

Abstract
Alzheimer’s disease (AD) is a debilitating neurodegenerative disorder affecting millions worldwide. Currently, there are only four approved treatments for AD, which improve symptoms modestly. AD is believed to be caused by the formation of intercellular plaques and intracellular tangles in the brain, but thus far all new drugs which target these pathologies have failed clinical trials. New research highlights the link between AD and Type II Diabetes (T2D), and some believe that AD is actually a brain specific form of it termed Type III Diabetes (T3D). Drugs which are currently approved for the treatment of T2D, such as metformin, have shown promising results in improving cognitive function and even preventing the development of AD in diabetic patients. Recent studies shed light on the relationship between the brain and cardiovascular system in which the brain and heart communicate with one another via the vasculature to regulate fluid and nutrient homeostasis. This line of research reveals how the brain–heart axis regulates hypertension and diabetes, both of which can impact cognitive function. In this review we survey past and ongoing research and clinical trials for AD, and argue that AD is a complex and systemic disorder which requires comprehensive approaches beyond the brain for effective prevention and/or treatment.
Keywords: Alzheimer’s disease, amyloid, diabetes, clinical trials, hypertension
Introduction
Alzheimer’s Disease Pathogenesis
Alzheimer’s disease (AD) is a fatal neurodegenerative disease thought to account for up to 70% of dementia cases.1 In the United States alone, nearly 6 million people are estimated to have AD, and this figure is projected to reach 13.8 million by 2050.2 The primary molecular hallmarks thought to drive the pathogenesis of AD are the generation of plaques comprised of amyloid β (Aβ) and neurofibrillary tangles (NFTs) formed within neuronal cell bodies due to hyperphosphorylation of the microtubule-associated protein, tau.1 Only four FDA approved therapies are currently used to treat AD; three central cholinesterase inhibitors (ChEIs) (donepezil, galantamine, and rivastigmine), and the N-methyl-d-aspartate (NMDA) receptor blocker memantine. While each drug, or a combination of memantine and ChEIs modestly improves cognitive scores in patients compared to placebo, no novel therapeutics have demonstrated efficacy in global trials to treat AD in over 15 years.3,4
Clinical Trials/Failures
Therapies designed to prevent Aβ accumulation (β-site amyloid precursor peptide cleaving enzyme 1 (BACE1) and γ-secretase inhibitors) and accelerate Aβ clearance (monoclonal antibodies) have largely failed in phase III clinical trials (summarized in Table 1). The BACE1 inhibition with Verubecestat failed to improve cognitive function5 while trials testing the BACE1 inhibitors lanabecestat and elenbecestat were halted for futility and unfavorable risk/benefit ratios according to recent press releases from AstraZeneca and Eisai/Biogen. γ-Secretase inhibitors Semagacestat and Avagacestat resulted in severe adverse outcomes including weight loss, infection, and skin cancer with no improvement in cognitive status; higher doses of Semagacestat even correlated to worsened cognitive function compared to placebo.6,7 Patients treated with the Notch-sparing γ-secretase inhibitor tarenflurbil experienced less severe adverse events than those taking nonselective inhibitors, but did not show improved cognition compared to placebo.8 Monoclonal antibodies bapineuzumab, solanezumab, crenezumab, and gantenerumab failed to show efficacy in phase III trials despite evidence of amyloid clearance in treatment groups.9−12 Notably, exploratory analyses from the Gantenerumab SCarlet RoAD trial suggested a potential need for higher dosages to attain clinical efficacy; as a result, two new phase III trials (NCT03443973, NCT03444870) have started to evaluate higher doses of gantenerumab in patients with early AD.12 Aducanumab phase III trials “Emerge” and “Engage” were halted in March 2019 as a futility analysis concluded that the trials were unlikely to meet their primary end points. Unexpectedly, Biogen recently filed for FDA approval for aducanumab. Biogen stated that the futility analysis was based on data from 1748 patients who completed the 18-month treatment; however, data from additional patients became available allowing Biogen to repeat the analysis with a total of 2066 patients who completed the treatment. With this expanded database, Biogen concluded that while the Engage trial still missed its primary end point, the Emerge trial met its primary end point, slowing cognitive decline by ∼23% compared to placebo in the high dose treatment group. Notably, higher doses of Aducanumab also positively correlated with amyloid related imaging abnormalities-edema (ARIA-E). In a Chinese cohort, GV-971, an oligosaccharide which targets Aβ, improved cognitive scores at 36 weeks of treatment compared to placebo in a phase III trial; Shanghai Green Valley Pharmaceutical is looking to test GV-971 in a global phase III trial in the near future.
Table 1. Outcomes of Clinical Trials Targeting Aβ and Tau To Treat AD.
