian approximation (FUBAR) models, implemented in the Datamonkey software (http://datamonkey.org). Based on the Bayesian information criterion and Akaike criterion obtained in Mega, v.5.1. [18], we used the generalized time-reversible nucleotide substitution model with gamma distribution and invariant sites (GTR+I+Г 4).
Rates of nucleotide substitutions (site/year) and TMRCA were estimated using the Bayesian Markov chain Monte Carlo (MCMC) method available in the BEAST v.1.7.5 [19]. As described above, we used the model GTR+I+Г 4 with parameters optimized during multiple runs. Based on the Bayes factor of likelihoods estimated in BEAST during preliminary runs, the exponential population growth and constant population size models were almost equally favored over the Bayesian skyline and Bayesian skyride models. The analysis was implemented under a relaxed uncorrelated log-normal molecular clock. Given that purifying selection was predominant in the previous studies [6, 7] and in our analysis, we linked substitution rates for the first and second codon positions (CP1+2) and allowed independent rates in CP3. In addition, Bayes factor favored this model over the model where partitioning in the codon positions was set to off. Two independent MCMC estimations were run for 70 million generations each to reach convergence, with samples from the posterior drawn every 7,000 generations following a burn-in of 20 %. The results from the two runs were combined to generate a maximum clade credibility tree and divergence time summaries.
Results
The RABV G sequences used in this study are listed in Table S1. Phylogenetic reconstruction (Fig. 1) revealed that we encompassed the majority of the previously described [15] RABV lineages associated with bats in North America. Each lineage was species-specific except the lineage MY associated with Myotis bats. Bats in this genus are difficult for conventional phenotypic identification, and to avoid potential misinterpretations we joined all RABV sequences originating from Myotis spp. bats in one lineage.
Fig. 1.
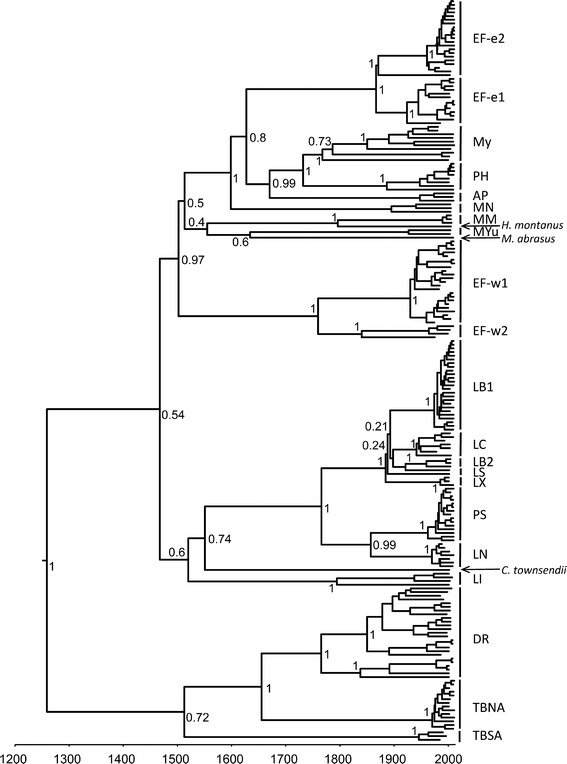
Bayesian tree of RABV G sequences analyzed in the present study. Posterior probabilities are shown for key nodes, and timeline (in years) is present at the bottom. Lineage abbreviations: EF Eptesicus fuscus (e1 and e2 represent two eastern lineages, whereas w1 and w2 represent two western lineages); My Myotis spp.; PH Parastrellus hesperus; AP Antrozous pallidus; MN Myotis nigricans; MM Molossus molossus; MYu Myotis yumanensis; LB Lasiurus borealis; LC Lasiurus cinereus; LS Lasiurus seminolus; LX Lasiurus xanthinus; PS Perimyotis subflavus; LN Lasionycteris noctivagans; LI Lasiurus intermedius; DR Desmodus rotundus; TBNA Tadarida brasiliensis (North America); TBSA T. brasiliensis (South America)
The mean substitution rate across the dataset was 1.56–1.78 × 10−4/site/year (95 % HPD 1.46–2.39 × 10−4), in general agreement with previously published data for evolution rates of RABV genes although it was somewhat in the low range [7–9, 16]. The mean ratio of non-synonymous to synonymous mutations (dN/dS) in the dataset was 0.139. No positive selection was detected under the SLAC, FEL, and FUBAR models. The majority of codons (~75 %) demonstrated strong purifying selection or evolved neutrally. The MEME model suggested that codons 240, 252, and 429 within the G ectodomain were subjected to statistically significant episodic positive selection (P < 0.05). However, for codons 240 and 252 non-synonymous substitutions were observed exclusively on the tips of the tree, indicative of false positives with respect to host adaptation. Only codon position 429 revealed the presence of non-synonymous substitutions along internal branches. Specifically, the majority of RABV isolates from lasiurine bats, such as red bat (Lasiurus borealis), hoary bat (L. cinereus), Seminole bat (L. seminolis), Western yellow bat (L. xanthinus; lineages LB, LC, LS, LX), and from the tri-colored bat (Perimyotis subflavus; lineage PS) harbored substitution G429D, whereas viruses from the silver-haired bat (Lasionycteris noctivagans; lineage LN) harbored substitution G429E. Nevertheless, since no further species-specific substitutions were identified for the viruses from lasiurine bats, and a more phylogenetically distanced lineage LI, associated with the Northern yellow bat (L. intermedius) did not harbor it at all, we consider this signal as false positive, likely resulted from a neutral point-mutation at the base of the lineage.
