

Abstract
The proglucagon gene encodes multiple structurally related peptides with overlapping actions promoting the absorption and assimilation of ingested energy. Notably, glucagon has been developed pharmaceutically to treat hypoglycemia, and glucagon-like peptide-1 (GLP-1) receptor agonists are used for the therapy of type 2 diabetes and obesity. Here I describe the discovery of glucagon-like peptide-2 (GLP-2), a 33 amino acid peptide cosecreted together with GLP-1 from gut endocrine cells. GLP-2 was found to exhibit robust intestinal growth-promoting activity, following serendipitous observations that proglucagon-producing tumors induced intestinal growth in mice. Key developments in the pharmaceutical development of GLP-2 included the cloning of the GLP-2 receptor, and the recognition of the importance of dipeptidyl peptidase-4 as a critical determinant of GLP-2 bioactivity. A therapeutic focus on short bowel syndrome, a serious medical disorder with compelling unmet medical need, enabled the pharmaceutical development of a simple GLP-2 analogue, teduglutide, suitable for once daily administration.
Keywords: intestinal failure, nutrition, glucagon-like peptides, G protein coupled receptors, hormones, inflammatory bowel disease
The discovery of glucagon-like peptide-2 (GLP-2) represents an example of curiosity-driven basic science serendipitously resulting in the identification of a new biological action for a small peptide with compelling therapeutic activity. At the time I started my studies of the proglucagon-derived peptides (PGDPs) in 1984, the 29 amino acid peptide glucagon was well established as an important islet hormone that regulates glycemia through control of hepatic glucose production.1 The cloning of the cDNAs and genes encoding mammalian proglucagon in the early 1980s revealed the sequences of two new glucagon-like peptides, glucagon-like peptide-1 (GLP-1) and glucagon-like peptide-2 (GLP-2), with unknown biological activity.2−5 A series of studies from multiple laboratories soon revealed that the truncated versions of GLP-1(1–37), principally GLP-1(7–37) and GLP-1(7–36amide), exhibited potent glucose-dependent insulinotropic activity when assessed in islet cells in vitro, perfused pancreata, and human subjects.6−9 Indeed, on the basis of studies carried out in our laboratory in Boston at the time, my supervisor, Joel Habener, filed the first United States patent describing the use of GLP-1 to treat diabetes. As a research fellow, I was unfamiliar with the concept of turning research observations into patents and intellectual property. Having my lab notebooks disappear into the lawyer’s offices for several weeks was my introduction to the concept and process. My postdoctoral fellowship studies also included the analysis of proglucagon post-translational processing, with which we demonstrated that GLP-2 and GLP-1 were simultaneously liberated from the same proglucagon precursor.10 Fortuitously, I also initiated studies examining the molecular control of islet proglucagon gene transcription. These experiments, carried out in collaboration with my colleague Jacques Philippe,11,12 started a series of investigations that would ultimately prove to be pivotal for discovering the actions of GLP-2.
Investigator-Initiated Serendipity
Upon my return to Toronto as an Assistant Professor in 1987, I was dismissively viewed, perhaps rightly, with suspicion by some established colleagues. My scant 3 years of research training and lack of a Ph.D. were legitimate reasons for doubting my scientific capability. Nevertheless, I was fortunate to be appointed to a clinical department and continued experiments directed at elucidation of the DNA sequences and transcription factors responsible for control of islet proglucagon gene transcription. Simultaneously, I cloned proglucagon cDNAs from human brain and rat intestine,13,14 and hence it seemed logical to extend our studies of proglucagon gene transcription beyond the islet. At that time, we had successfully used immortalized InR1-G9, RIN1056A, and αTC-1 islet cell lines to examine the molecular control of proglucagon gene expression in α-cells,10,11,15,16 however differentiated proglucagon-producing enteroendocrine cell lines were not available. My colleague in the Department of Physiology, Dr. Patricia Brubaker, had established primary cultures of fetal rat intestinal cells for studies of proglucagon gene expression;14 however, these cells were not suitable for extensive transfection studies and analysis of proglucagon gene transcription.
Accordingly I set out to generate a stable immortalized intestinal proglucagon-producing cell line, using a transgenic mouse approach that employed selective targeting of SV40 T antigen to PGDP-producing enteroendocrine cells.17 Dr. Ying Li, my first postdoctoral fellow, generated transgenic mice expressing SV40 T antigen sequences under the control of a 2.2 kilobase fragment of the rat proglucagon gene (Gcg) promoter.18 Transgene expression was detected in the brain, islet cells, and within some endocrine cells of the small and large intestine. After several months, Glucagon SV40 Tantigen (GLUTag) mice lost weight, stopped eating, and hence were euthanized. Upon necropsy it was evident that the majority of mice exhibited pancreatic endocrine cell hyperplasia, whereas all mice developed endocrine tumors in the large bowel.18 Histological analysis of these gut neoplasms revealed that they were immunopositive for GLP-1 and expressed high levels of proglucagon mRNA transcripts.
With this new mouse model providing a source of intestinal proglucagon-producing tumor cells, I set out to generate a stable immortalized intestinal PGDP-producing cell line, using, as starting material, the GLUTag tumors. Homogenized fragments of the primary GLUTag tumor were implanted subcutaneously in nude mice, resulting in reproducible tumor growth within the subcutaneous compartment19 and markedly elevated circulating levels of the PGDPs. After several weeks, GLUTag tumors were excised and dispersed into single cell suspensions for isolation of clonal PGDP-producing enteroendocrine cell lines in vitro.20 At that time, I was still working regularly at the bench, and did many of these experiments myself. The GLUTag cell line that emerged expressed high levels of PGDPs, including GLP-1 and GLP-2 secreted in a regulated manner,20,21 and has subsequently been widely utilized for studies of PGDP synthesis and secretion. Unexpectedly, during the course of propagating GLUTag tumors subcutaneously in nude mice, we observed inhibition of endogenous pancreatic proglucagon gene expression19 and marked enlargement of the small and large bowel. Moreover, we quickly determined that intestinal enlargement was reproducibly detected with subcutaneous passage of additional PGDP-producing cell lines, strengthening the link between PGDP production and intestinal growth.22
Critically, I was aware of published case reports describing massive intestinal enlargement in a few subjects presenting with glucagonomas.23,24 It may amuse some younger colleagues to learn that one actually had to physically go to a scientific library to manually search the literature and retrieve these older papers. Since our findings in mice overlapped considerably with related observations in humans (glucagonomas linked to gut growth), it seemed reasonable to pursue the long-standing hypothesis that one or more factors secreted by glucagonomas, possibly a peptide product of the proglucagon gene, was responsible for stimulation of bowel growth.
