

Abstract
Voltage-gated sodium (NaV) channels play a fundamental role in normal neurological function, especially via the initiation and propagation of action potentials. The NaV1.1 subtype is found in inhibitory interneurons of the brain and it is essential for maintaining a balance between excitation and inhibition in neuronal networks. Heterozygous loss-of-function mutations of SCN1A, the gene encoding NaV1.1, underlie Dravet syndrome (DS), a severe pediatric epilepsy. We recently demonstrated that selective inhibition of NaV1.1 inactivation prevents seizures and premature death in a mouse model of DS. Thus, selective modulators of NaV1.1 might be useful therapeutics for treatment of DS as they target the underlying molecular deficit. Numerous scorpion-venom peptides have been shown to modulate the activity of NaV channels, but little is known about their activity at NaV1.1. Here we report the isolation, sequence, three-dimensional structure, recombinant production, and functional characterization of two peptidic modulators of NaV1.1 from venom of the buthid scorpion Hottentotta jayakari. These peptides, Hj1a and Hj2a, are potent agonists of NaV1.1 (EC50 of 17 and 32 nM, respectively), and they present dual α/β activity by modifying both the activation and inactivation properties of the channel. NMR studies of rHj1a indicate that it adopts a cystine-stabilized αβ fold similar to known scorpion toxins. Although Hj1a and Hj2a have only limited selectivity for NaV1.1, their unusual dual mode of action provides an alternative approach to the development of selective NaV1.1 modulators for the treatment of DS.
Keywords: voltage-gated sodium channel, Dravet syndrome, antiepileptic drug, venom peptide, electrophysiology, gating modifier toxin, protein structure
Introduction
Voltage-gated sodium (NaV) channels are integral membrane proteins that underpin diverse fundamental processes, including electrical signaling in the nervous system, skeletal, and cardiac muscle contraction, and hormone secretion.1 In mammalian cells, NaV channels are composed of an α-subunit with four homologous but nonidentical domains (denoted DI–DIV) and one or two auxiliary β subunits. Each domain contains a voltage-sensing domain (VSD; S1–S4 segments) that functions as the primary sensor of changes in membrane potential, and a pore domain (S5–S6 segments) that mediates the channel’s selective permeability to sodium ions.2 Nine distinct NaV channel subtypes denoted NaV1.1–1.9 have been functionally characterized in humans, each of which displays unique functional profiles and tissue-specific expression patterns. Dysfunction in these proteins has been linked to many pathological conditions, including cardiac arrhythmias, muscular disorders, chronic pain, and epilepsy.1
NaV1.1 is predominantly expressed in inhibitory interneurons of the brain and it plays a major role in regulating brain rhythms and cognitive functions.3−5 Mutations in NaV1.1 have been characterized in patients suffering from various types of inherited epilepsy, most notably Dravet syndrome (DS).6 DS is a rare but severe form of pediatric epilepsy characterized by pharmacoresistant seizures, cognitive impairment, and a high incidence of premature death. NaV1.1 haploinsufficiency is the primary cause of DS,7,8 and this molecular deficit alters brain excitability by impairing action potential generation in inhibitory neurons. Recently, we demonstrated that inhibition of NaV1.1 inactivation using the spider-venom peptide Hm1a abolished seizures and premature mortality in a mouse model of DS,9 thereby providing proof-of-concept that pharmacological treatments that restore residual NaV1.1 function in DS patients will likely be a useful therapeutic strategy.
Venoms are the best natural source of ion channel modulators, and some of these have been developed as human therapeutics10 or are currently in clinical trials.11,12 However, it is currently not known whether venomous animals other than spiders contain compounds that potently target NaV1.1, with either similar or different modes of action to Hm1a, and thus hold potential for the treatment of DS. Scorpions are one of the most ancient arthropod Orders, and their venoms are a well-established source of disulfide-rich peptides that modulate the activity of NaV channels.10,13,14 These peptides adopt three main folds: the cystine-stabilized α-helix–loop–helix (CSαα) fold, cystine-stabilized α-helix/β-sheet (CSαβ) fold, and inhibitor cystine knot (ICK) motif.15 However, the vast majority of scorpion peptides adopt a CSαβ scaffold, which comprises one or two short α-helices connected to a double or triple-stranded antiparallel β-sheet, with cross-linking by 2–4 disulfide bridges.16 Scorpion toxins that act on NaV channels are conventionally classified into two pharmacological categories depending on their mode of action and binding properties: α-toxins and β-toxins.17 The voltage-sensor trapping mechanism, in which scorpion toxins stabilize particular conformational states of the NaV channel voltage sensors, has been proposed to account for their electrophysiological effects on NaV channels.18 In this model, α-toxins prevent the outward movement of the DIV S4 segment that is critical for inactivation,13 whereas β-toxins lock the DII S4 segment in its outward state to promote channel opening.19 Although this action does not represent classical agonism, we refer below to gating modifiers that increase NaV channel currents, either by promoting activation and/or delaying inactivation, as “agonists” for simplicity.
Here we describe the isolation, sequence determination, recombinant expression, and functional characterization of two novel NaV1.1 agonists, δ-buthitoxin-Hj1a and δ-buthitoxin-Hj2a (henceforth Hj1a and Hj2a, respectively), from venom of a previously unstudied scorpion Hottentotta jayakari. Complete amino acid sequences for each peptide were obtained by combining mass spectrometric sequencing techniques with venom-gland transcriptomic data. The three-dimensional (3D) structure of Hj1a that we elucidated using NMR spectroscopy revealed that it adopts a CSαβ fold. Hj1a and Hj2a are potent agonists of NaV1.1, but they have promiscuous activity across multiple human NaV channel subtypes. However, these peptides are unique among agonists of NaV1.1 in inducing a shift in both the voltage dependence of channel inactivation (α-toxin activity) and activation (β-toxin activity). Thus, this work expands the diversity of known scorpion-venom peptides and contributes to efforts to develop selective NaV1.1 agonists for the treatment of DS epilepsy.
Materials and Methods
Isolation of NaV1.1 Activators from Scorpion Venom
Lyophilized venom from H. jayakari (2 mg) was obtained by electrical stimulation20 and fractionated via reversed-phase high performance liquid chromatography (RP-HPLC) on a Prominence HPLC system (Shimadzu Scientific Instruments, Rydalmere, NSW, Australia). Venom was loaded onto a C18 RP-HPLC column (Phenomenex Jupiter; 250 mm × 4.6 mm, 5 μm), and fractionated using the following gradient: 5% solvent B (0.043% trifluoroacetic acid (TFA) in 90% MeCN) in solvent A (0.05% TFA in H2O) for 5 min, 5–20% solvent B over 5 min, 20–40% solvent B over 40 min, then 40–80% solvent B for 5 min (flow rate 1 mL/min). NaV1.1-agonist fractions selected on the basis of automated patch-clamp recordings (see below) were further purified using a Vision HT HILIC column (Grace; 150 mm × 4.6 mm, 5 μm), using a linear gradient of 95–75% solvent B in solvent A over 20 min (flow rate 1 mL/min), followed by a diphenyl column (Agilent; 150 mm × 3.0 mm, 3 μm) with a linear gradient of 20–40% solvent B in solvent A over 40 min (flow rate 0.6 mL/min), and finally a Promix MP column (SIELC; 250 mm × 4.6 mm, 5 μm) with a linear gradient of 20–25% solvent B in solvent A over 10 min (flow rate 0.75 mL/min). Peptide masses were determined using a 5600 Triple TOF mass spectrometer (AB SCIEX, Framingham, MA, USA). All reported masses are monoisotopic masses, unless otherwise stated.
