

Abstract
G protein-coupled receptors (GPCRs) form the largest family of membrane proteins involved in signal transduction. Because of their ability to regulate a wide range of cellular responses and their dysregulation being associated with many diseases, GPCRs remain a key therapeutic target for several clinical indications. In recent years, it has been demonstrated that ligands for a given receptor can engage distinct pathways with different relative efficacies, a concept known as biased signaling or functional selectivity. However, the structural determinants of this phenomenon remain poorly understood. Using the β2-adrenergic receptor as a model, we identified a linker residue (L1243.43) between the known PIF and NPxxY structural motifs, that plays a central role in the differential efficacy of biased ligands toward the Gs and β-arrestin pathways. Given the high level of conservation of this linker residue, the study provides structural explanations for biased signaling that can be extrapolated to other GPCRs.
Keywords: G protein-coupled receptors (GPCR), biased ligands, bioluminescence resonance energy transfer (BRET), biosensors, β2-adrenergic receptor (β2AR), mutagenesis, structural microswitches, signal transduction
Introduction
G protein-coupled receptors (GPCRs) constitute a family of membrane proteins that initiate signaling cascades in several biological processes. This family has been successfully targeted in several clinical indications. Recently, it has been demonstrated that ligands of a given receptor can preferentially engage some signaling pathways over others, a concept known as biased signaling.1,2 The emergence of this concept has raised the possibility of identifying ligands that selectively modulate therapeutically relevant pathways while avoiding the ones leading to side effects.3 The ability of different ligands to differentially bias signaling of a given receptor toward distinct pathways is believed to result from the stabilization of distinct conformational ensembles4−6 that may involve conserved residues forming microswitches.7,8 Consistent with this notion, distinct dynamic receptor conformations upon binding of different ligands were observed using resonance-energy transfer (RET) biosensors,9−11 solution-state nuclear magnetic resonance spectroscopy, and molecular dynamic simulations.12,13
The β2-adrenergic receptor (β2AR) is a prototypical receptor for which ligands with different propensity to activate different pathways have been identified.14,15 In particular, salbutamol (SALB) and salmeterol (SALM), which are partial agonists, have been shown to preferentially activate the stimulatory G protein, Gs, over promoting the recruitment of β-arrestin.11,16 Thus, these compounds can be qualified as partial biased agonists. The difference in the binding mode between one of these partial biased agonists, salmeterol, and the full balanced β2AR agonist, epinephrine, has recently been published,16 providing a first level of structural explanation for the different efficacies. However, how these different binding modalities are propagated from the binding pocket toward the structural elements involved in the engagement of Gs and β-arrestin remains unsolved.
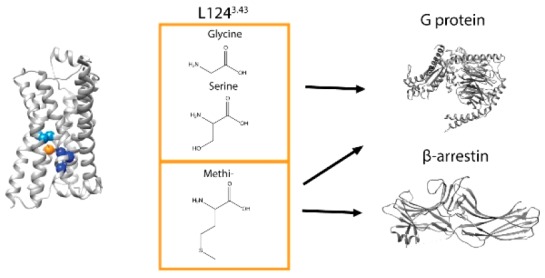
Indeed, although several microswitches such as the toggle switch, PIF/connector, NPxxY, and DRY motifs have been reported to be important for receptor activation, their specific roles in biased signaling remain poorly understood. The PIF/connector motif has been suggested to play an important role in connecting the agonist binding pocket to downstream conformational rearrangements required for receptor activation.17 The NPxxY motif for its part has been proposed as a stabilizing element of the active conformation.18 In a previous study, mutations of the residue L1243.43 (Ballesteros-Weinstein numbering19 in superscript) located between the PIF and NPxxY motifs of the β2AR (Figure 1a) resulted in a selective loss of isoproterenol (ISO)-stimulated β-arrestin recruitment.20 To test the hypothesis that this residue could represent a linker between the two microswitches and play a role in signal propagation and bias, we tested the impact of substituting L124 for M, G, and S on the activity of both balanced and biased ligands toward Gs activation and β-arrestin recruitment. These substitutions were selected based on evolutionary trace analysis20 for which the conservation of a given amino acid in class A GPCRs through evolution is considered, and suggested that these three substitutions could result in a distinct effect on receptor signaling.
