

Abstract

G-protein-coupled receptors (GPCRs) can bias signaling through distinct biochemical pathways that originate from G-protein/receptor and β-arrestin/receptor complexes. Receptor conformations supporting β-arrestin engagement depend on multiple receptor determinants. Using ghrelin receptor GHR1a, we demonstrate by bioluminescence resonance energy transfer and fluorescence microscopy a critical role for its second intracellular loop 2 (ICL2) domain in stabilizing β-arrestin/GHSR1a core interactions and determining receptor trafficking fate. We validate our findings in ICL2 gain- and loss-of-function experiments assessing β-arrestin and ubiquitin-dependent internalization of the CC chemokine receptor, CCR1. Like all CC and CXC subfamily chemokine receptors, CCR1 lacks a critical proline residue found in the ICL2 consensus domain of rhodopsin-family GPCRs. Our study indicates that ICL2, C-tail determinants, and the orthosteric binding pocket that regulates β-arrestin/receptor complex stability are sufficient to encode a broad repertoire of the trafficking fates observed for rhodopsin-family GPCRs, suggesting they provide the essential elements for regulating a large fraction of β-arrestin signaling bias.
Keywords: arrestin, beta-arrestin, β-arrestin, bias, Boolean, BRET, CCR1, chemokine, endocytosis, ghrelin, GPCR, GHSR1a, HIV, network, receptor, trafficking, ubiquitin
Plasma-membrane G-protein-coupled receptors (GPCRs) make ligand- and constitutive-based fate decisions that are observable as stochastic trafficking to different cell compartments. For hundreds of GPCRs in the rhodopsin family, arrestin proteins 2 and 3 (also known as β-arrestin-1 and β-arrestin-2, respectively) orchestrate many of these decisions. Receptors with a broad range of β-arrestin affinities support the idea that trafficking information is encoded in phosphorylation motifs located in the receptor C-terminal tails/third loops. These sites can modulate the affinity and spatial coordination of the agonist-bound and constitutively active receptor toward arrestins and vice versa. The structural decoding of the tail phosphorylation sites by arrestins has recently been described.1,3,4 Arrestins, however, can bind receptors independent of tail phosphorylation through other conformations. As early as 1993, a second hydrophobic-binding site was postulated for arrestin,5,6 and most recently, a receptor-core interaction was observed by cryo-EM,7 suggesting that information in the receptor C-tail is insufficient by itself to provide an accurate trafficking map. Determining how these separate areas complement one another in writing comprehensive instructions for receptor trafficking to and from the plasma membrane would provide a foundation for understanding a fundamental aspect of GPCR regulation.
Our studies of rhodopsin-family GPCRs suggest an important role for amino acid residues in the proximal portion of intracellular loop two (ICL2) following the (D/E)RY motif in managing β-arrestin binding.2 In rhodopsin-like GPCRs, the DRY motif aids in the formation of an ionic lock between receptor transmembranes III and VI.8,9 ICL2 typically originates at the DRY+3 position at the end of transmembrane III,8 where it contains a highly conserved segment with a proline at the DRY+6 position.2 The large chemokine receptor subfamily provides an exception to this rule in that the proline undergoes alanine substitution. Recent crystal structure data showing rhodopsin-ICL2 bound in a cleft formed in activated arrestin indicate that the proximal segment of ICL2 may also be a direct and important regulator of β-arrestin binding.2 Thus, GPCR ICL2 and the C-tail potentially contain separate incomplete pieces of the instructions directing β-arrestin/receptor trafficking at the plasma membrane and beyond,10 but a proof-of-concept demonstration of this is lacking.
Growth hormone secretagogue receptor GHSR1a is a rhodopsin-family GPCR that regulates food-seeking and reward-learning; for instance, GHSR1a antagonists reverse opioid-induced behaviors.11−14 The 28 amino acid peptide hormone ghrelin is the endogenous ligand for GHSR1a. It activates intracellular Ca2+ through a classical Gαq/phospholipase C/inositol phosphate 3-diacylglycerol signaling pathway. Ghrelin independently stimulates F-actin fiber formation through β-arrestin-2 signaling, a process associated with Rho activation and neuronal plasticity,15−17 and studies in rats suggest that β-arrestin also mediates ghrelin-induced appetite stimulation.18
GHSR1a binds β-arrestin with an affinity intermediate to those of other class A and B GPCRs that exhibit either very limited or lengthy post-internalization β-arrestin association.15,16,19 We have demonstrated that point mutations at GHSR1a ICL2 proline 148 (P148) and neighboring leucine 149 (L149) produce receptors with opposite, signaling-biased phenotypes. The proline mutant has reduced β-arrestin affinity and is biased toward G-protein-signaling, and the leucine mutant is biased toward β-arrestin.16 These studies together with arrestin-ICL2 spectroscopic20 and crystal structure data1 (Figure 1a,b) suggest that G-protein and β-arrestin cannot simultaneously engage ICL2 residues between DRY and DRY+7 and importantly support GHSR1a as a plausible prototype for deciphering β-arrestin/GPCR trafficking instructions. In the following study we show by fluorescence imaging of mutant GHSR1a receptors, bioluminescence resonance energy transfer (BRET) between GHSR1a and β-arrestin reporters, and gain-/loss-of-function experiments with the chemokine receptor CCR1 that a very broad range of β-arrestin-2 regulated trafficking fates are coded by combining independent sets of information contained in GHSR1a C-tail motifs and ICL2 core residues.
Figure 1.

Models of ICL2 arrestin structure for rhodopsin and GHSR1a substitution mutants. Graphics were generated from the crystal structure results of Kang et al.1 (a) Cartoon image of the visual-arrestin (in blue and green)/rhodopsin complex demonstrates the “core” conformation, in which both the receptor tail and the second intracellular loop (ICL2) are engaged. (b) Amino acid sequences of GHSR1a’s second intracellular loop (ICL2) and the C-tail. The D353ESS (DESS, green) and T360ESS (TESS, magenta) motifs in GHSR1a’s C-tail are putative regulatory sites for β-arrestin binding. In the ICL2 ERY motif, the +6 proline (PA, blue) and the +7 hydrophobic (leucine, LG, red) amino acid residues are putative β-arrestin and G-protein-binding sites, respectively. The two residues were mutated to alanine and glycine. (c) Complementary space filling model views of ICL2/arrestin engagement for rhodopsin (upper panels) and GHSR1a (lower panels). (d) Cartoon illustrating +6 proline N-terminal α helix capping of a short segment (smaller cylinder) of GHSR1a ICL2 in the active arrestin crevice. (e) Stick-figure image of the location of the GHSR1a (rhodopsin) ICL2+6 proline (green) relative to a C terminal arrestin middle loop hydrophobic patch containing leucine 250 (blue).
Results
During the desensitization process, G-protein-coupled receptor kinase (GRK) phosphorylated GPCRs undergo clathrin-mediated endocytosis as a result of β-arrestin-binding.21−23 It was initially hypothesized for endosomal recycling receptors, with the β2-adrenergic receptor (β2AR) serving as a role model for other GPCRs, that a downstream dephosphorylation/resensitization step was either initiated or accompanied by an acidic-induced loss of ligand binding. This view, however, was broadened to include the discovery that resensitization relevant phosphatases are active in nonendosomal cell compartments including those at the plasma membrane.24−27 Thus, β-arrestin-based receptor trafficking information appears susceptible to modification even before the receptor has an opportunity to internalize from the plasma membrane, and to assess it requires measurement techniques applicable over both short and long times. In addition, if we use the beta2-adrenergic receptor (β2AR) and rhodopsin as templates for characterizing the GHSR1a, then the principal locations to assess trafficking information are in its C-tail and ICL2 (Figure 1b–e).2,19,28 Generation of a GHSR1/arrestin active space-filling model (Figure 1c, lower panels) by corresponding residue substitution of its ICL2 into the rhodopsin ICL2 (Figure 1c, upper panels) suggests conservation of the ability to bind the active arrestin cleft as well as demonstrating conservation of a short N-capped proline α helix (Figure 1a, rhodopsin; Figure 1d, GHSR1a) within 2.4 Å of a hydrophobic patch containing arrestin leucine 250 (Figure 1e.)
We therefore began our study of GHSR1a mutants containing C-tail alanine substitutions, e.g., in the D353ESS or T360ESS serines and threonines that removed putative GRK-phosphorylation-related sites15 and point substitution mutants of ICL2 residues P148 and L149 distal to the E/DRY motif.2 Relative to concerns that a +6 proline mutation in the second loop may in general affect phosphorylation status in the C-tail of rhodopsin-like GPCRs, it is significant to note that mutation of the equivalent ICL2+6 proline to alanine in the β2AR-P138A did not produce a change in total isoproterenol-mediated phosphorylation or GRK-dependent phosphorylation of C-tail serines 355 and 356 between the wild type (WT) receptor and alanine mutant β2AR receptors (see Figure 4E,F of Marion et al.).2 Independent measures of receptor, β-arrestin, and receptor/β-arrestin cell locations were obtained over a broad temporal range using fluorogen-activating protein (FAP)-tagged GHSR1a variants, β-arrestin-2 fluorescent protein conjugates, and BRET sensors composed of GHSR1a/β-arrestin-2 pairs.29−32
G-Protein Signaling of ICL2 and C-Tail GHSR1a Mutants
GHSR1a is Gq-coupled and induces Ca2+ release to control anterior-pituitary growth hormone secretion, but it is also capable of recruiting other G protein subtypes.28,33,34Figure 2a shows the G-protein signaling response of GHSR1a ICL2 and C-tail mutant receptors to ghrelin stimulation using a luminescence reporter for Ca2+. The ICL2 mutants signal as previously reported,16 with the P148A receptor exhibiting G-protein bias and WT signaling efficacy (85 ± 9% of WT), and the β-arrestin biased L149G mutant showing almost no Ca2+ response (5 ± 2% of WT). GHSR1a mutant D353EAA also has WT signaling efficacy (85 ± 8% of WT), but mutant T360EAA has a reduced Ca2+ response equal to 46 ± 3% of WT efficacy. This result for T360EAA is surprising given that β-arrestin binding promotes desensitization and a phosphorylation-dependent reduction in β-arrestin binding affinity from loss of this particular GHSR1a phosphorylation motif15 would be expected to enhance G-protein-related signaling. However, the observation that the C-tail may directly either positively or negatively constrain the receptor conformation supporting G-protein signaling could provide a possible explanation.35,36
Figure 2.