Venom-Gland Transcriptome
Venom-gland mRNA was isolated from the telsons of two H. jayakari individuals according to a published protocol.21,22 Briefly, total RNA was purified using TRIzol, then mRNA was isolated using a NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs, Ipswich, MA, USA). Immediately following mRNA isolation, cDNA libraries were prepared for each of the two H. jayakari individuals using a NEBNext Ultra RNA Library Prep Kit (New England Biolabs, Ipswich, MA, USA). Following library preparation, the samples were sequenced on an Illumina HiSeq 2500 system (New England Biolabs, Ipswich, MA, USA) at the Translational Science Laboratory, College of Medicine, Florida State University. Adapters and low-quality bases were trimmed from the resulting reads using Trim Galore version 0.4.4.23 Reads were merged using PEAR version 0.9.10.24 Fifteen de novo assemblies were generated. The merged reads were assembled using Extender25 under the default settings with 1000 seeds, default settings with 2000 seeds, and increasing the default read overlap from 120 to 150 nucleotides. Assemblies were performed under default settings with Trinity version 2.4.0,26 BinPacker version 1.0, and SeqMan NGen version 14.0. We performed four assemblies with SOAPdenovo-Trans version 1.0327 using k-mer sizes of 31, 75, 97, and 127 and five assemblies with SPAdes version 3.10.128 using k-mer sizes of 31, 55, 75, 97, and 127. Contigs from all 15 assemblies were combined, and then we extracted and translated all open reading frames (ORFs) at least 90 nucleotides long using the getorf function from EMBOSS version 6.6.0.0.29 Exact duplicates were removed using cd-hit version 4.6.30 The transcriptomic data were not annotated; the data were used solely as a raw database to search for sequence matches to the proteomic data. A decoy database of 230 common mass spectrometry (MS) contaminants was added to the amino acid sequence database before use to prevent mis-assignment of contaminant spectra.
Sequencing of Hj1a and Hj2a
Purified peptides were reduced and alkylated using triethylphosphine and iodoethanol, respectively, as previously described.31,32 The samples were then processed by liquid chromatography coupled to tandem MS (LC–MS/MS) on either an AB SCIEX 5600 or 6600 Triple TOF mass spectrometer. Native and reduced/alkylated but untrypsinized samples were also prepared by omitting the digestion step and resuspending directly in 30 μL of 1% formic acid (FA) after reduction and alkylation. For the AB SCIEX 5600 mass spectrometer, samples were eluted using a Zorbax 300SB-C18 column (Agilent; 100 mm × 2.1 mm, 1.8 μm) with a linear gradient of 1–40% mobile phase B (0.1% FA in 90% MeCN) in mobile phase A (0.1% FA) over 25 min (flow rate 0.2 mL/min). MS1 scans were collected between 300 and 1800 m/z, and MS2 ions in the range m/z 80–1600. Precursor peptides with charge states between +2 and +5, and >100 counts/s, were selected for fragmentation using the dynamic “rolling collision energy” in SCIEX software.33 For the AB SCIEX 6600 mass spectrometer, samples were eluted using a ChromXP-C18–CL column (Eksigent; 150 mm × 0.3 mm, 3 μm) with a linear gradient of 1–30% mobile phase B in mobile phase A over 35 min (flow rate 5 μL/min). MS1 scans were collected between 350 and 1800 m/z, and precursor ions in the range m/z 80–1400. Mass spectra in WIFF format were compared to two libraries of translated venom gland cDNA (corresponding to two individual scorpions) using the Paragon and Protgroup algorithms implemented in ProteinPilot software (AB SCIEX, Framingham, MA, USA). The peptide sequences identified using the ProteinPilot search were compared manually to mass spectra using Analyst and Peakview software (AB SCIEX) and ProteinProspector Tools (http://prospector.ucsf.edu/prospector/mshome.htm).
Production of Recombinant Hj1a and Hj2a
Recombinant Hj1a and Hj2a (henceforth rHj1a and rHj2a, respectively) were produced using a previously described Escherichia coli periplasmic expression system.34,35 Synthetic genes encoding Hj1a and Hj2a, with codons optimized for high-level expression in E. coli, were produced and subcloned into a pLIC-MBP (maltose binding protein) expression vector by GeneArt (Invitrogen, Regensburg, Germany). Cultures were grown in Terrific Broth medium at 37 °C with shaking. Toxin gene expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at an OD600 of 1.2, and then cells were grown at 24 °C overnight before harvesting by centrifugation for 15 min at 6000 rpm. For production of uniformly 13C/15N-labeled rHj1a, cultures were grown in modified M9 minimal medium36 supplemented with 13C-glucose (4 g/L) and 15NH4Cl (1 g/L) as the sole carbon and nitrogen sources, respectively. Protein expression was induced with 1 mM IPTG, and then the culture was grown overnight at 16 °C with shaking. The His6-MBP-toxin fusion protein was extracted from the bacterial periplasm by cell disruption at 28 kpsi, followed by a second round at 32 kpsi (TS Series Cell Disrupter, Constant Systems Ltd., Northants, UK), then captured by passing the cell lysate (buffered in TN buffer: 20 mM Tris-HCl, 200 mM NaCl, pH 8.0) over Ni-NTA Superflow resin (Qiagen, Chadstone, Australia). Weakly bound proteins were removed by washing the column with TN buffer containing 15 mM imidazole (pH 8.0), and then the fusion protein was eluted with TN buffer containing 400 mM imidazole (pH 8.0).
After the fusion protein was concentrated, it was incubated at room temperature for 16 h on a shaker with 250 μL of His6-tagged tobacco etch virus (TEV) protease (1 mg/mL) in the presence of 5 mL of TN buffer (pH 8.0) containing reduced and oxidized glutathione (0.6 mM and 0.4 mM, respectively; Sigma-Aldrich). The redox potential of this buffer (−215 mV) is sufficient to maintain the active-site thiol of TEV protease in a reduced state without promoting reduction and shuffling of the preformed disulfide bonds in the venom peptides.34 The cleavage reaction was allowed to proceed at room temperature for 14 h. The cleaved His6-MBP and His6-TEV were precipitated by the addition of 1% TFA, and then the sample was centrifuged at 17000g. The supernatant was filtered through a 0.22 μm syringe-driven polyvinylidene fluoride (PVDF) membrane before purification of the liberated recombinant peptide using RP-HPLC. Peptide purification was performed using a semipreparative C4 column (Phenomenex Jupiter; 250 mm × 10 mm, 10 μm) with a linear gradient of 25–50% solvent B in solvent A over 30 min (flow rate 3 mL/min), followed by a Promix MP column (for rHj1a; SIELC; 250 mm × 4.6 mm, 5 μm) with a linear gradient of 25–40% solvent B in solvent A over 20 min (flow rate 0.8 mL/min) or a diphenyl column (for rHj2a; Agilent; 150 mm × 3.0 mm, 3 μm) with a linear gradient of 25–40% solvent B in solvent A over 20 min (flow rate 0.5 mL/min). Peptide masses were determined using a 5600 Triple TOF mass spectrometer (AB SCIEX, Framingham, MA, USA) using the protocol described above.