Figure 1.

Functional impacts of L124 mutations on the β2AR. (a) Position of the mutated residue (orange) relative to the position of the PIF and NPxxY motifs (light and dark blue) on the β2AR (pdb:2Rh1). (b) Cell surface expression of the receptors detected by ELISA. (c–d) Concentration–response curves for the WT and mutant forms of β2AR (L124M/G/S) upon ISO and SALB stimulation for Gs activation (c) and β-arrestin recruitment (d) detected using BRET-based sensors. Data are shown as the mean ± SEM of three or four independent experiments.
Results and Discussion
The impacts of L124M/G/S mutations were first assessed on Gs activation using a bioluminescence RET (BRET)-based sensor.21 As shown in Figure1b,c and Table S1, L124G/S substitutions greatly increase the ligand-independent (constitutive) activity of the receptor toward Gs while abolishing the ability of the agonists ISO and SALB to further activate Gs. This loss in agonist responsiveness does not result from reduced binding, since L124S substitution results in a 100-fold increased affinity for ISO.20 The L124 M substitution for its part results in a modest increase of the constitutive activation of the receptor toward Gs (40 ± 5% for WT to 68 ± 5% for L124M; Table S1) while maintaining an agonist-promoted response that reaches a similar maximal level as the wild-type (WT) receptor (99 ± 8% for WT and 102 ± 6% for L124 M upon ISO stimulation and 90 ± 6% for WT and 104 ± 8% for L124 M upon SALB stimulation; Figure 1c and Table S1).
A different outcome is observed for β-arrestin recruitment monitored by BRET.22 No constitutive activity to either WT or any of the mutant forms of the receptor was observed. In contrast, the mutations have residue- and ligand-specific impacts on the agonist promoted recruitment. L124G/S abrogate the ISO-promoted β-arrestin recruitment (Figure 1d), whereas L124 M resulted in an increased potency for the full and balanced agonists ISO and epinephrine (EPI) (Figures 1d and 2a and Table S2). For the partial and biased agonists SALB and SALM that only poorly promote β-arrestin recruitment to the WT receptor (Figures 1d and 2a), L124 M but not L124G/S led to a gain of function resulting in an increase in β-arrestin recruitment (Figures 1d and 2a). However, the mutation does not confer β-arrestin recruitment to antagonists (alprenolol, labetalol, propranolol, and xamoterol) or inverse agonists (metoprolol and timolol) (Figure 2c,b). These results suggest that the L124 M mutation enhances the ability of the receptor to transduce agonists signal toward β-arrestin engagement.
Figure 2.

Functional impacts of L124 M mutation on β-arrestin recruitment for different ligands. (a–c) Concentration–response curves for the WT (left) and L124 M (right)-promoted β-arrestin recruitment upon stimulation with agonists (ISO, EPI, NE, SALB, SALM) (a), antagonists (ALP, LAB, PRO, XAM) (b), and inverse agonists (MET, TIM) (c). Data are shown as the mean ± SEM of three or four independent experiments.
