

Abstract
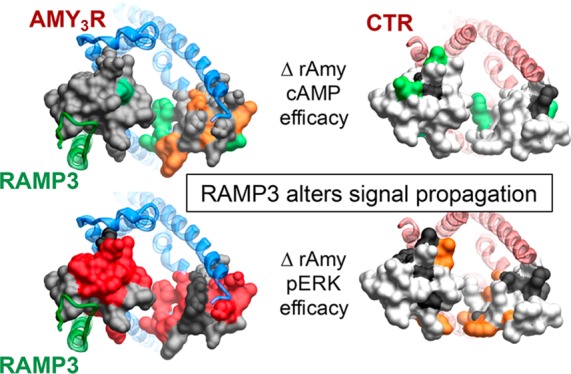
Amylin is coexpressed with insulin in pancreatic islet β-cells and has potent effects on gastric emptying and food intake. The effect of amylin on satiation has been postulated to involve AMY3 receptors (AMY3R) that are heteromers of the calcitonin receptor (CTR) and receptor activity-modifying protein 3 (RAMP3). Understanding the molecular control of signaling through the AMY3R is thus important for peptide drug targeting of this receptor. We have previously used alanine scanning mutagenesis to study the contribution of the extracellular surface of the CTR to binding and signaling initiated by calcitonin (CT) and related peptides (Dal Maso, E., et al. (2019) The molecular control of calcitonin receptor signaling. ACS Pharmacol. Transl. Sci.2, 31–51). That work revealed ligand- and pathway-specific effects of mutation, with extracellular loops (ECLs) 2 and 3 particularly important in the distinct propagation of signaling mediated by individual peptides. In the current study, we have used equivalent alanine scanning of ECL2 and ECL3 of the CTR in the context of coexpression with RAMP3 to form AMY3Rs, to examine functional affinity and efficacy of peptides in cAMP accumulation and extracellular signal-regulated kinase (ERK) phosphorylation (pERK). The effect of mutation was determined on representatives of the three major distinct classes of CT peptide, salmon CT (sCT), human CT (hCT), and porcine CT (pCT), as well as rat amylin (rAmy) or human α-CGRP (calcitonin gene-related peptide, hCGRP) whose potency is enhanced by RAMP interaction. We demonstrate that the dynamic nature of CTR ECL2 and ECL3 in propagation of signaling is fundamentally altered when complexed with RAMP3 to form the AMY3R, despite only having predicted direct interactions with ECL2. Moreover, the work shows that the role of these loops in receptor signaling is highly peptide dependent, illustrating that even subtle changes to peptide sequence may change signaling output downstream of the receptor.
Keywords: amylin receptor, calcitonin receptor, receptor activity-modifying protein, G protein-coupled receptor, receptor structure−function, cell signaling
Introduction
G protein-coupled receptors (GPCRs) are the largest superfamily of cell surface protein conduits of extracellular chemical information to the inside of cells.1 As such, understanding the molecular basis of how these extracellular signals are conformationally propagated through the GPCR to recruit and activate signal transducers is critically important to development of novel therapeutics that regulate this process. Moreover, GPCRs can recruit multiple different transducers and other regulatory proteins and this can be altered in a ligand-specific manner, leading to biased agonists that hold promise as precision medicines to treat various diseases.2
Class B1 GPCRs are an important subfamily for key physiological peptides that regulate diverse functions including energy homeostasis, bone metabolism, immune function, lymph and vascular formation, and the control of vascular tone.3 The calcitonin receptor (CTR) is a broadly expressed class B1 GPCR that is most recognized for its expression in bone resorbing osteoclasts, and its role in bone metabolism.4 However, CTRs also interact with a family of 3 receptor activity-modifying proteins (RAMPs) to yield high affinity receptors for amylin and calcitonin gene-related peptide (CGRP).5 These are termed AMY1, AMY2, and AMY3 receptors according to the interacting RAMP, i.e., RAMP1, RAMP2, and RAMP3, respectively. In addition to modifying the binding specificity of the CT family of receptors, a major consequence of GPCR-RAMP interaction is alteration to the signaling profile of the receptor,6 and this has been observed for the AMY receptors relative to CTR alone.7
Amylin is coexpressed with insulin in pancreatic islet β-cells and has potent effects on gastric emptying and food intake.8 Pramlintide, a nonamyloidogenic analogue of human amylin is approved for the treatment of type 1 diabetes in combination with insulin.8 However, amylin analogues also promote satiation and can lead to significant weight loss in overweight patients, and cause marked weight loss in animal models of obesity when coadministered with other agents that promote weight loss, such as glucagon-like peptide-1 (GLP-1) receptor agonists or leptin.9,10 Accordingly, there is significant interest in the development of new amylin analogues to better treat obesity.8
The amylin effect on satiation has been localized to amylin receptors in the area postrema and has been proposed to involve the AMY3R subtype (CTR:RAMP3 heteromer),11 although all three RAMPs are present in the area postrema.8 Understanding the molecular control of signaling through the AMY3R is thus crucial for peptide drug targeting of this receptor.
Recent advances in cryo-electron microscopy have allowed determination of the structures of active state class B GPCRs in complex with peptide agonists and the canonical Gs protein,12−15 including complexes of the CTR12,16 and the CGRP receptor.15 The latter is a hetromer of the related calcitonin receptor-like receptor (CLR) and RAMP1. These structures have allowed identification of the peptide binding domain within the receptor core and serve as a template for 3-dimensional (3D) mapping of the effect of mutation on receptor function. Importantly, the solution of the CGRP receptor (CGRPR) complex revealed a novel interface for RAMP interaction with transmembrane helices 4 and 5 that extended to parts of extracellular loop (ECL) 2,15 and allows for the first-time structure-based modeling of related RAMP-class B GPCR complexes.
We have previously used alanine scanning mutagenesis to study the contribution of the extracellular surface of the CTR to binding and signaling initiated by CT and related peptides.16,17 This work revealed ligand- and pathway-specific effects of mutation, with ECLs 2 and 3 particularly important in the distinct propagation of signaling mediated by individual peptides.17 In the current study, we have used equivalent alanine scanning of ECL2 and ECL3 of the CTR in the context of coexpression with RAMP3 to form AMY3Rs, in order to examine functional affinity and efficacy of peptides in cAMP accumulation and extracellular signal-regulated kinase (ERK) phosphorylation (pERK). The effect of mutation was determined on representatives of the three major distinct classes of CT peptide, salmon CT (sCT), human CT (hCT), and porcine CT (pCT), as well as rat amylin (rAmy) or human α-CGRP (hCGRP) whose potency is enhanced by RAMP interaction. The work illustrates that interaction with RAMP3 dynamically alters how ECL2 and ECL3 contribute to propagation of signaling through the CTR.
Results and Discussion
Importance of ECL2 and ECL3 in the Control of AMY3R Function
To gain insight into the role of RAMP3 in the AMY3R phenotype we performed alanine scanning mutagenesis on CTR ECL2 (I279-I300) and ECL3 (F356-M376) that play important roles in peptide binding and propagation of signaling at the CTR in the absence of RAMPs.17 Each of these mutants was analyzed for their effect on cell surface expression (Figure 1), binding affinity in competitive radioligand binding assays (Figure 2, Table 1, Figures S1–S6), and functional response (pERK and cAMP accumulation) for each peptide (Figures 3–8, Figures S7–S16, Tables 2–5). A homology model of the AMY3R complex was built from deposited structures of the CTR (6NIY16) and CGRPR (6E3Y;15 for initial positioning of RAMP3) active complexes and subjected to a short molecular dynamics (MD) simulation (200 ns) to resolve energetically unfavorable interactions. This model was used to map effects of mutation on the AMY3R and to enable comparison to the effects of previously published equivalent mutations on the CTR,16,17 mapped onto the recently published 3.3 Å structure (6NIY) that included ECL2 and ECL3 side chains16 (Figures 9–12).
Figure 1.
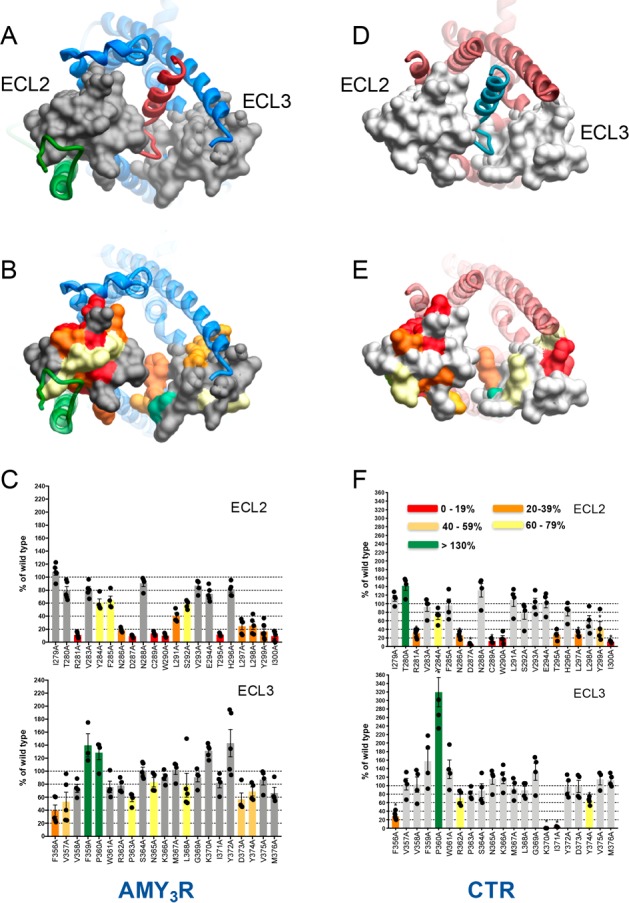
Effect of alanine mutation on the cell surface expression of the AMY3R and CTR. (A–C) AMY3R; (D–F) CTR (extracted from Dal Maso et al, 201817). (A, D) Top view of the active, sCT-bound, AMY3R (A) or CTR (6NIY) (D) model with the extracellular surface subject to alanine scanning depicted in gray (A) or off-white (D) (combined surface/cpk representation). The rest of the protein complex is shown in ribbon representation. CTR (blue, AMY3R; dark red, CTR), sCT peptide (dark red, AMY3R; aquamarine, CTR), RAMP3 (green). The receptor ECD is omitted for clarity. (B, E) Map of the effect of mutation on cell surface receptor expression colored according to the legend in panel F. (C, F) Effect of alanine mutation of ECL2 and ECL3 on CTR expression monitored by FACS of anti-c-Myc antibody binding to the N-terminal c-Myc epitope on the receptor. Data are normalized to the expression of the wild-type (WT) receptor (100%). Significant differences in the level of cell surface expression were determined by one-way ANOVA followed by Dunnett’s post-test comparison to the WT. P < 0.05 was used to denote significance, and colored according to the magnitude of change. Individual values (separate experiments) are shown within the bars.
Figure 2.