Securing Funding To Pursue the Discovery
Having established a reproducible and simple model of rapid gut growth associated with development of PGDP-producing tumors, I attempted to raise research funding from industry, based on my plan to precisely identify the putative intestinal growth factor. I prepared a concise two page grant application, and sent this unsolicited investigator-initiated research proposal, via regular mail or courier delivery service, to about 20 major pharmaceutical and biotechnology companies. Most did not acknowledge receipt of the application, a few politely declined interest, and several companies indicated they would discuss my proposal at the next meeting of their committee responsible for adjudication of external grant applications. None of the companies ultimately expressed any subsequent interest. Only one company agreed to meet with me, Allelix Biopharmaceuticals Inc., a local Canadian biotechnology company headquartered in Mississauga Ontario, with a lead program focused on recombinant parathyroid hormone. After two meetings and some productive discussions, Dr. Martin Sumner-Smith and Allelix colleagues agreed to provide me with an initial $100,000 grant in support of my gut growth factor discovery proposal.
Our initial approach was embarrassingly simple. On the basis of our PGDP-focused hypothesis, I would simply synthesize and inject the individual PGDPs (glicentin, GLP-1, GLP-2, glucagon, intervening peptides) into mice, following which one could easily assess intestinal growth in a matter of days (Figure 1). At the time, glucagon, GLP-1, and GLP-2 were commercially available, whereas intervening peptides, and glicentin, a much larger 69 amino acid protein was not. Given the quantities of peptides required for our chronic in vivo experiments in multiple mice, we ordered the custom syntheses of the individual PGDPs. Shortly before I commenced preparations for the experiments to inject the individual peptides in mice, Allelix sent me a copy of a just published provisional patent application, filed by colleagues in Japan, describing their discovery of glicentin as the PGDP with marked intestinotrophic activity, ultimately published a few years later.25 Upon reading the patent, I was simultaneously excited and disappointed. Clearly, I had hoped to be the first to identify the magic glucagonoma-related “growth factor” and file a patent, hence it seemed our pursuit of the mysterious factor was now pointless and the project should be closed down, before we had really made a serious attempt ourselves. On the other hand, I was pleased, and indirectly relieved, that the initial simple hypothesis, that a PGDP encoded within the GCG gene was in fact, the long sought after gut growth factor, had proven to be correct.
Figure 1.
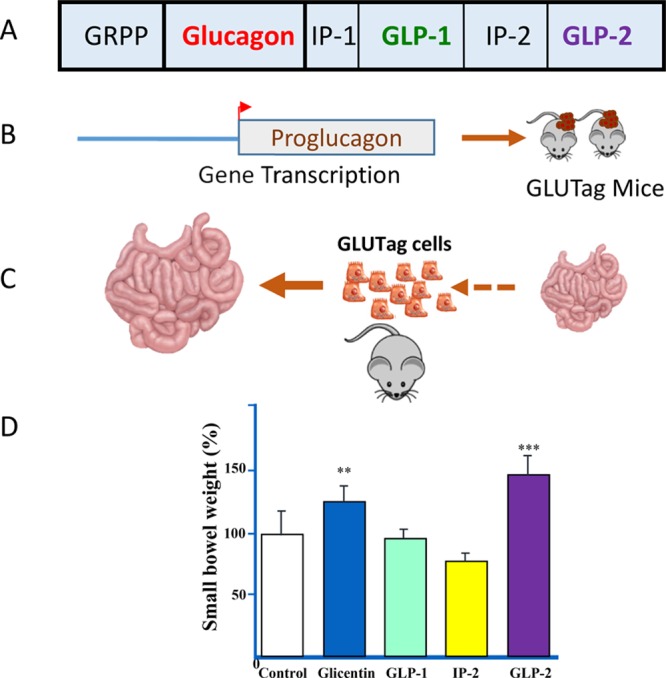
(A) Structure of proglucagon and the proglucagon-derived peptides (PGDPs); (B) studies of proglucagon gene transcription led to generation of transgenic proglucagon promoter-SV40T antigen transgenic mice with intestinal tumors; (C) small intestinal growth in mice with subcutaneous glucagon-producing tumors; and (D) identification of GLP-2 as the PGDP with the greatest intestinotrophic activity in mice.
A few weeks later, I received the ordered peptides and was tempted to simply abandon the project and store them in the freezer. Nevertheless, it also seemed reasonable, and little extra effort, to independently confirm the experimental findings from Japan identifying glicentin as the long sought after intestinotrophic PGDP. Accordingly, I injected each peptide, dissolved within a gelatin mixture to prolong bioavailability, subcutaneously in female CD1 mice, twice daily for 10 days. We confirmed the observation from Japan that glicentin administration produced a significant increase in small bowel weight.22 Remarkably, however, GLP-2, a small peptide with no clear biological function other than the ability to stimulate adenylate cyclase in hypothalamic and pituitary membranes ex vivo,26 was even more potent than glicentin (Figure 1) in producing intestinal growth.22 In fact, the intestinal enlargement was quite evident simply upon inspection of the contents of the abdominal cavity. For reasons that have never been explained to me, our Japanese colleagues had not simultaneously examined the putative actions of GLP-2 in their own studies.