To further explore the hypothesis that the effects of the L124 mutations result from a change in signal transduction affecting the equilibrium between conformational ensembles, the impact of the mutations on a previously described sensor (NY-β2AR) able to detect conformational rearrangements of the receptor by monitoring the BRET between probes located in the third cytoplasmic loop and the carboxyl terminal of the receptor11 was assessed. As shown in Figure 3a, mutations L124G/S result in a significant decrease in basal NY-β2AR BRET signal, reflecting a switch in the conformational ensemble that favors open conformations associated with active states. This is consistent with the increased constitutive activity of the receptor for Gs (Figure 1c). Furthermore, these mutations abolish the ISO-promoted conformational changes observed for the WT receptor (Figure 3b,c), consistent with the loss of agonist responsiveness for Gs activation (Figure 1c) and β-arrestin recruitment (Figure 1d) thus suggesting that these mutations disrupt the link between the binding pocket and the conformational changes leading to signal transduction. The uncoupling between the binding pocket and signaling is also supported by the lack of effect of the inverse agonist ICI 118,551 on the constitutive cAMP production promoted by L124G/S mutants (Figure S1). Disruption between the binding pocket and signaling has previously been reported for the adenosine A2a receptor.23 The increase constitutive activity and absence of responsiveness to ligands also indicate a shift of equilibrium of the unbound receptor toward the active states consistent with the increased affinity for ISO observed for the mutant L124S.20 The modest decrease in basal NY-β2AR BRET for the L124 M (Figure 3a) is also consistent with the small increase in the receptor constitutive activity for Gs (Figure 1c). However, in contrast with the effects of L124G/S, the L124 M mutation does not affect the ISO-promoted conformational changes of the receptor (Figure 3b). In contrast to the full and balanced agonist ISO, the biased and partial agonist SALB did not promote any detectable conformational rearrangement of NY-β2AR. Mutations L124G/S do not confer any detectable conformational changes upon SALB stimulation. However, the mutation L124 M results in a gain of SALB-induced NY-β2AR BRET change (Figure 3c), reflecting the occurrence of conformational changes upon activation. Such activation is consistent with the gain of SALB-promoted β-arrestin recruitment (Figure 1d).
Figure 3.

Conformational impacts of L124 mutations on the β2AR. (a) Effect of L124 mutations on the basal BRET level of the NY-β2AR conformational sensor. Statistical analysis was performed using ANOVA (* p-value < 0.05). (b,c) Effects of the L124 mutations on ISO- (b) and SALB- (c) promoted conformational changes of the NY-β2AR sensor. Data are shown as the mean ± SEM of four independent experiments.
To gain structural insight on the role of L124, impacts of the mutations were assessed using in silico modeling. For L124G/S, a complete loss of interactions between residue 124 and the PIF motif is observed in the inactive conformation derived from the crystal structures, PDB 2RH1 representing the carazolol bound β2AR (Figure 4). These changes should lead to a reduction of the structural constraints, allowing more flexibility, thus increasing the probability of the receptor to adopt an active conformation, leading to increased constitutive activation of Gs. Furthermore, this loss of interaction between the residue 124 and the PIF motif in the inactive conformation would decrease the stability of the inactive form shifting the equilibrium toward the active states, as suggested by the decrease in BRET level for the NY-β2AR sensor in the absence of the ligand (Figure 3a). The weakening of the interaction between the PIF and NPxxY microswitches can also explain the loss of response to ligand stimulation observed for L124G/S. The PIF motif has been described as playing a crucial role for signal transduction from the binding pocket24 and the NPxxY for active conformation stabilization.25 Therefore, disconnecting the two motifs by mutating the newly identified linker residue L124 most likely uncouples ligand binding from signal transduction. In addition to this reduction of structural constraints in the inactive conformation, the mutant L124S could form a hydrogen bond with Y326 of the NPxxY motif in the active conformation derived from the crystal structure, PDB 4LDE, representing the BI167107 bound β2AR. This additional hydrogen bond could stabilize the active state thus partially explaining the constitutive activity of this mutant for Gs activation. However, the mutant L124G has the same functional effects as L124S; therefore, the main structural explanation for constitutive activity is more likely to be due to a loss of interaction between residue 124 and the PIF motif.
Figure 4.

Modeling of the impacts of L124 mutant forms compared to the WT receptor using the crystal structure for the carazolol-bound inactive β2AR state (PDB: 2RH1) and the BI-167107-β2AR-NB80 active complex (PDB: 4LDE). The PIF motif (P211-I121-F282) is shown in orange and NPxxY (P323-N322-Y326) motif is shown in light blue, while the mutated residue is shown in purple. Black lines represent predicted stabilizing interactions.