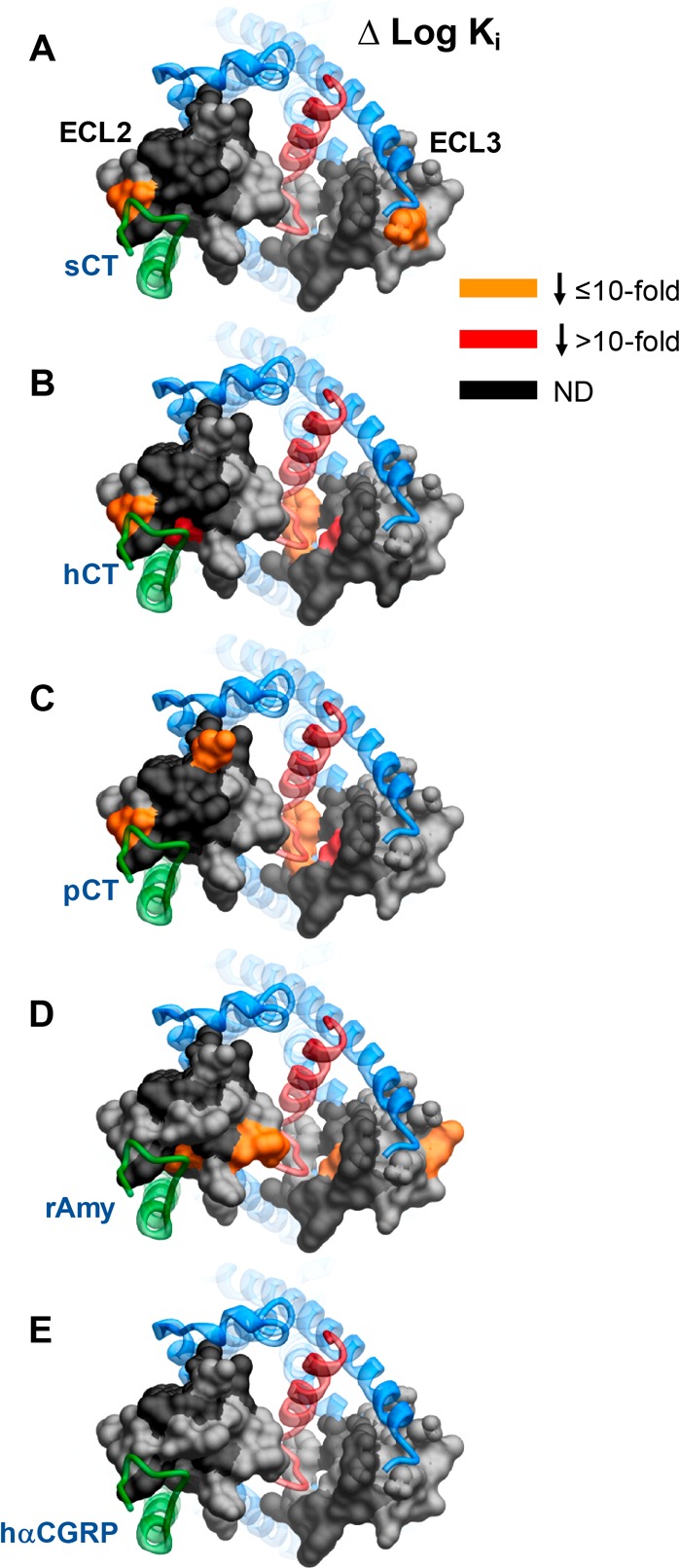
Identification of key amino acids of AMY3R ECL2 and ECL3 for peptide binding affinity (log Ki). (A) sCT; (B) hCT; (C) pCT; (D) rAmy; (E) hCGRP. Mutations that significantly decreased peptide affinity in radioligand competition assay are colored dark orange (≤10-fold effect) or red (>10-fold effect), with mutated amino acids without significant alteration to log Ki colored gray. Amino acid mutations where there was an insufficiently robust functional effect to quantify by radioligand competition binding are depicted in black. The receptor ECD is not shown for clarity, with the CTR TM bundle in blue ribbon and RAMP3 in green ribbon. Quantitative data are reported in Table 1.
Table 1. Effect of Single Alanine Mutation in AMY3R ECL2 or ECL3 on Binding Affinity (log Ki) of Peptides Derived from Competition Binding Isothermsa.

Log Ki values were derived for each ligand and mutant receptor from analysis of either homologous (rAmy) or heterologous (sCT, hCT, pCT, hαCGRP) competition of 125I-rAmy binding. Mean, S.E.M. and the individual experimental “n” values are reported. Significance of changes in log Ki of each ligand was determined by comparison of mutant receptors to WT values by a one-way ANOVA and Dunnett’s post-test (p < 0.05 denoted by bold coloured entries. Orange, significant decrease ≤10-fold; red, significant decrease >10-fold). Gray shading indicates mutants where robust radioligand binding was not detected. N.D. indicates that no value could be derived due to lack of robust competition and high data variance.
Figure 3.
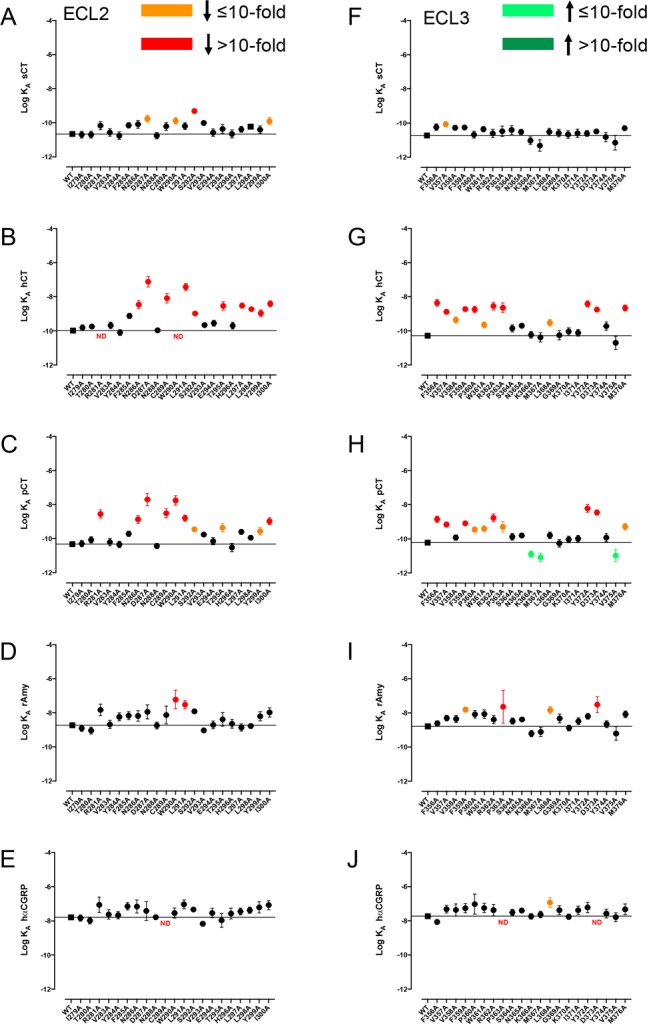
Alanine mutation of ECL2 and ECL3 of AMY3R alters cAMP functional affinity (log KA) in a peptide-specific manner. Functional affinities derived from operational fitting of concentration–response curves in cAMP accumulation for alanine mutation of ECL2 (A-E) and ECL3 (F-J) are displayed as log KA. Significance of changes was established by comparison of the WT to the other receptor mutants following one-way ANOVA and Dunnett’s post-test with P < 0.05 accepted as significant. Mutants that gave significant reductions between 3- and 10-fold are colored orange, and those with reductions greater than 10-fold are colored red. Mutants giving significant increases in log KA are colored green. Where data were insufficiently robust to derive a reliable value for log KA no symbol is shown (ND). Quantitative data are reported in Table 2.
Figure 8.
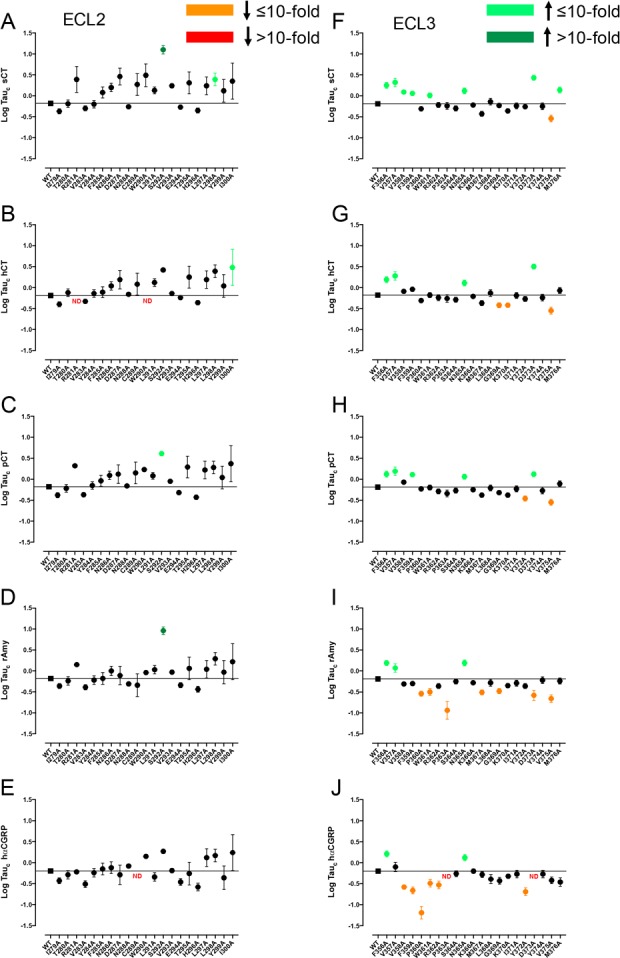
Alanine mutation of ECL2 and ECL3 of AMY3R alters cAMP efficacy (log τc) in a peptide-specific manner. Peptide efficacy (log τ) was derived from operational fitting of concentration–response curves in cAMP accumulation for alanine mutation of ECL2 (A–E) and ECL3 (F–J), and corrected for cell surface receptor expression to yield Log tauc. Significance of mutation effect was established by comparison of the WT to the other receptor mutants following one-way ANOVA and Dunnett’s post-test with P < 0.05 accepted as significant. Mutants that significantly reduced log τc are colored orange (≤10-fold change), or red (>10-fold change). Mutants that significantly increased log τc are colored green (≤10-fold change, light green; >10-fold, dark green). Where data were insufficiently robust to derive a reliable value for log τc no symbol is shown (ND). Quantitative data are reported in Table 4.
Figure 9.