Ligand-induced GHSR1a internalization. (a) The graph shows the ghrelin-mediated dose responses for the different receptor variants in a Ca2+ mobilization assay. The results are normalized to an ECmax of WT GHSR1a set at 100%. (b) Membrane expression of GHSR1a variants per cDNA plasmid transfection amount (per 48 well plates) used for panels c–e. (c) The curves depict 100 nM ghrelin-induced internalization of N-terminally tagged FAP-GHSR1a receptor variants standardized by a noninternalizing CD80 control. In panels d and e, results are presented as fold change following 60 min of 100 nM L585 treatment relative to membrane receptor expression at time 0. Vehicle treatment is set at 1. (#) indicates a significant difference between a receptor’s surface expression with L585 treatment versus vehicle treatment. (d) The bar graph shows the effects of Gq-protein and β-arrestin protein KO on GHSR1a WT, P148A, and L149G endocytosis. (*) indicates a significant difference in receptor surface expression with L585 treatment relative to results in WT control HEK293 cells. (e) The bar graph shows the effects of pcDNA empty vector control, dominant negative mutant K44A, or clathrin inhibitor protein AP180 transfection on GHSR1a WT, P148A, and L149G endocytosis. (*) indicates a significant difference in receptor surface expression with L585 treatment relative to pcDNA control transfection. (* or #, p < 0.05; ** or ##, p < 0.01; *** or ###, p < 0.001).
Regulation of GHSR1a Endocytosis
Since fluorogen quantum efficiency increases 4 orders of magnitude upon binding a cognate FAP, fluorogen fluorescence provides an excellent signal-to-noise ratio for observing FAP-tagged receptor distributions.32 To provide a time-dependent measure of surface receptor loss, plasma-membrane FAP-GHSR1a variants (Figure 2b) were exposed to a saturating concentration of ghrelin peptide followed by labeling with the cell-impermeable fluorogen (skc728).16,30 A single concentration of ghrelin (100 nM) was chosen for these receptor studies to address equi-effectiveness based upon efficacy determinations of β-arrestin-2 recruitment and calcium signaling to WT GHSR1a and L149G and P148A mutant GHSR1a.16Figure 2c shows the response to ghrelin and that GHSR1a P148A and L149G ICL2 mutants internalize to a similar degree over 2 h, more so than does the WT receptor. They also reach a steady state faster than does the WT receptor (WT = 31.8 ± 5.8 min; P148A = 18.3 ± 1.3 min; L149G = 14.4 ± 2.7 min). The D353EAA and T360EAA tail mutants internalize equally well and approach a steady state at a rate similar to that of WT receptor (D353EAA = 32.6 ± 11.9 min and T360EAA = 36.3 ± 12.6 min), they but internalize less well than does the WT receptor. Their absolute and relative percent loss of surface expression relative to WT receptor at steady state are as follows: WT (55 ± 4%, 1.0), P148A (67 ± 2%, 1.2 ± 0.06), L149G (58 ± 3%, 1.1 ± 0.07), D353EAA (37 ± 6%, 0.62 ± 0.06), and T360EAA (35 ± 5%, 0.61 ± 0.03).
We had expected and then observed, given the well-known role of β-arrestin binding in clathrin-mediated endocytosis,37 that the two tail mutants would internalize less well than WT receptor. However, given our previous findings that the ICL2 P148A and L149G receptors have opposite β-arrestin-2 recruitment phenotypes,16 their ability to internalize similarly to each other and WT receptor after 2 h was unexpected. We therefore examined endocytic behaviors using GHSR1a agonist L-692,585 (L585) in β-arrestin-1/-2 knockout and Gq-knockout HEK-293 cell lines (Figure 2d) as well as in the presence of known mediators of β-arrestin directed endocytosis, dominant negative dynamin-K44A,38,39 and the clathrin inhibitor C-terminal fragment of clathrin assembly protein 180 (AP180)40 (Figure 2e). The WT GHSR1a and ICL2 variants all internalized well in response to L585 in control cells (Figure 2d, WT: 0.62 ± 0.07, P148A: 0.47 ± 0.02, L149G: 0.55 ± 0.03), but under these conditions, L585-induced receptor internalization was impaired for all the receptors in β-arrestin-1/-2 knockout cells (Figure 2d, WT: 0.85 ± 0.03, P148A: 1.02 ± 0.06, L149G: 0.75 ± 0.06) and exposure to dynamin K44A (Figure 2e, WT: 1.03 ± 0.09, P148A: 0.88 ± 0.01, L149G: 0.83 ± 0.01) and AP180 (Figure 2e, WT: 0.99 ± 0.06, P148A: 0.87 ± 0.03, L149G: 0.88 ± 0.01) but not in Gq-knockout cells (Figure 2d, WT: 0.54 ± 0.03, P148A: . 0.78 ± 0.13, L149G: 0.70 ± 0.02). Thus, like WT GHSR1a, the ICL2 P148A and L149G mutants undergo agonist-mediated β-arrestin-, dynamin-, and clathrin-dependent endocytosis.
Visualizing β-Arrestin Recruitment to GHSR1a
The agonist-induced aggregation of β-arrestin-2–GFP with GPCRs provides a readout of the initiation, stability, and location of β-arrestin/receptor complex formation.29 L585 stimulation of WT GHSR1a produces numerous, long-lasting (before and after 20 min of treatment) β-arrestin-2–GFP aggregates on or near the plasma membrane (Figure 3a), a distribution pattern characteristic of class A GPCRs.19 Formation of β-arrestin-2–GFP aggregates with activation of the ICL2 L149G mutant is slightly delayed, but like WT, aggregates increase over the 20 min treatment (Figure 3b). The ICL2 P148A response is quite different. β-Arrestin-2–GFP aggregates are fewer and transient, with aggregates no longer evident at 20 min (Figure 3c). We observed a qualitative reduction in β-arrestin-2 recruitment for the two C-tail alanine substitution mutants (Figures 3d,e) that was consistent with their endocytic behavior (see Figure 2c). Together, these findings indicate that both the GHSR1a ICL2 and C-tail regulate GHSR1a/β-arrestin-2 recruitment but that the ICL2 P148A expresses a phenotypic defect for maintaining a stable β-arrestin complex. We elected to pursue this stability question using a quantitative BRET approach.
Figure 3.

Visualization of ligand-induced β-arrestin-2 recruitment to GHSR1a receptor mutants. Live human embryonic kidney (HEK-293)T cells stably expressing β-arrestin-2–green fluorescent protein (GFP) and transiently transfected with GHSR1a: (a) wild-type, (b) L149G (LG), (c) P148A (PA), (d) D353EAA (DEAA), and (e) T360EAA (TEAA) variants were treated with 1 μM L585 and imaged by confocal microscopy at the indicated times (columns).
BRET Signal Assessment of GHSR1a/β-Arrestin Isoform Binding
In general, β-arrestin-2 forms more stable complexes with Class A GPCRs in comparison to those of β-arrestin-1.19 The BRET study in HEK-293 cells (Figure 4a) shows that this generalization remains true for 100 nM ghrelin-activated GHSR1a, with the maximum BRET signal of β-arrestin-2 being approximately 2.5 times higher at 5 min. Additionally, the GHSR1a/β-arrestin-2 BRET signal is agonist-dependent (Figure 4b), being blocked by pretreatment with YIL781, a competitive GHSR1a antagonist.16
Figure 4.

BRET measurement of ligand-induced β-arrestin recruitment to WT GHSR1a. The individual signals were normalized to cells treated with vehicle to yield the delta net BRET. (a) Shown are curves for β-arrestin-1 and β-arrestin-2 recruitment to GHSR1a. Sum-of-squares F testing indicated efficacy differences between β-arrestin-1 and β-arrestin-2 (p < 0.0001; ECmax β-arrestin-1 = (12 ± 1) × 10–3, ECmax β-arrestin-2 = (33 ± 1) × 10–3. The EC50 values were similar (p = 0.16, log[EC50] β-arrestin-1 = −9.1 ± 0.2, log[EC50] β-arrestin-2 = −8.8 ± 0.1). (b) β-Arrestin-2 recruitment after 5 min of pretreatment with vehicle or 1 μM YIL781. F test revealed EC50 (p < 0.0001) and efficacy (p = 0.018) differences. (c) β-Arrestin-2 recruitment after 30 min of preincubation with kinase inhibitors. Compound 101 significantly reduced the maximum response. (d) Reversal of compound 101 inhibition of β-arrestin-2 recruitment by overexpression of GRK2. F test indicated variation in efficacy (p < 0.0001), whereas EC50 values showed no differences (p = 0.91). (e) Table summarizing ECmax and log[EC50] mean ± SEM values, and EC50 mean with 95% confidence intervals for curves in panels c and d. Shaded boxes highlight major treatment effects.
GHSR1a Binding of β-Arrestin Depends upon GRK2
β-Arrestin/receptor binding occurs at serine(S)/threonine(T) C-tail residues.22,23 Using phosphorylation prediction software (NetPhos), we identified putative GHSR1a tail phosphorylation sites for GRK, protein kinase C (PKC) and cyclin-dependent kinase (CDK). With the BRET assay, we evaluated the relative contribution of each of these kinases to ghrelin-induced GHSR1a recruitment of β-arrestin-2 using staurosporine (nonselective), bisindolylmaleimide II (for PKC), roscovitine (for CDK), and compound 101 (for GRK2).41 Only compound 101 reduced the GHSR1a/β-arrestin-2 BRET signal, and the inhibition was reversed by overexpression of GRK2 (Figures 4c,d). These data support GHSR1a binding of β-arrestin-2 being GRK2 phosphorylation-dependent.15,34
BRET Determination of the Time Dependence and Stability of GHSR1a/β-Arrestin Association
We assessed the time dependence of GHSR1a/β-arrestin-2 association in the ICL2 and C-tail mutants with kinetic BRET (Figures 5a–c). BRET signals had similar shapes with rising, peak, and variable-decaying portions over the 30 min measurement and differed in magnitude between receptor variants. We interpreted the curve shapes based upon a standard paradigm of receptor/β-arrestin trafficking, i.e. trafficking begins at the plasma membrane and evolves over time to involve different cell compartments.1,42 We generated time-dependent rate constants m = m(rate) to describe this evolution by computing linear approximations to the slopes of the BRET response curves at short, intermediate, and longer times (Figure 5d,e).
Figure 5.