Mammalian Cell Culture
Cell culture reagents were from Gibco (Life Technologies Corporation, Carlsbad, CA, USA) unless stated otherwise. HEK293 cells expressing human NaV1.1–1.7 along with the β1 auxiliary subunit (SB Drug Discovery, Glasgow, UK) were maintained in T75 flasks at 37 °C in a humidified 5% CO2 incubator in Dulbecco’s modified Eagle’s medium supplemented with 10% v/v heat inactivated fetal bovine serum (FBS; Assay Matrix, Melbourne, VIC, Australia), 1× GlutaMAX, 1× MEM Nonessential Amino Acid Solution, and concentrations of blasticidin, geneticin, and zeocin specified for each cell line in the supplier’s protocols. Cells were passaged when they reached 60–70% confluence. Cell culture processes were performed exclusively under sterile conditions. For passaging, the culture medium was discarded and cells were washed once with 1× Dulbecco’s phosphate buffered saline in order to remove residues of the old medium. For resuspension of cells, 1 mL of 0.05% w/v trypsin-EDTA was applied and the cells were incubated at 37 °C in 5% CO2 for 3 min to disrupt interactions between adherent cells and the surface of cell culture dishes, allowing the gentle removal of adherent cells. Subsequently, the cells were resuspended in fresh culture medium and transferred to a T75 flask (for maintenance) or T175 flask (for assays) in a ratio of 1:5 and 1:3, respectively. Cells with less than 25 passages were used for experiments.
Electrophysiological Characterization of rHj1a and rHj2a
The potency and selectivity of rHj1a and rHj2a on human NaV1.1–1.7 were determined using an automated QPatch-16X electrophysiology platform (Sophion Bioscience, Ballerup, Denmark) at room temperature (∼24 °C). All peptide effects were compared to baseline values obtained in vehicle (physiological buffer) in the same cell. The extracellular solution comprised (in mM): 2 CaCl2, 1 MgCl2, 10 HEPES, 4 KCl, 145 NaCl, and 10 sucrose, pH 7.4 with NaOH, and 305 mOsm. The intracellular solution comprised (in mM): 140 CsF, 1/5 EGTA/CsOH, 10 HEPES, 10 NaCl, pH 7.4 with CsOH, and 320 mOsm. Solutions were filtered using a 0.22 μm syringe-driven PVDF membrane filter. Before the recordings began, cells were detached from a T175 flask with Detachin (Genlantis, San Diego, CA, USA) when they reached 60–70% confluence and resuspended in culture medium. The cell suspension was centrifuged at 800 rpm for 5 min. The supernatant was discarded and cell suspension was formed with 4–6 million cells/mL of CD293 medium supplemented with 1× GlutaMAX and 25 mM HEPES.
For patch-clamp analysis, cells were maintained at a holding potential of −80 mV, followed by a prepulse of −120 mV for 200 ms and then a 50 ms step depolarization to 0 mV. The cell positioning and sealing parameters were as follows: positioning pressure, −60 mbar; minimum seal resistance, 0.1 GΩ; holding potential, −90 mV; and holding pressure, −20 mbar. Series resistance did not exceed 5 MΩ and was at least 80% compensated. Whole-cell currents were filtered at 4 kHz and digitized at 25 kHz. Leak-subtraction protocol was used with nonleak-subtracted currents acquired in parallel. Cells with peak amplitude less than 1 nA or greater than 5 nA were excluded. For concentration–response experiments, cells were incubated for at least 3 min with increasing concentrations of peptide, and the voltage protocol was routinely applied. Peptide stock solutions were made up to 10 μM and serial dilutions were prepared in extracellular solution containing 0.1% w/v bovine serum albumin. To determine EC50 values, channels were held at −80 mV in the presence of varying concentrations of peptide, then stepped to −120 mV for 200 ms, followed by 50 ms test depolarization to 0 mV. To quantify the magnitude of the effect, data points from each cell were fitted with the following Hill equation:
where Isustained denotes the sustained current at the end of a 20 ms depolarizing pulse, [P] denotes the peptide concentration, Ipeak denotes the peak current, Imax is the current in the presence of peptide at 1 μM, EC50 is the concentration of agonist that induces 50% of the maximal current, and nH is the slope (Hill) factor. All reported values are given as mean ± s.e.m. for the number of independent measurements indicated (n).
Mode of Action of rHj1a and rHj2a
The current–voltage (I–V) relationship for NaV.1.1 in the absence and presence of 1 μM rHj1a or rHj2a was obtained using 5-mV incremental depolarizing steps from a holding potential of −120 mV, up to +30 mV for 100 ms. The conductance–voltage (G–V) relationship was determined by measuring the peak sodium conductance, which was determined using
where Ipeak denotes the peak current during the step voltage and Vrev denotes the calculated reversal potential for sodium ions, which is 67.55 mV. The calculated conductance value at each voltage step was normalized to maximum peak conductance (Gmax) and fitted with the Boltzmann equation:
where Vhalf, act denotes the half-maximal activation potential and kact denotes the slope factor of activation describing voltage sensitivity of the channel.
To examine the effect of rHj1a and rHj2a (1 μM) on steady-state fast inactivation, cells were held at a prepulse potential ranging from −120 mV to +30 mV in 5-mV increments for 500 ms and then subjected to a 0 mV test pulse for 20 ms. The peak current amplitude during the test depolarization was normalized to the maximum current amplitude (Imax) and plotted as a function of the prepulse potential. Data were fitted with the Boltzmann function:
where Vhalf, inact denotes the half-maximal inactivation potential and kinact denotes the slope factor of inactivation.
Recovery from fast inactivation was studied using a series of paired pulses in the absence and presence of 1 μM rHj1a or rHj2a. For each paired pulse, NaV1.1 channels were inactivated from a holding potential of −120 mV to 0 mV for 30 ms (conditioning pulse) and then repolarized to −120 mV for an interval of variable durations (Δt) followed by a 30 ms test pulse to 0 mV to assay the fraction of current recovered. The ratio of the peak amplitude evoked by the test pulse to that evoked by the conditioning pulse (Itest/Iconditioning) was normalized to the maximum amplitude (Imax) and plotted as a function of duration time between the two pulses. Data were fitted with the double-exponential function:
where t denotes time, I0 denotes the noninactivating component, and Afast and Aslow denote the components for the fast and slow recovery time constants (τfast and τslow, respectively).
NMR Structure Determination of Hj1a
NMR spectra were acquired at 25 °C using a 900 MHz NMR spectrometer (Avance III, BrukerBioSpin, Germany) equipped with a cryogenically cooled triple resonance probe. Sequence specific resonance assignments were obtained using a combination of 2D 1H–15N HSQC, 2D 1H–13C HSQC, 3D HNCACB, 3D CBCA(CO)NH, 3D HNCO, and 3D HBHA(CO)NH spectra. The 3D spectra used for resonance assignments were acquired using nonuniform sampling and transformed using maximum entropy reconstruction with the Rowland NMR Toolkit.37 Side chain assignments and interproton restraints were obtained from 3D 13C-aliphatic NOESY, 3D 13C-aromatic NOESY and 15N-edited HSQC-NOESY spectra. The NOESY spectra were acquired using uniform sampling with a mixing time of 200 ms. Spectra were analyzed using CcpNmr v2.4.1.38 Dihedral-angle restraints were derived from chemical shift data using TALOS-N,39 and the restraint range used in structure calculations was set to twice the estimated standard deviation. The NOESY spectra were manually peak-picked and integrated, then CYANA v3.98.540 was used to automatically assign the peak lists, extract distance restraints, and calculate an ensemble of structures. A total of 200 structures were calculated from random starting conformations based on 1189 interproton distance restraints, 12 disulfide-bond distance restraints, and 94 dihedral angle restraints. The 30 structures with the lowest penalty function values as judged by CYANA (target function) were analyzed using MolProbity.41 The 20 conformers with best stereochemical quality as judged by MolProbity were selected to represent the solution structure of rHj1a. Atomic coordinates are available from the Protein Data Bank (accession code 6OHX).