| class | therapy | target | trial outcome | associated trials |
|---|---|---|---|---|
| APP cleaving enzyme inhibitors | verubecestat (MK-8931) | BACE1 | failed in phase III – lack of efficacy5 | NCT01953601 |
| lanabecestat (LY3314814, AZD3293) | BACE1 | stopped for futility in phase III | NCT02783573 | |
| NCT02245737 | ||||
| elenbecestat | BACE1 | discontinued by Eisai/Biogen in phase III–unfavorable risk-benefit ratio | NCT02956486 | |
| NCT03036280 | ||||
| Semagacestat (LY450139) | γ-secretase | failed in phase III–lack of efficacy and serious adverse events6 | NCT00594568 - IDENTITY1 | |
| NCT00762411 - IDENTITY2 | ||||
| Avagacestat (BMS-708163) | γ-secretase | failed in phase II–lack of efficacy and serious adverse events7 | NCT00890890 | |
| tarenflurbil | γ-secretase selective | failed in phase III–lack of efficacy8 | NCT00105547 | |
| (flurizan, r-flurbiprofen, MPC-7869) | NCT00322036 | |||
| NCT00380276 | ||||
| monoclonal antibodies | bapineuzumab | Aβ oligomers/plaques | failed in phase III–lack of efficacy9,50 | NCT00667810 |
| NCT00575055 | ||||
| NCT00574132 | ||||
| solanezumab (LY2062430) | monomeric Aβ | failed in phase III–lack of efficacy10,11 | NCT00905372 - EXPEDITION1 | |
| NCT00904683 - EXPEDITION2 | ||||
| NCT01900665 - EXPEDITION3 | ||||
| crenezumab | Aβ monomers/oligomers/fibrils | stopped for futility in phase III | NCT02670083 - CREAD | |
| NCT03114657 - CREAD2 | ||||
| gantenerumab | Aβ fibrils | SCarlet RoAD and Marguerite RoAD were stopped for futility in phase III. Two more phase III trials are still ongoing.12 | NCT01224106 (SCarlet RoAD) | |
| NCT02051608 (Marguerite RoAD) | ||||
| NCT03443973 | ||||
| NCT03444870 | ||||
| aducanumab | Aβ oligomers/fibrils | stopped for futility in phase IIIa | NCT02484547 - EMERGE | |
| NCT02477800 - ENGAGE | ||||
| BAN2401 | Aβ protofibrils | ongoing in phase II and phase III | NCT01767311 | |
| NCT03887455 | ||||
| CAD106 | Aβ amyloid formation | ongoing in phase II | NCT02565511 | |
| Tau aggregate inhibitor | LMTM (TRx0237) | Tau aggregates | failed in phase III–lack of efficacy | NCT01689233 |
| a new phase III trial evaluating LMTM as a monotherapy is ongoing13 | NCT01689246 | |||
| NCT03539380 | ||||
| Tau PTM inhibitor | lithium | GSK3 (α and β) | failed in phase II–lack of efficacy14 | ISRCTN72046462 |
| tideglusib | GSK3β | failed in phase II–lack of efficacy15 | NCT01350362 | |
| tau immunotherapy | AADvac1 | vaccine to generate antibodies against tau | safe and tolerable in phase II and promising trends in biomarker and cognitive readouts vs placebo | NCT02579252 |
| anti-amyloid | GV-971 (sodium oligo-mannurarate) | preclinical data suggests the compound inhibits Aβ fibril formation, is anti-inflammatory, and normalizes gut microbiome changes connected to AD | met primary and secondary end points in Chinese phase III, looking to expand to global population | NCT02293915 |
Biogen recently filed for FDA approval for Aducanumab.
Tau has also been targeted therapeutically. The tau aggregate inhibitor LMTM failed to demonstrate efficacy in phase III clinical trial; however, a posthoc subgroup analysis suggested LMTM may be salutary as a monotherapy.13 A new trial (NCT03539380) is underway to evaluate the efficacy of LMTM in patients with early and mild-moderate AD who are not currently taking approved AD drugs. Phase II trials targeting the glycogen synthase kinase 3 (GSK3) failed to reduce phosphorylated levels of its downstream target, tau, in cerebrospinal fluid (CSF) or improve cognitive performance.14,15 The tau vaccine AADvac1 demonstrated safety and tolerability in a phase II clinical trial with promising trends in reduced phosphorylated tau and improved cognitive function; thus, phase III trials will begin soon.
While some therapies targeting Aβ and tau remain underway, most therapies to date have failed to meet primary end points in phase II/III trials. It has been hypothesized that many of these treatments have been administered too late; once plaques and NFTs form, cytotoxicity and neurodegeneration may be irreversible.3 However, recent trials testing crenezumab, gantenerumab, and LMTM in patients with early disease states also failed to show efficacy. Moreover, transgenic mouse models overexpressing Aβ42 did not recapitulate memory deficits or neurodegeneration seen in amyloid precursor peptide (APP) transgenic models, but still generated amyloid plaques suggesting amyloid aggregation is not inherently cytotoxic.16 In addition, PET scans have established that subgroups of patients with cognitive impairment may have little to no amyloid accumulation, whereas elderly individuals with normal cognition may have extensive amyloid accumulation as seen in dementia patients.17 Thus, it has been argued that AD pathology is multifactorial rather than driven by a single process.4,17
Diabetes Exacerbates Alzheimer’s Disease
Growing evidence suggests a link between diabetes and AD. Patients with diabetes mellitus have a 65% increased risk of developing AD.18 Among AD patients within the Mayo Clinic Alzheimer’s Disease Patient Registry, 35% also had type II diabetes mellitus (T2D) and another 46% had impaired fasting glucose.19 Glucose utilization, as well as insulin and insulin-like growth factor (IGF) signaling are impaired in the brains of patients with AD.20 However, mice challenged with high fat diet did not exhibit AD histopathology, amyloid plaque formation, tau hyperphosphorylation, or impaired IGF signaling despite increased insulin resistance in the brain and peripheral tissues, increased tau protein levels, and mild brain atrophy, suggesting that T2D does not cause AD, but may contribute to disease progression.21 Furthermore, the Honolulu–Asia aging study established that patients with T2D had elevated risks for vascular brain damage and neurodegeneration.22
Intriguingly, a single intracerebral injection of streptozotocin (STZ) in rats markedly impaired insulin signaling, caused substantial brain atrophy, activated GSK-3β, increased tau phosphorylation, and increased Aβ accumulation without elevating blood glucose or insulin levels.21 Remarkably, treatment with peroxisome proliferator-activated receptor (PPAR)-δ or PPAR-γ agonists partially prevented STZ-mediated neurodegeneration and cell loss, tau phosphorylation, learning and memory impairments, and partially restored insulin signaling, suggesting that treatment with insulin sensitizing agents may be neuroprotective in patients. Thus, the de la Monte group has proposed that AD is a distinct, “brain specific” form of diabetes, “type 3 diabetes” (T3D).21
Using Diabetic Therapies To Treat AD
Due to the epidemiological link observed between T2D and AD as well as preclinical studies suggesting AD is a brain specific form of diabetes itself, diabetic therapies have been tested in clinical trials to treat AD in nondiabetics (summarized in Table 2). Thus far, PPAR-γ agonists pioglitazone and rosiglitazone and inhaled insulin have failed to demonstrate efficacy in phase III trials.23−25 However, the device used to administer inhaled insulin was subject to malfunctioning and was switched out for a new device partway through the study. According to the authors, posthoc analyses on the group using the original device showed improved cognitive scores after 6 months of treatment compared to placebo. Intriguingly, the incidence of dementia and cognitive impairment was significantly lower in diabetics taking metformin than in diabetics not taking metformin, suggesting it may be effective at preventing the development of AD in diabetics.26 A phase II crossover study indicated improved executive function with trends toward improved learning, memory, and attention in patients treated with metformin, compared to placebo.27 Due to the link between T2D and AD, it is possible that effective treatment for T2D in turn reduces the development of AD. Metformin may have additional neuroprotective properties independent from its role in controlling T2D.