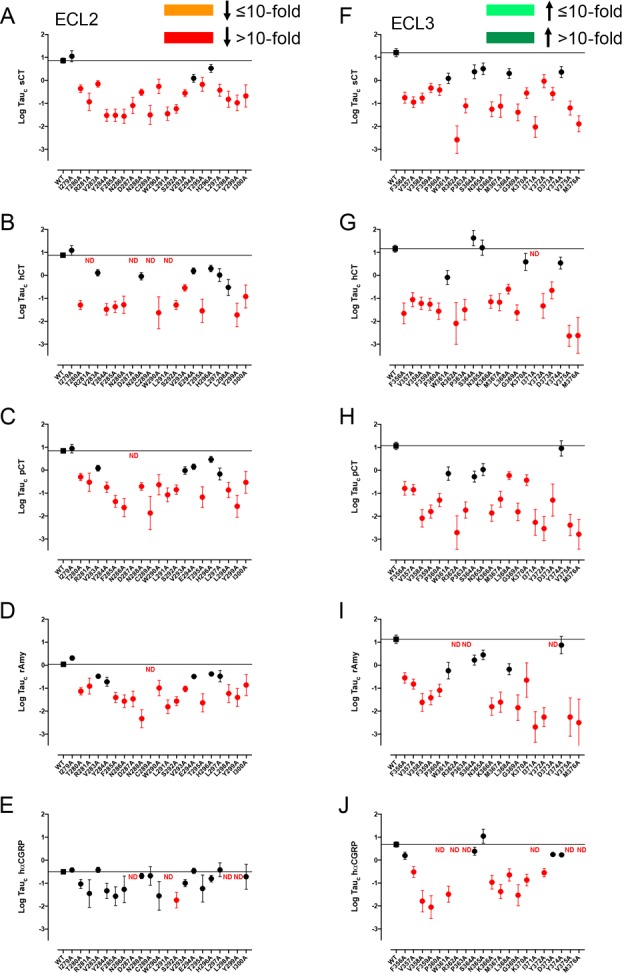
Alanine mutation of ECL2 and ECL3 of AMY3R alters pERK efficacy (log τc) in a peptide-specific manner. Peptide efficacy (log τ) was derived from operational fitting of concentration–response curves in pERK for alanine mutation of ECL2 (A–E) and ECL3 (F–J), and corrected for cell surface receptor expression to yield log τc. Significance of mutation effect was established by comparison of the wild-type to the other receptor mutants following one-way ANOVA and Dunnett’s post-test with P < 0.05 accepted as significant. Mutants that significantly reduced log τc are colored orange (≤10-fold change), or red (>10-fold change). Where data were insufficiently robust to derive a reliable value for log τc no symbol is shown (ND). Quantitative data are reported in Table 5.
Figure 12.
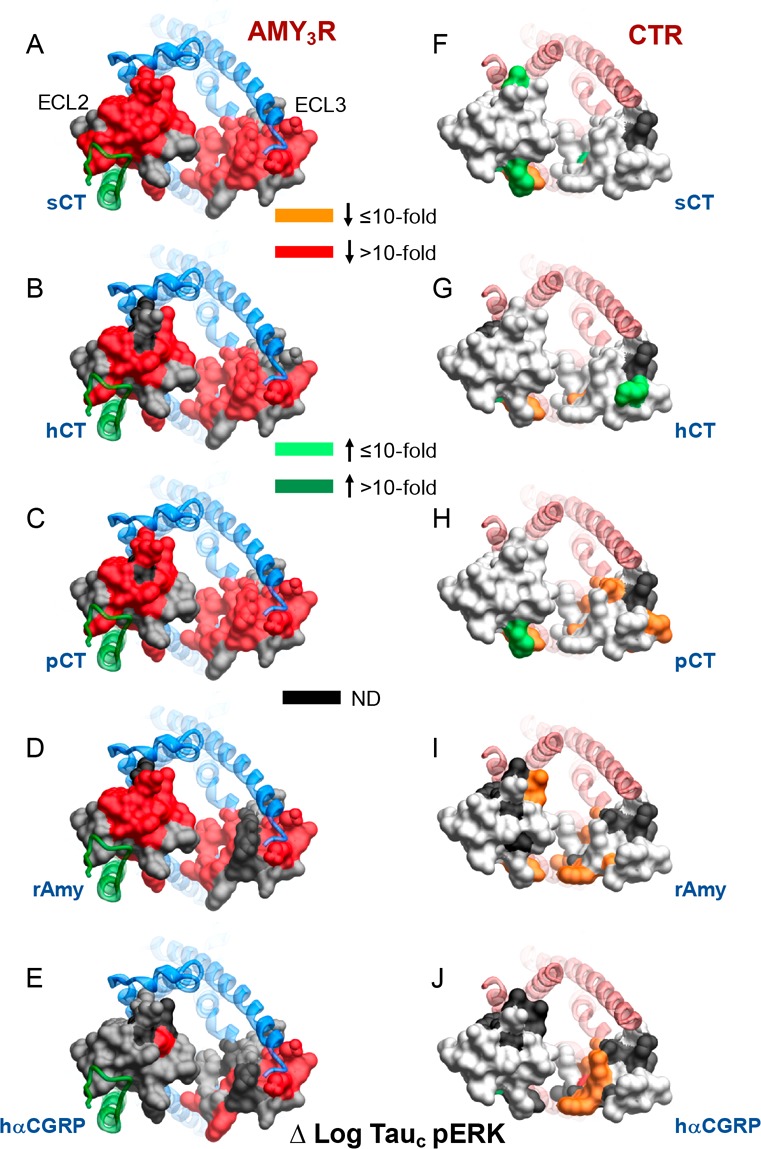
Alanine mutation of ECL2 and ECL3 has distinct effects on pERK peptide efficacy for AMY3R and CTR. Peptide efficacy, derived from operational fitting of concentration–response curves in pERK, are displayed as Δlog τc from wild-type for AMY3R (A–E) and CTR (F–J). Illustrated are top views of the receptors with the extracellular surface subject to alanine scanning depicted (combined surface/cpk representation). Mutations that significantly alter peptide functional log τc are colored according to the magnitude of effect, with mutated amino acids without significant alteration to log τc colored gray. Amino acid mutations for which there was an insufficiently robust functional effect to quantify by operational modeling are depicted in black. The receptor ECD and peptide are not shown for clarity, with the CTR TM bundle in blue ribbon and RAMP3 in green ribbon in A–E, and CTR TM bundling in red ribbon in F–J. Data for CTR peptide efficacy are from Dal Maso et al., 2018.17
Receptor Expression
AMY3Rs were expressed using a Flp-In bicistronic vector of N-terminally tagged cMyc-CTR and RAMP3, where the RAMP is overexpressed relative to the CTR, and stable cell lines of each mutant receptor generated by isogenic integration into Flp-In CV-1 cells. Anti-cMyc antibody binding to the CTR was measured by FACS as a marker of the cell surface expression of the AMY3R (Figure 1).
There was a marked decrease in surface expression of CTR in the AMY3R for the R281A, N286A, D287A, C289A, W290A, T295A, L297A, L298A, Y299A, and I300A mutants within ECL2, with decreased expression to a lesser extent also seen with Y284A, F285A, L291A, and S292A mutants within this loop (Figure 1B,C). In general, the pattern of effect was similar to that seen with the CTR expressed alone17 (Figure 1E,F) although greater loss of expression was seen for the L291A and S292A mutants when the receptor was coexpressed with RAMP3. Alanine mutation within ECL3 had less overall impact relative to ECL2, with moderate, significant, decreases in expression for F356A, V357A, P363A, N365A, L286A, D373A, and Y374A, and increases for F359A and P360A (Figure 1B,C). The lack of effect of K370A and I371A mutation of the AMY3R was in marked contrast to the effect of these mutants in the absence of RAMP3, where there was almost undetectable levels of CTR expression, and very high levels of expression observed for the P360A mutation (Figure 1E,F), although overall there was only limited impact of mutation in ECL3. Although RAMP3 is overexpressed relative to CTR in the bicistronic vector, it is likely that both AMY3R and CTR alone forms of the receptor are formed. Nonetheless, the marked difference in surface expression of the K370A and I371A mutants (100% versus <5% for AMY3R and CTR, respectively) supports that the cocomplex with RAMP3 accounts for the majority of receptors in the CV-1 cells.
Peptide Affinity (Competition Binding)
To specifically examine the impact of mutation on the affinity of peptide ligands for the AMY3R, radioligand competition binding studies were performed with 125I-rAmylin. We have previously demonstrated that there is no measurable specific binding to CTR alone at the concentrations of radioligand used.18 No specific binding was detected for the R281A, Y284A, N286A, D287A, C289A, W290A, L291A, T295A and I300A mutants in ECL2, or for the V357A, P360A, W361A R362A, P363A, L368A, D373A, and M376A mutants in ECL3 (Table 1, Figure 2). Many of these within ECL2 also had low cell surface expression (Figure 1C). In contrast, there was moderate to strong cell surface expression of most ECL3 mutants indicating that the loss of binding was likely due to alterations to binding affinity of the radioligand. Of those mutants with a robust specific binding window, there was a subset that exhibited loss of affinity, in a peptide specific manner (Table 1, Figure 2, Figures S1–S6). hCGRP was least impacted, with no significant change in observed affinity (Figure S1E). There was a selective loss of rAmy affinity for the E294A and G369A mutants (Figure S1D), of hCT for the T280A, L298A, and S364A mutants (Figure S1B), of sCT for the K366A mutant, and pCT for the N288A mutant (Figures S1A and S1C, respectively). F285A and Y299A displayed a selective loss of affinity for CT peptides with no significant effect on rAmy or hCGRP. Similarly, there was loss of affinity for all CT peptides for the V358A and F359A mutants; rAmy affinity was also decreased at V358A (Table 1, Figures S1A–D). There was selective loss of affinity for sCT and pCT at the V283A mutant, hCT, pCT, and rAmy at the L297A and Y372A mutants, and of hCT and pCT at the F356A and M367A mutants (Table 1, Figures S1A–D). The effect of mutations within ECL2 appeared to coincide with residues known to contribute to packing of ECL2 in CTR16 or were in close proximity to the predicted RAMP3 interface (Figure 2). In contrast, the effect of most mutations within ECL3 were consistent with potential peptide binding interfaces and differential strength of interaction for individual peptides (Figure 2).
Peptide Functional Affinity
For each peptide, concentration–response isotherms were established in assays of cAMP accumulation and pERK1/2 (Figures S7–S16), and data were analyzed by operational modeling to derive estimates of functional affinity (log KA) (Figures 3 and 4, Tables 2 and 3) and efficacy (log τ) for each pathway; the latter were corrected for differences in cell surface expression (Figures 6 and 7, Tables 4 and 5).
Figure 4.