Determination of Disulfide-Bond Connectivity of rHj1a
Approximately 50 μg of rHj1a was digested using a 1:12.5 w/w ratio of proteomics-grade trypsin (Sigma-Aldrich) to peptide in 10% v/v MeCN, 50 mM NH4HCO3, pH 8.0. Samples were incubated at 37 °C for 18 h. The digestion was quenched by adding 50 μL of extraction reagent (5% v/v FA, 50% v/v MeCN), and then the sample was vacuum-dried. The samples were then resuspended in 25 μL of 1% FA and centrifuged at 25000g for 10 min at 4 °C. Twenty μL of supernatant was transferred into a low-volume insert in a LC–MS vial and 10 μL of sample was processed by LC–MS/MS on an AB SCIEX 5600 Triple TOF mass spectrometer. The sample was eluted using a Zorbax 300SB-C18 column (Agilent; 100 mm × 2.1 mm, 1.8 μm) with a linear gradient of 2–50% mobile phase B in mobile phase A over 6.5 min (flow rate 0.18 mL/min). MS1 scans were collected between 300 and 1800 m/z. Precursor peptides with charge states between +2 and +5, and >100 counts/s, were selected for fragmentation using the dynamic “rolling collision energy” in SCIEX software.33 Mass spectra in WIFF format were analyzed using Peakview software (AB SCIEX).
Results
Discovery of Hj1a and Hj2a
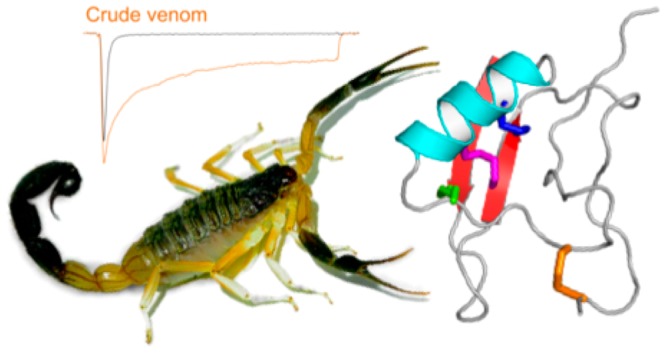
In a search for venom components that might potentiate the activity of the antiepileptic drug target NaV1.1, we noted that crude venom from the scorpion H. jayakari (Figure 1a) strongly delayed inactivation of human NaV1.1 expressed in HEK293 cells (Figure 1b). Fractionation of the venom using RP-HPLC revealed numerous components eluting at 20–40% solvent B, consistent with an abundance of peptide toxins (Figure 1c). Forty fractions corresponding to individual peaks were manually collected, then these were pooled into four groups of 10 fractions that were then diluted 100-fold for electrophysiological screening. Only pooled fraction II (shaded orange in Figure 1c) inhibited NaV1.1 inactivation. Following three subsequent purification steps (Figure 1d), each subfraction was tested against NaV1.1 using the same electrophysiological protocol. This led to isolation of two “hit” peptides that we named Hj1a and Hj2a, with monoisotopic masses of 7476.30 Da and 7112.20 Da, respectively (Figure 1e). The purified peptides had minimal effect on peak current amplitude but they both robustly inhibited fast inactivation of NaV1.1, resulting in sustained currents at the end of the depolarizing pulse (Figure 1f).
Figure 1.

Identification of scorpion-venom peptides that agonize human NaV1.1. (a) Hottentotta jayakari, a buthid scorpion native to Iran, Oman, Saudi Arabia, United Arab Emirates, and Yemen85 (image courtesy of M. Hogan at the Florida State University). (b) Representative current traces for HEK293 cells expressing NaV1.1 in the presence of vehicle (gray) or H. jayakari venom (200 ng/μL; orange). Inward sodium currents were evoked using the voltage protocol shown above the traces. (c) Four groups of pooled fractions were screened against NaV1.1 using automated whole-cell patch clamp electrophysiology, and only group II (shaded orange) contained NaV1.1-active fractions. (d) Chromatograms resulting from subsequent purification of active peptides using a VisionHT HILIC column (left panel), followed by a diphenyl column (middle panel), and a Promix MP column (right panel). Asterisks denote active fractions. Native Hj1a (shaded red) and Hj2a (shaded blue) were purified to homogeneity. The acetonitrile gradient is indicated by a dotted line. (e) MS analysis revealed that these venom peptides have monoisotopic masses of 7476.30 Da (Hj1a, top panel) and 7112.20 Da (Hj2a, bottom panel). (f) Each of the isolated peptides recapitulated the NaV1.1-modulating activity of crude H. jayakari venom.
Peptide Sequence Determination
For primary structure determination, both untrypsinized and trypsinized samples of reduced and alkylated Hj1a and Hj2a were analyzed using LC–MS/MS. The mass of each peptide was found to increase by 360 Da after reduction/alkylation, corresponding to the addition of eight ethanolyl groups (Supporting Information, Figure S1). These data suggest that each peptide contains four disulfide bonds. To obtain the complete amino acid sequences of Hj1a and Hj2a, tandem MS data were compared to an amino acid sequence database containing translated ORFs from the H. jayakari venom-gland transcriptome using the Paragon algorithm in ProteinPilot software.33
For Hj1a, the top sequence match identified in both trypsinized and untrypsinized samples was EEVRDAYIAQPHNCVYHCFRDSYCNDLCIKHGAESGECKWFTSSGNACWCVKLPKSEPIKVPGKCH. The identification was extremely confident, with ProteinPilot scores of 79.2 and 14.5 for digested and undigested samples, respectively (ProteinPilot scores of >1.3 correspond to >95% confidence identification). A total of 296 peptide variants originating from this sequence were detected from the trypsinized sample. The identified sequence contains eight cysteine residues, consistent with the observed mass change upon reduction and alkylation. The y-ion series (Figure 3a) indicates that the C-terminal His residue has a carboxyl terminus, consistent with the lack of an amidation signal in the contig encoding this ORF. The product of this ORF has a predicted monoisotopic mass of 7477.40 Da, which is very close to the measured monoisotopic mass of the peptide purified from venom, 7476.30 Da.
Figure 3.

Overview of LC–MS/MS analysis of Hj1a. (a) Mass spectrum of reduced, alkylated, and undigested Hj1a. The Cys residues were alkylated with iodoethanol (indicated by C*). (b) MS1 spectrum of reduced and alkylated trypsin-digested Hj1a over the m/z range 300–1500. (c) Comparison of observed and theoretical monoisotopic masses for the ions observed, their corresponding residue positions, and fragment sequences. (d) MS2 spectrum of the doubly charged peptide ion at m/z 788.9 supports the presence of the sequence WFTSSGNACWCVK in Hj1a.
For Hj2a, the top sequence match identified in the trypsinized sample (ProteinPilot score 24.5, 18 peptide variants) was GRDAYIADDKNCVYTCAKNSYCNNECTKNGAESGYCQWLGKYGNGCWCKNLPDKVPIRIPGPCRGK. The top hit for the untrypsinized sample (ProteinPilot score 7.6) was for the same sequence with an Asp in position 24 instead of Asn. The m/z values of the y-ion series (Figure 4a) indicate the C-terminal Arg residue is amidated, consistent with the amidation signal (GK) that follows the final Arg residue encoded in the ORF and precedes the stop codon. Again, eight cysteine residues are present and the predicted monoisotopic mass (with C-terminal amide, 7113.22 Da) is close to the measured mass of the purified peptide, 7112.20 Da.