Experimental Validation and Reproducibility of GLP-2 action
Although we were uncertain whether other colleagues had already discovered actions for GLP-2 perhaps described in unpublished provisional patent applications, I pushed ahead, filed a patent describing the GLP-2 discovery, and begin further studies to learn as much as I could about the physiology and pharmacology of GLP-2 action in vivo. Many of these experiments were carried out in collaboration with my colleague in the Department of Physiology, Dr. Patricia Brubaker. In an experiment that seems trivial by current lofty scientific standards, we first tested whether GLP-2 increased intestinal growth in both male and female mice with varying genetic backgrounds. Reassuringly, GLP-2 promoted gut growth in all mouse strains analyzed, although the relative increase varied across mouse lines.27 Mindful of the enhanced proliferative activity demonstrated for numerous cell types in young mice, we also assessed GLP-2 action in mice from 4 weeks to 24 months of age. Importantly, GLP-2 retained its intestinotrophic actions in mice of all ages.27 The intestinotrophic actions of GLP-2 were dose-dependent, independent of changes in food intake, evident even with peptide administration every other day, and preserved whether the peptide was administered via subcutaneous, intramuscular, or intraperitoneal administration.27,28
To ensure we had not simply discovered a mouse-specific bowel growth factor, we next carried out a series of experiments in rats. To my enormous disappointment, we did not detect the same robust increase in the mass of the gastrointestinal tract in rats (relative to findings in mice) following native GLP-2 administration. Intriguingly, GLP-2 did increase crypt and villus height in rats, hinting that at least some intestinotrophic activity was preserved. Upon reflection we wondered whether the pronounced differential efficacy of GLP-2 in mice vs rats reflected greater stability of the peptide in murine plasma. At the time, there was emerging recognition that some peptides, including GLP-1, were cleaved and enzymatically inactivated by the ubiquitous protease dipeptidyl peptidase-4.29,30 As GLP-2 was highly structurally related to GLP-1 and both peptides contained a conserved position 2 alanine, a target for DPP-4 cleavage, we hypothesized that the diminished intestinotrophic activity of native GLP-2 in rats reflected more rapid degradation due to enhanced DPP-4 activity. To test this hypothesis, we carried out two complementary experiments. First, we administered native GLP-2 to Fischer 344 rats, a strain with markedly reduced DPP-4 activity secondary to a naturally occurring mutation in the Dpp4 gene.31 In parallel, we synthesized and tested the biological activity of a degradation-resistant DPP-4 insensitive analogue, r[Gly2]-GLP2, in wildtype rats. To our great relief, both native GLP-2 and the GLP-2 analogue robustly activated gut growth in the respective rat studies, as we had hoped (Figure 2).32 These findings highlighted the significance of recognizing the importance of DPP-4 for the biological activity of GLP-2 early in the discovery process, and hastened the subsequent rapid development of a series of DPP-4-resistant GLP-2 analogues for optimization of pharmacokinetic and pharmacodynamic activity.
Figure 2.

(A) Native GLP-2 does not increase small bowel growth in rats. (B) Amino acid sequences of human, rat, and mouse GLP-2, with amino acid differences underlined, and the cleavage by dipeptidyl peptidase-4 (DPP-4) at the position 2 alanine designated by an arrowhead. (C) Left panel, native GLP-2 and [Gly2}-GLP-2 stimulate intestinal growth in Fischer 344 rats with a Dpp4 mutation. Right panel, [Gly2}-GLP-2 stimulates bowel growth in control DPP-4+ Fischer 344 rats. Adapted from ref (32).
While assessing the physiology and pharmacology of GLP-2 in normal animals, we also carried out multiple experiments in animal models of gut epithelial injury. GLP-2 invariably improved intestinal structure and function and consistently attenuated disease activity in commonly utilized mouse models of experimental intestinal injury.33−35 Importantly, the regenerative and cytoprotective actions of GLP-2 were largely confined to the gut, and broadly reproducible in independent laboratories.36 Furthermore, GLP-2 rapidly enhanced nutrient absorption in normal mice,37 as well as in rats with extensive experimental bowel resection maintained on parenteral nutrition,38 highlighting the therapeutic potential of GLP-2 in the context of short bowel syndrome (SBS).
Collaboration with Industrial Partners
An important contribution to our efforts directed at identification of an optimal GLP-2 analogue suitable for pharmaceutical development was the results of peptide structure function studies undertaken to identify the critical determinants of GLP-2 bioactivity. A key to these efforts was the cloning of the rat and human GLP-2 receptors,39 an effort spearheaded by Dr. Donald Munroe at Allelix Biopharmaceuticals. The GLP-2R was a member of the Class B GPCR family, related in structure (and signal transduction) to existing receptors for glucagon, GLP-1, and GIP.40 Importantly, the expression of the GLP-2R, assessed using RNase protection assays, was predominantly localized to the gastrointestinal tract, with limited detection of GLP-2 receptor mRNA transcripts in other peripheral tissues.39 Subsequent studies using a combination of Northern blotting and semiquantitative RT-PCR, demonstrated robust expression of the murine Glp2r in stomach and small and large intestine, with Glp2r mRNA transcripts also detectable in lung, hypothalamus, and brainstem by RT-PCR.41 This relatively restricted receptor distribution contrasted with the much wider tissue expression of receptors for growth hormone/IGF-1 and keratinocyte growth factor, molecules that also exhibited intestinotrophic activity, and raised the possibility that sustained GLP-2R agonism might be associated with fewer unexpected side effects in peripheral tissues.
The cloning of the GLP-2 receptor, coupled with the availability of a reproducible bioassay (murine gut growth), enabled us to interrogate the functional importance of individual residues within GLP-2, in collaboration with Martin Sumner Smith and Anna Crivici, scientists at Allelix Biopharmaceuticals Inc. Collectively, we designed and tested the in vitro stability, receptor binding, signal transduction activity, and bioactivity, of more than a hundred GLP-2 analogues, including several dozen peptides characterized and reported together with my colleague Patricia Brubaker.42 These and related biochemical studies at Allelix enabled us to select a lead clinical candidate, [hGly2]-GLP-2, later designated teduglutide, for more detailed characterization, including toxicology studies supporting an investigational new drug application for clinical testing. It is remarkable that simply changing a single amino acid in the native peptide, without other modifications that would enable extensive prolongation of the circulating t1/2, would ultimately prove to be sufficient for induction of reasonable therapeutic efficacy in humans.