Table 2. Outcomes of Clinical Trials Using Diabetic Therapies to Treat AD.
| therapy | target | trial outcome | associated trials |
|---|---|---|---|
| metformin | AMPK, cAMP and others | improved executive function and trends toward increased learning, memory, and attention in phase II crossover study26 | NCT01965756 |
| inhaled insulin | blood glucose | failed in phase II/III–lack of efficacy | NCT01767909 |
| pioglitazone | PPARγ | terminated in phase III–lack of efficacy | NCT01931566 |
| rosiglitazone | PPARγ | failed in phase III–lack of efficacy23 | NCT00428090 (REFLECT-1) |
| NCT00348309 (REFLECT-2) | |||
| NCT00550420 | |||
| liraglutide | GLP-1 analogue | ongoing in phase II | NCT01843075 (ELAD) |
Recent studies suggest that targeting the incretin hormone glucagon-like peptide-1 (GLP-1), which promotes insulin secretion, shows promise for AD treatment. GLP-1 receptors are present in the brain and natural circulating GLP-1 receptor agonists can cross the blood-brain-barrier (BBB), thus opening the possibility to target this pathway in AD patients.28,29 GLP-1 agonists prevented cognitive decline in preclinical models and liraglutide, a GLP-1 analogue currently used to treat T2D is being tested in a phase II clinical trial for mild AD (NCT01843075).30,31 Moreover, a combination of GLP-1 and another incretin hormone, glucose independent insulinotropic polypeptide (GIP), has shown increased neuroprotective effects compared to GLP-1 alone.30
Inhibitors of the protease that degrades GLP-1, dipeptidyl peptidase-4 (DPP-4), such as vildagliptin and sitagliptin are also currently used to treat T2D. Brain GLP-1 and GLP-1 receptor levels are decreased both in human AD patients and in mouse models of AD. Furthermore, DPP4 inhibition restored brain expression of GLP-1 and GLP-1 receptor, improved learning and memory, and decreased tau phosphorylation and Aβ accumulation in mice suggesting a promising avenue for treating AD.32
Finally, sodium–glucose cotransporter 2 (SGLT2) inhibitors including empagliflozin, dapagliflozin, and canagliflozin, which are widely utilized to treat T2D, are also being explored for the treatment of AD.33 Diabetic mice treated with empagliflozin exhibited significant improvement in cognitive abilities which was linked to decreased cerebral oxidative stress and DNA damage.34 A new phase I clinical trial (NCT03852901) will evaluate the effect of empagliflozin on ketone levels and cognitive function.
Alzheimer’s Disease Is Systemic
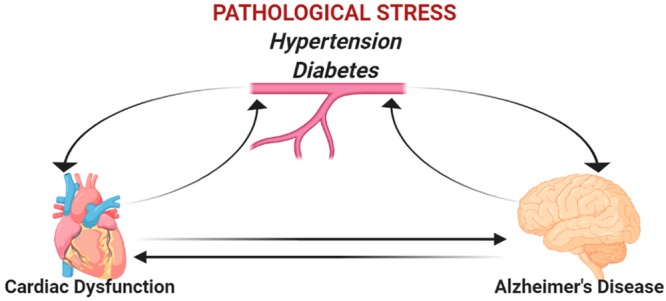
The origin of Aβ accumulation in the brain is somewhat controversial; Aβ and APP are produced both in the brain and in peripheral tissues.35 Circulating Aβ can cross the BBB via the receptor for advanced glycation end products (RAGE)36 and is sufficient to drive AD pathology in mice.35 In addition, hypertension induces RAGE expression, thereby promoting Aβ accumulation in the brain.37 The RAGE inhibitor azeliragon was tested in patients with mild AD but failed to show efficacy in a phase III clinical trial (NCT02080364). Moreover, it has been observed that Aβ also accumulates and may contribute to pathogenesis in peripheral tissues.38 Recently, Aβ accumulation was observed within cardiomyocytes from AD patient hearts, which coincided with trends toward cardiac diastolic dysfunction in early onset AD populations, thickening of the left ventricle walls in late-onset AD populations, and calcium homeostasis defects in isolated cardiomyocytes.39 Despite the limited sample size, this study identifies an intriguing link between AD and cardiac dysfunction warranting further investigation.