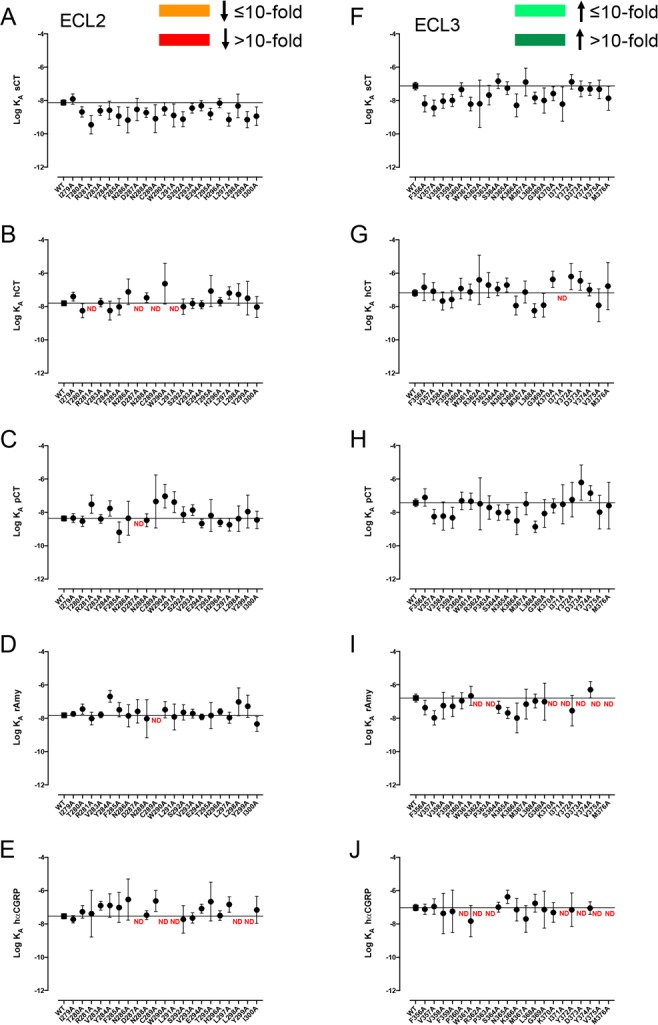
Alanine mutation of ECL2 and ECL3 of AMY3R has limited effect on pERK functional affinity (log KA). Functional affinities derived from operational fitting of concentration–response curves in ERK phosphorylation for alanine mutation of ECL2 (A–E) and ECL3 (F–J) are displayed as log KA. No significant changes in log KA from WT were seen for receptor mutants following one-way ANOVA and Dunnett’s post-test with P < 0.05 accepted as significant. Where data were insufficiently robust to derive a reliable value for log KA no symbol is shown (ND). Quantitative data are reported in Table 3.
Table 2. Effect of Single Alanine Mutation in AMY3R ECL2 or ECL3 on cAMP Functional Affinity (log KA) of Peptidesa.

For each receptor mutant and ligand, concentration–response data for each pathway were fit with the Black and Leff operational model to derive an affinity-independent measure of efficacy and functional affinity (log KA). Mean, S.E.M., and the individual experimental “n” values are reported. Significance of changes in log KA of each ligand was determined by comparison of mutant receptors to WT values by a one-way ANOVA and Dunnett’s post-test (p < 0.05 denoted by bold coloured entries. Orange, significant decrease ≤ 10-fold; red, significant decrease > 10-fold; Light green, significant increase ≤ 10-fold; Dark green, significant increase > 10-fold). ND, data were not able to be reliably determined.
Table 3. Effect of Single Alanine Mutation in AMY3R ECL2 or ECL3 on pERK Functional Affinity (log KA) of Peptidesa.

For each receptor mutant and ligand, concentration–response data for each pathway were fit with the Black and Leff operational model to derive an affinity-independent measure of efficacy and functional affinity (log KA). Mean, S.E.M. and the individual experimental “n” values are reported. ND, data were not able to be reliably determined. Where quantitative data could be derived, no significant differences from WT values were observed, as assessed by a one-way ANOVA and Dunnett’s post-test (significance set at p < 0.05).
Figure 6.

Alanine mutation of ECL2 and ECL3 has distinct effects on AMY3R and CTR cAMP functional affinity (log KA). Functional affinities derived from operational fitting of concentration–response curves in cAMP accumulation are displayed as Δlog KA from wild-type for AMY3R (A–E) and CTR (F–J). Illustrated are top views of the receptors with the extracellular surface subject to alanine scanning depicted (combined surface/cpk representation). Mutations that significantly alter peptide functional log KA are colored according to the magnitude of effect, with mutated amino acids without significant alteration to log KA colored gray. Amino acid mutations where there was an insufficiently robust functional effect to quantify by operational modeling are depicted in black. The receptor ECD and peptide are not shown for clarity, with the CTR TM bundle in blue ribbon and RAMP3 in green ribbon in panels A–E, and CTR TM bunding in red ribbon in panels F–J. Data for CTR functional affinity are from Dal Maso et al., 2018.17
Figure 7.
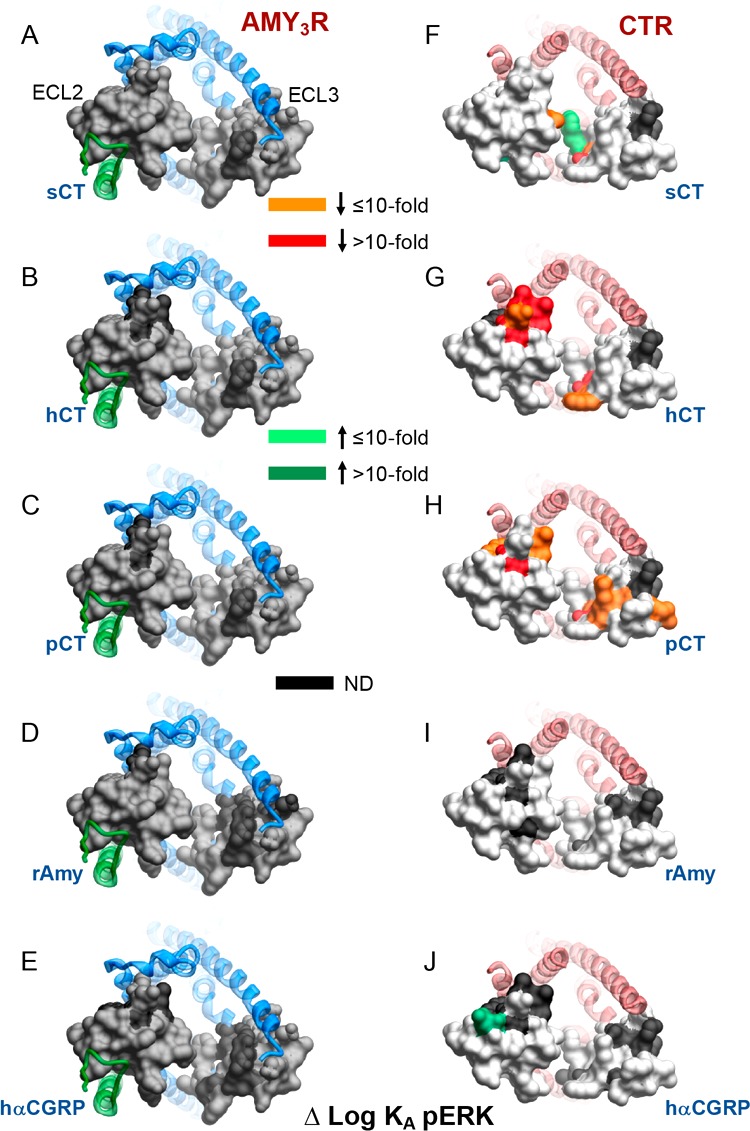
Alanine mutation of ECL2 and ECL3 has distinct effects on AMY3R and CTR pERK functional affinity (log KA). Functional affinities derived from operational fitting of concentration–response curves in pERK are displayed as Δlog KA from wild-type for AMY3R (A–E) and CTR (F–J). Illustrated are top views of the receptors with the extracellular surface subject to alanine scanning depicted (combined surface/cpk representation). Mutations that significantly alter peptide functional log KA are colored according to the magnitude of effect, with mutated amino acids without significant alteration to log KA colored gray. Amino acid mutations where there was an insufficiently robust functional effect to quantify by operational modeling are depicted in black. The receptor ECD and peptide are not shown for clarity, with the CTR TM bundle in blue ribbon and RAMP3 in green ribbon in A-E, and CTR TM bunding in red ribbon in F-J. Data for CTR functional affinity are from Dal Maso et al., 2018.17
Table 4. Effect of Single Alanine Mutation in AMY3R ECL2 or ECL3 on cAMP Signaling Efficacy (log τc) of Peptidesa.

For each receptor mutant and ligand, concentration–response data for each pathway were fit with the Black and Leff operational model to derive an affinity-independent measure of efficacy (log τ) and functional affinity. These data were corrected for changes in cell surface expression from FACS to yield Log τc. Mean, S.E.M., and the individual experimental “n” values are reported. Significance of changes in log τc of each ligand was determined by comparison of mutant receptors to WT values by a one-way ANOVA and Dunnett’s post-test (p < 0.05 denoted by bold coloured entries. Orange, significant decrease ≤ 10-fold; red, significant decrease > 10-fold; light green, significant increase ≤ 10-fold; dark green, significant increase > 10-fold). ND, data were not able to be reliably determined.
Table 5. Effect of Single Alanine Mutation in AMY3R ECL2 or ECL3 on pERK Signaling Efficacy (log τc) of Peptidesa.

For each receptor mutant and ligand, concentration–response data for each pathway were fit with the Black and Leff operational model to derive an affinity-independent measure of efficacy (log τ) and functional affinity. These data were corrected for changes in cell surface expression from FACS to yield log τc. Mean, S.E.M., and the individual experimental “n” values are reported. Significance of changes in log τc of each ligand was determined by comparison of mutant receptors to WT values by a one-way ANOVA and Dunnett’s post-test (p < 0.05 denoted by bold coloured entries. Red, significant decrease > 10-fold). ND, data were not able to be reliably determined.
There was a marked peptide dependence in the effect of mutation on cAMP functional affinity, with the greatest impact on hCT and pCT across both ECL2 and ECL3 (Figure 3, Table 2). hCGRP functional affinity was minimally affected by mutation with significant loss of affinity for L368A, but no detectable response (ND) for C289A, P363A, and D373A (Figures 3A,B; and 5E). Similarly, there was only limited effect on rAmy functional affinity, with loss of affinity for W290A and L291A in ECL2 and F359A, P363A, L368A, and D373A in ECL3 (Figures 3D,I; 5D). For sCT, only V357A in ECL3 altered affinity, with greater impact in ECL2 with decreased functional affinity for D287A, W290A, S292A and I300A (Figures 3A,B; 5A). In distinction to the limited effects of mutations on responses to these peptides, there was very marked, extended impact on hCT and pCT (Figures 3B,C,G,H; 5B,C). Within ECL2 and the TM5 proximal segment of ECL3, there was very similar impact on cAMP functional affinity for both peptides with attenuated affinity for R281A (ND for hCT), N286A, D297A, C289A, W290A (ND for hCT), L291A, S292A, T295A, and L297A-I300A within ECL2, and F359A-P363A in ECL3, with the exception of L297A, L298A, and V358A that had no significant effect on pCT affinity. Similarly, there was parallel loss of affinity for Y372A, D373A, and M376A for both peptides. Nonetheless, divergent effects were seen for K366A, M367A, and V375A (selective increased affinity for pCT), and L368A (selective decreased affinity for hCT) (Figures 3B,C,G,H; 5B,C; Table 2).
Figure 5.