Commercialization of the GLP-2 Discovery
The discovery of GLP-2 action in 1995 enabled me, together with the University of Toronto and the University Health Network, to negotiate a licensing deal with Allelix Biopharmaceuticals Inc. outlining terms supporting the licensing of GLP-2 intellectual property, and the commercialization of GLP-2 receptor agonists. After several years of preclinical validation, we were set to scale up the development program and embark on human studies. The initial indication selected was SBS, in part reflecting the known actions of GLP-2 to expand mucosal surface area and enhance nutrient absorption, with demonstrated efficacy in multiple independent preclinical models of SBS.36 Furthermore, SBS represented a burdensome condition with clear unmet medical need, for which no previous clinical therapy had produced compelling results or received an indication for chronic administration. With the ramping up of a GLP-2 clinical program we (Allelix & Drucker) explored external partnering opportunities with established firms in the pharmaceutical industry to expedite clinical development and help defray the considerable costs of an international clinical trial program. The reaction of potential “big pharma partners” to our entreaties followed a similar pattern. Initially, colleagues were intrigued by the discovery and impressed by the therapeutic potential for GLP-2 as a novel gut growth factor with therapeutic efficacy in preclinical proof of concept studies. Once the discussion turned to the pharmaceutical market for SBS, our lead clinical indication, the enthusiasm rapidly waned. There were only a few thousand individuals with SBS worldwide, and the concept of orphan drug development (and potential pricing/reimbursement strategies) was not yet universally established or embraced. Once business development colleagues crunched the numbers on the SBS market opportunity, our discussions usually terminated and meetings often ended early. In September of 1999, Allelix entered into a merger agreement with NPS Pharmaceuticals Inc. and the newly merged entity, first NPS Allelix, then ultimately NPS Pharmaceuticals, assumed ongoing responsibility for teduglutide development.
The initial clinical testing of native GLP-2 in human subjects with SBS was organized by Allelix and subsequently NPS, engaging many academic partners, including Dr. Palle Jeppesen, a leading expert in the pathophysiology and management of human SBS. The management of SBS is extremely challenging for individuals, ranging from small babies to older adults, often characterized by problems with maintaining adequate hydration, restrictions of ingestion of food, weight loss, and excess rectal or stomal loss of energy and enhanced fluid output (Figure 3).43,44 Although hydration and delivery of energy can be managed through parenteral nutrition (PN), long-term PN use is often associated with intermittent line infections, sepsis, and the risk of developing chronic liver disease, sometimes resulting in hepatic failure.43,44 Moreover, the requirement for PN, from 1 to 7 nights per week, is time-consuming, burdensome, greatly restricts mobility and travel, and is associated with an impaired quality of life.
Figure 3.

Clinical challenges and conditions associated with short bowel syndrome (left panel) and the consequences of teduglutide therapy (right panel) in human subjects with short bowel syndrome.
Native GLP-2 (400 μg injected subcutaneously twice a day) was administered for 35 days to eight subjects with SBS without a colon in continuity.45 The results of these early proof of concept studies demonstrated enhanced energy absorption, increased lean body mass, and modest weight gain, with intestinal biopsies revealing histological evidence for increased crypt depth and villus height.45 A subsequent dose-ranging pilot study of h[Gly2]-GLP-2, teduglutide, was carried out in 16 subjects with SBS, with similar increases observed in energy absorption and reduced fecal energy excretion, without major adverse safety events.46
Toward Regulatory Approval of Teduglutide
Following additional dose-ranging studies, the first Phase 3 study of teduglutide was carried out over several years, in human subjects with SBS randomized to placebo, 0.05 or 0.1 mg/kg/day of teduglutide, for an initial duration of 24 weeks. Eligible trial participants needed to be at least 18 years of age, be treated with PN for at least 12 months prior to study entry, and require intravenous PN at least 3 nights per week.47 These studies were highly demanding from a participant perspective. Study subjects were required to demonstrate unequivocal and stable parenteral nutrition (PN)-dependence, and to undergo additional run in periods before randomization to ensure clinical stability and adequate hydration. The actual trial protocol included home collections of urine output, careful attention to food and beverage intake, daily recording of PN infusion, and multiple patient visits, initially every 2 weeks, to trial sites. The complexity of the international multicenter trial is partly reflected in the time required to recruit study subjects, which was more than 3 years. The difference in the primary trial end point (20% reduction in PN fluid volume) was not statistically significant in subjects treated with 0.1 mg/kg of teduglutide, but was highly significant in the cohort treated with the lower dose, 0.05 mg/kg/day.47 Importantly, subjects treated with teduglutide also exhibited stable to increased urine output, despite a reduction in PN fluid volume.
From a regulatory perspective, the prespecified primary end point outlined in the formal statistical analysis plan was the clinical result achieved with the higher dose, 0.1 mg/kg/day cohort. Hence the favorable results obtained in the lower dose group, albeit highly encouraging and clear evidence for drug efficacy, were formally viewed as simply “hypothesis generating”. After consultation with regulatory authorities, it was determined that an entirely new Phase 3 trial was required for regulatory approval, comparing a single daily dose of teduglutide (0.05 mg/kg/) with placebo. Enrollment and completion of this second Phase 3 trial also took more than 2 years, eroding considerable time from the patent estate and decreasing the corresponding commercial value of the teduglutide franchise. Once again, the results achieved in the 0.05 mg/kg/day teduglutide group were highly statistically significant, with 63% of the teduglutide-treated subjects, vs 30% of the placebo-treated group, achieving the primary outcome of 20% reduction in parenteral nutrition.48 Notably, compliance with teduglutide therapy was very high during both Phase 3 clinical trials. Individuals with SBS generally report that reducing the nights required for TPN, as achieved with teduglutide therapy, is associated with an improved quality of life.47,49,50
A reproducible feature of teduglutide therapy is the observation that a small subset of PN-dependent subjects with SBS is able to completely discontinue PN. This is perhaps the most meaningful end point for SBS subjects, as it means they are no longer dependent on a strict nightly and weekly routine, are able to eat and drink more liberally, free to travel, and enjoy a more flexible life style. A posthoc analysis of the teduglutide clinical trial program, including the two Phase 3 trials and their extension studies, revealed that 16/134 individuals gained oral or parenteral autonomy from nutritional support (after a median of 5 years of previous PN dependence), after a mean duration of 89 weeks of teduglutide treatment.51 Intriguingly from a mechanistic perspective, PN-independence may occur later in the course of tedulgutide therapy, even after 1–2 years of teduglutide administration. Although the experience with durability of intestinal rehabilitation following discontinuation of teduglutide therapy is limited, a small subset of patients may maintain body weight and adequate nutrition and hydration after cessation of teduglutide therapy.52
Collectively, the consistent results obtained from the two independent Phase 3 trials examining the 0.05 mg/kg/day dose were unprecedented for medicinal approaches to SBS therapy, and supported filing of a new drug application with the European Medicine Authority and the Food and Drug Administration for the use of teduglutide in the chronic therapy of SBS. Notably, although oral glutamine and parenteral growth hormone administration exhibit efficacy in some subjects with SBS, no previous therapy had been approved for chronic administration in this patient population. Nevertheless, I was uncertain how the regulatory authorities would perceive the benefit/risk balance for teduglutide, and had very little expectations of the ultimate regulatory review outcome. Teduglutide causes stomal irritation in some subjects, is associated with reports of acute gallbladder disease, and there was conflicting preclinical data surrounded its effect on neoplastic growth in the rodent gastrointestinal tract. In formal two species, two year toxicology studies, there was no evidence that sustained teduglutide promoted neoplastic transformation or tumorigenesis. On the other hand, teduglutide enhanced tumor growth in some but not all genetically or chemically sensitized rodent models of intestinal tumorigenesis.36,53 In June of 2012, while riding an exercise bike in the gym of my hotel (I was attending the American Diabetes Association meeting) I noticed an email sent by an investment analyst who covered NPS Pharmaceuticals, congratulating me on the EMA recommendation released earlier that day recommending teduglutide approval in the European Union.