Numerous studies have revealed a clear link between cardiovascular and neurodegenerative diseases. It has been observed that cardiovascular disease decreases cerebral blood flow (CBF) resulting in oxidative stress and neurodegeneration; moreover, decreased CBF is a substantial risk factor for AD and promotes Aβ deposition.20,38 Furthermore, heart failure is positively correlated to cognitive decline.38 It has also been established that genetic mutations within presenilin (PSEN) 1 and 2 genes are linked to both AD and dilated cardiomyopathy, suggesting common pathogenesis for both diseases.40,41 With this in mind we propose to take a closer look at the brain–heart axis and its role in AD.
Renin-Angiotensin System
The renin-angiotensin system (RAS) is a key regulator of blood pressure and vasoconstriction. In this system, renin catalyzes the conversion of angiotensinogen (AGT) into angiotensin I (ANGI), which is subsequently converted to angiotensin II (ANGII) via the angiotensin converting enzyme 1 (ACE1). ANGII binds to angiotensin receptors (ATRs) which stimulate a wide variety of signaling cascades resulting in increased blood pressure. Renin also stimulates the conversion of AGT to ANGII via activation of the pro-renin receptor (PRR), which increases the enzymatic activity of pro-renin. While activation of RAS in the short term can have beneficial effects, chronic RAS activation leads to hypertension with deleterious effects on the renal and cardiovascular systems including hypertrophy and fibrosis.42
Hypertension and diabetes are two of the most common risk factors for cardiovascular disease and are often observed together.43 Many pathways activated by ANGII are also deregulated in patients with diabetes, including oxidative stress and inflammation. Further, increased reactive oxygen species triggered by chronic ANGII activation can lead to insulin resistance and dyslipidemia thereby causing diabetes, and can lead to increased sodium retention thereby contributing to hypertension.43 In support of their interconnected role ACE and ATR inhibitors reduce mortality in patients with both hypertension and diabetes.
It has been more recently established that all of the major RAS players are expressed in astrocytes, glial cells, and neurons, and they are most abundant in areas of the brain which regulate the heart and fluid homeostasis44 (Figure 1). This brain RAS system appears to play a major role in regulating systemic blood pressure. Brain specific knockout of AGT, PRR, or ATRs can blunt hypertension, and brain specific overexpression of genes which promote ANGII production in the brain can induce hypertension in rodents.42
Figure 1.
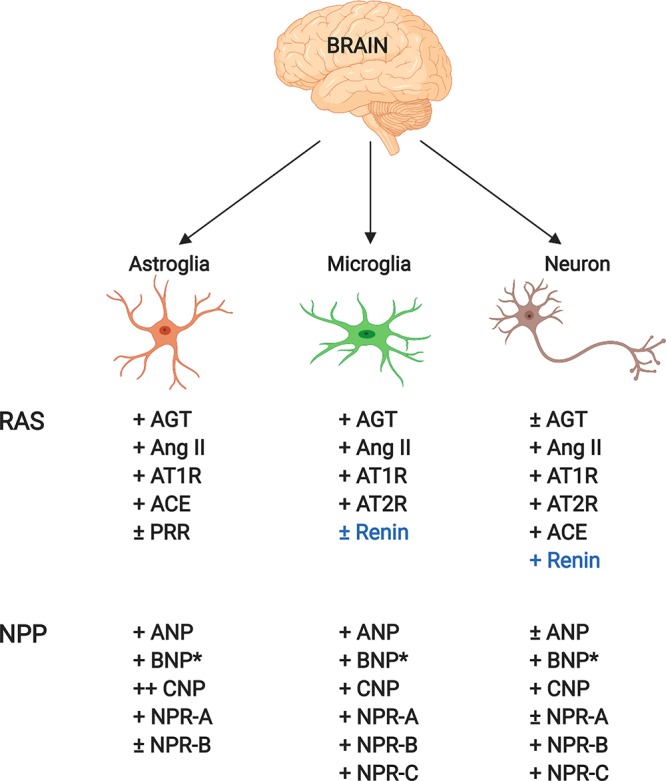
Depiction of renin angiotensin (RAS) and natriuretic peptide (NP) players and their expression levels in the brain. Blue text refers to the controversial nature of whether renin is expressed in the brain: (∗) only protein detected, not mRNA; (+) expressed; (±) expressed at lower levels compared to other cell types.
Increased RAS activity in the brain is associated with increased resting metabolic rate, and inhibition of ATRs in the brain can impair weight loss by preventing sympathetic activation of brown adipose tissue.43 Angiotensin and leptin hormone receptors can be found on the same neurons in the brain suggesting that the two are interconnected. Given the link between obesity and hypertension/diabetes, this suggests that brain RAS may be a major regulator of metabolism and that alterations could lead to metabolic syndrome and its associated cardiovascular and neurological disorders.
Intriguingly, brain ACE activity is increased in patients with cardiovascular disease, and ACE inhibitors have been demonstrated to reduce cognitive decline in elderly patients.44 Thus, this axis could be an important way in which the brain and heart can interact with one another and could be a novel therapeutic target.45 For example, ∼25% of the 80 million Americans who are hypertensive do not respond to classical RAS inhibitors and have been classified as having “resistant hypertension”.46 Xu et al., propose using novel brain PRR antagonists which block pathological activity of PRRs and inhibit hypertension.47 This avenue is currently limited as existing peptide antagonists cannot pass the BBB, but remains a promising target to reduce hypertension and thereby decrease the risk of both T2D and “T3D”.