Characterization of ligand-induced β-arrestin-2 recruitment by kinetic BRET. GHSR1a variants were assessed for β-arrestin-2 activity. (a–c) Data were assessed as described in the “Materials and Methods” section by nonlinear least-squares analysis followed by parametric modeling. (d) Table summarizing means and 95% CI values for panels a–c. (e) Multistage β-arrestin-2 recruitment of panels a–c modeled at different stages by linear approximation to the interaction curve. β-Arrestin-2 recruitment can be described using a three-stage model of association (left panel), steady-state (middle panel), and dissociation (right panel). Association is characterized by a linear slope greater than zero, while the temporary steady-state phase is characterized by a slope of approximately zero. Dissociation is characterized by a slope less than zero. Results are illustrated with lines of best fit (solid lines) and 95% confidence intervals (dotted lines).
The magnitudes of the BRET signals were reduced for the P148A, D353EAA, and T360EAA mutants compared to that of the WT receptor, whereas the L149G mutant and WT receptor had nearly identical BRET profiles (Figure 5a). The initial rate of association of β-arrestin-2 with WT GHSR1a (left panel, association constant m > 0) was marginally reduced in the L149G mutant and was significantly slower in the P148A, D353EAA, and T360EAA (Figure 5d). After approximately 2 min, a relative steady state was temporarily reached, i.e., the number of complexes relatively constant, by the WT, L149G, D353EAA, and T360EAA mutants but not by the P148A variant whose BRET signal is already decaying. At 15 min after ghrelin addition, the BRET signals for the WT and L149G receptors begin to decay, most plausibly reflecting increased separation between the components of the BRET sensors (m < 0). The BRET signals of the D353EAA and T360EAA tail variants remain plateaued for nearly 15 min longer and begin to show small signs of decay at about 30 min.
We also tested double GHSR1a mutants composed of an ICL2 mutation and tail mutation. These double mutants displayed profound deficits in β-arrestin-2 recruitment. Minimal to no appreciable recruitment was observed for mutants P148A/D353EAA and P148A/T360EAA (Figure 5b) and mutant L149G/D353EAA (Figure 5c). The L149G/T360EAA double mutant retained the ability to recruit β-arrestin, and like the T360EAA single mutant, it produced a BRET curve that lacked a dissociation phase (Figure 5d). In light of the finding by Bouzo-Lorenzo et al.10 that T360ESS is the principal ghrelin phosphorylation motif, our data indicate that there is some redundancy in the phosphorylation sites in the tail that are available if T360ESS is unavailable.
Notably, both the native receptor tail and ICL2 were necessary for WT-like GHSR1a/β-arrestin-2 interaction kinetics. The BRET data indicate that the receptor C-tail is the primary regulator of the early GHSR1a/β-arrestin-2 association and that ICL2 is necessary for sustaining the interaction. The lack of complex dissociation detected in the C-tail mutants suggests this region may also play a positive critical role in enabling termination of the receptor/β-arrestin interaction.
GHSR1a Trafficking in Early and Recycling Endosomes
The P148A and L149G receptors internalized similarly in response to ghrelin exposure despite their differences in β-arrestin-2 binding. It was somewhat unexpected that the P148A ghrelin receptor would internalize well because the analogous mutation in ICL2 of the β2AR, as well as that of other Class A receptors, reduces internalization.31 To look for phenotypical trafficking differences, we examined their ghrelin-mediated recompartmentalizing in HEK-293 cells expressing Rab5 and Rab4 vesicle markers (Figure 6).43,44 A 30 min incubation of the HEK cells with 1 μM ghrelin resulted in robust localization in Rab5- and Rab4-labeled vesicles for WT GHSR1a as well as the L149G and P148A mutants. Because of the vesicle overlap, we turned to an alternative model provided by chemokine receptors to identify a less fuzzy phenotypic consequence of β-arrestin affinity change arising in ICL2.
Figure 6.
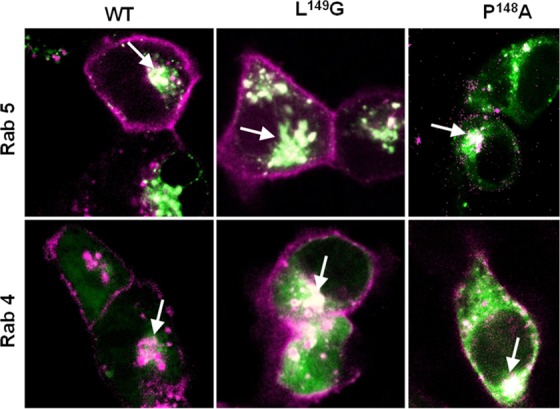
Endosome localization of ICL2 FAP-GHSR1a variants. Merged fluorescence confocal images of agonist-treated WT, P148A, and L149G ghrelin receptors (in purple) expressed in HEK-293 cells containing a GFP–Rab construct (in green). Colocalization (white hue) of receptors with either Rab4 or Rab5 endosomes are indicated by the arrows.
Restoration of Function Trafficking Phenotype in CCR1 Receptors with an ICL2 Proline Substitution
The previous data show that over the first 30 min in response to ghrelin, the second-loop GHSR1a-P148A substitution mutant is lost from the plasma membrane to the same extent as the WT receptor. This result is unexpected for receptors with a loss of β-arrestin affinity that as a consequence should be internalizing less well or recycling from endosomes more rapidly, and it suggests a competing trafficking mechanism may be responsible for maintaining the internalized pool. We investigated this idea in a restoration of function paradigm with CC chemokine receptor-1 (CCR1) and its ICL2 mutant CCR1-A138P, because chemokine receptors as a family express a second-loop alanine at the DRY motif +6 position and CCR1 has been shown to undergo β-arrestin mediated internalization both constitutively and with agonist.45
The CCR1 tail GRK phosphorylation motif, sstspst, with its run of serine/threonines is characteristic of a class B GPCR that exhibits higher agonist-induced affinity for β-arrestin than that of the prototypical class A β2AR. Class B receptors and β-arrestins traffic together in doughnut-shaped endosomes that are easily distinguished with fluorescent β-arrestin sensors.46 We show (Figures 7a–d) that agonist-treated WT and CCR1-A138P have characteristic and similar Class B appearances when imaged for β-arrestin–GFP. In the basal state, where the absence of agonist would be expected to reduce β-arrestin affinity for either receptor, we are now able to uncover differences in their constitutive trafficking behaviors in the context of the findings of Gilliland et al. of constitutive β-arrestin-mediated CCR1 internalization.37 The assay we are using is a gain-of-function determination between the CCR1 WT alanine receptor and what is now the ICL2 proline mutant. The cells presented in Figure 7e–h are imaged for the receptor rather than for β-arrestin and show two striking phenotypic differences. In the absence of ligand, the WT CCR1 decorates the nuclear membrane (Figure 7e, inset and arrow), weakly labels the plasma membrane, and is observed in cytosolic vesicles. Conversely (Figure 7f), CCR1-A138P has pronounced plasma membrane expression that contrasts with its weak to absent cytosolic and perinuclear membrane expression. This loss of perinuclear localization is demonstrated by receptor intensity tracings (yellow) across the cells in Figure 7e,f. In Figure 7e, there are additional peaks (up arrow) representing the location of the nuclear membrane that are absent in the proline mutant image in Figure 7f.
Figure 7.

β-arrestin and CCR1 Distributions in HEK293 Cells. Fluorescence images of β-arrestin-2–GFP in vehicle-treated cells containing (a) wild-type CCR1 or (b) CCR1-A138P. (c, d) Cells treated in parallel to panels a and b with 100 nM CCl14. Boxes provide 2× magnification of areas indicated by arrows demonstrating doughnut-shaped vesicles. Fluorescence images of vehicle-treated (e) HA-tagged wild-type CCR1 or (f) HA-tagged CCR1-A138P. Boxes show enlargements of perinuclear areas identified by corresponding arrows. The amplitude tracings across the images provide relative intensity measurements of the fluorescence from corresponding baseline intersected pixels and can be used to identify edges of plasma membrane or nuclear membrane (right arrow in panel e). (g, h) Fluorescence images of receptors from cells treated in parallel to those in panels e and f with 100 nM CCL14 for 30 min. (i) Western blot of total ubiquinated CCR1 or its variant from cells permanently expressing transfected WT CCR1, transfected CCR1-A138P, and no added receptor (from left to right). (j) Scatter plot of ubiquitinated CCR1 as in panel i normalized by total receptor (WT: 1.86 ± 0.01, A138P: 1.28 ± 0.17), N = 3 independent experiments. Bars represent Mean ± SEM; *, p < 0.05. Sequences of regions in human receptors for (k) ICL2 of the two predominant chemokine families, (l) ICL2 of the two remaining one member chemokine families and the atypical chemokines, and (m) the ICL2 DRY motif and +6 position of the CCR1 and GHSR1a compared to the rhodopsin family consensus motif (upper, yellow and blue highlights),2 and a C-tail alignment from the NPXXY motif for comparison of phosphorylation sites on CCR1 and GHSR1a (lower, in yellow).
Mechanistically, ubiquitination-dependent perinuclear endosome localization of targets is a common regulatory process affecting GPCRs that can provide a simple, rational explanation for the dichotomy in basal CCR1 trafficking observations.47,48 The blot and corresponding graph (Figure 7i,j) show that there is a 33% reduction in total basal ubiquitination of the CCR1A138P mutant receptor relative to its WT counterpart. This suggests that the CC, CXC, and CX3C chemokine receptors, all lacking ICL2+6 proline (Figure 7k–m), may as a group prefer lower β-arrestin affinity basal states and ubiquitin-directed trafficking mechanisms that relatively bias them toward intracellular signaling and degradation pathways.49 Thus, it is reasonable to hypothesize that the chemokine family of receptors does not express the ICL2+6 proline in order to mitigate salvage recycling to the plasma membrane and to facilitate compartmentalization at the nuclear membrane, a fate decision that can be associated with β-arrestin activity but with relatively less β-arrestin affinity. It would be of interest to examine this compartmentalization phenomenon regarding cellular HIV entry mediated by CCR5.50−54
Discussion
Our functional data support a model where an overwhelming majority of β-arrestin affinity for GPCRs can be explained by concurrent regulation from three distinct factors: (1) ligands, (2) GRK-phosphorylated receptor sites in the C-tail/third loop, and (3) putative sensor sites in the receptor core that involve ICL2 in the proximity of the DRY motif. We have specifically demonstrated here by mutagenesis that the GHSR1a C-tail and second-loop-regulated core components bind β-arrestin-2 independently. The tail appears crucial for an initial β-arrestin recruitment, whereas the proximal segment of ICL2 is necessary to maintain a stable, sustained β-arrestin-2 interaction that by analogy to the rhodopsin/arrestin complex would require formation of an ICL2 binding cleft in activated β-arrestin. Moreover, our data in combination with the crystal structure analysis suggest that the ICL2+6 proline actively participates in the binding and stabilization of a receptor arrestin complex through a combination of the following two mechanisms. First, the +6 proline forms an N-terminal α-helix cap for a contiguous short segment of ICL2, and the proline would serve to stabilize the helix versus an alanine-substituted variant, particularly in a membrane environment.55,56 Second, the rhodopsin proline sits within 2.5–3.5 Å from a hydrophobic patch containing arrestin leucine 250, as predicted for the arrestin hydrophobic contribution to receptor binding.6 Thus, a directed interaction via the proline and proximate residues is more than likely, though a theoretical understanding of hydrophobic interactions remains incomplete for protein folding and the use of the term “hydrophobic bond” may therefore be imprecise.57,58 Nevertheless, the above two considerations would lead to a prediction of receptor/arrestin behavior consistent with our observations.