Figure 4.

Overview of LC–MS/MS analysis of Hj2a. (a) Mass spectrum of reduced, alkylated, and undigested Hj2a. The Cys residues were alkylated with iodoethanol (indicated by C*). (b) MS1 spectrum of reduced and alkylated trypsin-digested Hj2a over the m/z range 400–1600. (c) Comparison of observed and theoretical monoisotopic masses for the ions observed, their corresponding residue positions and fragment sequences. (d) MS2 spectrum of doubly charged peptide ion at m/z 632.2 supports the presence of the sequence NSYCNNECTK in Hj2a.
Peptide mass fingerprints of Hj1a and Hj2a (with ethanolylated Cys residues) were manually examined, revealing four and seven major digestion fragments, respectively (Figures 3b, and 4b). All ions observed from the tryptic digestion matched the theoretical digest values from the sequences above (Figures 3c and 4c). MS/MS fragmentation of doubly charged precursor ions at m/z 788.9 and 632.2 confirmed the amino acid sequence WFTSSGNACWCVK at positions 40–52 for Hj1a (Figure 3d), and NSYCNNECTK at positions 19–28 for Hj2a (Figure 4d), respectively. The observed +1.0 Da higher values of predicted compared to measured monoisotopic masses of the undigested peptides could not be explained by amidation or amino acid substitutions present in our sequence database. Comparison of isotopic distributions of Hj2a showed better correspondence to a mass of 7113.20 Da than the previously measured monoisotopic mass of 7112.20 Da, suggesting that the discrepancy in both cases is artifactual. Taken together, the data provide strong support that the sequences presented above and in Figures 3 and 4 correspond the mature forms of Hj1a and Hj2a present in H. jayakari venom.
The sequences of Hj1a and Hj2a are 53% identical (Figure 2). The peptides are homologous (i.e., >60% sequence identity and BLAST e < 10–20) to classical α-toxins (e.g., Lqh4 and Lqq4), insect α-toxins (e.g., Lqq3 and LqhαIT), and α-like toxins (e.g., BmKM1 and Bom4) from buthid scorpions. Hj2a is 89% identical to Lqq4, whereas Hj1a is more divergent from known scorpion venom peptides and shows the most similarity to BmkM1 (67% identity).
Figure 2.

Sequence alignment of Hj1a and Hj2a with closely related α-scorpion toxins from Buthus martensii Karsch (BmKM1 and BmKm2), Buthus occitanus mardochei (Bom4), Leiurus quinquestriatus hebraeus (Lqh4 and LqhαIT), Leiurus quinquestriatus quinquestriatus (Lqq3 and Lqq4), Odonthobuthus doriae (OD1), and Mesobuthus eupeus (MeuNaTx-4 and MeuNaTx-5). Cysteine residues are shadowed in yellow and asterisks denote C-terminal amidation.
NaV Channel Selectivity of rHj1a and rHj2a
We used an E. coli periplasmic expression system to produce recombinant Hj1a and Hj2a (rHj1a and rHj2a, respectively) for structure–function characterization. rHj1a contains a non-native N-terminal Gly residue to facilitate TEV protease cleavage of rHj1a from the His6-MBP fusion tag, whereas rHj2a contains a native N-terminal Gly residue but it lacks the C-terminal amide group present in native Hj2a (Figure S2). Initial expression trials for both peptides conducted on a small scale indicated a high yield of soluble fusion protein expression at an induction temperature of 24 °C (Figure S2). Large-scale expression of rHj1a and rHj2a using the previously outlined protocol yielded >1 mg of purified peptide per liter of bacterial cell culture (Figure S3).
We evaluated the activity of rHj1a and rHj2a against human NaV1.1–1.7 expressed in HEK293 cells using automated whole-cell patch-clamp electrophysiology. rHj1a (1 μM) slowed fast inactivation for all NaV channel subtypes examined, although the effects on NaV1.2, NaV1.3, and NaV1.7 were modest (Figure 5a,b). In addition to slowing channel inactivation, rHj1a prevented transition to the inactivated state in a subtype-specific manner, leading to sustained currents at the end of the test pulse (Figure 5a). rHj1a potentiated NaV1.1 and induced a sustained current to a lesser extent than for NaV1.4 and NaV1.5 (both p < 0.0005) and NaV1.6 (p < 0.05) (Figure 5c). Detailed concentration–response analysis showed that rHj1a is an equipotent agonist of NaV1.4 and NaV1.5 with EC50 values of 7.5 ± 1.2 nM and 9.2 ± 0.8 nM, respectively (Figure 5d and Table 1), and a slightly weaker agonist of NaV1.1 (EC50 17.0 ± 1.9 nM) and NaV1.6 (EC50 37.3 ± 5.9 nM). EC50 values were not determined for NaV1.2, NaV1.3, and NaV1.7 due to their low sensitivity to the toxin. Overall, rHj1a recapitulates the action of native Hj1a against NaV1.1. In addition, rHj1a was found to be a potent agonist of NaV1.4, NaV1.5, and NaV1.6.
Figure 5.

Effect of rHj1a on human NaV1.1–1.7 expressed in HEK293 cells. (a) Representative currents for NaV1.1–1.7 in the presence of vehicle (gray) or 1 μM rHj1a (red). The shaded orange region indicates a sustained current at the end of a depolarizing pulse. (b) Relative size of NaV1.1–1.7 currents at 5 ms after the peak current following addition of 1 μM rHj1a (n = 5–8). (c) The sustained NaV1.4 and NaV1.5 currents after treatment with 1 μM rHj1a (22.1 ± 2.0% and 27.2 ± 2.5%, respectively) were 2.5-fold higher than NaV1.1 (8.8 ± 1.4%) (P < 0.0005, both). (d) Fitting the Hill equation to concentration–response curves for the sustained currents yielded EC50 values of 17.0 ± 1.9 nM (n = 6) for NaV1.1, 7.5 ± 1.2 nM (n = 8) for NaV1.4, 9.2 ± 0.8 nM (n = 6) for NaV1.5, and 37.3 ± 5.9 nM (n = 5) for NaV1.6. Sustained current (30 ms from peak current) was normalized to peak current to quantify the magnitude of the effect. Data are mean ± s.e.m. *P < 0.05, **P < 0.005, ***P < 0.0005, using one-way ANOVA followed by post hoc analysis using Tukey’s multiple comparisons test.
Table 1. Pharmacological Properties of NaV1.1 Agonists.
| EC50 (nM; mean ± s.e.m.)a | ||||||||
|---|---|---|---|---|---|---|---|---|
| peptide | NaV1.1 | NaV1.2 | NaV1.3 | NaV1.4 | NaV1.5 | NaV1.6 | NaV1.7 | ref |
| rHj1a | 17.0 ± 1.9 (n = 6) | ND | ND | 7.5 ± 1.2 (n = 8) | 9.2 ± 0.8 (n = 6) | 37.3 ± 5.9 (n = 5) | ND | current study |
| rHj1b | 52.8 ± 2.5 (n = 5) | NE | NE | 32.0 ± 7.5 (n = 6) | 116.7 ± 23.5 (n = 8) | 46.3 ± 6.2 (n = 5) | 147.4 ± 20.6 (n = 4) | current study |
| MeuNaTxα-12 | 910b | >10 μMb | ND | >10 μMb | NE | >10 μMc | ND | (50) |
| MeuNaTxα-13 | 2500b | >10 μMb | ND | >10 μMb | NE | >10 μMc | ND | (50) |
ND = not determined; NE, no effect.