Several months later, I was fortunate to participate in the scientific preparation for the FDA advisory committee hearing as well as attend the actual FDA committee meeting October 16 2012, acting in the capacity of a scientific expert in GLP-2 biology, on behalf of the company. As an individual more familiar with FDA reviews of new medications for diabetes and obesity, I was quite accustomed to and prepared for a sometimes confrontational set of discussions, where the agency often took a different and more critical view of the data. I was pleasantly surprised when the FDA assessment of the teduglutide submission largely agreed with the company’s interpretation of the relative efficacy and adverse event profile for teduglutide. Most memorable were the dozen patient testimonials, describing how beneficial and life-altering teduglutide therapy could be for the subset of individuals with terrific responses, including major reduction of the number of nights on PN and in some individuals, discontinuation of PN administration. The FDA advisory committee voted unanimously to approve chronic teduglutide therapy for the sustained treatment of SBS, with formal FDA approval following shortly thereafter on December 21 2012. Importantly, the initiation of teduglutide therapy is accompanied by a Risk Evaluation and Mitigation Strategy (REMS) that includes education of health care providers, a screening colonoscopy within 6 months of starting therapy, and appropriate monitoring for liver and gallbladder function.
Lessons Learned
A few reflections on the story of teduglutide with the benefit of several decades of hindsight. First, we did not set out to identify a new bowel growth factor. The discovery of GLP-2 was an accidental byproduct of curiosity-driven basic science, focused on understanding the molecular control of proglucagon gene transcription. The knowledge of the earlier case reports linking glucagonomas to bowel growth in humans immediately suggested we had rediscovered in mice a highly conserved mechanism of PGDP action that would likely hold up in subsequent human studies. Many grant programs today are structured to require a multidisciplinary group of scientists, with knowledge and skill sets far outside the immediate area of focused expertise. Our own narrow line of investigation of peptide hormone action has generally never qualified my lab to be an eligible applicant for such grand funding programs, and it remains unclear how many important discoveries are driven by top down agency-selected requests for applications in areas of science that come and go in regard to popularity.
Another recurring feature of GLP-2 science is the virtual absence of papers published in “the top journals” and few if any breathtaking press releases heralding the “breakthrough” actions of GLP-2 in animals or humans. Most of our papers were published in respectable society journals, with modest impact factors, and featured experiments with tedious dose–responses, time courses, and careful pharmacological studies. We spent a lot of time determining that one could give GLP-2 in a number of very different experimental regimens with consistent therapeutic success; however, a precise reductionist mechanistic description of GLP-2 action has generally eluded us. Two of the most important papers, the original description of GLP-2 action and the cloning of the GLP-2 receptor were published in Proceedings of the National Academy of Sciences,22,39 after multiple rejections from “high impact” journals. Understandably, it is very clear why our GLP-2 work has never been that attractive to major Journals in the field. It is simply solid pharmacology and physiology, often devoid of contrived overly simplistic mechanistic pathways, without exaggerated claims and promises. Of the very few GLP-2 papers published by other groups in top Nature and Cell Journals, these have generally turned out to not be reproducible and have no clinical relevance.
It is often stated that successful drug development needs multiple champions, and teduglutide is no exception. My colleague Dr. Patricia Brubaker has worked tirelessly for several decades, both independently and collaboratively, to unravel the mechanisms of GLP-2 action. Several dozen students and fellows in my lab have made major contributions, led by research scientists Drs. Bernardo Yusta and Jacqueline Koehler. Dr. Martin Sumner-Smith greenlighted the GLP-2 discovery program at Allelix, and subsequently Drs. Anna Crivici and Lydia Demchyshyn shepherded the GLP-2 program within Allelix and later, NPS Pharmaceuticals Inc., respectively. The academic leadership of Professor Palle Jeppesen and many dedicated SBS investigators contributed enormously to the successful conduction of rigorous multicenter clinical trials testing the efficacy and safety of teduglutide. The organization and completion of the teduglutide clinical trial program, and the successful NDA filing required a sustained effort from dozens of colleagues at NPS Pharmaceuticals, leading to FDA approval of teduglutide under the leadership of Dr. Francois Nader. Most importantly, hundreds of individuals with SBS from multiple countries volunteered their time and made enormous contributions to the ultimate success of the teduglutide clinical trial program.
In today’s research environment, there is a tremendous emphasis on complex fancy science, big data, and sophisticated omics-driven investigation, often underpinned by millions of data points generated in a single experiment. It is worthwhile reflecting that the discovery of GLP-2 bioactivity required intuition, synthesis of a few milligrams of peptide, injection of peptides into several dozen mice, for about a week, and then weighing of individual mouse organs using a Mettler balance. Proposing a similar grant program today would lead to immediate triage, and some degree of snickering among colleagues. The story of GLP-2 and teduglutide reminds us that simple, careful curiosity-driven research, however unpredictable, often pays enormous dividends, that cannot be preordained by top down-driven research mandates, that stipulate the formation of networks, consortium, or centers of excellence. Indeed, the stories underlying the discovery and development of GLP-1 for diabetes and obesity, and DPP-4 inhibitors for diabetes, yield similar insights and lessons supporting the singular importance of investigator-initiated research. Notwithstanding the clear value of collaborative science, it seems reasonable to always ensure we allocate a substantial proportion of research funding for individual curious scientists to pursue simple yet important questions, unencumbered by numerous prespecified conditions for envisioned success.