We can use a Boolean network analysis59,60 in place of a series of differential equations to provide an intuitive, visual means to display the receptor/arrestin reaction scheme, its inputs, and outputs and at what mesh or branch points regulators enter or leave the pathway. While this approach is not nearly as powerful to explain behaviors as solving a series of coupled equations, it is both simple and sufficient to enable general predictions about reaction outcomes. Moreover, with this analysis we can also illustrate why the use of a single letter code or determinant for predicting arrestin/receptor behavior may frequently break down due to the contribution of the ICL2 component to the C-tail β-arrestin-dependent trafficking outcomes (Figure 8). We find that the independence of the two nonligand β-arrestin instruction sets with their flexible coding of trafficking instructions are sufficient to support a general set of 6–8 β-arrestin/GPCR cell-trafficking phenotypes. These phenotypic fates can be classified using a parameter Bji (capital Bji with full agonist versus Pji partial agonist versus lower case bji without agonist) that signifies the source and magnitude of the β-arrestin affinity due to the ligand (letter body), available tail (superscript), and ICL2 (subscript) interactions. For example, endocytosis trafficking classifications, B2–31, B11, and B01 respectively correspond to what was formerly defined as Class B, vasopressin-receptor type II (V2R)-like, Class A β2AR-like, and to very limited noninteracting, Class Ø β3AR-like61 receptors. Thus, the fuzzy nature of the β-arrestin–GPCR interaction supports a broad range of ligand-dependent and -independent receptor fates that either leaves receptors at or near the plasma membrane or moves them along to other membrane lipid environments or internal endocytic compartments. Information theory tells us that these finite possible outcomes can be coded by sequences of letters that may be subject in the decoding process to alternative reads,62,63 which like the encoding may be a reflection of some local system bias. Thus, at a minimum, attempts to define the β-arrestin-mediated fates of receptors on the basis of one interaction site alone, such as C-tail GRK phosphorylation motifs, would by analogy be similar to assigning meaning based on only one letter, X, to a three-letter message, WXZ, without considering the impact of the other two letters on the message and message entropy.63 This is especially apparent when each position may have multiple values with unequal probabilities. For example, if W equals B or b, X equals a, e, i, o, or u, and Z equals G or g, then for every value of X there are four possible outcomes, i.e., BiG, Big, biG, or big.
Figure 8.

Boolean network analysis of β-arrestin-receptor interactions for determination of receptor trafficking fate. This type of mesh analysis in which the state of a protein changes according to logical rules and the state of precursors provides a simplification in modeling biological processes in comparison to more demanding mathematically rigorous methods. Mesh nodes represent components participating in the biochemical process and edges connecting nodes the processes. Cartoons of the different receptor complexes corresponding to distinct nodes are shown juxtaposed to corresponding nodes. (a) The leftmost network represents the transition of inactive plasma membrane receptor C1 through multiple stages to C2 that are driven by interactions with ligand, G-protein, β-arrestin, and phosphatase. The upper network between cuts at positions I and II delineate the plasma membrane receptor fates C1 through the transition to GRK-phosphorylated receptor G2 residing in a lipid-raft signaling compartment. The lower half of the cut network represents what happens to the receptor subsequent to interaction with β-arrestin. The fate of receptor C3 is determined by the affinity of receptor B2 for β-arrestin and by the compartment in which β-arrestin dissociation from the receptor occurs. The cube on the right with its multiple faces represents fates where trafficking has occurred to compartments other than the plasma membrane. (b) Boolean logic table describing the relationships occurring at each receptor node. (c) Probability based fate determinations, P(i, j), for trafficking receptors Cij resulting from affinity and dissociation differences of β-arrestin from receptor B2. The table below the cartoon shows six trafficking fates for ligand bound receptors due to the differential binding occurring at ICL2 (0 and 1) or the tail (0, 1, 2–3). Conventional class A and B receptors are now designated in the table by A1 and B1, and the corresponding β-arrestin affinity for the ligand bound receptor due to the ICL2 and the C-tail is indicated by the superscript and subscripts of β-arrestin as Bij. The new class designation Ø corresponds to an absent to very weak ICL2 core interaction permissive for a β-arrestin dissociating receptor remaining at the plasma membrane.
The chemokine receptors represent a large subfamily of exceptions to the rule of an ICL2 proline at DRY+6 for rhodopsin family GPCRs. Studies of the CCR1 indicate that the associated β-arrestin-2 constitutive internalization is critical to chemokine-peptide scavenging, a function that is postulated to maintain a chemokine gradient and receptor responsiveness during inflammation.37 CCR1, unlike GHSR1a, has an alanine residue in place of proline in the ICL2 DRY+6 position. We have observed with GHSR1a that in the absence of a sustained core interaction but with preserved tail binding GHSR1a internalization is surprisingly maintained by an alternative sorting mechanism. Conversely, a strategy for stabilization of the CCR1 β-arrestin-2 interaction for recycling purposes using ICL2 proline substitution leads to better plasma-membrane localization of CCR1 at the expense of its perinuclear expression. While the reduction of CCR1 ubiquitination in the absence of an alteration of a lysine residue is unexpected, a plausible explanation is rendered by prior observations that have shown ubiquitination of GPCRs and β-arrestin to correlate with protein conformation, which can affect accessibility and transfer of ubiquitins by cognate E3 ligases.48 Extrapolation of this observation for CCR1 to the chemokine family as a whole suggests that this may be a common GPCR regulatory phenomenon because ubiquitination marks many GPCRs for lysosomal transport or proteasomal degradation in lieu of plasma-membrane recycling. Thus, while still surprising to observe, a restoration of CCR1-A138P to the plasma membrane by reducing ubiquitination is predictable, and in the context of antagonist-rescue therapy, it may be desirable because it provides an independent downstream mechanism to restore receptor populations at the plasma membrane.64,65
An additional interesting observation arising from the A to P substitution CCR1 data in Figure 7 is that CCl14 agonist produces “β-arrestin doughnut-shaped” vesicles in both the WT A and mutant P+6 receptors. CCR1 has a single C-tail internal serine/threonine cluster whose phosphorylation would be expected to produce this exact type of phenotypical response only if this cluster is well-phosphorylated in both instances (Oakley et al.).46 Thus, this result is consistent with the β2AR P to A substitution data (Marion et al.)2 showing the mutation does not affect initial GRK2/3-mediated receptor phosphorylation and lends support to the notion that at least for some rhodopsin-family GPCRs the proline +6 position in ICl2 can modulate β-arrestin activity independently of the C-tail while remaining phosphorylation-neutral with regard to substitution. While our current data do not rule out exceptions, these observations suggest a general neutrality rule that will require further confirmation with many other receptors and that may by necessity be a logical consequence of β-arrestin-C-tail engagement occurring prior to core engagement.
WT GHSR1a normally expresses constitutive activity and internalization in the absence of agonist.34 Our BRET data show almost zero β-arrestin recruitment to ghrelin receptors with impaired tail phosphorylation combined with impaired ICL2 affinity even with agonist presence. The BRET data also indicate that in the presence of agonist and a GHSR1a tail mutation that reduces the probability of a β-arrestin-C-tail induced interaction a β-arrestin-core interaction may still occur albeit relatively more slowly than if the tail were optimally phosphorylated. Our data suggest that this core interaction is not well supported by alternative tail motifs that participate in β-arrestin-2 coupling other than commonly recognized serine and threonine phosphorylation sites. The observations that β-arrestin-mediated GHSR1a recruitment may occur without significant receptor-tail phosphorylation and that almost no β-arrestin recruitment occurs if ICL2 activity is also correspondingly reduced by point mutation in ICL2 supports three-motif binding model (including the ligand orthosteric site) would be sufficient to accommodate almost all β-arrestin-regulated GPCR behaviors originating at the plasma membrane.
In summary, we provide experimental evidence that GPCRs couple to β-arrestin via both the C-terminal tail and ICL2. Our data demonstrate that a functional consequence of the greater conformational variability supplied by two major β-arrestin regulatory regions versus one is an expanded repertoire of trafficking fates. Thus, because of the presence of this other fundamental β-arrestin regulatory site in ICL2, the data provide an important explanation as to why there will be an inherent difficulty in constructing a primer for decoding receptor trafficking based upon an analysis of C-tail resident β-arrestin trafficking motifs alone.4,66,67 Our findings support designing future GPCR studies directing GPCR/β-arrestin complexes to different signaling compartments by considering the strengths of the ligand-dependent and -independent core and tail interactions to deduce where β-arrestin separation from the receptor occurs. Understanding how GPCRs recruit and maintain interactions with β-arrestins in making fate determinations should facilitate the development of better functionally selective therapeutics and address important issues associated with receptor expression and ligand pharmacology.
Materials and Methods
Plasmids and Constructs
The 3xHA-GHSR1a plasmid and the 3xHA-CCR1 (also referred to as HA-CCR1) plasmid were purchased from the cDNA Resource Center (Bloomsberg, PA) and were used to generate receptor mutants by QuikChange site-directed mutagenesis (Agilent Technologies, Santa Clara, CA). The GHSR1a-Rluc constructs for BRET assays were generated by fusing Renilla luciferase (Rluc) to the C-terminal tails of WT and receptor mutants. β-Arrestin-2-YFP and β-arrestin-2-Venus68 plasmids as well as dynamin-dominant negative (dynamin K44A) plasmids were available in our laboratory and have been described previously.31,69,70 The clathrin inhibitor AP180-C-terminal fragment (SNAP91) was a gift from Dr. Harvey T. McMahon (University of Cambridge, UK). The mitochondrion-targeting apoaequorin expression vector71 was a gift from Dr. Stanley Thayer (University of Minnesota).