Rat NaV channel subtypes.
Mouse NaV subtype expressed in Xenopus oocytes.
rHj2a was found to have a similar selectivity profile to rHj1a (Figure 6). The most striking difference between rHj1a and rHj2a is that the latter has no effect on NaV1.2 or NaV1.3 (Figure 6a and Table 1). At a saturating concentration of 1 μM, rHj2a inhibited fast inactivation of all other NaV channel subtypes examined (Figure 6b). As for rHj1a, rHj2a had a pronounced effect on NaV1.4–1.6, and to a lesser degree on NaV1.1 and NaV1.7, inducing a sustained current at the end of a 50 ms depolarization (Figure 6a). On average, the sustained currents for NaV1.4 and NaV1.6 were 5.5-fold greater than that of NaV1.1 in the presence of 1 μM rHj2a (P < 0.0005; Figure 6c). The sustained NaV1.1 current induced by rHj1a (8.8 ± 1.4% of peak; Figure 5c) was higher than that induced by rHj2a (2.5 ± 0.4% of peak; Figure 6c). As for rHj1a, NaV1.4 is the channel that is most sensitive to the toxin (EC50 32.0 ± 7.5 nM; Figure 6d), followed by NaV1.6 (EC50 46.3 ± 6.2 nM) and NaV1.1 (EC50 52.8 ± 2.5 nM).
Figure 6.

Effect of rHj2a on human NaV1.1–1.7 expressed in HEK293 cells. (a) Representative currents for NaV1.1–1.7 in the presence of vehicle (gray) or 1 μM rHj2a (blue). (b) Relative size of NaV1.1–1.7 currents at 5 ms after the peak current following addition of 1 μM rHj2a (n = 4–8). (c) The sustained NaV1.4 and NaV1.6 currents after treatment with 1 μM rHj1a (14.0 ± 1.9% and 14.0 ± 1.0%, respectively) were 5.5-fold higher than for NaV1.1 (2.5 ± 0.4%) (P < 0.0005, both). (d) Fitting the Hill equation to concentration–response curves for the sustained currents induced by rHj2a yielded EC50 values of 52.8 ± 2.5 nM (n = 5) for NaV1.1, 32.0 ± 7.5 nM (n = 6) for NaV1.4, 116.7 ± 23.5 nM (n = 8) for NaV1.5, 46.3 ± 6.2 nM (n = 5) for NaV1.6, and 147.4 ± 20.6 nM (n = 4) for NaV1.7. Sustained current (30 ms from peak current) was normalized to peak current to quantify the magnitude of the effect. Data are mean ± s.e.m. ***P < 0.0005, using one-way ANOVA followed by post hoc analysis using Tukey’s multiple comparisons test.
Mode of Action of rHj1a and rHj2a
We explored the mechanism of action of rHj1a and rHj2a by investigating their effects on the gating properties of NaV1.1 (Figure 7a,b). One μM rHj1a induced a significant hyperpolarizing shift in the voltage-dependence of channel activation (Vhalf, act vehicle, −26.6 ± 0.6 mV; Vhalf, act rHj1a, −33.2 ± 0.5 mV, p < 0.005, n = 6; paired t-test) as well as a depolarizing shift in the voltage dependence of steady-state inactivation (Vhalf, inact vehicle, −56.2 ± 0.6 mV; Vhalf, inact rHj1a, −50.9 ± 0.4 mV, p < 0.0005, n = 6; paired t-test). Likewise, 1 μM rHj2a shifted the voltage-dependence of channel activation in a hyperpolarizing direction (Vhalf, act vehicle, −22.9 ± 0.6 mV; Vhalf, act rHj1a, −25.4 ± 0.4 mV, p < 0.005, n = 6; paired t-test) and the voltage-dependence of inactivation in a depolarizing direction (Vhalf, inact vehicle, −58.5 ± 0.2 mV; Vhalf, act rHj1a, −55.6 ± 0.3 mV, p < 0.005, n = 6; paired t-test). The opposing shifts that rHj1a and rHj2a cause in the activation and inactivation curves generate a small window current (indicated by orange shading; Figure 7a) within the physiologically relevant membrane potential range. Within this window, NaV1.1 channels will be activated but the membrane potential will not be sufficient to allow complete inactivation. This results in a greater probability that any resting channel may spontaneously open and produce a sustained current in response to depolarization. Neither peptide altered the rate at which channels recovered from fast inactivation (Figure 7d); although there is a trend toward slower recovery in the case of rHj2a, it was not statistically significant.
Figure 7.

Effects of rHj1a and rHj2a on the biophysical properties of human NaV1.1. (a) Voltage dependence of normalized peak conductance (circles) and steady-state inactivation (squares) in the presence of vehicle (gray; n = 6), 1 μM rHj1a (red; n = 6) and 1 μM rHj2a (blue; n = 6). The shaded orange area represents the window current in the presence of peptide. The activation and inactivation protocols are shown in the inset. (b) Both peptides (1 μM) caused a significant hyperpolarizing shift in the voltage dependence of channel activation as well as a depolarizing shift in the voltage dependence of steady-state inactivation. (c) Neither peptide induced a change in the activation and inactivation slope factors. (d) Ratio of peak amplitude evoked by the test pulse to that evoked by the conditioning pulse, versus duration of interpulse interval (n = 5, both). The voltage protocol is shown in the upper panel. Curves represent a double-exponential fit, generating fast and slow recovery time constants (inset). Neither peptide (at 1 μM) induced a statistically significant change in either the fast or slow recovery time constants. Data are mean ± s.e.m. **P < 0.005, ***P < 0.0005; n.s., not significant, paired two-tailed Student’s t-test.
Insect Toxicity
To explore the ecological role of these toxins, we examined whether rHj1a and rHj2a are insecticidal, since insects are the primary prey of most scorpions. Insecticidal activity was assessed by injection of the toxins into two dipteran insects, blowflies (Lucilia cuprina) and fruit flies (Drosophila melanogaster), using previously described methods.20 Only rHj1a displayed insecticidal activity, inducing irreversible paralysis in blowflies at 5 nmol/g (Table S1) and lethal effects in fruit flies (LD50 = 745 ± 108 pmol/g; Figure S5). Native Hj1a showed similar activity to rHj1a (LD50 = 914 ± 138 pmol/g in fruit flies; Figure S5); an unpaired t-test comparison with Welch’s correction revealed no significant difference between the LD50 values of native and recombinant Hj1a (p = 0.3903). Neither native nor recombinant Hj2a induced insecticidal effects in either dipteran species.
The insecticidal potency of Hj1a against fruit flies is only moderate compared to venom peptides that are considered leads for bioinsecticide development, which have significantly lower LD50 values (9–240 pmol/g).20 However, this does not exclude this possibility that Hj1a is more potent against native prey of H. jayakari, as the insecticidal potency of some venom peptides varies dramatically between prey taxa.42
Solution Structure of rHj1a
The 3D structure of rHj1a was determined using heteronuclear NMR spectroscopic analysis of 15N/13C-labeled peptide. Preliminary structure calculations performed without disulfide-bond restraints provided support for a Cys29–Cys51 disulfide bond and eliminated several possible disulfide linkages including Cys15–Cys39, but we were unable to unequivocally assign all disulfide linkages. Thus, tryptic digestion combined LC–MS/MS analysis (without disulfide reduction) was employed to unambiguously determine the disulfide-bond connectivities in rHj1a.