In contrast with L124G/S, L124 M mutation places the sulfur atom of the methionine in a position to interact with the π-electrons of the aromatic ring of F282 in the inactive and active conformations, and with the π electrons of the Y326 in the active conformation (Figure 4) providing a more general stabilization of the receptor with a greater increase being seen in the active one. These gains of interactions most likely stabilize active conformation ensembles, as such interactions have been demonstrated to display stabilizing effects on different protein structures,26 thus increasing the efficiency of signal transduction and resulting in a gain of β-arrestin recruitment. This is supported by the gain of SALB-promoted NY-β2AR opening by the L124 M mutation. It is noteworthy that ligands biased against β-arrestin, such as SALB and SALM, cannot on their own promote a detectable conformational change of WT NY-β2AR.11 This could indicate that balanced and biased ligands stabilize distinct conformations (Figure 5a) as previously suggested by a single molecule FRET study9 and by the recently obtained crystal structure of the SALM-β2AR complex.16 Another possibility is that the differences in equilibrium are caused by the difference in overall stability of active conformations, where balanced ligands would stabilize them for a longer time as compared with biased ligands (Figure 5b). The strengthening of the link between the PIF and NPxxY motifs by the L124 M mutation would increase the time spent in active conformations for all agonists, through methionine–aromatic interaction, leading to increased β-arrestin recruitment. Such differences in conformational equilibrium would not affect Gs activation since G protein engagement is faster than β-arrestin recruitment.22,27 The observation that Gs on its own can promote conformational changes11 associated with activation and several studies indicating the existence of a precoupling between receptors and G proteins28,29 would be compatible with the faster activation rate of Gs compared with β-arrestin. For L124G/S mutants, the main effect would be a reduced stabilization of the inactive states, thus increasing Gs activation at the basal level, while the absence of additional stabilization by the ligands would prevent β-arrestin recruitment. The difference in the active conformations’ half-life that would be required to engage Gs versus β-arrestin agrees with studies showing that the residency time of the ligand in the binding pocket of the receptor is important for β-arrestin recruitment but does not affect G protein activation.30
Figure 5.

Schematic representation of proposed models. (a) Different active conformations are stabilized by balanced and full agonists (green) and partial and biased agonists (orange). (b) Balanced and biased ligands stabilize similar conformations but for different time lengths as represented by the equilibrium arrow and color intensity.
In conclusion, L1243.43 plays a major role in receptor activation by interacting with the PIF and NPxxY motifs. Whether the mutations at this position modify the receptor conformation or the equilibrium between active and inactive states remains to be further investigated, but it is clear that this region plays a crucial role in dictating the selective engagement of G protein vs β-arrestin and linking ligand binding to effectors engagement. Given the conservation (73%) of this linker residue among class A GPCRs (GPCRdb.org), we can propose that it represents a key element in signal propagation and biased signaling. This opens the possibility of rationally designing allosteric ligands that would target this region to generate biased ligands.
Methods
Reagents
(−)-Isoproterenol hydrochloride (ISO), (−)-epinephrine (EPI), (−)-norepinephrine (NE), alprenolol hydrochloride (ALP), labetalol hydrochloride (LAB), (±)-propranolol hydrochloride (PRO), metoprolol tartrate (MET), and timolol maleate (TIM) were purchased from Sigma-Aldrich. Salbutamol hemisulfate (SALB) and xamoterol hemifumarate (XAM) were purchased from Tocris Bioscience. Salmeterol xinofolate was purchased from Selleckchem. Coelenterazine 400a (Coel400a) was purchased from NanoLight Technology.
Plasmids
The β-arrestin-RlucII,22 HA-β2AR WT and mutant forms,20 Gαs-117-RlucII,21 Gβ1,31 Gγ1-GFP10,31 β2AR-GFP10,32 and NY-β2AR11 have been previously described. Point mutations in the NY-β2AR and β2AR-GFP10 were introduced by PCR using QuickChange Site-directed Mutagenesis Kit (Agilent Technologies) following the manufacturer’s instructions. All variants were verified by sequencing.