Acknowledgments
Daniel J Drucker (D.J.D.) is the sole guarantor of this work and takes responsibility for its content. D.J.D. is supported by the Banting and Best Diabetes Center-Novo Nordisk Chair in Incretin biology, by CIHR Grant 154321, and by investigator-initiated operating grants to Mt. Sinai Hospital from Merck Inc., Novo Nordisk Inc, and Shire/Takeda Inc.
Glossary
Abbreviations:
- Glucagon-like peptide-2
GLP-2
- glucagon-like peptide-1
GLP-1
- proglucagon-derived peptides
PGDPs
- glucagon SV 40 T antigen
GLUTag
- dipeptidyl peptidase-4
DPP-4
- reverse transcription polymerase chain reaction
RT-PCR
- short bowel syndrome
SBS
- parenteral nutrition
PN
- Food and Drug Administration
FDA
- glucagon-like peptide-2 receptor
GLP-2R
The author declares the following competing financial interest(s): Dr. Drucker has served as an advisor or consultant or speaker within the past 12 months to Forkhead Biotherapeutics, Heliome Inc, Intarcia Therapeutics, Kallyope, Eli Lilly, Merck Research Laboratories, Novo Nordisk Inc., and Pfizer Inc. Neither Dr. Drucker or his family members hold stock directly or indirectly in any of these companies. GLP-2 is the subject of a patent license agreement between Takeda Inc and the University of Toronto, Toronto General Hospital (UHN) and Daniel Drucker.
This article is made available for a limited time sponsored by ACS under the ACS Free to Read License, which permits copying and redistribution of the article for non-commercial scholarly purposes.
References
- Unger R. H. (1971) Glucagon physiology and pathophysiology. N. Engl. J. Med. 285 (8), 443–9. 10.1056/NEJM197108192850806. [DOI] [PubMed] [Google Scholar]
- Bell G. I.; Sanchez-Pescador R.; Laybourn P. J.; Najarian R. C. (1983) Exon duplication and divergence in the human preproglucagon gene. Nature 304, 368–371. 10.1038/304368a0. [DOI] [PubMed] [Google Scholar]
- Bell G. I.; Santerre R. F.; Mullenbach G. T. (1983) Hamster preproglucagon contains the sequence of glucagon and two related peptides. Nature 302, 716–718. 10.1038/302716a0. [DOI] [PubMed] [Google Scholar]
- Heinrich G.; Gros P.; Habener J. F. (1984) Glucagon gene sequence. Four of six exons encode separate functional domains of rat pre-proglucagon. J. Biol. Chem. 259 (22), 14082–7. [PubMed] [Google Scholar]
- Heinrich G.; Gros P.; Lund P. K.; Bentley R. C.; Habener J. F. (1984) Pre-proglucagon messenger ribonucleic acid: Nucleotide and encoded amino acid sequences of the rat pancreatic complementary deoxyribonucleic acid. Endocrinology 115, 2176–2181. 10.1210/endo-115-6-2176. [DOI] [PubMed] [Google Scholar]
- Drucker D. J.; Philippe J.; Mojsov S.; Chick W. L.; Habener J. F. (1987) Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc. Natl. Acad. Sci. U. S. A. 84, 3434–3438. 10.1073/pnas.84.10.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojsov S.; Weir G. C.; Habener J. F. (1987) Insulinotropin: Glucagon-like peptide I (7–37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J. Clin. Invest. 79, 616–619. 10.1172/JCI112855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst J. J.; Orskov C.; Nielsen O. V.; Schwartz T. W. (1987) Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Lett. 211, 169–174. 10.1016/0014-5793(87)81430-8. [DOI] [PubMed] [Google Scholar]
- Kreymann B.; Ghatei M. A.; Williams G.; Bloom S. R. (1987) Glucagon-like peptide-1 7–36: A physiological incretin in man. Lancet 330, 1300–1304. 10.1016/S0140-6736(87)91194-9. [DOI] [PubMed] [Google Scholar]
- Drucker D. J.; Philippe J.; Mojsov S. (1988) Proglucagon gene expression and posttranslational processing in a hamster islet cell line. Endocrinology 123, 1861–1867. 10.1210/endo-123-4-1861. [DOI] [PubMed] [Google Scholar]
- Drucker D. J.; Philippe J.; Jepeal L.; Habener J. F. (1987) Glucagon gene 5′-flanking sequences promote islet cell-specific glucagon gene transcription. J. Biol. Chem. 262, 15659–15665. [PubMed] [Google Scholar]
- Philippe J.; Drucker D. J.; Habener J. F. (1987) Glucagon gene transcription in an islet cell line is regulated via a protein kinase C-activated pathway. J. Biol. Chem. 262, 1823–1828. [PubMed] [Google Scholar]
- Drucker D. J.; Asa S. (1988) Glucagon gene expression in vertebrate brain. J. Biol. Chem. 263, 13475–13478. [PubMed] [Google Scholar]
- Drucker D. J.; Brubaker P. L. (1989) Proglucagon gene expression is regulated by a cyclic AMP-dependent pathway in rat intestine. Proc. Natl. Acad. Sci. U. S. A. 86, 3953–3957. 10.1073/pnas.86.11.3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe J.; Drucker D. J.; Knepel W.; Jepeal L.; Misulovin Z.; Habener J. F. (1988) Alpha-cell-specific expression of the glucagon gene is conferred to the glucagon promoter element by the interactions of DNA-binding proteins. Mol. Cell. Biol. 8, 4877–4888. 10.1128/MCB.8.11.4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe J.; Mojsov S.; Drucker D. J.; Habener J. F. (1986) Proglucagon processing in rat islet cell line resembles phenotype of intestine rather than pancreas. Endocrinology 119, 2833–2839. 10.1210/endo-119-6-2833. [DOI] [PubMed] [Google Scholar]