We hypothesized that Hj1a has the same connectivity as related scorpion-venom peptides such as BmKM1 (PDB 1SN1). If true, tryptic digestion of rHj1a would be expected to yield fragments with monoisotopic masses of 3106.268 Da and 2656.095, with each peptide fragment containing two intact disulfide bonds (Figure S4). After trypsin digestion, fragments with monoisotopic masses of 3106.277 and 2656.094 Da were among the most abundant peaks detected, closely matching this prediction. Taken together with the NMR-based assignment of the Cys29–Cys51 disulfide bond and elimination of the Cys15–Cys39 linkage, these data allowed the disulfide-bond connectivity of Hj1a to be unambiguously assigned as Cys15–Cys66, Cys19–Cys39, Cys25–Cys49, and Cys29–Cys51, which corresponds to the disulfide pattern in BmKM1 (Figure 8a). Incorporation of disulfide-bond restraints corresponding to these connectivities in the structure calculations allowed a high-resolution structure of Hj1a to be determined.
Figure 8.

NMR solution structure of rHj1a. (a) Alignment of Hj1a with closely related CSαβ venom peptides from the centipede Scolopendra morsitans (Sm2) and the scorpions L. quinquestriatus hebraeus (agitoxin 2 and LqhαIT), Tityus discrepans (discrepin), and L. quinquestriatus quinquestriatus (Lqq3). Cysteine residues characteristic of CSαβ peptides are shown in bold. The secondary structural elements of rHj1a are shown above the sequence alignment. Disulfide-bond connectivities are colored coded and illustrated below the sequences. (b) Stereoview of the ensemble of 20 rHj1a structures (PDB code 6OHX) overlaid for best fit over the backbone atoms of residues 3–66. The N- and C-termini are labeled. (c) Schematic of the rHj1a structure determined in this study with the corresponding secondary structure topology on the right. The disulfide-bond connectivities are colored according to that in part a. (d) Structural variations of the CSαβ motif. Ribbon representations of centipede toxin Sm2 (PDB 6BL9), two short-chain scorpion toxins agitoxin 2 (PDB 1AGT) and discrepin (PDB 2AXK), and two long-chain scorpion toxins Lqq3 (PDB 1LQQ) and LqhαIT (PDB 1LQH), with the corresponding secondary structure topology and disulfide connectivities shown at right.
rHj1a adopts a Csαβ fold similar to peptides with which it has sequence homology43,44 (Figure 8b–d). The final 20 conformers chosen to represent the solution structure of Hj1a have very high precision, with a backbone RMSD of 0.27 Å over the structurally ordered region of the peptide (residues 3–66) (Table 2). The mean MolProbity score of 1.84 places the ensemble in the 82th percentile (100th being the best) relative to all other structures ranked by MolProbity (Table 2). rHj1a consists of an α-helix (Asp22–His32; highlighted in cyan; Figure 8b,c) and an antiparallel β-sheet comprising Glu35–Lys40 (β1) and Ala48–Val52 (β2) (highlighted in red; Figure 8b,c). A short loop of only two residues (Gly33–Ala34) links the helix to β1 and the two β-strands are connected by a well ordered β-hairpin loop (Trp41–Asn47). The long N-terminal (Gly1–Arg21) and C-terminal (Lys53–His67) sections are devoid of regular secondary structure but are well ordered and connected via the Cys15–Cys66 disulfide bond. The tertiary structure of Hj1a conforms to the canonical Csαβ motif, which is characterized by an α-helix anchored to a double-stranded antiparallel β-sheet via two disulfide bonds (highlighted in red and blue; Figure 8c).45
Table 2. Structural Statistics for the Ensemble of Hj1a Structures (PDB 6OHX)a.
| PDB ID | 6OHX |
| Experimental Restraints | |
| interproton distance restraints | |
| total | 1189 |
| intraresidue (i = j) | 278 |
| sequential (|i - j| = 1) | 344 |
| medium range (1 < |i–j| < 5) | 188 |
| long range (|i–j| ≥ 5) | 379 |
| disulfide bond restraints | 12 |
| dihedral-angle restraints (ϕ, ψ) | |
| total | 94 |
| ϕ dihedral angle restraints | 50 |
| ψ dihedral angle restraints | 44 |
| total number of restraints per residue | 19.3 |
| Violations of Experimental Restraints | 0 |
| RMSD from Mean Coordinate Structure (Å)b | |
| all backbone atoms | 0.34 ± 0.06 |
| all heavy atoms | 0.72 ± 0.07 |
| backbone atoms (residue 3–25, 32–75) | 0.27 ± 0.07 |
| heavy atoms (residue 3–25, 32–75) | 0.62 ± 0.06 |
| Stereochemical Qualityc | |
| residues in most favored Ramachandran region (%) | 88.2 ± 3.4 |
| residues in disallowed Ramachandran regions (%) | 0.0 ± 0.0 |
| unfavorable side chain rotamers (%) | 9.7 ± 2.1 |
| clashscore, all atomsd | 0.0 ± 0.0 |
| overall MolProbity score | 1.84 ± 0.11 |
All statistics are given as mean ± SD.
Mean RMSD calculated over the entire ensemble of 20 structures.
Sterochemical quality according to MolProbity (http://helix.research.duhs.duke.edu).
Clashscore is defined the number of steric overlaps >0.4 Å per 1000 atoms.
Discussion
Hj1a and Hj2a Are Novel CSαβ Scorpion Toxins
In this study, we identified two peptides that have dual agonistic actions on NaV1.1, the ion channel that is deficient in Dravet syndrome epilepsy. To the best of our knowledge, these are the first peptides isolated from venom of the Old World scorpion H. jayakari. Complete primary structures were obtained for both peptides by combining de novo MS sequencing with a venom-gland transcriptome.
Recombinant versions of the peptides recapitulated the effect of the native toxins, despite minor differences compared to their venom-derived counterparts: rHj1a has an additional N-terminal Gly residue, while rHj2a has a C-terminal carboxylate group instead of the native C-terminal amide. C-terminal amidation plays an important role in the biological activity of some scorpion toxins. For instance, the native amidated version of the 66-residue scorpion-venom peptide CssII, isolated from venom of buthid scorpion Centruroides suffusus suffusus, inhibits human NaV1.6 with ∼15-fold higher potency than a recombinant version with a C-terminal carboxylate group.46 It was not possible to pharmacologically characterize the venom-derived peptides in detail due to the small quantities available, but given the similar activity of native and recombinant rHj2a it seems unlikely that amidation has a profound effect on the ability of this peptide to modulate NaV channels.
NMR analysis revealed that rHj1a is a new member of the CSαβ family, which also includes defensins from invertebrates, fungi, and plants.45 As the name suggests, the CSαβ motif consists of two disulfides linking an α-helix and β-sheet. The motif is characterized by a CXXXCXnCXC pattern with Cys1–Cys3 and Cys2–Cys4 connectivity, as exemplified by venom peptide U-SLPTX15-Sm2a (Sm2) isolated from venom of the centipede Scolopendra morsitans (Figure 8a,d).47 The majority of CSαβ scorpion toxins contain three disulfide bonds as exemplified by agitoxin 2 and discrepin from venom of the scorpions L. quinquestriatus hebraeus and Tityus discrepans, respectively.48,49 In these prototypical structures (but not Sm2), one additional disulfide bond connects the N-terminal region of the α-helix to the second β-strand (highlighted in green; Figure 8d), resulting in a Cys1–Cys4, Cys2–Cys5, Cys3–Cys6 connectivity. Comparison of the structure of rHj1a with other CSαβ peptides revealed that it is most similar to Lqq3 and LqhαIT, two long-chain insect α-toxins isolated from the venom of L. quinquestriatus quinquestriatus and L. quinquestriatus hebraeus, respectively.50,51 Hj1a has high sequence identity with Lqq3 and LqhαIT (70% and 67%, respectively), and all three toxins contain a fourth disulfide bridge (highlighted in orange; Figure 8d) linking the N-terminal region (i.e., after the first β-strand) and the C-terminal cap region.