Cell Culture and Transfection
HEK293T cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% newborn calf serum (NCS) at 37 °C with 5% CO2. For transfection, cells were detached with trypsin, diluted at a density of 500 000 cells/mL and transfected with 2.5 μg of total DNA per 106 cells using linear polyethylenimine (PEI, Polysciences) as transfecting agent with a PEI/DNA ratio of 3:1. Directly after transfection, cells were plated in white 96-wells culture plates (Greiner) coated with poly-l-ornithine (Sigma-Aldrich) at a density of 50 000 cells per well and incubated for 48 h before experiments. Cells were regularly tested for mycoplasma contamination (PCR Mycoplasma Detection kit, abm).
BRET Measurements
At 48 h after transfection, cells were washed with PBS, and stimulation buffer (Hank’s balanced salt solution, HBSS) was added. Concentrated ligand (10×) was added 5 min (Gs) or 15 min (NY-β2AR and β-arrestin assays) before BRET measurement while Coel400a was added to a final concentration of 2.5 μM, 5 min before reading. BRET was monitored with a TriStar2 LB942 microplate reader (Berthold) equipped with a donor filter of 410/80 nm and an acceptor filter of 515/40 nm (Gs and β-arrestin assays) or a donor filter of 485/20 nm and an acceptor filter of 530/25 nm (NY-β2AR assay). BRET ratios were calculated by dividing the acceptor emission by the donor emission.
Conformational Sensor
HEK293T cells were transfected with the conformational biosensor (NY-β2AR).11 BRET was then monitored as described above. The NY-β2AR consists of a β2AR construct where Nluc was fused to the ICL3 between positions 251 and 252 and YFP at position 369 of a truncated receptor. These positions allow the detection of conformational changes resulting from the TM5 rotation and TM6 outward movement upon receptor activation.
β-Arrestin Recruitment
HEK293T cells were cotransfected with the BRET-based biosensors β2AR-GFP10 (WT or mutants) and βarr2-RlucII, as described above. BRET was then monitored as described above. After recruitment of β-arrestin to the receptor, the increase proximity between the two proteins leads to an increase in BRET ratio.
Gs Activation
HEK293T cells were cotransfected with the β2AR receptor (WT or mutants) along with Gαs-117-RlucII, Gβ1, Gγ1-GFP10 (G protein activation BRET biosensor21), and BRET monitored as described above. The dissociation of the Gα and Gβ/Gγ subunits upon G protein activation leads to a decrease in BRET ratio.
Cell Surface ELISA
HEK293T cells transfected with the different receptor constructs were washed with PBS, then fixed with 3% PFA diluted in PBS. Fixed cells were washed with WashB solution (0.5% BSA in PBS). The monoclonal anti-HA-HRP (3f10, Roche Diagnostics) was added at a dilution of 1/2000 and cells were incubated at RT for 1 h. After incubation, cells were washed with WashB solution. HBSS was added in the wells, and 2 min before the reading, ECL (PerkinElmer) was added. Total luminescence was monitored with a Mithras LB 940 microplate reader (Berthold).
BRET Signal Analysis
Concentration–response curves were analyzed using GraphPad Prism 6 software (version 6, GraphPad Software). The data were normalized to WT curves. The maximal response upon ISO stimulation was used as 100% and the values of unstimulated mock conditions were used as 0%. This normalization allows the detection of changes in constitutive activity, which correspond to the lower asymptotes of the concentration–response curves. Cell surface ELISA was performed as described above to control receptors expression.