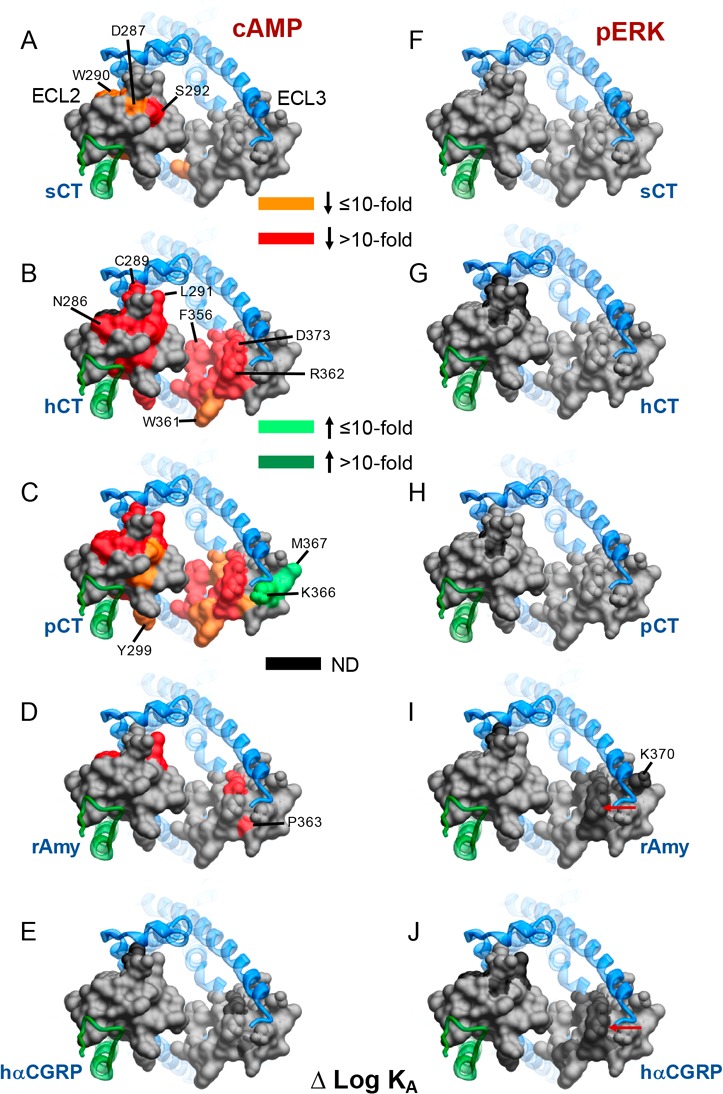
Alanine mutation of ECL2 and ECL3 of AMY3R alters functional affinity (log KA) in a peptide- and pathway-specific manner. Functional affinities derived from operational fitting of concentration–response curves in cAMP accumulation (A–E) and pERK (F–J) are displayed as Δlog KA from wild-type. Illustrated is a top view of the AMY3R model with the extracellular surface subject to alanine scanning depicted (combined surface/cpk representation). Mutations that significantly alter peptide functional log KA are colored according to the magnitude of effect, with mutated amino acids without significant alteration to log KA colored gray. Amino acid mutations for which there was an insufficiently robust functional effect to quantify by operational modeling are depicted in black. The receptor ECD and peptide are not shown for clarity, with the CTR TM bundle in blue ribbon and RAMP3 in green ribbon. Red arrows in panels I and J indicate residues at the apex of ECL3 that are affected for Amy and CGRP but not CT peptides.
In contrast to the dramatic effect on cAMP functional affinity, there was no significant effect on measured pERK functional affinity for any of the peptides (Figures 4 and 5, Table 3). However, robust responses were not seen for the following mutants and peptides; R281A (hCT), D287A (hCT, pCT, hCGRP), C289A (hCT, rAmy), W290A (hCGRP), L291A (hCT, hCGRP), L298A, I300A, P360A (hCGRP), R362A, P363A (rAmy, hCGRP), K370A (rAmy), I371A (hCT, rAmy, hCGRP), D373A, V375A, and M376A (hCGRP) and thus the nature of the loss of response could not be determined.
These data revealed marked differences in how ECL2 and ECL3 contribute to functional affinity across the two pathways at the AMY3R (Figure 5). The most notable differences were seen for hCT and pCT for which there was broad importance of the amino acids in the core of ECL2, and peptide proximal residues of ECL3, in cAMP but not pERK functional affinity (Figure 5B,C versus Figure 5G,H). Alanine mutants that selectively increased cAMP functional affinity for pCT clustered away from the peptide binding site and were located on the periphery of the receptor transmembrane domain. This region would be predicted to interact with the membrane bilayer, suggesting that these candidate receptor-membrane interactions constrain the receptor in a way that limits pCT functional affinity when RAMP3 is present, as the effect of mutation was not seen with mutants of CTR alone17 (Figure 6C versus Figure 6H). For the other peptides, there was limited effect of loop mutation on functional affinity for either pathway, with the quantifiable effects primarily occurring within residues involved in packing of the ECL2 in the active structures (Figure 5). While no quantifiable effect was seen on pERK functional affinity, there was a cluster of residues at the apex of ECL3 that adversely affected rAmy and hCGRP responses, but not those of CT peptides, suggesting that they are important for initiation or propagation of pERK signaling (Figure 5I,J, see red arrows).
The equivalent amino acids in CTR did not impact on pERK functional affinity of these peptides (Figure 7D,E versus Figure 7I,J), consistent with RAMP3 allosterically altering interaction of rAmy and hCGRP with this segment of the receptor, potentially through effects on engagement of the receptor with the midregion of the agonist peptide α-helix.
In general, the effect of ECL2 and ECL3 mutation was similar for AMY3R and CTR for cAMP functional affinity (Figure 6), for most peptides, although RAMP3 appeared to impart increased sensitivity to mutation for hCT and pCT. The exception to this was hCGRP in which a greater effect of mutation was seen for CTR relative to AMY3R (Figure 6E versus Figure 6J), and this might reflect increased strength of interaction of this peptide at the AMY3R such that individual mutation of amino acids had lesser effect.
Intriguingly, while overall there was relatively limited impact of ECL2 or ECL3 mutation on pERK functional affinity for either AMY3R or CTR, there was a greater effect of mutation on CTR, particularly for CT peptides and within ECL3 (Figure 7A-C versus 7F–H). This greater effect on CTR mutation occurred for select amino acids deep in the peptide binding pocket, with additional effects on pCT for residues that extended, in 3D space, from the peptide proximal residues.
Peptide Efficacy
The operationally derived efficacy parameter, τ, is a measure of pathway-specific coupling efficiency that relates the number of receptors occupied to response.19 Peptide efficacy for cAMP accumulation was largely unaffected by mutation to ECL2 residues (Figure 8A–E, Table 4), albeit that the relatively high variance may have limited those effects that achieved statistical significance.
Overall, ECL2 mutation tended to lead to increased measures of peptide efficacy for CT peptides (Figure 8A–C), with effects achieving significance for S292A mutation (sCT, pCT), L298A (sCT) and I300A (hCT). Increased efficacy of rAmy was also observed with the S292A mutant (Figure 8D). In contrast, both increased and decreased efficacy was observed following mutation of amino acids in ECL3, in a peptide-dependent manner (Figure 8F–J). Greater numbers of mutations had significant effects within ECL3; however, this was partially attributable to more robust expression of ECL3 mutants (Figure 1, Table 1) and greater precision in the calculation of log τ (Table 4).
The pattern of effect was similar for CT peptides, with increased efficacy for each peptide observed with the F356A, V358A, N365A, and D373A mutants and loss of efficacy for the V375A mutant (Figure 8F–H). Nonetheless, peptide specific effects were also observed with increased efficacy at the V358A (sCT), F359A (sCT, pCT), W361A (sCT), and M376A (sCT) mutants. Selective attenuation of efficacy was seen at the G369A and K370A (hCT) and Y372A (pCT) mutants (Figure 8F–H). While there were parallels in the effect of the F356A and N365A mutation for rAmy and hCGRP, the pattern of effect was generally distinct (Figure 8I,J), and more similar for these two peptides than between them and the CT peptides (Figure 8F–J). Of all the ECL3 mutants, only the F356A and N365A mutants had equivalent effect (increased efficacy) on rAmy, hCGRP, and CT peptides, while the effects of increased efficacy (V357A) and decreased efficacy (V375A) were also observed for rAmy but not hCGRP. Similar effects, distinct from those for CT peptides, were observed for both rAmy and hCGRP for P360A, W361A, P363A, and D373A (decreased efficacy or ND), with the nature of effect of the D373A mutant opposite to that seen for all CT peptides (Figure 8F–J). Peptide-specific loss of efficacy was seen for V358A, F359A, R362A, Y372A (hCGRP), M367A, and G369A (rAmy) (Figure 8I,J).
Remarkably, there was a dramatic loss of peptide efficacy for pERK for most individual mutants within both ECL2 and ECL3 (Figure 9, Table 5), although there was a lesser effect of ECL2 mutation on hCGRP efficacy (Figure 9E). Within ECL2, only I279A did not have any negative effect on CT or Amy peptide efficacy, albeit that the loss of efficacy did not achieve significance for V283A (hCT, pCT, rAmy), Y284A (rAmy), N288A (hCT), V293A (pCT), E294A, H296A (sCT, hCT, pCT, rAmy), L297A (hCT, pCT, rAmy), and L298A (hCT) (Figure 9A–D, Table 5). As noted above, for hCGRP, no robust response was observed for D287A, L291A, L298A, and Y299A (Tables 3, 5). Of the other ECL2 mutants, only the S292A mutant produced a significant loss of hCGRP efficacy (Figure 9E). Like ECL2, alanine mutation of ECL3 broadly led to loss of peptide efficacy (Figure 9F–J). For this loop, the pattern of effect was also mirrored for hCGRP (Figure 9J). Of the ECL3 residues, only S364A, N365A, and Y284A did not significantly reduce efficacy of any of the peptides (Table 5). For W361A and L368A, while loss of efficacy occurred for all peptides, this was not statistically significant for some of the peptides. Of the other mutants only K370A (hCT), F357A, and D373A (hCGRP) did not have significant effects on pERK peptide efficacy (Figure 9F–J).
Mapping the effects of ECL2 and ECL3 mutation onto the 3D AMY3R model revealed major differences in the effect of alanine mutation on the two signaling pathways (Figure 10). Mutations through the core of ECL2 dramatically reduced CT peptide and rAmy efficacy for pERK but had very limited impact on the efficacy in cAMP assays (Figure 10).
Figure 10.