The TMRCA estimates of bat-associated RABV lineages under different demographic tree priors were distinct, although significantly overlapping within the 95 % HPD (Table 1). As the most conservative interpretation, we consider the result obtained under the exponential population growth model which suggested TMRCA of 732 years with 95 % HPD 436–1107 years. The TMRCA for the cluster that joins RABV lineages associated with vampire bats (Desmodus rotundus; lineage DR) and Mexican free-tailed bats (Tadarida brasiliensis) from North America (lineage TBNA) did not appear older than the TMRCA for other bat RABV lineages, in contrast to an earlier suggestion [7]. The lineage associated with T. brasiliensis from South America was ancestral to this cluster, with an estimated divergence time of approximately 500 years ago. Other RABV lineages associated with different insectivorous bats segregated in two large clusters of approximately the same age. Lineages from North and South American bats were intermixed (including both migratory and non-migratory species), suggesting that geographic distribution and compartmentalization of viruses into particular bat species occurred significantly earlier. From phylogenetic structure of the extant lineages it is impossible to infer which viruses from which geographic area were ancestral.
Table 1.
Evolution rates and times of the most recent common ancestors (TMRCA in years; the 95 % HPD is shown in the brackets) of bat rabies virus lineages analyzed in the present study (only the lineages where number of sequences ≥4, and years span ≥6 are included)
| RABV lineage | Number of sequences | Years span | Constant population size | Exponential population growth | Bayesian skyline | Bayesian skyride | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Substitution rates | TMRCA | Substitution rates | TMRCA | Substitution rates | TMRCA | Substitution rates | TMRCA | |||
| LB1 | 25 | 2004–2011 | 2.93E-4 | 37 (23–54) | 3.78E-4 | 36 (20–56) | 2.59E-4 | 39 (22–64) | 2.57E-4 | 37 (21–58) |
| LC | 7 | 1979–2010 | 2.24E-4 | 74 (51–100) | 2.92E-4 | 68 (47–97) | 1.95E-4 | 80 (50–117) | 1.98E-4 | 80 (51–115) |
| LN | 7 | 2003–2011 | 1.17E-4 | 42 (27–60) | 1.27E-4 | 40 (26–61) | 1.03E-4 | 41 (25–64) | 1.07E-4 | 40 (25–61) |
| LI | 4 | 2002–2008 | 1.74E-4 | 237 (108–392) | 1.99E-4 | 206 (92–367) | 1.80E-4 | 272 (110–446) | 1.85E-4 | 258 (113–439) |
| PS | 15 | 2005–2011 | 1.54E-4 | 49 (27–75) | 1.80E-4 | 47 (25–77) | 1.39E-4 | 54 (25–89) | 1.44E-4 | 53 (27–86) |
| TBNA | 14 | 2002–2012 | 1.36E-4 | 41 (25–61) | 1.56E-4 | 39 (23–63) | 1.23E-4 | 43 (23–70) | 1.27E-4 | 42 (23–67) |
| DR | 25 | 1986–2009 | 2.07E-4 | 267 (165–386) | 2.64E-4 | 236 (141–368) | 1.90E-4 | 309 (172–484) | 2.00E-4 | 300 (160–463) |
| EF-w1 | 23 | 1975–2012 | 1.40E-4 | 84 (58–117) | 1.69E-4 | 80 (55–115) | 1.23E-4 | 94 (59–137) | 1.26E-4 | 91 (57–132) |
| EF-w2 | 4 | 1976–2010 | 1.26E-4 | 193 (101–309) | 1.29E-4 | 165 (80–285) | 1.14E-4 | 226 (105–385) | 1.15E-4 | 220 (100–368) |
| EF-e1 | 13 | 1986–2011 | 1.87E-4 | 69 (63–100) | 6.48E-5 | 70 (41–103) | 1.66E-4 | 78 (42–118) | 1.65E-4 | 76 (44–117) |
| EF–e2 | 21 | 1984–2012 | 1.89E-4 | 286 (168–426) | 1.38E-4 | 270 (151–412) | 1.66E-4 | 339 (163–552) | 1.68E-4 | 332 (158–534) |
| MY | 10 | 1981–2012 | 1.59E-4 | 307 (182–434) | 1.80E-4 | 288 (173–418) | 1.44E-4 | 366 (202–581) | 1.45E-4 | 355 (159–550) |
| PH | 8 | 2002–2012 | 1.93E-4 | 138 (73–213) | 2.60E-4 | 119 (57–199) | 1.71E-4 | 161 (73–267) | 1.71E-4 | 154 (72–249) |
| The total tree | 1976–2012 | 1.78E-4 | 795 (503–1143) | 1.60E-4 | 732 (436–1107) | 1.56E-4 | 813 (461–1252) | 1.62E-4 | 786 (460–1212) | |
Discussion
Although the phylogeny of bat RABV has been studied relatively well, evolutionary history of the virus remains elusive. Interpretations of changes in viral genome are somewhat controversial [5, 7–9, 16, 20]. The observation that most codons in the RABV genes are subjected to purifying selection and a more limited number of codons evolve neutrally has been supported in all studies. In contrast, positive selection was detected inconsistently. Even when it was, different studies suggested different codons subjected to positive selection. The most recent finding was an episodic positive selection detected via MEME method in various RABV genes, particularly in the G [16]. Our results corroborate only one of these codons in the position 429 (counted as 448 in [16] because the first 19 amino acids of signal peptide were not removed). We still consider this result inconclusive as no further species-specific substitutions were detected in this codon.