- Hanahan D. (1985) Heritable formation of pancreatic B-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 315, 115–122. 10.1038/315115a0. [DOI] [PubMed] [Google Scholar]
- Lee Y. C.; Asa S. L.; Drucker D. J. (1992) Glucagon gene 5′-flanking sequences direct expression of SV40 large T antigen to the intestine producing carcinoma of the large bowel in transgenic mice. J. Biol. Chem. 267, 10705–10708. [PubMed] [Google Scholar]
- Drucker D. J.; Lee Y. C.; Asa S. L.; Brubaker P. L. (1992) Inhibition of pancreatic glucagon gene expression in mice bearing a subcutaneous glucagon-producing GLUTag transplantable tumor. Mol. Endocrinol. 6, 2175–2184. 10.1210/mend.6.12.1491697. [DOI] [PubMed] [Google Scholar]
- Drucker D. J.; Jin T.; Asa S. L.; Young T. A.; Brubaker P. L. (1994) Activation of proglucagon gene transcription by protein kinase A in a novel mouse enteroendocrine cell line. Mol. Endocrinol. 8, 1646–1655. 10.1210/mend.8.12.7535893. [DOI] [PubMed] [Google Scholar]
- Brubaker P. L.; Schloos J.; Drucker D. J. (1998) Regulation of glucagon-like peptide-1 synthesis and secretion in the GLUTag enteroendocrine cell line. Endocrinology 139 (10), 4108–14. 10.1210/endo.139.10.6228. [DOI] [PubMed] [Google Scholar]
- Drucker D. J.; Erlich P.; Asa S. L.; Brubaker P. L. (1996) Induction of intestinal epithelial proliferation by glucagon-like peptide 2. Proc. Natl. Acad. Sci. U. S. A. 93, 7911–7916. 10.1073/pnas.93.15.7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson M. H.; Bloom S. R.; Polak J. M.; Henry K.; Dowling R. H. (1971) Endocrine tumour in kidney affecting small bowel structure, motility, and absorptive function. Gut 12, 773–782. 10.1136/gut.12.10.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B.; Fishman E. K.; Bayless T. M.; Siegelman S. S. (1983) Villous hypertrophy of the small bowel in a patient with glucagonoma. J. Comput. Assist Tomogr 7 (2), 334–7. [PubMed] [Google Scholar]
- Myojo S.; Tsujikawa T.; Sasaki M.; Fujiyama Y.; Bamba T. (1997) Trophic effects of glicentin on rat small-intestinal mucosa in vivo and in vitro. J. Gastroenterol. 32 (3), 300–5. 10.1007/BF02934484. [DOI] [PubMed] [Google Scholar]
- Hoosein N. M.; Gurd R. S. (1984) Human glucagon-like peptides 1 and 2 activate rat brain adenylate cyclase. FEBS Lett. 178, 83–86. 10.1016/0014-5793(84)81245-4. [DOI] [PubMed] [Google Scholar]
- Tsai C.-H.; Hill M.; Asa S. L.; Brubaker P. L.; Drucker D. J. (1997) Intestinal growth-promoting properties of glucagon-like peptide 2 in mice. American journal of physiology 273, E77–E84. 10.1152/ajpendo.1997.273.1.E77. [DOI] [PubMed] [Google Scholar]
- Tsai C.-H.; Hill M.; Drucker D. J. (1997) Biological determinants of intestinotrophic properties of GLP-2 in vivo. American journal of physiology 272, G662–G668. 10.1152/ajpgi.1997.272.3.G662. [DOI] [PubMed] [Google Scholar]
- Mentlein R.; Gallwitz B.; Schmidt W. E. (1993) Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7–36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur. J. Biochem. 214, 829–835. 10.1111/j.1432-1033.1993.tb17986.x. [DOI] [PubMed] [Google Scholar]
- Kieffer T. J.; McIntosh C. H.; Pederson R. A. (1995) Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 136 (8), 3585–96. 10.1210/endo.136.8.7628397. [DOI] [PubMed] [Google Scholar]
- Watanabe Y.; Kojima T.; Fujimoto Y. (1987) Deficiency of membrane-bound dipeptidyl aminopeptidase IV in a certain rat strain. Experientia 43, 400–401. 10.1007/BF01940426. [DOI] [PubMed] [Google Scholar]
- Drucker D. J.; Shi Q.; Crivici A.; Sumner-Smith M.; Tavares W.; Hill M.; DeForest L.; Cooper S.; Brubaker P. L. (1997) Regulation of the biological activity of glucagon-like peptide 2 in vivo by dipeptidyl peptidase IV. Nat. Biotechnol. 15 (7), 673–7. 10.1038/nbt0797-673. [DOI] [PubMed] [Google Scholar]
- Boushey R. P.; Yusta B.; Drucker D. J. (1999) Glucagon-like peptide 2 decreases mortality and reduces the severity of indomethacin-induced murine enteritis. American journal of physiology 277, E937–E947. 10.1152/ajpendo.1999.277.5.E937. [DOI] [PubMed] [Google Scholar]
- Boushey R. P.; Yusta B.; Drucker D. J. (2001) Glucagon-like peptide (GLP)-2 reduces chemotherapy-associated mortality and enhances cell survival in cells expressing a transfected GLP-2 receptor. Cancer Res. 61 (2), 687–93. [PubMed] [Google Scholar]
- Drucker D. J.; Yusta B.; Boushey R. P.; Deforest L.; Brubaker P. L. (1999) Human [Gly2]-GLP-2 reduces the severity of colonic injury in a murine model of experimental colitis. American journal of physiology 276, G79–G91. 10.1152/ajpgi.1999.276.1.G79. [DOI] [PubMed] [Google Scholar]
- Drucker D. J.; Yusta B. (2014) Physiology and pharmacology of the enteroendocrine hormone glucagon-like peptide-2. Annu. Rev. Physiol. 76, 561–83. 10.1146/annurev-physiol-021113-170317. [DOI] [PubMed] [Google Scholar]
- Brubaker P. L.; Izzo A.; Hill M.; Drucker D. J. (1997) Intestinal function in mice with small bowel growth induced by glucagon-like peptide-2. American journal of physiology 272, E1050–E1058. 10.1152/ajpendo.1997.272.6.E1050. [DOI] [PubMed] [Google Scholar]