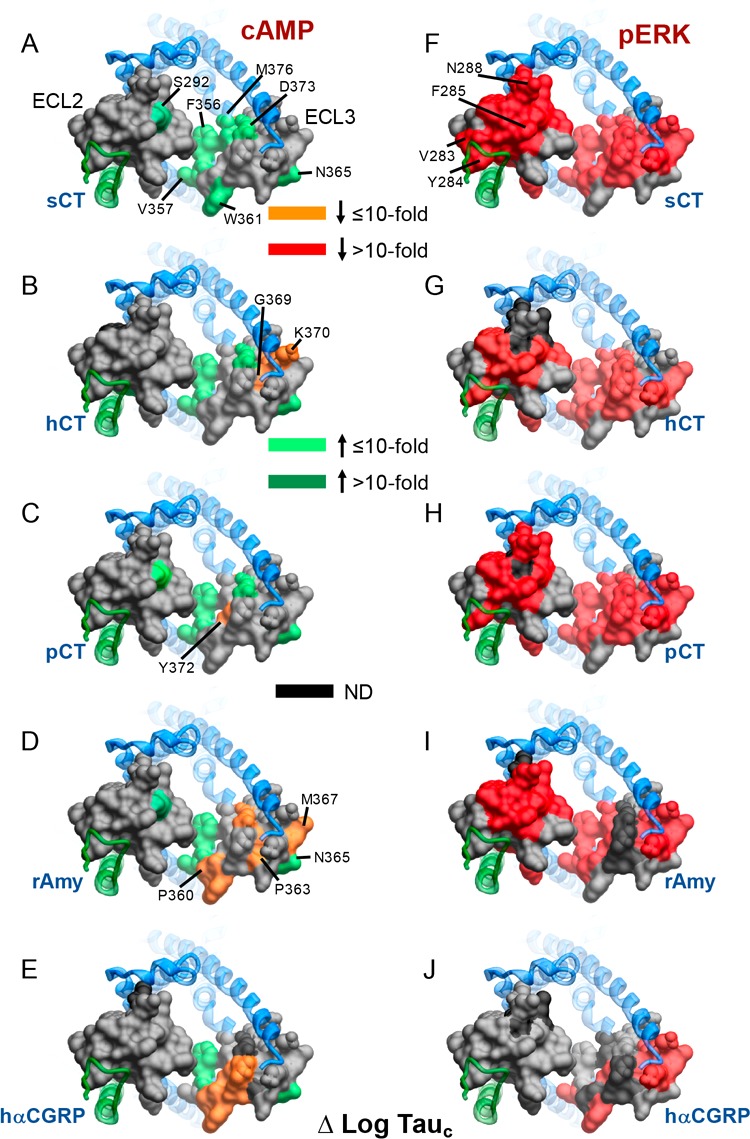
Alanine mutation of ECL2 and ECL3 of AMY3R alters peptide efficacy (log τc) in a pathway-specific manner. Efficacy values derived from operational fitting of concentration–response curves in cAMP accumulation (A–E) and pERK (F–J) are displayed as Δlog τc from wild-type. Illustrated is a top view of the AMY3R model with the extracellular surface subject to alanine scanning depicted (combined surface/cpk representation). Mutations that significantly alter peptide log τc are colored according to the magnitude of effect, with mutated amino acids without significant alteration to log τc colored gray. Amino acid mutations for which there was an insufficiently robust functional effect to quantify by operational modeling are depicted in black. The receptor ECD and peptide are not shown for clarity, with the CTR TM bundle in blue ribbon and RAMP3 in green ribbon.
Within ECL2, the most notable effect on cAMP efficacy was increased efficacy with the S292A mutation. This contrasts to the decreased functional affinity of the CT peptides for this pathway. S292 is capable of forming polar interactions and, in the CTR, contributes to packing of ECL2 in the active state.16 Nonetheless, in CTR the S292A mutation does not alter either affinity or efficacy of peptides for this pathway.17 This suggests that RAMP3 allosterically alters CTR ECL2 conformation, potentially allowing this residue to interact with rAmy and CT peptides when coupled to Gs.
Within ECL3, the mutation of residues at the proximal end of TM6 that are located deep in the peptide binding pocket led to enhanced cAMP efficacy for all peptides (Figure 10A–E). Interestingly, N365, whose alanine mutation also enhanced cAMP efficacy for all peptides, is located at the external face of the receptor (Figure 10A–E), and thus may make polar interactions with lipid head groups that constrain conformational propagation for Gs engagement in the context of the AMY3R. This amino acid was one of very few that did not adversely affect pERK efficacy (Figure 10A–E versus Figure 10F–J), suggesting that such a constraint does not affect non-Gs pathways. We have previously shown that pERK is independent of cAMP-dependent PKA activity, and PTX-sensitive Gi/o proteins for the AMY3R7 indicating that, like CTR, the effect of mutations on the two measured pathways reveal distinct conformational propagation pathways. Comparison of the effect of mutation on AMY3R and CTR pointed to peptide-specific influence on cAMP efficacy (Figure 11).
Figure 11.
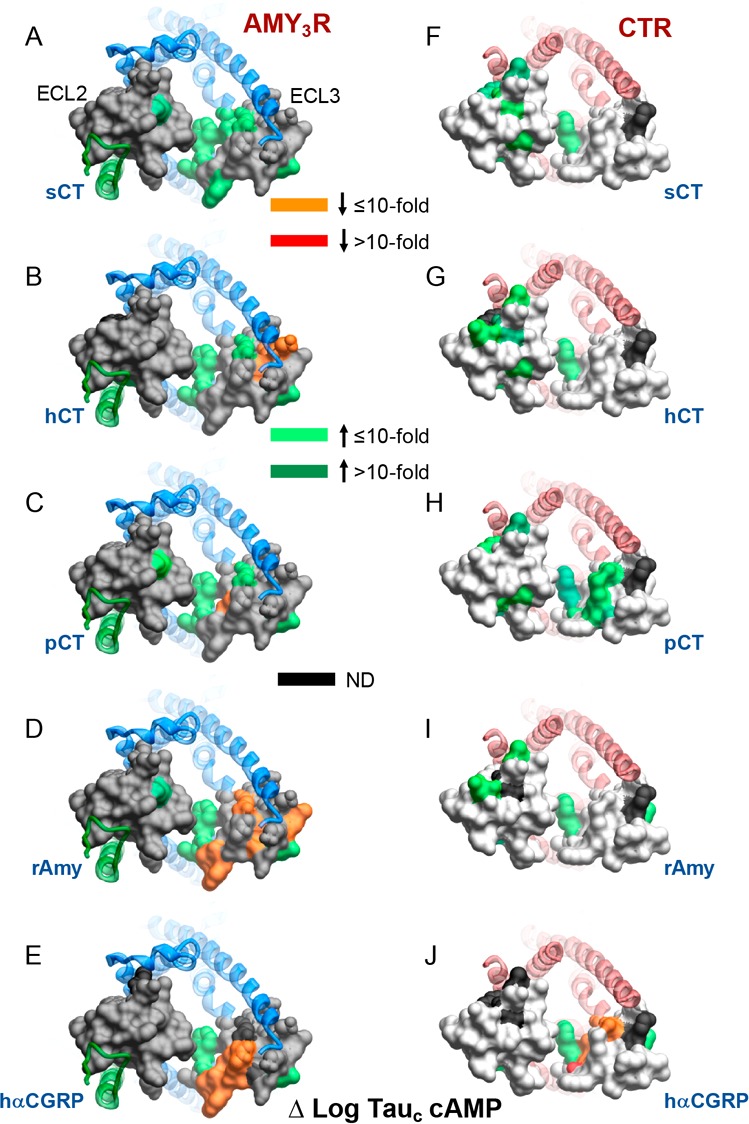
Alanine mutation of ECL2 and ECL3 has distinct effects on cAMP peptide efficacy for AMY3R and CTR. Peptide efficacy, derived from operational fitting of concentration–response curves in cAMP accumulation, are displayed as Δlog τc from wild-type for AMY3R (A–E) and CTR (F–J). Illustrated are top views of the receptors with the extracellular surface subject to alanine scanning depicted (combined surface/cpk representation). Mutations that significantly alter peptide functional log τc are colored according to the magnitude of effect, with mutated amino acids without significant alteration to log τc colored gray. Amino acid mutations for which there was an insufficiently robust functional effect to quantify by operational modeling are depicted in black. The receptor ECD and peptide are not shown for clarity, with the CTR TM bundle in blue ribbon and RAMP3 in green ribbon in A–E, and CTR TM bunding in red ribbon in F–J. Data for CTR peptide efficacy are from Dal Maso et al., 2018.17
Intriguingly, while the pattern of effect was similar, alanine mutation in ECL3 had greater impact on sCT efficacy at AMY3R compared to CTR, with mutation of the deeper, peptide-proximal residues within ECL3 enhancing cAMP efficacy (Figure 11A versus Figure 11F), implying a role for RAMP3 in directing the conformation change required for activation of the Gs pathway for this peptide. For hCT, RAMP3 in AMY3R appeared to shift the path of change from ECL2 to ECL3, based on the shift in mutational sensitivity between the two receptors (Figure 11B,G), while, in 3D representation, the effect of mutation of pCT cAMP response was similar across AMY3R and CTR (Figure 11C,H).
The largest divergence between the effect of mutation for AMY3R and CTR, on peptide-mediated cAMP efficacy, occurred for rAmy, in which, outside of deep pocket residues described above, mutation in ECL3 was generically associated with loss of efficacy for AMY3R but had limited effect on CTR (Figure 11D,I). This is consistent with the selective enhancement of rAmy affinity and potency seen at the AMY3R. While hCGRP potency is also enhanced at the AMY3R, this effect is less prominent than that induced by RAMP1.20 As such, it is perhaps unsurprising that the effect of mutation on hCGRP cAMP efficacy at the AMY3R was generally similar to that observed for CTR (Figure 11E,J).
Among the most profound differences in the effect of mutation on AMY3R versus CTR was the effect on pERK efficacy (Figure 12). For the CTR, there was minimal observed effect of ECL2 and ECL3 mutation on CTR-mediated pERK (Figure 12F–J). In contrast, there was broad loss of pERK efficacy for mutation of AMY3R across both ECL2 and ECL3 (Figure 12A–E), supporting a model in which RAMP3 causes a switch in the intracellular transducers engaged by CTR that are linked to the pERK pathway. While there has not been much investigation into the pathways linked to pERK downstream of AMY3R and CTR, the use of pathway inhibitors has implicated PKC, PI3K, and PLC in the phosphorylation of ERK.7 While those studies suggested subtle differences in the effect of inhibitors between AMY3R and CTR,7 the mechanistic basis for the major changes to sensitivity of mutants for AMY3R versus CTR remains to be elucidated. Nonetheless, they suggest that RAMP3 alters conformational propagation at least through ECL2 and ECL3. The exception to this was hCGRP that exhibited a similar pattern of mutational effect for both receptor phenotypes (Figure 12E,J). This lack of effect may reflect the limited induction of hCGRP binding and signaling that occurs with RAMP3, relative to CTR alone, when compared to the phenotypic induction by RAMP1.20
Conclusion
Biased agonism is a key pharmacological behavior of both endogenous GPCR ligands and drugs that target these receptors. Class B GPCRs are important physiological targets that display biased signaling in response to both endogenous and exogenous agonists, although the mechanistic basis for these differential effects is unclear. In this study, we demonstrate that the dynamic nature of CTR ECL2 and ECL3 in propagation of signaling is fundamentally altered when complexed with RAMP3 to form the AMY3R, despite only having predicted direct interactions with ECL2. Moreover, the work shows that the role of these loops in receptor signaling is highly peptide dependent, illustrating that even subtle changes to peptide sequence may change signaling output downstream of the receptor. The work further supports the allosteric role proposed for RAMPs in altering GPCR function21−25 with these changes, as assessed in the current study, extending well beyond the RAMP-CTR interface. While full understanding of these findings will likely require solution of structures of CTR:RAMP3 along with individual agonist peptides and transducer proteins, the current work advances our understanding of peptide control of class B GPCR signaling and the molecular basis for RAMP modulation of receptor function.