The results obtained in our analysis differed from the results of several previous studies [5, 7–9, 16], suggesting a longer evolutionary history for bat RABV lineages, although marginal overlaps are present for 95 % HPD. The reason for this discrepancy is difficult to explain. One likely reason is the specific gene used for the analysis. The N gene sequences, used in the majority of previous estimations, are more conservative as the result of strong purifying selection pressure. This may involve constraints applied by viral structural or functional properties (such as the need for tight association with RNA and efficient interaction of the N protein with the phosphoprotein and polymerase during all stages of infection). The G is one of the most variable RABV genes [5]. More importantly, the G is not as hyper-variable as non-coding intergenic regions of RABV, and not as constrained by purifying selection as the N. Both these extremes were shown to affect negatively evolutionary estimates for various viruses [10, 21]. Of particular importance, strong purifying selection can mask ancient origins of recently sampled pathogens [10].
Previous estimates based on G sequences incorporated very limited numbers of bat RABV samples, and population models were not specified. These issues might lead to reduced resolution of models and broader TMRCA HPD [5, 9]. A comparative estimate of TMRCA for mongoose RABV based on the N and G sequences [22] provided different results for the two genes however, estimates based on the G resulted in a more recent TMRCA than estimates based on the N. Unfortunately, authors did not describe whether they used a partitioned model, and did not compare results obtained from different population demographic models. Therefore, it is hard to compare their results with ours. We believe that our model [(1 + 2), 3] is most appropriate for estimates on a protein coding region, and it was strongly favored by Bayes factor. It also accommodates constraints from purifying selection, at least partly. Of note, a limited analysis performed on P gene sequences by Davis et al. [8] provided results similar to ours, but those were not taken into account by authors because of dataset limitations and extremely broad HPD range. Furthermore, our results are in agreement with the estimated TMRCA for several RABV lineages associated with big brown bats (Eptesicus fuscus) in Canada (~500 years), performed on a significant number of P gene sequences [20]. We would like to note that if purifying selection can significantly mask ancient origins of pathogens [10], the age of bat RABV should be even greater than estimated in our study.
An accurate age estimation for RABV, particularly for the American bat RABV lineages, is important for better understanding of the natural history of these viruses. For example, the earlier estimated origin of these viruses during the seventeenth to eighteenth centuries [7–9] was associated with European colonization of the Americas, and two major alternative hypotheses were discussed: either the virus could be introduced by Europeans with companion mammals and shifted from these to bats (with vampire bats as the first affected species), or the RABV was already present in the vampire bats, and European colonization followed with deforestation and livestock population growth facilitated rapid expansion of the vampire bats, increasing the likelihood for RABV shifts to other bat hosts [2, 6]. However, according to our estimates, RABV circulated in New World bats long before the European colonization, and the RABV lineage associated with vampire bats cannot be considered as a common ancestor to the majority of RABV lineages associated with insectivorous bats.