Structure Prediction and Analysis
Protein structure prediction was performed as previously described.20 Briefly, MOE structure-based design package was used. Automated structure preparation protocol Protonate3D was run on both inactive and active receptor templates (2RH1 and 4LDE, respectively), then the mutations were inserted using the residue scanning in the protein design panel. The conformation of the side chain was determined by a selection from a rotamer library followed by a force field energy minimization-based protocol, using AMBER12EHT force field. Structure predictions were visualized and analyzed using chimera visualization system. Interactions between mutated residues and the microswitches have been determined by a distance inferior to 0.4 Å between the van der Waals radii of the different atoms.
Acknowledgments
This work was supported by grants from the Canadian Institutes for Health Research (CIHR), [MOP11215] and [FDN148431]. L.-P.P. received scholarships from CIHR and the Fonds de la Recherche du Quebec—Santé (FRQ-S). A.M.S. received a postdoctoral fellowship from FRQ-S. M.B. holds a Canada Research Chair in Signal Transduction and Molecular Pharmacology. We are grateful to Dr. Brian Kobilka and Monique Lagacé for their critical reading of the manuscript.
Glossary
Abbreviations
- BRET
bioluminescence resonance-energy transfer
- RET
resonance-energy transfer
- GPCR
G protein-coupled receptors
- β2AR
β2-adrenergic receptor
- ISO
isoproterenol
- SALB
salbutamol
- SALM
salmeterol
- WT
wild-type.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsptsci.9b00012.
cAMP production of WT and mutants β2AR; efficacies, potencies and errors for Gs activation and β-arrestin recruitment essay (PDF)
Author Contributions
L.-P.P., A.M.S., and M.B. designed the study. L.-P.P. and A.M.S. performed the experiments, and the analysis of the data. L.-P.P. and M.B. interpreted the data and wrote the manuscript.
The authors declare no competing financial interest.
This article is made available for a limited time sponsored by ACS under the ACS Free to Read License, which permits copying and redistribution of the article for non-commercial scholarly purposes.
Supplementary Material
References
- Pupo A. S.; Duarte D. A.; Lima V.; Teixeira L. B.; Parreiras E. S. L. T.; Costa-Neto C. M. (2016) Recent updates on GPCR biased agonism. Pharmacol. Res. 112, 49–57. 10.1016/j.phrs.2016.01.031. [DOI] [PubMed] [Google Scholar]
- Rankovic Z.; Brust T. F.; Bohn L. M. (2016) Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg. Med. Chem. Lett. 26, 241–250. 10.1016/j.bmcl.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. (2012) The potential for selective pharmacological therapies through biased receptor signaling. BMC Pharmacol. Toxicol. 13, 3. 10.1186/2050-6511-13-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zocher M.; Fung J. J.; Kobilka B. K.; Muller D. J. (2012) Ligand-specific interactions modulate kinetic, energetic, and mechanical properties of the human beta2 adrenergic receptor. Structure 20, 1391–1402. 10.1016/j.str.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror R. O.; Arlow D. H.; Maragakis P.; Mildorf T. J.; Pan A. C.; Xu H.; Borhani D. W.; Shaw D. E. (2011) Activation mechanism of the beta2-adrenergic receptor. Proc. Natl. Acad. Sci. U. S. A. 108, 18684–18689. 10.1073/pnas.1110499108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. S.; Lefkowitz R. J.; Rajagopal S. (2018) Biased signalling: from simple switches to allosteric microprocessors. Nat. Rev. Drug Discovery 17, 243–260. 10.1038/nrd.2017.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tehan B. G.; Bortolato A.; Blaney F. E.; Weir M. P.; Mason J. S. (2014) Unifying family A GPCR theories of activation. Pharmacol. Ther. 143, 51–60. 10.1016/j.pharmthera.2014.02.004. [DOI] [PubMed] [Google Scholar]