Ligands, Inhibitors, and Antibodies
Ghrelin peptide (#1463), L-692,585 (#2261), and YIL781 (#3959) were purchased from Tocris Biosciences (Ellisville, MI). PKC inhibitors bisindolylmaleimide (ab144207) and staurosporine (ab120056) and CDK inhibitor roscovitine (ab144231) were purchased from Abcam (Cambridge, MA). The GRK 2/3 inhibitor Cmpd101 (#2840) was purchased from HelloBio (Princeton, NJ), and the CCR1 agonist CCL 14 (SRP3054) was from Sigma-Aldrich (St. Louis, MO). For detection of ubiquitination, anti-ubiquitin rabbit polyclonal Ab(CST; Cat No. 3933) and the secondary antibody, anti-rabbit IgG (CST 7074), were purchased from Cell Signaling Technology (Danvers, MA).
Molecular Visualization
All protein structure images were produced using coordinates available in NCBI PubMed Structure Summary (Protein Databank ID: 4ZWJ, MMDB ID: 131321). Space-filling images of activated rhodopsin/arrestin complexes were computed and drawn using the computer program PyMOL version 2.3.0 (Schrodinger, LLC, Cambridge, MA 02142). Space-filling images of the GHSR1a/arrestin complexes were computed using the protein mutagenesis function of PyMOL and drawn with the same program. Images of hydrophobic patches expressed in the arrestin/rhodopsin figure were computed and drawn using the Swiss-PDB Viewer 4.10 DeepView (Swiss Institute of Bioinformatics, Lausanne, Switzerland) and the ray trace software POV-Ray 3.7 (http://www.povray.org/). The cartoon image of the GHSR1a/arrestin ICL2 proline capped α-helix complex was drawn using ChimeraX72 (Resource for Biocomputing, Visualization, and Informatics, University of California, San Francisco, CA, grant support from NIH R01-GM129325 and P41-GM103311).
Receptor Internalization
On-cell, FAP-receptor internalization assays were performed in HEK-T cells at 2 × 106 cells/well, β-arrestin-1/-2 KO cells (arrestin KO) at 1 × 106 cells/well, and in Gq KO cells at 1 × 106 cells/well plated in 6-well plates for 24 h prior to Lipofectamine 2000 transfection (Thermo Fischer Scientific). Proximity increase transfections were done in accordance with the manufacturer’s directions as follows. Briefly, in polypropylene tube 1 was added 200 μL of OptiMEM plus a cDNA plasmid for a ghrelin receptor or endocytosis inhibitor (GHSR1a-WT = 500 ng, Pro148 = 2.5 μg, Leu149 = 250 ng; dynamin-K44A = 500 ng, AP180-C-term = 500 ng) and up to 3 μg of empty vector. In a second polypropylene tube (1.7 mL) was added 200 μL pf OptiMEM + 7.5 μL of Lipofectamine 2000. The two tubes were mixed and incubated for 25 min before being added to freshly plated cells. Following 6 h of transfection, fresh media was applied, and cells were grown overnight. The next day, the cells were split to poly-d-lysine-coated 96-well plates (Corning Costar, Corning, NY) at 1 × 105 cells per well in phenol-red-free MEM (cMEM, Gibco, 51200038, Gaithersburg, MD) containing 2% FBS and incubated overnight. On the day of the experiment, growth media was removed, and the cells were treated with either 100 nM ghrelin peptide or 100 nM small-molecule ghrelin-receptor agonist L-692,585 in 100 μL of cMEM or vehicle for different periods of time (5, 10, 15, 20, 30, 40, 60, 90, and 120 min) in a 37 °C CO2 incubator. Each well then received a 50 μL aliquot of ice-cold cMEM containing 1:5000 skc728 fluorogen and was incubated for an additional 5 min in a 4 °C cold room before imaging on a LI-COR Odyssey (Lincoln, NE) at the settings (channel, 700 nm; intensity, 5.5; focal offset, 3.0 mm). Intensity corrections were made by subtracting the signal from nontransfected cells. The expression of membrane-localized CD80 protein was used to normalize data where indicated. The membrane-impermeable FAP dye skc728 was used to selectively assess plasma membrane receptors. L585 induced internalization was normalized to vehicle treated wells at each time point. Each experiment was comprised of three replicates at each time point. A minimum of three independent experiments were performed and analyzed.
β-Arrestin-2 Translocation
HEK cells were transfected with ghrelin receptor variants and GFP-tagged β-arrestin-2 (β-arrestin-2–GFP) and plated on 35 mm glass-bottomed dishes (#P35G-0–10C MatTek Corporation, Ashland, MA)) treated with 75 μg/mL fibronectin for 1 h at room temperature. The plates were incubated overnight at 37 °C in 5% CO2, the media changed to serum- and phenol-red-free MEM, returned to the incubator for 4 h, and then imaged at the indicated times using a Zeiss LSM 510 META (Carl Zeiss Microimaging, Thornwood, NY).
Bioluminescence Resonance Energy Transfer
Assays were conducted using a modified version of a previously described protocol.31 Briefly, HEK-293T cells were transiently transfected 24 h after cell seeding using a calcium phosphate protocol.73 The ability of the β-arrestin-2–Venus construct to support the equal expression of WT GHSR1a and GHSR1a mutants at the membrane as well as the specificity of its interaction with GHSR–Rluc were established. Assays were started by removing phenol-red-free medium from cells and replacing it with PBS. BRET readings were obtained 2 min after manual addition of Rluc substrate, coelenterazine-h (2 μg/mL), using 440–480 and 520–550 nm filters, to respectively monitor Rluc and Venus (stable YFP) emissions. The experiments and reads were performed at 37 °C using a Mithras LB 940 reader (Berthold Technologies, Germany) equipped with a microinjector that allowed automatic delivery of the vehicle or GHSR1a agonist. Ghrelin (100 nM) was injected 5 min after the Rluc substrate (Promega, Madison, WI), and BRET data were obtained every 8 s for 33 min. The BRET signal generated by each sample was determined by calculating the ratio of light emitted by Venus (measured at 530 nm) over the light emitted by Rluc (measured at 485 nm). BRET values were then corrected by subtracting the background signal (detected when the Rluc-tagged construct was expressed without acceptor) from the BRET ratio detected in cells coexpressing both Rluc and Venus (net BRET). Agonist-induced BRET values (delta net BRET) were calculated by subtracting net BRET values of nonstimulated conditions from net BRET values corresponding to the stimulated conditions.74
Ca2+ Mobilization
The Ca2+ responses of the different GHSR1a mutants were compared using HEK-293T cells that permanently expressed mitochondrial apoaequorin.11,71 Stable cells were plated at 1 × 106 per well in a 6-well plate. After 24 h, cells were transiently transfected with 2.5 μg of either the WT or mutant GHSR1a.75 To promote cell recovery, media was changed after 4–6 h of transfection. On the test day, growth media (OptiMEM, Gibco, 51985034) was replaced with cMEM/HEPES/GlutaMAX (Gibco Thermo Fisher) for 2–4 h before adding at 2.5 μL of a 1 μM stock solution of coelenterazine-h (Promega, Madison, WI) per 1 mL of media. Following incubation (1–2h) cells were washed, lifted from the plate, and dispensed (30 μL/well) into ghrelin-containing (at 2× concentration) wells of a 96-well white flat- and clear-bottomed microplate (Corning Costar 3903, Corning NY). Luminescence was recorded using a Mithras LB 940 instrument (Berthold Technologies, Oak Ridge, TN) for 20 s/well immediately afterward. Subsequently, 80 μL of calcium lysis buffer (100 mM CaCl + 0.2% Triton-X) was added to each well, and luminescence was recorded for 5 s (total signal). To control for variations in cell numbers, the agonist-stimulated response (net Aeq) was normalized by dividing the agonist-induced response (L) by the total response value (L, the agonist-stimulated response, plus the response resulting from calcium buffer cell lysis). Each experiment comprised three technical replicates for each time point and construct tested. A minimum of three independent experiments were performed.
Endosomal Trafficking
HEK-293T cells were transiently transfected with FAP-tagged ghrelin receptor variants GFP-tagged Rab4 or Rab5 endosomal marker proteins and plated on fibronectin-coated 35 mm glass-bottomed dishes (MatTek Corporation, Ashland, MA, P35G-0–10C). The plates were incubated overnight at 37 °C in 5% CO2, and on the following day, the media was changed to clear MEM without serum. After 4 h of serum starvation, cells were treated with skc728 (1:5000) and 500 nM ghrelin and were then returned to the incubator for 20 min. Cells were washed 4 times with cold cMEM and fixed with 4% paraformaldehyde. The cells were imaged using a Zeiss LSM-510 META confocal microscope and the images were processed and merged using ImageJ software.
Chemokine Receptor 1 Microscopy
Permanent lines of HEK-293 cells expressing HA-CCR1 receptor or the HA-CCR1-A138P substitution mutant were prepared as below (“Chemokine Receptor-1 Ubiquitination”) and plated into MatTek 35 mm glass-bottomed dishes (Ashland, MA). For assessing β-arrestin activity the receptor-expressing cell lines were transiently transfected with plasmid containing β-arrestin-2–GFP. Prior to imaging, cells were fixed in 5% formaldehyde. Receptors in fixed cells were labeled with a polyclonal rabbit anti-HA antibody, washed, and counterstained with a secondary Alexa 488 goat anti-rabbit antibody. Imaging was performed on a Zeiss LSM 510 META, and intensity analysis was performed using the accompanying Zeiss software.