Natriuretic Peptides
The natriuretic peptides (NPs) are a group of hormones which regulate fluid volume and blood pressure by the stimulation of sodium excretion, in direct opposition to RAS. The three NPs are atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and C-type natriuretic peptide (CNP). NPs bind to the three natriuretic peptide receptors (NPRs)-NPRA (binds ANP/BNP), NPRB (binds CNP), and NPRC which binds to all three and is responsible for their turnover. It is well-known that NPs are expressed in cardiomyocytes during early development but are also re-expressed during cardiac remodeling in response to stress. As mentioned, hypertension is a major risk factor for T2D, which in turn affects brain function. Thus, just as the heart–brain relationship of RAS could be targeted therapeutically, perhaps so could the NPs.
NPRA and NPRB are expressed in the brain, with B being the more abundant variant which is expressed ubiquitously throughout the brain48 (Figure 1). All three NPs are also detected in the brain, but the lack of BNP mRNA indicates that it is synthesized elsewhere. In vitro experiments suggest that NPs have neuroprotective effects given that they increase cortical spreading depression, which can protect the brain from ischemia. In vivo studies suggest that large doses of ANP in the brain can decrease blood pressure, but only at supraphysiological levels. In contrast, intracerebroventricular delivery of CNP in sheep depressed blood pressure, suggesting that it may be the more potent player in the brain.
Human patients with brain injuries and stroke show increased levels of BNP. Additionally, the AGES study established a significant correlation between increased abundance of a precursor form of BNP and brain atrophy.49 Given that the brain cannot create its own BNP, this implies that another tissue, possibly the heart, must respond to brain injuries resulting in BNP activation, and further, that changes in the heart directly affect the brain. In a post-mortem study of AD vs healthy patients an increased number of NPRA receptors were observed in the brains of Alzheimer’s patients, and a decrease in NPRB receptors were observed in the CSF suggesting that there is a dynamic regulation of NP activity in the central nervous system of patients with AD. Complicating things, inhibitors of NP degrading peptidases such as neprilysin, which are currently used to combat hypertension, have recently come under scrutiny for potential side-effects in the brain. Neprilysin can also inhibit the endopeptidases which degrade Aβ, which could increase Aβ accumulation in the brain, thereby increasing the risk of AD. Thus, elucidating additional mechanisms to promote the NP pathways in the brain may be necessary for effective treatment.
Perspectives
Alzheimer’s disease is a complex, multifactorial disease that we do not fully understand. Brain-centric therapies have not succeeded clinically, as monotherapies or in conjunction with existing therapies including ChEIs and memantine thus far. Diabetes and AD are closely linked, and diabetic therapies (metformin specifically) have been demonstrated to prevent the development of AD suggesting that strategies to target insulin and IGF resistance in the brain may be the way of the future. Both AD and T2D have a strong connection to hypertension, and the pathways which regulate blood pressure and sodium retention have a strong relationship between the brain and the heart (Figure 2). Therefore, combination therapies that target T2D, hypertension, and AD may provide additional benefit to patients. We propose that future studies should take a more holistic approach to understanding the effects of AD and think outside the brain to uncover novel druggable targets to prevent or treat Alzheimer’s disease.
Figure 2.

Diagram depicting the interconnection of diabetes, hypertension, cardiovascular disease, and AD and how disease progression has been targeted clinically thus far. Causative factors driving increased circulating Aβ and IGF resistance in the brain (blue text) remain incompletely understood despite being tightly linked to AD pathogenesis.
Acknowledgments
We thank Ashley Bourke (University of Colorado Anschutz Medical Campus) for valuable insight on this manuscript. R.A.B. and J.L.M. were supported by fellowships from the Canadian Institutes of Health Research (FRN-216927) and (FRN-395620). A.S.R. was supported by a predoctoral award from the American Heart Association (18PRE34030030). Figures were created with Biorender.com.
Glossary
List of Abbreviations
- Aβ
amyloid beta
- ACE1
angiotensin converting enzyme I
- AD
Alzheimer’s disease
- AGT
angiotensinogen
- ANGI
angiotensin I
- ANGII
angiotensin II
- ANP
atrial natriuretic peptide
- APP
amyloid precursor peptide
- ATR
angiotensin receptor
- BACE1
β-site amyloid precursor peptide cleaving enzyme 1
- BBB
blood brain barrier
- BNP
brain natriuretic peptide
- CBF
cerebral blood flow
- ChEI
central cholinesterase inhibitor
- CNP
C-type natriuretic peptide
- CSF
cerebrospinal fluid
- DPP4
dipeptidyl peptidase-4
- GIP
glucose independent insulinotropic polypeptide
- GLP-1
glucagon-like peptide-1
- GSK3
glycogen synthase kinase 3
- IGF
insulin-like growth factor
- LMTM
leuco- methylthioninium bis (hydromethanesulfonate)
- NFT
neurofibrillary tangles
- NMDA
N-methyl-d-aspartate
- NP
natriuretic peptide
- NPR
natriuretic peptide receptor
- PPAR
peroxisome proliferator-activated receptor
- PRR
pro-renin receptor
- PSEN
presenilin
- RAGE
receptor for advanced glycation end products
- RAS
renin angiotensin system
- SGLT2
sodium–glucose cotransporter 2
- STZ
streptozotocin
- T2D
type 2 diabetes
- T3D
type 3 diabetes
Author Contributions
# Equal contribution.
The authors declare no competing financial interest.
This article is made available for a limited time sponsored by ACS under the ACS Free to Read License, which permits copying and redistribution of the article for non-commercial scholarly purposes.