- Deupi X.; Standfuss J.; Schertler G. (2012) Conserved activation pathways in G-protein-coupled receptors. Biochem. Soc. Trans. 40, 383–388. 10.1042/BST20120001. [DOI] [PubMed] [Google Scholar]
- Gregorio G. G.; Masureel M.; Hilger D.; Terry D. S.; Juette M.; Zhao H.; Zhou Z.; Perez-Aguilar J. M.; Hauge M.; Mathiasen S.; Javitch J. A.; Weinstein H.; Kobilka B. K.; Blanchard S. C. (2017) Single-molecule analysis of ligand efficacy in beta2AR-G-protein activation. Nature 547, 68–73. 10.1038/nature22354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner S.; Ambrosio M.; Hoffmann C.; Lohse M. J. (2010) Differential signaling of the endogenous agonists at the beta2-adrenergic receptor. J. Biol. Chem. 285, 36188–36198. 10.1074/jbc.M110.175604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard L.-P.; Schönegge A. M.; Lohse M. J.; Bouvier M. (2018) Bioluminescence resonance energy transfer-based biosensors allow monitoring of ligand- and transducer-mediated GPCR conformational changes. Communications Biology 1, 106. 10.1038/s42003-018-0101-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard R.; Zou Y.; Dror R. O.; Mildorf T. J.; Arlow D. H.; Manglik A.; Pan A. C.; Liu C. W.; Fung J. J.; Bokoch M. P.; Thian F. S.; Kobilka T. S.; Shaw D. E.; Mueller L.; Prosser R. S.; Kobilka B. K. (2013) The dynamic process of beta(2)-adrenergic receptor activation. Cell 152, 532–542. 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. J.; Horst R.; Katritch V.; Stevens R. C.; Wuthrich K. (2012) Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science 335, 1106–1110. 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisler J. W.; DeWire S. M.; Whalen E. J.; Violin J. D.; Drake M. T.; Ahn S.; Shenoy S. K.; Lefkowitz R. J. (2007) A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc. Natl. Acad. Sci. U. S. A. 104, 16657–16662. 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Westhuizen E. T.; Breton B.; Christopoulos A.; Bouvier M. (2014) Quantification of ligand bias for clinically relevant beta2-adrenergic receptor ligands: implications for drug taxonomy. Mol. Pharmacol. 85, 492–509. 10.1124/mol.113.088880. [DOI] [PubMed] [Google Scholar]
- Masureel M.; Zou Y.; Picard L.-P.; van der Westhuizen E.; Mahoney J. P.; Rodrigues J. P. G. L. M.; Mildorf T. J.; Dror R. O.; Shaw D. E.; Bouvier M.; Pardon E.; Steyaert J.; Sunahara R. K.; Weis W. I.; Zhang C.; Kobilka B. K. (2018) Structural insights into binding specificity, efficacy and bias of a β2AR partial agonist. Nat. Chem. Biol. 14, 1059–1066. 10.1038/s41589-018-0145-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S. G.; DeVree B. T.; Zou Y.; Kruse A. C.; Chung K. Y.; Kobilka T. S.; Thian F. S.; Chae P. S.; Pardon E.; Calinski D.; Mathiesen J. M.; Shah S. T.; Lyons J. A.; Caffrey M.; Gellman S. H.; Steyaert J.; Skiniotis G.; Weis W. I.; Sunahara R. K.; Kobilka B. K. (2011) Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555. 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Manglik A.; Venkatakrishnan A. J.; Laeremans T.; Feinberg E. N.; Sanborn A. L.; Kato H. E.; Livingston K. E.; Thorsen T. S.; Kling R. C.; Granier S.; Gmeiner P.; Husbands S. M.; Traynor J. R.; Weis W. I.; Steyaert J.; Dror R. O.; Kobilka B. K. (2015) Structural insights into micro-opioid receptor activation. Nature 524, 315–321. 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J. A.; Weinstein H. (1995) [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428. 10.1016/S1043-9471(05)80049-7. [DOI] [Google Scholar]