- Scott R. B.; Kirk D.; MacNaughton W. K.; Meddings J. B. (1998) GLP-2 augments the adaptive response to massive intestinal resection in rat. American journal of physiology 275, G911–G921. 10.1152/ajpgi.1998.275.5.G911. [DOI] [PubMed] [Google Scholar]
- Munroe D. G.; Gupta A. K.; Kooshesh F.; Vyas T. B.; Rizkalla G.; Wang H.; Demchyshyn L.; Yang Z. J.; Kamboj R. K.; Chen H.; McCallum K.; Sumner-Smith M.; Drucker D. J.; Crivici A. (1999) Prototypic G protein-coupled receptor for the intestinotrophic factor glucagon-like peptide 2. Proc. Natl. Acad. Sci. U. S. A. 96 (4), 1569–73. 10.1073/pnas.96.4.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo K. E.; Miller L. J.; Bataille D.; Dalle S.; Goke B.; Thorens B.; Drucker D. J. (2003) International Union of Pharmacology. XXXV. The Glucagon Receptor Family. Pharmacol. Rev. 55 (1), 167–94. 10.1124/pr.55.1.6. [DOI] [PubMed] [Google Scholar]
- Yusta B.; Huang L.; Munroe D.; Wolff G.; Fantaske R.; Sharma S.; Demchyshyn L.; Asa S. L.; Drucker D. J. (2000) Enteroendocrine localization of GLP-2 receptor expression. Gastroenterology 119 (3), 744–755. 10.1053/gast.2000.16489. [DOI] [PubMed] [Google Scholar]
- DaCambra M. P.; Yusta B.; Sumner-Smith M.; Crivici A.; Drucker D. J.; Brubaker P. L. (2000) Structural Determinants for Activity of Glucagon-like Peptide-2. Biochemistry 39 (30), 8888–8894. 10.1021/bi000497p. [DOI] [PubMed] [Google Scholar]
- Carroll R. E.; Benedetti E.; Schowalter J. P.; Buchman A. L. (2016) Management and Complications of Short Bowel Syndrome: an Updated Review. Curr. Gastroenterol Rep 18 (7), 40. 10.1007/s11894-016-0511-3. [DOI] [PubMed] [Google Scholar]
- Duggan C. P.; Jaksic T. (2017) Pediatric Intestinal Failure. N. Engl. J. Med. 377 (7), 666–675. 10.1056/NEJMra1602650. [DOI] [PubMed] [Google Scholar]
- Jeppesen P. B.; Hartmann B.; Thulesen J.; Graff J.; Lohmann J.; Hansen B. S.; Tofteng F.; Poulsen S. S.; Madsen J. L.; Holst J. J.; Mortensen P. B. (2001) Glucagon-like Peptide 2 Improves Nutrient Absorption and Nutritional Status in Short-Bowel Patients With No Colon. Gastroenterology 120 (4), 806–815. 10.1053/gast.2001.22555. [DOI] [PubMed] [Google Scholar]
- Jeppesen P. B.; Sanguinetti E. L.; Buchman A.; Howard L.; Scolapio J. S.; Ziegler T. R.; Gregory J.; Tappenden K. A.; Holst J.; Mortensen P. B. (2005) Teduglutide (ALX-0600), a dipeptidyl peptidase IV resistant glucagon-like peptide 2 analogue, improves intestinal function in short bowel syndrome patients. Gut 54 (9), 1224–31. 10.1136/gut.2004.061440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen P. B.; Gilroy R.; Pertkiewicz M.; Allard J. P.; Messing B.; O’Keefe S. J. (2011) Randomised placebo-controlled trial of teduglutide in reducing parenteral nutrition and/or intravenous fluid requirements in patients with short bowel syndrome. Gut 60 (7), 902–14. 10.1136/gut.2010.218271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen P. B.; Pertkiewicz M.; Messing B.; Iyer K.; Seidner D. L.; O’Keefe J.; Forbes A.; Heinze H.; Joelsson B. (2012) Teduglutide reduces need for parenteral support among patients with short bowel syndrome with intestinal failure. Gastroenterology 143 (6), 1473–1481. 10.1053/j.gastro.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Jeppesen P. B.; Lund P.; Gottschalck I. B.; Nielsen H. B.; Holst J. J.; Mortensen J.; Poulsen S. S.; Quistorff B.; Mortensen P. B. (2009) Short bowel patients treated for two years with glucagon-like Peptide 2: effects on intestinal morphology and absorption, renal function, bone and body composition, and muscle function. Gastroenterol Res. Pract 2009, 616054. 10.1155/2009/616054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen P. B.; Lund P.; Gottschalck I. B.; Nielsen H. B.; Holst J. J.; Mortensen J.; Poulsen S. S.; Quistorff B.; Mortensen P. B. (2009) Short bowel patients treated for two years with glucagon-like peptide 2 (GLP-2): compliance, safety, and effects on quality of life. Gastroenterol Res. Pract 2009, 425759. 10.1155/2009/425759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer K. R.; Kunecki M.; Boullata J. I.; Fujioka K.; Joly F.; Gabe S.; Pape U. F.; Schneider S. M.; Virgili Casas M. N.; Ziegler T. R.; Li B.; Youssef N. N.; Jeppesen P. B. (2017) Independence From Parenteral Nutrition and Intravenous Fluid Support During Treatment With Teduglutide Among Patients With Intestinal Failure Associated With Short Bowel Syndrome. JPEN, J. Parenter. Enteral Nutr. 41, 946. 10.1177/0148607116680791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compher C.; Gilroy R.; Pertkiewicz M.; Ziegler T. R.; Ratcliffe S. J.; Joly F.; Rochling F.; Messing B. (2011) Maintenance of parenteral nutrition volume reduction, without weight loss, after stopping teduglutide in a subset of patients with short bowel syndrome. JPEN, J. Parenter. Enteral Nutr. 35 (5), 603–9. 10.1177/0148607111414431. [DOI] [PubMed] [Google Scholar]
- Koehler J. A.; Harper W.; Barnard M.; Yusta B.; Drucker D. J. (2008) Glucagon-like peptide-2 does not modify the growth or survival of murine or human intestinal tumor cells. Cancer Res. 68, 7897–7904. 10.1158/0008-5472.CAN-08-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]