References
- Kametani F.; Hasegawa M. (2018) Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 12, 25. 10.3389/fnins.2018.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert L. E.; Weuve J.; Scherr P. A.; Evans D. A. (2013) Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 80, 1778–1783. 10.1212/WNL.0b013e31828726f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D.; Jackson R.; Paul G.; Shi J.; Sabbagh M. (2017) Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Invest. Drugs 26, 735–739. 10.1080/13543784.2017.1323868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J.; Hou J.; Ping J.; Cai D. (2018) Advances in developing novel therapeutic strategies for Alzheimer’s disease. Mol. Neurodegener. 13, 64. 10.1186/s13024-018-0299-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan M. F.; Kost J.; Voss T.; Mukai Y.; Aisen P. S.; Cummings J. L.; Tariot P. N.; Vellas B.; van Dyck C. H.; Boada M.; Zhang Y.; Li W.; Furtek C.; Mahoney E.; Harper Mozley L.; Mo Y.; Sur C.; Michelson D. (2019) Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N. Engl. J. Med. 380, 1408–1420. 10.1056/NEJMoa1812840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doody R. S.; Raman R.; Farlow M.; Iwatsubo T.; Vellas B.; Joffe S.; Kieburtz K.; He F.; Sun X.; Thomas R. G.; Aisen P. S.; Siemers E.; Sethuraman G.; Mohs R. (2013) A phase 3 trial of Semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 369, 341–350. 10.1056/NEJMoa1210951. [DOI] [PubMed] [Google Scholar]
- Coric V.; Salloway S.; van Dyck C. H.; Dubois B.; Andreasen N.; Brody M.; Curtis C.; Soininen H.; Thein S.; Shiovitz T.; Pilcher G.; Ferris S.; Colby S.; Kerselaers W.; Dockens R.; Soares H.; Kaplita S.; Luo F.; Pachai C.; Bracoud L.; Mintun M.; Grill J. D.; Marek K.; Seibyl J.; Cedarbaum J. M.; Albright C.; Feldman H. H.; Berman R. M. (2015) Targeting Prodromal Alzheimer Disease With Avagacestat: A Randomized Clinical Trial. JAMA Neurol 72, 1324–1333. 10.1001/jamaneurol.2015.0607. [DOI] [PubMed] [Google Scholar]
- Green R. C.; Schneider L. S.; Amato D. A.; Beelen A. P.; Wilcock G.; Swabb E. A.; Zavitz K. H.; (2009) Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA 302, 2557–2564. 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloway S.; Sperling R.; Fox N. C.; Blennow K.; Klunk W.; Raskind M.; Sabbagh M.; Honig L. S.; Porsteinsson A. P.; Ferris S.; Reichert M.; Ketter N.; Nejadnik B.; Guenzler V.; Miloslavsky M.; Wang D.; Lu Y.; Lull J.; Tudor I. C.; Liu E.; Grundman M.; Yuen E.; Black R.; Brashear H. R. (2014) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 322–333. 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doody R. S.; Thomas R. G.; Farlow M.; Iwatsubo T.; Vellas B.; Joffe S.; Kieburtz K.; Raman R.; Sun X.; Aisen P. S.; Siemers E.; Liu-Seifert H.; Mohs R. (2014) Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 311–321. 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]
- Honig L. S.; Vellas B.; Woodward M.; Boada M.; Bullock R.; Borrie M.; Hager K.; Andreasen N.; Scarpini E.; Liu-Seifert H.; Case M.; Dean R. A.; Hake A.; Sundell K.; Poole Hoffmann V.; Carlson C.; Khanna R.; Mintun M.; DeMattos R.; Selzler K. J.; Siemers E. (2018) Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med. 378, 321–330. 10.1056/NEJMoa1705971. [DOI] [PubMed] [Google Scholar]
- Ostrowitzki S.; Lasser R. A.; Dorflinger E.; Scheltens P.; Barkhof F.; Nikolcheva T.; Ashford E.; Retout S.; Hofmann C.; Delmar P.; Klein G.; Andjelkovic M.; Dubois B.; Boada M.; Blennow K.; Santarelli L.; Fontoura P. (2017) A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimer's Res. Ther. 9, 95. 10.1186/s13195-017-0318-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier S.; Feldman H. H.; Schneider L. S.; Wilcock G. K.; Frisoni G. B.; Hardlund J. H.; Moebius H. J.; Bentham P.; Kook K. A.; Wischik D. J.; Schelter B. O.; Davis C. S.; Staff R. T.; Bracoud L.; Shamsi K.; Storey J. M.; Harrington C. R.; Wischik C. M. (2016) Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 388, 2873–2884. 10.1016/S0140-6736(16)31275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H.; Ewers M.; Burger K.; Annas P.; Mortberg A.; Bogstedt A.; Frolich L.; Schroder J.; Schonknecht P.; Riepe M. W.; Kraft I.; Gasser T.; Leyhe T.; Moller H. J.; Kurz A.; Basun H. (2009) Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 70, 922–931. 10.4088/JCP.08m04606. [DOI] [PubMed] [Google Scholar]
- Lovestone S.; Boada M.; Dubois B.; Hull M.; Rinne J. O.; Huppertz H. J.; Calero M.; Andres M. V.; Gomez-Carrillo B.; Leon T.; del Ser T. (2015) A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimer's Dis. 45, 75–88. 10.3233/JAD-141959. [DOI] [PubMed] [Google Scholar]