- Schonegge A. M.; Gallion J.; Picard L. P.; Wilkins A. D.; Le Gouill C.; Audet M.; Stallaert W.; Lohse M. J.; Kimmel M.; Lichtarge O.; Bouvier M. (2017) Evolutionary action and structural basis of the allosteric switch controlling beta2AR functional selectivity. Nat. Commun. 8, 2169. 10.1038/s41467-017-02257-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen A. R.; Plouffe B.; Cahill T. J. 3rd; Shukla A. K.; Tarrasch J. T.; Dosey A. M.; Kahsai A. W.; Strachan R. T.; Pani B.; Mahoney J. P.; Huang L.; Breton B.; Heydenreich F. M.; Sunahara R. K.; Skiniotis G.; Bouvier M.; Lefkowitz R. J. (2016) GPCR-G Protein-beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell 166, 907–919. 10.1016/j.cell.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzi M.; Charest P. G.; Angers S.; Rousseau G.; Kohout T.; Bouvier M.; Pineyro G. (2003) Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc. Natl. Acad. Sci. U. S. A. 100, 11406–11411. 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G.; Warne T.; Edwards P. C.; Bennett K.; Langmead C. J.; Leslie A. G.; Tate C. G. (2011) Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474, 521–525. 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Wang C.; Katritch V.; Han G. W.; Huang X. P.; Vardy E.; McCorvy J. D.; Jiang Y.; Chu M.; Siu F. Y.; Liu W.; Xu H. E.; Cherezov V.; Roth B. L.; Stevens R. C. (2013) Structural features for functional selectivity at serotonin receptors. Science 340, 615–619. 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak L. S.; Menard L.; Ferguson S. S.; Colapietro A. M.; Caron M. G. (1995) The conserved seven-transmembrane sequence NP(X)2,3Y of the G-protein-coupled receptor superfamily regulates multiple properties of the beta 2-adrenergic receptor. Biochemistry 34, 15407–15414. 10.1021/bi00047a003. [DOI] [PubMed] [Google Scholar]
- Valley C. C.; Cembran A.; Perlmutter J. D.; Lewis A. K.; Labello N. P.; Gao J.; Sachs J. N. (2012) The methionine-aromatic motif plays a unique role in stabilizing protein structure. J. Biol. Chem. 287, 34979–34991. 10.1074/jbc.M112.374504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gales C.; Rebois R. V.; Hogue M.; Trieu P.; Breit A.; Hebert T. E.; Bouvier M. (2005) Real-time monitoring of receptor and G-protein interactions in living cells. Nat. Methods 2, 177–184. 10.1038/nmeth743. [DOI] [PubMed] [Google Scholar]
- Nobles M.; Benians A.; Tinker A. (2005) Heterotrimeric G proteins precouple with G protein-coupled receptors in living cells. Proc. Natl. Acad. Sci. U. S. A. 102, 18706–18711. 10.1073/pnas.0504778102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevheroglu O.; Becker J. M.; Son C. D. (2017) GPCR-Galpha protein precoupling: Interaction between Ste2p, a yeast GPCR, and Gpa1p, its Galpha protein, is formed before ligand binding via the Ste2p C-terminal domain and the Gpa1p N-terminal domain. Biochim. Biophys. Acta, Biomembr. 1859, 2435–2446. 10.1016/j.bbamem.2017.09.022. [DOI] [PubMed] [Google Scholar]
- Wacker D.; Wang S.; McCorvy J. D.; Betz R. M.; Venkatakrishnan A. J.; Levit A.; Lansu K.; Schools Z. L.; Che T.; Nichols D. E.; Shoichet B. K.; Dror R. O.; Roth B. L. (2017) Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 168, 377–389. 10.1016/j.cell.2016.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gales C.; Van Durm J. J.; Schaak S.; Pontier S.; Percherancier Y.; Audet M.; Paris H.; Bouvier M. (2006) Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat. Struct. Mol. Biol. 13, 778–786. 10.1038/nsmb1134. [DOI] [PubMed] [Google Scholar]
- Mercier J. F.; Salahpour A.; Angers S.; Breit A.; Bouvier M. (2002) Quantitative assessment of beta 1- and beta 2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J. Biol. Chem. 277, 44925–44931. 10.1074/jbc.M205767200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.