Methods
Reagents
All peptides were purchased from Mimotopes. Dulbecco’s Modified Eagle’s Medium (DMEM) was purchased from Invitrogen. Fetal bovine serum (FBS) was purchased from ThermoFisher Scientific. AlphaScreen reagents, Lance cAMP kit, and 384-well Optiplates were purchased from PerkinElmer. SureFire ERK1/2 reagents were obtained from TGR Biosciences and PerkinElmer. Antibodies were purchased from R&D Systems and ThermoFisher. All other reagents were purchased from Sigma-Aldrich or BDH Merck and were of an analytical grade.
Mutagenesis
Desired mutations were introduced to N-terminally c-Myc tagged human CTR in pENTER11 (Invitrogen) as previously described,17 then LR recombination reactions were conducted to transfer mutated and wild-type (WT) receptor into the pEF5/FRT/Rluc-8/IRES/RAMP3/Venus destination vector using Gateway Technology (Invitrogen), similar to our previously reported design of bicistronic constructs.26 Mutants were confirmed by automated-sequencing.
Stable Cell Line Generation and Cell Culture
The mutant or WT receptor genes were integrated into FlpIn-CV1 cells using Flp-In system (Invitrogen). Stable Flp-In expression cell lines were generated through polyclonal selection and screening and maintained in DMEM supplemented with 5% (v/v) FBS, 300 μg/mL hygromycin B (Invitrogen) at 37 °C in 5% CO2. Independent WT controls were established for each of the ECL2 and ECL3 mutants, as these were created at different times.
Whole Cell Competition Binding Assay
Radioligand competition binding was performed as previously described17 on whole cells seeded into 96-well plates and cultured overnight, except that 125I-rAmy was used as the radioligand. For homologous competition binding experiments, cells were incubated overnight at 4 °C with ∼100 pM 125I-rAmy (specific activity, 2000 Ci/mmol) and serial dilutions of non-iodinated rAmy, while heterologous competition was in the presence of increasing concentrations of unlabeled peptide. Non-bound ligand was removed and bound ligand activity was measured using a γ counter (Wallac Wizard 1470 Gamma Counter, PerkinElmer, 78% counter efficiency). Values were normalized against nonspecific binding, defined by the presence of 1 μM of unlabeled rAmy, and total ligand bound radioligand.
Cell Surface Expression Assessment by FACS
Surface expression of AMY3R mutants stably expressed in CV-1 cells was quantified by flow cytometry of antibody binding to the c-Myc tagged CTR subunit of the receptor using standard methods. Cells were grown in 6-well plates at ∼5 × 105 cells per well the day before assay. Cells were harvested in the presence of versene. All staining steps were conducted in ice cold Hank’s Balanced Salt Solution (HBSS) with 0.1% bovine serum albumin (BSA) and 20 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.4). Blocking was conducted in 5% BSA. Primary antibody staining was performed with 5 μg/mL 9E10 (anti-c-Myc) antibody. The secondary antibody was 1 μg/mL goat antimouse AF647 (ThermoFisher). Sytox blue was used for live/dead discrimination. Data were collected on a FACS CantosII (BD Biosciences) with at least 20 000 live cells collected per sample. WT stained CTR sample and stained parental CV-1 cells were collected at the beginning and the end of each run. Data were analyzed using FlowJo. The mean AF647 fluorescence intensity from each sample for a particular experiment was normalized against parental (0%) and WT CTR (100%) controls.
cAMP Accumulation Assay
Cells (2.5 × 104 cells/well) were seeded into 96-well plates and incubated overnight. Complete media was replaced with phenol red-free DMEM containing 0.5 mM 3-isobutyl-1-methylxanthine (IBMX) and 0.1% BSA and preincubated for 30 min. Cells were stimulated with increasing concentrations of ligands for 30 min in the presence of 0.5 mM 3-isobutyl-1-methylxanthine (IBMX). The media was discarded, changed to absolute ethanol and volatilized to dryness at room temperature. Samples were then lysed and intracellular cAMP was detected using the PerkinElmer Lance kit as previously described.14 Data were normalized to the maximal response of each peptide.
ERK1/2 Phosphorylation
Cells (2.5 × 104 cells/well) were seeded into 96-well culture plates and incubated overnight. Initially, pERK1/2 time-course experiments were performed over 30 min to identify the time point when the pERK1/2 response is maximal (6–8 min). Subsequently, this time point was selected to generate concentration response curves for different agonists with ligand addition performed after overnight serum starvation with DMEM. FBS was used as a positive control. pERK1/2 was detected using an AlphaScreen assay as previously described.27 Data were normalized to the maximal response elicited by each peptide.
Pharmacological Data Analysis
IC50 and Bmax values were estimated from competitive inhibition of 125I-rAmy binding using a three-parameter logistic equation [Log(inhibitor versus response)] in Prism (v7 or v8; GraphPad). The concentration of the radioligand was ≤ 5% of the KD values. Under these conditions, the IC50 approximates Ki, and such data are reported as log Ki. The Black and Leff operational model of partial agonism19,27 was applied to separate effects on pathway-specific efficacy (defined by the value tau, τ) from those that modify ligand functional affinity (log KA). Derived τ values were normalized to experimentally determined levels of cell surface expression to provide a measure of efficacy (τc) that is independent of affinity and altered cell surface receptor expression.28 pKi, pKA, and log τc values for mutant receptors were statistically compared to those of the respective WT receptor using a one-way analysis of variance (ANOVA) and Dunnett’s post-test. Significance was accepted at P < 0.05.
Computational Methods
System Preparation
The CTR:RAMP3 complex was built using the Modeler comparative modeling program from the full CGRPR15 and CTR16 3.3 Å cryo-EM receptor structures, which included the missing loops, and the 1.76 Å CLR:RAMP2 X-ray crystal structure of the extracellular domain of the adrenomedullin receptor29 (PDB codes 6E3Y, 6NIY, and 4RWF, respectively). The disulfide bond between RAMP3 residues Cys 28 and Cys 72 was included. The structure with the best discrete optimized protein energy (DOPE) score30 out of 1000 generated models was prepared for molecular dynamics simulation using a combination of python htmd31 and tcl (Tool Command Language) scripts. Hydrogen atoms were added using pdb 2pqr;32 the protonation state of titratable side chains was determined using propka33 (run at pH 7) coupled with visual inspection. The systems were embedded in a pre-existing 1-palmitoyl-2-oleyl-sn-glycerol-3-phospho-choline (POPC) bilayer using an insertion method,34 with overlapping lipids removed. The receptor orientation was determined from the Calcitonin receptor-Gs complex (PDB ID: 5UZ7) entry in the OPM database.35 TIP3P water molecules36 were added to the 106 Å × 106 Å × 141 Å simulation box using the VMD Solvate plugin version 1.5 (http://www.ks.uiuc.edu/Research/vmd/plugins/solvate/). Sodium and chloride ions were added to mimic an ionic strength of 0.150 M and to obtain overall charge neutrality, using the VMD Autoionize plugin 1.3 (Autoionize Plugin, Version 1.3. at < http://www.ks.uiuc.edu/Research/vmd/plugins/autoionize/).
Systems Equilibration and MD Settings
The MD engine ACEMD37 was employed for both the equilibration and productive simulations, which employed the CHARMM36 force field.38 Equilibration was achieved in isothermal–isobaric conditions (NPT) using the Langevin thermostat39 (target: 300 K) with a low damping of 1 ps–1 and the Berendsen barostat40 (target: 1 atm) over a three-stage procedure using an integration time step of 2 fs. First, clashes between protein and lipid atoms were reduced through 2500 conjugate-gradient minimization steps followed by a 2 ns long MD simulation with a positional constraint of 1 kcal mol–1 Å–2 on protein and lipid phosphorus atoms. Second, 33 ns of MD simulation was performed with only the protein atoms constrained. Third, positional constraints were applied only to the protein backbone alpha carbons for a further 35 ns.
A 1 μs simulation was run in the canonical ensemble (NVT) at 300 K, using a thermostat damping of 0.1 ps–1. The M-SHAKE algorithm41 was used to constrain the covalent bonds involving hydrogen atoms, enabling a time step of 4 fs. A 9 Å cutoff distance was used for the electrostatic interactions, with a switching function applied beyond 7.5 Å; long-range Coulomb interactions were handled using the particle mesh Ewald summation method (PME)42 with a mesh spacing to 1.0 Å. The mutagenesis results were plotted on the 200 ns structure, since this was deemed sufficient to remove any strain within the initial structure.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia (NHMRC) program grant (1055134) and project grants (1061044, 1159006), the National Key R&D Program of China grant (2018YFA0507000), the National Mega R&D Program for Drug Discovery grants (2018ZX09711002-002-005 and 2018ZX09735-001), the National Natural Science Foundation of China grants (81573479 and 81773792), Shanghai Science and Technology Development Fund grant (15DZ2291600 and 16ZR1407100), Novo Nordisk-CAS Research Fund grant (NNCAS-2017-1-CC), and the Biotechnology and Biological Sciences Research Council (BBSRC; BB/M006883/1). S.G.B.F. is an ARC Future Fellow. D.W. is a Senior Research Fellow, and A.C. and P.M.S. are Senior Principal Research Fellows, of the NHMRC. D.L.H. is a James Cook Research Fellow of the Royal Society of New Zealand. C.A.R. is a Royal Society Industrial Fellow. S.A. is supported by the Peter Nicholls bequest.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsptsci.9b00010.
Effect of alanine mutation of ECL2 and ECL3 of AMY3R on peptide affinity; competition binding isotherms at WT and mutant AMY3R; concentration response curves in cAMP accumulation assays and in ERK1/2 phosphorylation assays at WT and mutant AMY3R (PDF)
Author Contributions
Contributed to experimental design (E.d.M., D.L.H., D.Y., M.-W.W., D.W., P.M.S.); performed experiments (V.P., E.d.M., YZ, C.A.H.); contributed to conceptual development of the project (S.G.B.F., D.L.H., A.C., D.Y., M.-W.W., D.W., P.M.S.); performed molecular modeling (G.D., S.A., C.A.R.); performed pharmacological data analysis (V.P., Y.Z., E.d.M., D.W., P.M.S.); contributed to production of figures (Y.Z., E.d.M., D.W., P.M.S.); provided oversight of the project (M.-W.W., D.W., P.M.S.); drafted the manuscript (Y.Z., P.M.S.).
Author Contributions
# These authors contributed equally to this work.
The authors declare no competing financial interest.
This article is made available for a limited time sponsored by ACS under the ACS Free to Read License, which permits copying and redistribution of the article for non-commercial scholarly purposes.