- Kim J.; Chakrabarty P.; Hanna A.; March A.; Dickson D. W.; Borchelt D. R.; Golde T.; Janus C. (2013) Normal cognition in transgenic BRI2-Abeta mice. Mol. Neurodegener. 8, 15. 10.1186/1750-1326-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetelat G.; La Joie R.; Villain N.; Perrotin A.; de La Sayette V.; Eustache F.; Vandenberghe R. (2013) Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer’s disease. Neuroimage Clin 2, 356–365. 10.1016/j.nicl.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanitakis Z.; Wilson R. S.; Bienias J. L.; Evans D. A.; Bennett D. A. (2004) Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol. 61, 661–666. 10.1001/archneur.61.5.661. [DOI] [PubMed] [Google Scholar]
- Janson J.; Laedtke T.; Parisi J. E.; O’Brien P.; Petersen R. C.; Butler P. C. (2004) Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 53, 474–481. 10.2337/diabetes.53.2.474. [DOI] [PubMed] [Google Scholar]
- Ribaric S. (2016) The Rationale for Insulin Therapy in Alzheimer’s Disease. Molecules 21, 689. 10.3390/molecules21060689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte S. M.; Wands J. R. (2008) Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2, 1101–1113. 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf E. S.; White L. R.; Scheltens P.; Launer L. J. (2006) Brain aging in very old men with type 2 diabetes: the Honolulu-Asia Aging Study. Diabetes Care 29, 2268–2274. 10.2337/dc06-0243. [DOI] [PubMed] [Google Scholar]
- Risner M. E.; Saunders A. M.; Altman J. F.; Ormandy G. C.; Craft S.; Foley I. M.; Zvartau-Hind M. E.; Hosford D. A.; Roses A. D. (2006) Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 6, 246–254. 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- Harrington C.; Sawchak S.; Chiang C.; Davies J.; Donovan C.; Saunders A. M.; Irizarry M.; Jeter B.; Zvartau-Hind M.; van Dyck C. H.; Gold M. (2011) Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimer’s disease: two phase 3 studies. Curr. Alzheimer Res. 8, 592–606. 10.2174/156720511796391935. [DOI] [PubMed] [Google Scholar]
- Gold M.; Alderton C.; Zvartau-Hind M.; Egginton S.; Saunders A. M.; Irizarry M.; Craft S.; Landreth G.; Linnamagi U.; Sawchak S. (2010) Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dementia Geriatr. Cognit. Disord. 30, 131–146. 10.1159/000318845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig A. M.; Mechanic-Hamilton D.; Xie S. X.; Combs M. F.; Cappola A. R.; Xie L.; Detre J. A.; Wolk D. A.; Arnold S. E. (2017) Effects of the Insulin Sensitizer Metformin in Alzheimer Disease: Pilot Data From a Randomized Placebo-controlled Crossover Study. Alzheimer Dis. Assoc. Disord. 31, 107–113. 10.1097/WAD.0000000000000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell J. M.; Stephenson M. D.; de Courten B.; Chapman I.; Bellman S. M.; Aromataris E. (2018) Metformin Use Associated with Reduced Risk of Dementia in Patients with Diabetes: A Systematic Review and Meta-Analysis. J. Alzheimer's Dis. 65, 1225–1236. 10.3233/JAD-180263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst J. J. (2007) The physiology of glucagon-like peptide 1. Physiol. Rev. 87, 1409–1439. 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- Hansen H. H.; Fabricius K.; Barkholt P.; Niehoff M. L.; Morley J. E.; Jelsing J.; Pyke C.; Knudsen L. B.; Farr S. A.; Vrang N. (2015) The GLP-1 Receptor Agonist Liraglutide Improves Memory Function and Increases Hippocampal CA1 Neuronal Numbers in a Senescence-Accelerated Mouse Model of Alzheimer’s Disease. J. Alzheimer's Dis. 46, 877–888. 10.3233/JAD-143090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holscher C. (2018) Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer’s and Parkinson’s disease models. Neuropharmacology 136, 251–259. 10.1016/j.neuropharm.2018.01.040. [DOI] [PubMed] [Google Scholar]
- Batista A. F.; Forny-Germano L.; Clarke J. R.; Lyra E. S. N. M.; Brito-Moreira J.; Boehnke S. E.; Winterborn A.; Coe B. C.; Lablans A.; Vital J. F.; Marques S. A.; Martinez A. M.; Gralle M.; Holscher C.; Klein W. L.; Houzel J. C.; Ferreira S. T.; Munoz D. P.; De Felice F. G. (2018) The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J. Pathol 245, 85–100. 10.1002/path.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Zhou M.; Sun J.; Guo A.; Fernando R. L.; Chen Y.; Peng P.; Zhao G.; Deng Y. (2019) DPP-4 inhibitor improves learning and memory deficits and AD-like neurodegeneration by modulating the GLP-1 signaling. Neuropharmacology 157, 107668. 10.1016/j.neuropharm.2019.107668. [DOI] [PubMed] [Google Scholar]
- Heerspink H. J.; Perkins B. A.; Fitchett D. H.; Husain M.; Cherney D. Z. (2016) Sodium Glucose Cotransporter 2 Inhibitors in the Treatment of Diabetes Mellitus: Cardiovascular and Kidney Effects, Potential Mechanisms, and Clinical Applications. Circulation 134, 752–772. 10.1161/CIRCULATIONAHA.116.021887. [DOI] [PubMed] [Google Scholar]