Chemokine Receptor-1 Ubiquitination
Selection
HEK-293 cells were obtained from the American Type Culture Collection and cultured in minimal essential medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. To select HEK cells for the stable expression of human HA-CCR1-WT or HA-CCR1-A138P mutant receptors, cells at a confluency of 40–50% were transfected with the respective plasmid using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA) as per manufacture’s protocol. Stably expressing cell lines were initially selected by culturing in growth medium supplemented with 1 mg/mL of G418 and later maintained in 400 μg/mL of G418-containing growth media.76
Immunoprecipitation and Immunoblotting
HEK-293 cells stably expressing WT HA-CCR1 or HA-CCR1-A138P mutant receptor were serum-starved for 1 h prior to solubilization in ice-cold lysis buffer containing 50 mM HEPES (pH 7.5), 2 mM EDTA (pH 8.0), 250 mM NaCl, 10% (v/v) glycerol, and 0.5% (v/v) IGEPAL CA-630 (9002–93–1, Sigma-Aldrich, St Louis, MO) supplemented with phosphatase and protease inhibitors (1 mM sodium orthovanadate, 10 mM sodium fluoride, 1 mM phenylmethylsulfonyl fluoride, 5 μg/mL leupeptin, 5 μg/mL aprotinin, 1 μg/mL pepstatin A, and 100 μM benzamidine; all from Sigma-Aldrich) and with 10 mM N-ethylmaleimide (NEM). NEM helps in preserving receptor ubiquitination by inhibiting cellular deubiquitinase activity. Lysates obtained from these cells were centrifuged at 13 000 rpm for 10 min at 4 °C, and the concentration was measured using Bradford reagent. A 800–900 μg sample of solubilized protein was immunoprecipitated using Pierce Anti-HA magnetic beads Cat No. 88837, Thermo Fisher Scientific, Waltham, MA). Immunoprecipitation samples were kept for end–overend rotation at 4 °C for overnight and subsequently washed 3 times with lysis buffer to remove the nonspecific binding, and bound protein was eluted in 2 volumes of sample buffer. The proteins were resolved on a 4–20% gradient gel and then transferred onto nitrocellulose membrane for Western blotting. Blocking and incubation of membrane with an antibody was done in 5% (w/v) dried skim milk powder dissolved in TTBS [(0.2% (v/v) Tween 20, 10 mM Tris-Cl, (pH 8.0), and 150 mM NaCl)], while TTBS used for washing the immunoblot. For chemiluminescence detection of immunoblotted proteins, Super Signal West Pico plus reagent (Pierce Cat No. 1863097) was used. Chemiluminescence signals were observed and acquired with the charge-coupled device camera system (Bio-Rad Chemidoc-XRS, Bio-Rad, Hercules, CA). Ubiquitination and receptor signals were quantified with Image-Lab software (Bio-Rad). For all experiments, ubiquitination levels were normalized to receptor expression.77
Confocal Microscopy
HEK cells stably expressing HA-CCR1-WT or HA-CCR1A138P receptor were seeded on poly d-lysine-coated 20 mm glass-bottomed plates (NEST Scientific, Rahway, NJ, Cat No. 801001). The next day, the cells were starved in serum-free media for 1 h before stimulation. After stimulation, cells were fixed with 5% formaldehyde diluted in Dulbecco’s phosphate-buffered saline (DPBS) containing calcium and magnesium for 20 min. Permeabilization of fixed cells was done for 25 min using 0.1% Triton in phosphate-buffered saline containing 2% bovine serum albumin, incubated with primary antibody for the HA tag (Cell Signaling Technology, Danvers, MA, catalog number 3724S) at 4 °C for overnight, followed by secondary antibody incubation at room temperature for 1–2 h. During secondary antibody incubation, cells were also stained with 0.5 μM 4′,6-diamidino-2-phenylindole (DAPI) for nuclei staining. Antibody and DAPI were diluted in 2% bovine serum albumin diluted in phosphate-buffered saline. Cells were washed after cell fixation, and antibody incubations were carried out in DPBS. Confocal images were captured with a Zeiss LSM-510 META confocal microscope.
Data Analysis
Statistical analyses and data plotting were performed using GraphPad Prism software versions 5 and 6. Data requiring multiple comparisons were analyzed using two-way ANOVA followed by Dunnett’s or Newman–Keuls multiple comparison tests. Data requiring comparison between curves for multiple parameters were assessed in models requiring either parameter sharing or parameter independence, and the most appropriate model was chosen using the Prism sum-of-squares F test to determine the best fit. Values of p < 0.05 were accepted as statistically significant, and data were presented as the mean ± SEM or as the mean with 95% confidence interval (CI). All data are from at least three or more independent experiments.
Acknowledgments
This work was supported by National Institutes on Drug Abuse grants R21-DA35421 (K.T., L.S.B.) and P30 DA029925 (M.G.C., L.S.B.), F32DA043931 (L.M.S.) and by the Mandel Center for Hypertension and Atherosclerosis Research (S.K., S.K.S.).
Glossary
Abbreviations
- AP180
clathrin assembly protein 180
- β-arrestin
beta-arrestin
- β2AR
beta 2 adrenergic receptor
- β3AR
beta 3 adrenergic receptor
- BRET
bioluminescence resonance energy transfer
- CCR1
CC chemokine receptor 1
- CCR5
CC chemokine receptor 5
- CDK
cyclin-dependent kinase
- DPBS
Dulbecco’s phosphate-buffered saline
- FAP
fluorogen-activating protein
- GHSR1a
growth hormone secretagogue receptor 1a
- GFP
green fluorescent protein
- GPCR
G-protein coupled receptor
- GRK2
G-protein-coupled receptor kinase 2
- ICL2
second intracellular loop 2
- HIV
human immunodeficiency virus
- L585
L-692,585
- PKC
protein kinase C
- Rluc
renilla luciferase
- WT
wild type.
Author Present Address
K.T.: Campbell University, Pharmaceutical Sciences, Buies Creek, NC 27506.
Author Present Address
K.N.: College of Medicine, Member of QU Health, Qatar University, P.O. Box 2713, Doha, Qatar.
Author Contributions
# K.T. and K.N. contributed equally to this work. All laboratory work was performed at Duke University. K.T., K.N., L.S., and L.S.B. were responsible for experimental design, data analysis, and composing the manuscript. M.G.C. aided with experimental design. K.T. and K.N. performed BRET studies. K.T., L.R., and C.R. made receptor mutants, and C.R. made BRET constructs. L.R. and K.N. carried out internalization and Ca2+ mobilization assays. S.K. and S.K.S. designed and carried out CCR1 ubiquitination experiments and developed permanent cell lines.
The authors declare no competing financial interest.
This article is made available for a limited time sponsored by ACS under the ACS Free to Read License, which permits copying and redistribution of the article for non-commercial scholarly purposes.
References
- Kang Y.; Zhou X. E.; Gao X.; He Y.; Liu W.; Ishchenko A.; Barty A.; White T. A.; Yefanov O.; Han G. W.; Xu Q.; de Waal P. W.; Ke J.; Tan M. H.; Zhang C.; Moeller A.; West G. M.; Pascal B. D.; Van Eps N.; Caro L. N.; Vishnivetskiy S. A.; Lee R. J.; Suino-Powell K. M.; Gu X.; Pal K.; Ma J.; Zhi X.; Boutet S.; Williams G. J.; Messerschmidt M.; Gati C.; Zatsepin N. A.; Wang D.; James D.; Basu S.; Roy-Chowdhury S.; Conrad C. E.; Coe J.; Liu H.; Lisova S.; Kupitz C.; Grotjohann I.; Fromme R.; Jiang Y.; Tan M.; Yang H.; Li J.; Wang M.; Zheng Z.; Li D.; Howe N.; Zhao Y.; Standfuss J.; Diederichs K.; Dong Y.; Potter C. S.; Carragher B.; Caffrey M.; Jiang H.; Chapman H. N.; Spence J. C.; Fromme P.; Weierstall U.; Ernst O. P.; Katritch V.; Gurevich V. V.; Griffin P. R.; Hubbell W. L.; Stevens R. C.; Cherezov V.; Melcher K.; Xu H. E. (2015) Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523 (7562), 561–7. 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion S.; Oakley R. H.; Kim K. M.; Caron M. G.; Barak L. S. (2006) A beta-arrestin binding determinant common to the second intracellular loops of rhodopsin family G protein-coupled receptors. J. Biol. Chem. 281 (5), 2932–8. 10.1074/jbc.M508074200. [DOI] [PubMed] [Google Scholar]
- Gurevich V. V.; Gurevich E. V. (2004) The molecular acrobatics of arrestin activation. Trends Pharmacol. Sci. 25 (2), 105–11. 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Zhou X. E.; He Y.; de Waal P. W.; Gao X.; Kang Y.; Van Eps N.; Yin Y.; Pal K.; Goswami D.; White T. A.; Barty A.; Latorraca N. R.; Chapman H. N.; Hubbell W. L.; Dror R. O.; Stevens R. C.; Cherezov V.; Gurevich V. V.; Griffin P. R.; Ernst O. P.; Melcher K.; Xu H. E. (2017) Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell 170 (3), 457–469.e13. 10.1016/j.cell.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleicher A.; Kuhn H.; Hofmann K. P. (1989) Kinetics, binding constant, and activation energy of the 48-kDa protein-rhodopsin complex by extra-metarhodopsin II. Biochemistry 28 (4), 1770–5. 10.1021/bi00430a052. [DOI] [PubMed] [Google Scholar]
- Gurevich V. V.; Benovic J. L. (1993) Visual arrestin interaction with rhodopsin. Sequential multisite binding ensures strict selectivity toward light-activated phosphorylated rhodopsin. J. Biol. Chem. 268 (16), 11628–11638. [PubMed] [Google Scholar]
- Shukla A. K.; Westfield G. H.; Xiao K.; Reis R. I.; Huang L. Y.; Tripathi-Shukla P.; Qian J.; Li S.; Blanc A.; Oleskie A. N.; Dosey A. M.; Su M.; Liang C. R.; Gu L. L.; Shan J. M.; Chen X.; Hanna R.; Choi M.; Yao X. J.; Klink B. U.; Kahsai A. W.; Sidhu S. S.; Koide S.; Penczek P. A.; Kossiakoff A. A.; Woods V. L. Jr.; Kobilka B. K.; Skiniotis G.; Lefkowitz R. J. (2014) Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 512 (7513), 218–22. 10.1038/nature13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider E. H.; Schnell D.; Strasser A.; Dove S.; Seifert R. (2010) Impact of the DRY motif and the missing ″ionic lock″ on constitutive activity and G-protein coupling of the human histamine H4 receptor. J. Pharmacol. Exp. Ther. 333 (2), 382–92. 10.1124/jpet.109.163220. [DOI] [PubMed] [Google Scholar]