As in other studies, the obvious limitation of our analysis was the amount of data available. It is hard to make a definitive conclusion on the duration of viral evolution using isolates from the most latter 6–36 years only even if appropriate model is selected. Our results may be adjusted if additional data, or more robust approaches for the estimation of viral evolution, become available. And likely such approaches would show even longer evolutionary history of RABV as was recently suggested for coronaviruses [23]. Rabies is the oldest recorded bat zoonosis, and introspection of when, where, and how lyssaviruses evolved should provide insight to the emergence of other infectious diseases associated with this highly diverse mammalian order.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank staff in the CDC Rabies Program for their support during the study, and the U.S. State Veterinary diagnostic laboratories and Health Departments for submission of rabid animal samples.
Disclaimer
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the CDC or other host institutions. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Baer GM, Neville J, Turner GS. Rabbis and Rabies: A Pictorial History Through the Ages. Mexico City: Laboratorios Baer; 1996. p. 133. [Google Scholar]
- 2.Rupprecht CE, Turmelle A, Kuzmin IV. Curr. Opin. Virol. 2011;1:662–670. doi: 10.1016/j.coviro.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 3.Dietzgen RG, Calisher CH, Kurath G, Kuzmin IV, Rodriguez LL, Stone DM, Tesh RB, Tordo N, Walker PJ, Wetzel T. A.E. Whitfield. In: King AM, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus Taxonomy: Classification and Nomenclature of Viruses. Ninth Report of the International Committee on Taxonomy of Viruses. San Diego: Elsevier; 2011. pp. 686–713. [Google Scholar]
- 4.Shope RE. Yale J. Biol. Med. 1982;55:271–275. [PMC free article] [PubMed] [Google Scholar]
- 5.Badrane H, Tordo N. J. Virol. 2001;75:8096–8104. doi: 10.1128/JVI.75.17.8096-8104.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuzmin IV, Rupprecht CE. In: Rabies. 2. Jackson AC, Wunner WH, editors. London: Elsevier; 2007. pp. 259–307. [Google Scholar]
- 7.Hughes GJ, Orciari LA, Rupprecht CE. J. Gen. Virol. 2005;86:1467–1474. doi: 10.1099/vir.0.80710-0. [DOI] [PubMed] [Google Scholar]
- 8.Davis PL, Bourhy H, Holmes CE. Infect. Genet. Evol. 2006;6:464–473. doi: 10.1016/j.meegid.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 9.Bourhy H, Reynes JM, Dunham EJ, Dacheux L, Larrous F, Huong VT, Xu G, Yan J, Miranda ME, Holmes EC. J. Gen. Virol. 2008;89:2673–2681. doi: 10.1099/vir.0.2008/003913-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wertheim JO, Kosakovsky Pond SL. Mol. Biol. Evol. 2011;28:3355–3365. doi: 10.1093/molbev/msr170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuzmin IV, Hughes GJ, Botvinkin AD, Gribencha SG, Rupprecht CE. Epidemiol. Infect. 2008;136:509–519. doi: 10.1017/S095026880700903X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuzmin IV, Niezgoda M, Franka R, Agwanda B, Markotter W, Beagley JC, Urazova OY, Breiman RF, Rupprecht CE. J. Clin. Microbiol. 2008;46:1451–1461. doi: 10.1128/JCM.00016-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuzmin IV, Shi M, Orciari LA, Yager PA, Velasco-Villa A, Kuzmina NA, Streicker DG, Bergman DL, Rupprecht CE. PLoS Pathog. 2012;8:e1002786. doi: 10.1371/journal.ppat.1002786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lafon M. J. Neurovirol. 2005;11:82–87. doi: 10.1080/13550280590900427. [DOI] [PubMed] [Google Scholar]
- 15.Wunner WH. In: Rabies. 2. Jackson AC, Wunner WH, editors. London: Elsevier; 2007. pp. 23–68. [Google Scholar]
- 16.Streicker DG, Altizer SM, Velasco-Villa A, Rupprecht CE. Proc. Natl. Acad. Sci. USA. 2012;109:19715–19720. doi: 10.1073/pnas.1203456109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall TA. Nucl. Acids. Symp. 1999;41:95–98. [Google Scholar]
- 18.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drummond AJ, Rambaut A. BMC Evol. Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nadin-Davis SA, Feng Y, Mousse D, Wandeler AI, Aris-Brosou S. Mol. Ecol. 2010;19:2120–2136. doi: 10.1111/j.1365-294X.2010.04630.x. [DOI] [PubMed] [Google Scholar]
- 21.Purdy MA, Khudyakov YE. PLoS One. 2010;5:e14376. doi: 10.1371/journal.pone.0014376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Zyl N, Markotter W, Nel LH. Virus Res. 2010;150:93–102. doi: 10.1016/j.virusres.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 23.Wertheim JO, Chu DKW, Peiris JSM, Kosakovsky Pond SL, Poon LLM. J. Virol. 2013;87:7039–7045. doi: 10.1128/JVI.03273-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.