- Lin B.; Koibuchi N.; Hasegawa Y.; Sueta D.; Toyama K.; Uekawa K.; Ma M.; Nakagawa T.; Kusaka H.; Kim-Mitsuyama S. (2014) Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc. Diabetol. 13, 148. 10.1186/s12933-014-0148-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu X. L.; Xiang Y.; Jin W. S.; Wang J.; Shen L. L.; Huang Z. L.; Zhang K.; Liu Y. H.; Zeng F.; Liu J. H.; Sun H. L.; Zhuang Z. Q.; Chen S. H.; Yao X. Q.; Giunta B.; Shan Y. C.; Tan J.; Chen X. W.; Dong Z. F.; Zhou H. D.; Zhou X. F.; Song W.; Wang Y. J. (2018) Blood-derived amyloid-beta protein induces Alzheimer’s disease pathologies. Mol. Psychiatry 23, 1948–1956. 10.1038/mp.2017.204. [DOI] [PubMed] [Google Scholar]
- Deane R.; Du Yan S.; Submamaryan R. K.; LaRue B.; Jovanovic S.; Hogg E.; Welch D.; Manness L.; Lin C.; Yu J.; Zhu H.; Ghiso J.; Frangione B.; Stern A.; Schmidt A. M.; Armstrong D. L.; Arnold B.; Liliensiek B.; Nawroth P.; Hofman F.; Kindy M.; Stern D.; Zlokovic B. (2003) RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 9, 907–913. 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Carnevale D.; Mascio G.; D’Andrea I.; Fardella V.; Bell R. D.; Branchi I.; Pallante F.; Zlokovic B.; Yan S. S.; Lembo G. (2012) Hypertension induces brain beta-amyloid accumulation, cognitive impairment, and memory deterioration through activation of receptor for advanced glycation end products in brain vasculature. Hypertension 60, 188–197. 10.1161/HYPERTENSIONAHA.112.195511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tublin J. M.; Adelstein J. M.; Del Monte F.; Combs C. K.; Wold L. E. (2019) Getting to the Heart of Alzheimer Disease. Circ. Res. 124, 142–149. 10.1161/CIRCRESAHA.118.313563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troncone L.; Luciani M.; Coggins M.; Wilker E. H.; Ho C. Y.; Codispoti K. E.; Frosch M. P.; Kayed R.; Del Monte F. (2016) Abeta Amyloid Pathology Affects the Hearts of Patients With Alzheimer’s Disease: Mind the Heart. J. Am. Coll. Cardiol. 68, 2395–2407. 10.1016/j.jacc.2016.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.; Parks S. B.; Kushner J. D.; Nauman D.; Burgess D.; Ludwigsen S.; Partain J.; Nixon R. R.; Allen C. N.; Irwin R. P.; Jakobs P. M.; Litt M.; Hershberger R. E. (2006) Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am. J. Hum. Genet. 79, 1030–1039. 10.1086/509900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni D.; Li A.; Tesco G.; McKay K. M.; Moore J.; Raygor K.; Rota M.; Gwathmey J. K.; Dec G. W.; Aretz T.; Leri A.; Semigran M. J.; Anversa P.; Macgillivray T. E.; Tanzi R. E.; del Monte F. (2010) Protein aggregates and novel presenilin gene variants in idiopathic dilated cardiomyopathy. Circulation 121, 1216–1226. 10.1161/CIRCULATIONAHA.109.879510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta P. K.; Griendling K. K. (2007) Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol Cell Physiol 292, C82–97. 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- Cheung B. M.; Li C. (2012) Diabetes and hypertension: is there a common metabolic pathway?. Curr. Atheroscler. Rep. 14, 160–166. 10.1007/s11883-012-0227-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa P.; Sigmund C. D. (2017) How Is the Brain Renin-Angiotensin System Regulated?. Hypertension 70, 10–18. 10.1161/HYPERTENSIONAHA.117.08550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester S. J.; Booz G. W.; Sigmund C. D.; Coffman T. M.; Kawai T.; Rizzo V.; Scalia R.; Eguchi S. (2018) Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 98, 1627–1738. 10.1152/physrev.00038.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun D. A.; Jones D.; Textor S.; Goff D. C.; Murphy T. P.; Toto R. D.; White A.; Cushman W. C.; White W.; Sica D.; Ferdinand K.; Giles T. D.; Falkner B.; Carey R. M. (2008) Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 51, 1403–1419. 10.1161/HYPERTENSIONAHA.108.189141. [DOI] [PubMed] [Google Scholar]
- Xu Q.; Jensen D. D.; Peng H.; Feng Y. (2016) The critical role of the central nervous system (pro)renin receptor in regulating systemic blood pressure. Pharmacol. Ther. 164, 126–134. 10.1016/j.pharmthera.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodes A.; Lichtstein D. (2014) Natriuretic hormones in brain function. Front. Endocrinol. (Lausanne, Switz.) 5, 201. 10.3389/fendo.2014.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabayan B.; van Buchem M. A.; Sigurdsson S.; Zhang Q.; Meirelles O.; Harris T. B.; Gudnason V.; Arai A. E.; Launer L. J. (2016) Cardiac and Carotid Markers Link With Accelerated Brain Atrophy: The AGES-Reykjavik Study (Age, Gene/Environment Susceptibility-Reykjavik). Arterioscler., Thromb., Vasc. Biol. 36, 2246–2251. 10.1161/ATVBAHA.116.308018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe R.; Rinne J. O.; Boada M.; Katayama S.; Scheltens P.; Vellas B.; Tuchman M.; Gass A.; Fiebach J. B.; Hill D.; Lobello K.; Li D.; McRae T.; Lucas P.; Evans I.; Booth K.; Luscan G.; Wyman B. T.; Hua L.; Yang L.; Brashear H. R.; Black R. S. (2016) Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimer's Res. Ther. 8, 18. 10.1186/s13195-016-0189-7. [DOI] [PMC free article] [PubMed] [Google Scholar]