- Palczewski K.; Kumasaka T.; Hori T.; Behnke C. A.; Motoshima H.; Fox B. A.; Le Trong I.; Teller D. C.; Okada T.; Stenkamp R. E.; Yamamoto M.; Miyano M. (2000) Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289 (5480), 739–45. 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Cahill T. J. III; Thomsen A. R.; Tarrasch J. T.; Plouffe B.; Nguyen A. H.; Yang F.; Huang L. Y.; Kahsai A. W.; Bassoni D. L.; Gavino B. J. (2017) Distinct conformations of GPCR-beta-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc. Natl. Acad. Sci. U. S. A. 114, 2562–2567. 10.1073/pnas.1701529114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labarthe A.; Fiquet O.; Hassouna R.; Zizzari P.; Lanfumey L.; Ramoz N.; Grouselle D.; Epelbaum J.; Tolle V. (2014) Ghrelin-Derived Peptides: A Link between Appetite/Reward, GH Axis, and Psychiatric Disorders?. Front. Endocrinol. 5, 163. 10.3389/fendo.2014.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller T. D.; Nogueiras R.; Andermann M. L.; Andrews Z. B.; Anker S. D.; Argente J.; Batterham R. L.; Benoit S. C.; Bowers C. Y.; Broglio F.; Casanueva F. F.; D’Alessio D.; Depoortere I.; Geliebter A.; Ghigo E.; Cole P. A.; Cowley M.; Cummings D. E.; Dagher A.; Diano S.; Dickson S. L.; Dieguez C.; Granata R.; Grill H. J.; Grove K.; Habegger K. M.; Heppner K.; Heiman M. L.; Holsen L.; Holst B.; Inui A.; Jansson J. O.; Kirchner H.; Korbonits M.; Laferrere B.; LeRoux C. W.; Lopez M.; Morin S.; Nakazato M.; Nass R.; Perez-Tilve D.; Pfluger P. T.; Schwartz T. W.; Seeley R. J.; Sleeman M.; Sun Y.; Sussel L.; Tong J.; Thorner M. O.; van der Lely A. J.; van der Ploeg L. H.; Zigman J. M.; Kojima M.; Kangawa K.; Smith R. G.; Horvath T.; Tschop M. H. (2015) Mol. Metab. 4 (6), 437–60. 10.1016/j.molmet.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittekind D. A.; Kluge M. (2015) Ghrelin in psychiatric disorders - A review. Psychoneuroendocrinology 52, 176–94. 10.1016/j.psyneuen.2014.11.013. [DOI] [PubMed] [Google Scholar]
- Engel J. A.; Nylander I.; Jerlhag E. (2015) A ghrelin receptor (GHS-R1A) antagonist attenuates the rewarding properties of morphine and increases opioid peptide levels in reward areas in mice. Eur. Neuropsychopharmacol. 25 (12), 2364–71. 10.1016/j.euroneuro.2015.10.004. [DOI] [PubMed] [Google Scholar]
- Bouzo-Lorenzo M.; Santo-Zas I.; Lodeiro M.; Nogueiras R.; Casanueva F. F.; Castro M.; Pazos Y.; Tobin A. B.; Butcher A. J.; Camina J. P. (2016) Distinct phosphorylation sites on the ghrelin receptor, GHSR1a, establish a code that determines the functions of ss-arrestins. Sci. Rep. 6, 22495. 10.1038/srep22495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evron T.; Peterson S. M.; Urs N. M.; Bai Y.; Rochelle L. K.; Caron M. G.; Barak L. S. (2014) G Protein and beta-arrestin signaling bias at the ghrelin receptor. J. Biol. Chem. 289 (48), 33442–55. 10.1074/jbc.M114.581397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz D. M.; Sun H.; Lobo M. K.; Cahill M. E.; Chadwick B.; Gao V.; Koo J. W.; Mazei-Robison M. S.; Dias C.; Maze I.; Damez-Werno D.; Dietz K. C.; Scobie K. N.; Ferguson D.; Christoffel D.; Ohnishi Y.; Hodes G. E.; Zheng Y.; Neve R. L.; Hahn K. M.; Russo S. J.; Nestler E. J. (2012) Rac1 is essential in cocaine-induced structural plasticity of nucleus accumbens neurons. Nat. Neurosci. 15 (6), 891–6. 10.1038/nn.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chebani Y.; Marion C.; Zizzari P.; Chettab K.; Pastor M.; Korostelev M.; Geny D.; Epelbaum J.; Tolle V.; Morisset-Lopez S.; Pantel J. (2016) Enhanced responsiveness of Ghsr Q343X rats to ghrelin results in enhanced adiposity without increased appetite. Sci. Signaling 9 (424), ra39. 10.1126/scisignal.aae0374. [DOI] [PubMed] [Google Scholar]
- Oakley R. H.; Laporte S. A.; Holt J. A.; Caron M. G.; Barak L. S. (2000) Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 275 (22), 17201–10. 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- Konig B.; Arendt A.; McDowell J. H.; Kahlert M.; Hargrave P. A.; Hofmann K. P. (1989) Three cytoplasmic loops of rhodopsin interact with transducin. Proc. Natl. Acad. Sci. U. S. A. 86 (18), 6878–82. 10.1073/pnas.86.18.6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov R. R.; Premont R. T.; Bohn L. M.; Lefkowitz R. J.; Caron M. G. (2004) Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci. 27, 107–44. 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Butcher A. J.; Kong K. C.; Prihandoko R.; Tobin A. B. (2012) Physiological role of G-protein coupled receptor phosphorylation. Handb. Exp. Pharmacol. 208, 79–94. 10.1007/978-3-642-23274-9_5. [DOI] [PubMed] [Google Scholar]
- Gurevich E. V.; Tesmer J. J.; Mushegian A.; Gurevich V. V. (2012) G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol. Ther. 133 (1), 40–69. 10.1016/j.pharmthera.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly E. (2006) G-protein-coupled receptor dephosphorylation at the cell surface. Br. J. Pharmacol. 147 (3), 235–6. 10.1038/sj.bjp.0706553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer A.; Reinscheid R. K.; Schulz S. (2017) Emerging Paradigms of G Protein-Coupled Receptor Dephosphorylation. Trends Pharmacol. Sci. 38 (7), 621–636. 10.1016/j.tips.2017.04.002. [DOI] [PubMed] [Google Scholar]
- Murphy J. E.; Roosterman D.; Cottrell G. S.; Padilla B. E.; Feld M.; Brand E.; Cedron W. J.; Bunnett N. W.; Steinhoff M. (2011) Protein phosphatase 2A mediates resensitization of the neurokinin 1 receptor. Am. J. Physiol Cell Physiol 301 (4), C780–91. 10.1152/ajpcell.00096.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poll F.; Doll C.; Schulz S. (2011) Rapid dephosphorylation of G protein-coupled receptors by protein phosphatase 1beta is required for termination of beta-arrestin-dependent signaling. J. Biol. Chem. 286 (38), 32931–6. 10.1074/jbc.M111.224899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson S. S.; Downey W. E. 3rd; Colapietro A. M.; Barak L. S.; Ménard L.; Caron M. G. (1996) Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 271 (5247), 363–366. 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- Barak L. S.; Ferguson S. S.; Zhang J.; Caron M. G. (1997) A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J. Biol. Chem. 272 (44), 27497–500. 10.1074/jbc.272.44.27497. [DOI] [PubMed] [Google Scholar]
- Chakraborty S. K., Zhang M., and Waggoner A. S. (2014) Near infrared fluorogen and fluorescent activating proteins for in vivo imaging and live-cell biosensin. US20140243509A1.
- Masri B.; Salahpour A.; Didriksen M.; Ghisi V.; Beaulieu J. M.; Gainetdinov R. R.; Caron M. G. (2008) Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc. Natl. Acad. Sci. U. S. A. 105 (36), 13656–61. 10.1073/pnas.0803522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szent-Gyorgyi C.; Schmidt B. F.; Creeger Y.; Fisher G. W.; Zakel K. L.; Adler S.; Fitzpatrick J. A.; Woolford C. A.; Yan Q.; Vasilev K. V.; Berget P. B.; Bruchez M. P.; Jarvik J. W.; Waggoner A. (2008) Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat. Biotechnol. 26 (2), 235–40. 10.1038/nbt1368. [DOI] [PubMed] [Google Scholar]
- Bennett K. A.; Langmead C. J.; Wise A.; Milligan G. (2009) Growth hormone secretagogues and growth hormone releasing peptides act as orthosteric super-agonists but not allosteric regulators for activation of the G protein Galpha(o1) by the Ghrelin receptor. Molecular pharmacology 76 (4), 802–11. 10.1124/mol.109.056101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst B.; Holliday N. D.; Bach A.; Elling C. E.; Cox H. M.; Schwartz T. W. (2004) Common structural basis for constitutive activity of the ghrelin receptor family. J. Biol. Chem. 279 (51), 53806–17. 10.1074/jbc.M407676200. [DOI] [PubMed] [Google Scholar]
- Schroder R.; Merten N.; Mathiesen J. M.; Martini L.; Kruljac-Letunic A.; Krop F.; Blaukat A.; Fang Y.; Tran E.; Ulven T.; Drewke C.; Whistler J.; Pardo L.; Gomeza J.; Kostenis E. (2009) The C-terminal tail of CRTH2 is a key molecular determinant that constrains Galphai and downstream signaling cascade activation. J. Biol. Chem. 284 (2), 1324–36. 10.1074/jbc.M806867200. [DOI] [PubMed] [Google Scholar]
- Cronshaw D. G.; Nie Y.; Waite J.; Zou Y. R. (2010) An essential role of the cytoplasmic tail of CXCR4 in G-protein signaling and organogenesis. PLoS One 5 (11), e15397. 10.1371/journal.pone.0015397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X.; Kang D. S.; Benovic J. L. (2014) beta-arrestins and G protein-coupled receptor trafficking. Handb. Exp. Pharmacol. 219, 173–86. 10.1007/978-3-642-41199-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte S. A.; Oakley R. H.; Zhang J.; Holt J. A.; Ferguson S. S.; Caron M. G.; Barak L. S. (1999) The beta2-adrenergic receptor/betaarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc. Natl. Acad. Sci. U. S. A. 96 (7), 3712–7. 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Bliek A. M.; Redelmeier T. E.; Damke H.; Tisdale E. J.; Meyerowitz E. M.; Schmid S. L. (1993) Mutations in human dynamin block an intermediate stage in coated vesicle formation. J. Cell Biol. 122 (3), 553–63. 10.1083/jcb.122.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan L. S.; Moshkanbaryans L.; Xue J.; Graham M. E. (2014) The approximately 16 kDa C-terminal sequence of clathrin assembly protein AP180 is essential for efficient clathrin binding. PLoS One 9 (10), e110557. 10.1371/journal.pone.0110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs N. M.; Gee S. M.; Pack T. F.; McCorvy J. D.; Evron T.; Snyder J. C.; Yang X.; Rodriguiz R. M.; Borrelli E.; Wetsel W. C.; Jin J.; Roth B. L.; O’Donnell P.; Caron M. G. (2016) Distinct cortical and striatal actions of a beta-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc. Natl. Acad. Sci. U. S. A. 113 (50), E8178–E8186. 10.1073/pnas.1614347113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung Y.; Le Gouill C.; Lukashova V.; Kobayashi H.; Hogue M.; Khoury E.; Song M.; Bouvier M.; Laporte S. A. (2016) Monitoring G protein-coupled receptor and beta-arrestin trafficking in live cells using enhanced bystander BRET. Nat. Commun. 7, 12178. 10.1038/ncomms12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorvel J. P.; Chavrier P.; Zerial M.; Gruenberg J. (1991) rab5 controls early endosome fusion in vitro. Cell 64 (5), 915–25. 10.1016/0092-8674(91)90316-Q. [DOI] [PubMed] [Google Scholar]