Supplementary Material
References
- Alexander S. P.; Christopoulos A.; Davenport A. P.; Kelly E.; Marrion N. V.; Peters J. A.; Faccenda E.; Harding S. D.; Pawson A. J.; Sharman J. L.; Southan C.; Davies J. A. (2017) CGTP Collaborators. The concise guide to pharmacology 2017/18: G protein-coupled receptors. Br. J. Pharmacol. 174 (Suppl. 1), S17–S129. 10.1111/bph.13878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootten D.; Christopoulos A.; Marti-Solano M.; Babu M. M.; Sexton P. M. (2018) Mechanisms of signalling and biased agonism in G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 19, 638–653. 10.1038/s41580-018-0049-3. [DOI] [PubMed] [Google Scholar]
- Wootten D.; Miller L. J.; Koole C.; Christopoulos A.; Sexton P. M. (2017) Allostery and Biased Agonism at Class B G Protein-Coupled Receptors. Chem. Rev. 117, 111–138. 10.1021/acs.chemrev.6b00049. [DOI] [PubMed] [Google Scholar]
- Ostrovskaya A.; Findlay D. M.; Sexton P. M.; Furness S. G. B. (2017) Calcitonin. Reference Module in Neuroscience and Biobehavioral Psychology 1–12. 10.1016/B978-0-12-809324-5.03223-5. [DOI] [Google Scholar]
- Hay D. L.; Garelja M. L.; Poyner D. R.; Walker C. S. (2018) Update on the pharmacology of calcitonin/ CGRP family of peptides: IUPHAR Review 25. Br. J. Pharmacol. 175, 3–17. 10.1111/bph.14075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routledge S. J.; Ladds G.; Poyner D. R. (2017) The effects of RAMPs upon cell signalling. Mol. Cell. Endocrinol. 449, 12–20. 10.1016/j.mce.2017.03.033. [DOI] [PubMed] [Google Scholar]
- Morfis M.; Tilakaratne N.; Furness S. G. B.; Christopoulos G.; Werry T. D.; Christopoulos A.; Sexton P. M. (2008) Receptor activity modifying proteins differentially modulate the G protein-coupling efficiency of amylin receptors. Endocrinology 149, 5423–5431. 10.1210/en.2007-1735. [DOI] [PubMed] [Google Scholar]
- Hay D. L.; Chen S.; Lutz T. A.; Parkes D. G.; Roth J. D. (2015) Amylin: pharmacology, physiology and clinical potential. Pharmacol. Rev. 67, 564–600. 10.1124/pr.115.010629. [DOI] [PubMed] [Google Scholar]
- Gydesen S.; Andreassen K. V.; Hjuler S. T.; Hellgren L. I.; Karsdal M. A.; Henriksen K. (2017) Optimization of tolerability and efficacy of the novel dual amylin and calcitonin receptor agonist KBP-089 through dose escalation and combination with a GLP-1 analog. Am. J. Physiol Endocrinol Metab 313, E598–E607. 10.1152/ajpendo.00419.2016. [DOI] [PubMed] [Google Scholar]
- Levin B. E.; Lutz T. A. (2017) Amylin and Leptin: Co-Regulators of Energy Homeostasis and Neuronal Development. Trends Endocrinol. Metab. 28, 153–164. 10.1016/j.tem.2016.11.004. [DOI] [PubMed] [Google Scholar]
- Liberini C. G.; Boyle C. N.; Cifani C.; Venniro M.; Hope B. T.; Lutz T. A. (2016) Amylin receptor components and the leptin receptor are co-expressed in single rat area postrema neurons. Eur. J. Neurosci 43, 653–661. 10.1111/ejn.13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.-L.; Khoshouei M.; Radjainia M.; Zhang Y.; Glukhova A.; Tarrasch J.; Thal D. M.; Furness S. G. B.; Christopoulos G.; Coudrat T.; Danev R.; Baumeister W.; Miller L. J.; Christopoulos A.; Kobilka B. K.; Wootten D.; Skiniotis G.; Sexton P. M. (2017) Phase-plate cryo-EM structure of a class B GPCR-G protein complex. Nature 546, 118–123. 10.1038/nature22327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Sun B.; Feng D.; Hu H.; Chu M.; Qu Q.; Tarrasch J. T.; Li S.; Kobilka T. S.; Kobilka B. K.; Skiniotis G. (2017) Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546, 248–253. 10.1038/nature22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.-L.; Khoshouei M.; Glukhova A.; Furness S. G. B.; Zhao P.; Clydesdale L.; Koole C.; Truong T. T.; Thal D. M.; Lei S.; Radjainia M.; Danev R.; Baumeister W.; Wang M.-W.; Miller L. J.; Christopoulos A.; Sexton P. M.; Wootten D. (2018) Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature 555, 121–125. 10.1038/nature25773. [DOI] [PubMed] [Google Scholar]
- Liang Y.-L.; Khoshouei M.; Deganutti G.; Glukhova A.; Koole C.; Peat T. S.; Radjainia M.; Plitzko J. M.; Baumeister W.; Miller L. J.; Hay D. L.; Christopoulos A.; Reynolds C. A.; Wootten D.; Sexton P. M. (2018) Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 561, 492–497. 10.1038/s41586-018-0535-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Maso E.; Glukhova A.; Zhu Y.; Garcia-Nafria J.; Tate C. G.; Atanasio S.; Reynolds C. A.; Ramirez-Aportela E.; Carazo J.-M.; Hick C. A.; Furness S. G. B.; Hay D. L.; Liang Y.-L.; Miller L. J.; Christopoulos A.; Wang M.-W.; Wootten D.; Sexton P. M. (2019) The molecular control of calcitonin receptor (CTR) signaling. ACS Pharmacol Transl Sci. 2, 31–51. 10.1021/acsptsci.8b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Maso E.; Zhu Y.; Pham V.; Reynolds C. A.; Deganutti G.; Hick C. A.; Yang D.; Christopoulos A.; Hay D. L.; Wang M.-W.; Sexton P. M.; Furness S. G. B.; Wootten D. (2018) Extracellular loops 2 and 3 of the calcitonin receptor selectively modify agonist binding and efficacy. Biochem. Pharmacol. 150, 214–244. 10.1016/j.bcp.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos G.; Perry K. J.; Morfis M.; Tilakaratne N.; Gao Y.; Fraser N. J.; Main M. J.; Foord S. M.; Sexton P. M. (1999) Multiple amylin receptors arise from receptor-activity modifying protein interaction with the calcitonin receptor gene product. Mol. Pharmacol. 56, 235–242. 10.1124/mol.56.1.235. [DOI] [PubMed] [Google Scholar]
- Black J. W.; Leff P. (1983) Operational models of pharmacological agonism. Proc. R Soc. Lond B Biol. Sci. 220, 141–162. 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Hay D. L.; Christopoulos G.; Christopoulos A.; Poyner D. R.; Sexton P. M. (2005) Pharmacological discrimination of calcitonin receptor:receptor activity-modying protein complexes. Mol. Pharmacol. 67, 1655–1665. 10.1124/mol.104.008615. [DOI] [PubMed] [Google Scholar]
- Woolley M. J.; Reynolds C. A.; Simms J.; Walker C. S.; Mobarec J. C.; Garelja M. L.; Conner A. C.; Poyner D. R.; Hay D. L. (2017) Receptor activity-modifying protein dependent and independent activation mechanisms in the coupling of calcitonin gene-related peptide and adrenomedullin receptors to Gs. Biochem. Pharmacol. 142, 96–110. 10.1016/j.bcp.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingell J. J.; Simms J.; Barwell J.; Poyner D. R.; Watkins H. A.; Pioszak A. A.; Sexton P. M.; Hay D. L. (2016) An allosteric role for receptor activity-modifying proteins in defining GPCR pharmacology. Cell Discov 2, 16012. 10.1038/celldisc.2016.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins H. A.; Chakravarthy M.; Abhayawardana R. S.; Gingell J. J.; Garelja M.; Pardamwar M.; McElhinney J. M.; Lathbridge A.; Constantine A.; Harris P. W.; Yuen T. Y.; Brimble M. A.; Barwell J.; Poyner D. R.; Woolley M. J.; Conner A. C.; Pioszak A. A.; Reynolds C. A.; Hay D. L. (2016) Receptor activity-modifying proteins 2 and 3 generate adrenomedullin receptor subtypes with distinct molecular properties. J. Biol. Chem. 291, 11657–11675. 10.1074/jbc.M115.688218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. M.; Hay D. L.; Pioszak A. A. (2016) Calcitonin and amylin receptor peptide interaction mechanisms: Insights into peptide-binding modes and allosteric modulation of the calcitonin receptor by receptor activity-modifying proteins. J. Biol. Chem. 291, 8686–8700. 10.1074/jbc.M115.713628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booe J. M.; Warner M. L.; Roehrkasse A. M.; Hay D. L.; Pioszak A. A. (2018) Probing the Mechanism of receptor activity-modifying protein modulation of GPCR ligand selectivity through rational design of potent adrenomedullin and calcitonin gene-related peptide antagonists. Mol. Pharmacol. 93, 355–367. 10.1124/mol.117.110916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage E. E.; Wootten D.; Christopoulos A.; Sexton P. M.; Furness S. G. (2013) A simple method to generate stable cell lines for the analysis of transient protein-protein interactions. BioTechniques 54, 217–221. 10.2144/000114013. [DOI] [PubMed] [Google Scholar]
- Koole C.; Wootten D.; Simms J.; Valant C.; Sridhar R.; Woodman O. L.; Miller L. J.; Summers R. J.; Christopoulos A.; Sexton P. M. (2010) Allosteric ligands of the glucagon-like peptide 1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner; implications for drug screening. Mol. Pharmacol. 78, 456–465. 10.1124/mol.110.065664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koole C.; Wootten D.; Simms J.; Miller L. J.; Christopoulos A.; Sexton P. M. (2012) The second extracellular loop of the human glucagon-like peptide-1 receptor (GLP-1R) has a critical role in GLP-1 peptide binding and receptor activation. J. Biol. Chem. 287, 3642–3648. 10.1074/jbc.M111.309328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booe J. M.; Walker C. S.; Barwell J.; Kuteyi G.; Simms J.; Jamaluddin M. A.; Warner M. L.; Bill R. M.; Harris P. W.; Brimble M. A.; Poyner D. R.; Hay D. L.; Pioszak A. A. (2015) Structural basis for receptor activity-modifying protein-dependent selective peptide recognition by a G protein-coupled receptor. Mol. Cell 58, 1040–1052. 10.1016/j.molcel.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M.; Sali A. (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524. 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerr S.; Harvey M. J.; Noé F.; De Fabritiis G. (2016) HTMD: High-Throughput Molecular Dynamics for Molecular Discovery. J. Chem. Theory Comput. 12, 1845–1852. 10.1021/acs.jctc.6b00049. [DOI] [PubMed] [Google Scholar]
- Dolinsky T. J.; Nielsen J. E.; McCammon J. A.; Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667. 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson M. H. M.; Søndergaard C. R.; Rostkowski M.; Jensen J. H. (2011) PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537. 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- Sommer B. (2013) Membrane packing problems: A short review on computational membrane modeling methods and tools. Comput. Struct. Biotechnol. J. 5, e201302014 10.5936/csbj.201302014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize M. A.; Lomize A. L.; Pogozheva I. D.; Mosberg H. I. (2006) OPM: orientations of proteins in membranes database. Bioinformatics 22, 623–625. 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Harvey M. J.; Giupponi G.; De Fabritiis G. (2009) ACEMD: Accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput. 5, 1632–1639. 10.1021/ct9000685. [DOI] [PubMed] [Google Scholar]
- Huang J.; MacKerell A. D. (2013) CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145. 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loncharich R. J.; Brooks B. R.; Pastor R. W. (1992) Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N’-methylamide. Biopolymers 32, 523–535. 10.1002/bip.360320508. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Kräutler V.; Van Gunsteren W. F.; Hünenberger P. H. (2001) A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 22, 501–508. . [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.