- van der Sluijs P.; Hull M.; Webster P.; Male P.; Goud B.; Mellman I. (1992) The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell 70 (5), 729–40. 10.1016/0092-8674(92)90307-X. [DOI] [PubMed] [Google Scholar]
- Gilliland C. T.; Salanga C. L.; Kawamura T.; Trejo J.; Handel T. M. (2013) The chemokine receptor CCR1 is constitutively active, which leads to G protein-independent, beta-arrestin-mediated internalization. J. Biol. Chem. 288 (45), 32194–210. 10.1074/jbc.M113.503797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley R. H.; Laporte S. A.; Holt J. A.; Barak L. S.; Caron M. G. (1999) Association of beta-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J. Biol. Chem. 274 (45), 32248–57. 10.1074/jbc.274.45.32248. [DOI] [PubMed] [Google Scholar]
- Berthouze M.; Venkataramanan V.; Li Y.; Shenoy S. K. (2009) The deubiquitinases USP33 and USP20 coordinate beta2 adrenergic receptor recycling and resensitization. EMBO J. 28 (12), 1684–96. 10.1038/emboj.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean-Charles P. Y.; Snyder J. C.; Shenoy S. K. (2016) Chapter One - Ubiquitination and Deubiquitination of G Protein-Coupled Receptors. Prog. Mol. Biol. Transl Sci. 141, 1–55. 10.1016/bs.pmbts.2016.05.001. [DOI] [PubMed] [Google Scholar]
- Borroni E. M.; Mantovani A.; Locati M.; Bonecchi R. (2010) Chemokine receptors intracellular trafficking. Pharmacol. Ther. 127 (1), 1–8. 10.1016/j.pharmthera.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Flanagan C. A. (2014) Receptor conformation and constitutive activity in CCR5 chemokine receptor function and HIV infection. Adv. Pharmacol. 70, 215–63. 10.1016/B978-0-12-417197-8.00008-0. [DOI] [PubMed] [Google Scholar]
- Jin J.; Colin P.; Staropoli I.; Lima-Fernandes E.; Ferret C.; Demir A.; Rogee S.; Hartley O.; Randriamampita C.; Scott M. G.; Marullo S.; Sauvonnet N.; Arenzana-Seisdedos F.; Lagane B.; Brelot A. (2014) Targeting spare CC chemokine receptor 5 (CCR5) as a principle to inhibit HIV-1 entry. J. Biol. Chem. 289 (27), 19042–52. 10.1074/jbc.M114.559831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagane B.; Ballet S.; Planchenault T.; Balabanian K.; Le Poul E.; Blanpain C.; Percherancier Y.; Staropoli I.; Vassart G.; Oppermann M.; Parmentier M.; Bachelerie F. (2005) Mutation of the DRY motif reveals different structural requirements for the CC chemokine receptor 5-mediated signaling and receptor endocytosis. Molecular pharmacology 67 (6), 1966–76. 10.1124/mol.104.009779. [DOI] [PubMed] [Google Scholar]
- Venuti A.; Pastori C.; Pennisi R.; Riva A.; Sciortino M. T.; Lopalco L. (2016) Class B beta-arrestin2-dependent CCR5 signalosome retention with natural antibodies to CCR5. Sci. Rep. 6, 39382. 10.1038/srep39382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramori I.; Ferguson S. S.; Bieniasz P. D.; Zhang J.; Cullen B.; Caron M. G. (1997) Molecular mechanism of desensitization of the chemokine receptor CCR-5: receptor signaling and internalization are dissociable from its role as an HIV-1 co-receptor. EMBO J. 16 (15), 4606–16. 10.1093/emboj/16.15.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurora R.; Rosee G. D. (1998) Helix capping. Protein Sci. 7 (1), 21–38. 10.1002/pro.5560070103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. C.; Goto N. K.; Williams K. A.; Deber C. M. (1996) Alpha-helical, but not beta-sheet, propensity of proline is determined by peptide environment. Proc. Natl. Acad. Sci. U. S. A. 93 (13), 6676–81. 10.1073/pnas.93.13.6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilloni C.; Bonetti D.; Morrone A.; Giri R.; Dobson C. M.; Brunori M.; Gianni S.; Vendruscolo M. (2016) Towards a structural biology of the hydrophobic effect in protein folding. Sci. Rep. 6, 28285. 10.1038/srep28285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer E. E.; Rosenberg K. J.; Israelachvili J. (2006) Recent progress in understanding hydrophobic interactions. Proc. Natl. Acad. Sci. U. S. A. 103 (43), 15739–46. 10.1073/pnas.0606422103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M. K.; Saez-Rodriguez J.; Clarke D. C.; Sorger P. K.; Lauffenburger D. A. (2011) Training signaling pathway maps to biochemical data with constrained fuzzy logic: quantitative analysis of liver cell responses to inflammatory stimuli. PLoS Comput. Biol. 7 (3), e1001099. 10.1371/journal.pcbi.1001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrell J.; Socolar J. E. (2009) Boolean modeling of collective effects in complex networks. Phys. Rev. E Stat Nonlin Soft Matter Phys. 79, 061908. 10.1103/PhysRevE.79.061908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W.; Luttrell L. M.; Medvedev A. V.; Pierce K. L.; Daniel K. W.; Dixon T. M.; Lefkowitz R. J.; Collins S. (2000) Direct binding of activated c-Src to the beta 3-adrenergic receptor is required for MAP kinase activation. J. Biol. Chem. 275 (49), 38131–4. 10.1074/jbc.C000592200. [DOI] [PubMed] [Google Scholar]
- Shannon C. E., and Weaver W. (1949) The Mathematical Theory of Communication; University of Illinois Press: Urbana, IL; p v (i.e., vii). [Google Scholar]
- Shannon C. E. (1948) A Mathematical Theory of Communication. Bell Syst. Tech. J. 27 (3), 379–423. 10.1002/j.1538-7305.1948.tb01338.x. [DOI] [Google Scholar]
- Morello J. P.; Petaja-Repo U. E.; Bichet D. G.; Bouvier M. (2000) Pharmacological chaperones: a new twist on receptor folding. Trends Pharmacol. Sci. 21 (12), 466–9. 10.1016/S0165-6147(00)01575-3. [DOI] [PubMed] [Google Scholar]
- Morello J. P.; Salahpour A.; Laperriere A.; Bernier V.; Arthus M. F.; Lonergan M.; Petaja-Repo U.; Angers S.; Morin D.; Bichet D. G.; Bouvier M. (2000) Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J. Clin. Invest. 105 (7), 887–95. 10.1172/JCI8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobles K. N.; Xiao K.; Ahn S.; Shukla A. K.; Lam C. M.; Rajagopal S.; Strachan R. T.; Huang T. Y.; Bressler E. A.; Hara M. R.; Shenoy S. K.; Gygi S. P.; Lefkowitz R. J. (2011) Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci. Signaling 4 (185), ra51. 10.1126/scisignal.2001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy S. K.; Lefkowitz R. J. (2011) beta-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 32 (9), 521–33. 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T.; Ibata K.; Park E. S.; Kubota M.; Mikoshiba K.; Miyawaki A. (2002) A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 20 (1), 87–90. 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Peterson S. M.; Pack T. F.; Caron M. G. (2015) Receptor, Ligand and Transducer Contributions to Dopamine D2 Receptor Functional Selectivity. PLoS One 10 (10), e0141637. 10.1371/journal.pone.0141637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Ferguson S. S.; Barak L. S.; Menard L.; Caron M. G. (1996) Dynamin and beta-arrestin reveal distinct mechanisms for G protein-coupled receptor internalization. J. Biol. Chem. 271 (31), 18302–5. 10.1074/jbc.271.31.18302. [DOI] [PubMed] [Google Scholar]
- Rizzuto R.; Simpson A. W.; Brini M.; Pozzan T. (1992) Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358 (6384), 325–7. 10.1038/358325a0. [DOI] [PubMed] [Google Scholar]
- Goddard T. D.; Huang C. C.; Meng E. C.; Pettersen E. F.; Couch G. S.; Morris J. H.; Ferrin T. E. (2018) UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27 (1), 14–25. 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagi K.; Shenoy S. K. (2019) Detection of beta-Arrestin-Mediated G Protein-Coupled Receptor Ubiquitination Using BRET. Methods Mol. Biol. 1957, 93–104. 10.1007/978-1-4939-9158-7_6. [DOI] [PubMed] [Google Scholar]
- Nagi K.; Charfi I.; Pineyro G. (2015) Kir3 channels undergo arrestin-dependant internalization following delta opioid receptor activation. Cell. Mol. Life Sci. 72 (18), 3543–57. 10.1007/s00018-015-1899-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston R. E.; Chen C. A.; Rose J. K. (2003) Calcium phosphate transfection. Curr. Protoc. Mol. Biol. 63, 9.1.1. 10.1002/0471142727.mb0901s63. [DOI] [PubMed] [Google Scholar]
- Shenoy S. K.; Drake M. T.; Nelson C. D.; Houtz D. A.; Xiao K.; Madabushi S.; Reiter E.; Premont R. T.; Lichtarge O.; Lefkowitz R. J. (2006) beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J. Biol. Chem. 281 (2), 1261–73. 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- Yu S. M.; Jean-Charles P. Y.; Abraham D. M.; Kaur S.; Gareri C.; Mao L.; Rockman H. A.; Shenoy S. K. (2019) The deubiquitinase ubiquitin-specific protease 20 is a positive modulator of myocardial beta1-adrenergic receptor expression and signaling. J. Biol. Chem. 294 (7), 2500–2518. 10.1074/jbc.RA118.004926. [DOI] [PMC free article] [PubMed] [Google Scholar]