Hj1a and Hj2a Have Dual Modulatory Activity on Nav1.1
To date, very few scorpion toxins have been shown to potentiate NaV1.1. MeuNaTxαs are a large family of 13 α-toxins isolated from the scorpion Mesobuthus eupeus. Recombinant versions of MeuNaTxα-12 and MeuNaTxα-13 were shown to preferentially modulate rat NaV1.1 with EC50 values of 0.9 and 2.5 μM, respectively (Table 1),52 but their potency is orders of magnitude weaker than rHj1a (EC50 17 nM). Moreover, a key finding of this study is that rHj1a and rHj2a are very different to the MeuNaTxα toxins and the previously described spider toxin Hm1a that agonizes NaV1.19 in that they have dual modulatory activity. These toxins not only cause a shift in the voltage-dependence of NaV1.1 inactivation (α-toxin activity) but also induce a hyperpolarizing shift in the voltage-dependence of channel activation (β-toxin activity). Such dual α/β activity has been reported for a few scorpion-venom peptides, such as OD1 from the Iranian scorpion Odontobuthus doriae(53) and Ts2 from the Brazilian scorpion Tityus serrulatus,54 but it has not been described for toxins that modulate the activity of NaV1.1. Our electrophysiological experiments suggest that rHj1a and rHj2a most likely inhibit inactivation of NaV1.1 by interacting with the DIV VSD, as previously demonstrated for Hm1a,9 but the question remains as to whether their β-like effects on channel activation result from binding to other VSDs. Dual sites of interaction have been reported for other NaV channel toxins, such as the spider-venom peptides ProTx-I and ProTx-II which bind to both VSD II and IV of mammalian NaV channels.55,56 In any case, the complex pharmacological effects of these peptides suggest that they are best understood as dual α/β-type NaV channel activators.
Hj1a and Hj2a as Potential Leads for Development of a NaV1.1-Directed Therapeutic for DS
Almost 20% of small-molecule therapeutics target ion channels, making them the third most common human drug target after kinases and G-protein-coupled receptors.57 Venoms are a prolific source of ion channel modulators,10 and disulfide-rich venom peptides are being widely pursued as ion channel drug leads because of their exceptional potency and stability, and better subtype selectivity than small molecules.58−64 While conducting a screen to identify venom peptides that activate pain-sensing neurons, Osteen et al. discovered two disulfide-rich peptides (Hm1a and Hm1b) in venom of the tarantula Heteroscodra maculata that activate NaV1.1 with a high degree of selectivity over other NaV channel subtypes.65 Unlike Hj1a and Hj2a, these toxins are members of the knottin family of peptides with C1–C4, C2–C5, C3–C6 disulfide architecture.66,67 Continuous intracerebroventricular infusion of Hm1a in a mouse model of DS eliminated seizures and premature death, suggesting that pharmacological potentiation of NaV1.1 might be a good strategy for treating DS.9
In contrast with Hm1a and Hm1b, most scorpion-venom peptides that target NaV channels are not useful therapeutic leads due to their lack of subtype selectivity.68 Similarly, despite their high potency toward NaV1.1, rHj1a and rHj2a lack the subtype selectivity required for a NaV1.1-directed antiepileptic drug (AED). However, the differential tissue distribution and function of NaV channel subtypes69 suggest that some off-target effects of Hj1a and Hj2a could be ameliorated by route of administration and dosage. For example, rHj1a and rHj2a have agonistic activity on NaV1.4, which is found in smooth and skeletal muscle, and NaV1.5, which is localized to the heart. These channels would be critical off-targets for a peripherally administered peptide drug, as activation of these channels would enhance contractility of smooth, skeletal, and cardiac muscle, with potentially fatal consequences. However, since these subtypes are restricted to the peripheral nervous system, activity at these channels may not be a serious concern for a CNS-restricted AED.
A more serious off-target subtype is NaV1.2, which is potently agonized by rHj1a, but not rHj2a. NaV1.2 is one of four NaV channel subtypes found in the CNS, along with NaV1.1, NaV1.3, and NaV1.6, and it is predominantly expressed in the axon initial segment of most neocortical pyramidal cells in the cerebral cortex.70 Gain-of-function mutations in the SCN2A gene encoding NaV1.2, which are likely to have similar effects to pharmacological activation, underlie early infantile-onset severe epileptic encephalopathies, whereas loss-of-function mutations lead to autism spectrum disorder and intellectual disability.71 Thus, selectivity for NaV1.1 over NaV1.2 is paramount in the development of NaV1.1-directed AEDs for treatment of DS. Given their contrasting effects on NaV1.2, insights into avoiding off-target effects on NaV1.2 may be achieved by comparative structure–function relationship (SAR) studies of Hj1a and Hj2a. Detailed atomic-level insights are likely to require detailed structural analysis, such as determination of a high-resolution structure of rHj1a or rHj2a in complex with a functional human NaV channel. Such structures are increasingly achievable, as evidenced by recently reported cryoelectron microscopy structures of human NaV1.2,72 NaV1.4,73 and NaV1.7.56 Structures of Hj1a and/or Hj2a in complex with a human NaV channel would provide important insights into the molecular mechanism by which these peptides impede channel activation, and they would facilitate rational design of analogues with enhanced selectivity for NaV1.1.
What Are the Biological Roles of Hj1a and Hj2a?
In contrast with mammals, insects possess only a single NaV channel subtype,74 and consequently this channel is the most common target of arachnid venom toxins75,76 and chemical insecticides.77 We therefore surmised that Hj1a and Hj2a might be used in prey capture. However, surprisingly, Hj1a was found to be only moderately insecticidal, whereas Hj2a was devoid of insecticidal activity (Figure S5 and Table S1). This suggests that theses toxins might have a different ecological role.
Even venoms that evolved primarily for prey capture are also used to deter predators, and the venoms of some animals, such as bees, are used solely for this purpose. Many diverse venoms contain peptide toxins that activate vertebrate sensory neurons in order to elucidate immediate noxious pain as a deterrence mechanism,78,79 including ants,80 centipedes,81 snakes,82 scorpions,83 and spiders.65 Indeed, we recently demonstrated that NaV1.1 is found in peripheral sensory neurons, where it plays a key role in transduction of mechanical pain, and that this channel is targeted by algogenic spider toxins.65 Thus, agonism of NaV1.1 by Hj1a and Hj2a is likely to activate sensory neurons and induce pain in vertebrate predators,65 consistent with the pain reported after human envenomations by H. jayakari.(84) Thus, we speculate that these toxins are used primarily for predator deterrence rather than prey capture. The fact that Hj1a and Hj2a have different sequences and slightly different pharmacology on human NaV channels suggests they might have evolved to defend against different predator species.
Summary
We report the discovery and characterization of two scorpion-venom peptides, Hj1a and Hj2a, which are among the most potent known activators of the antiepileptic drug target NaV1.1. Neither peptide in its present form would be a clinically useful AED due to a lack of subtype selectivity, in contrast to more subtype-selective NaV1.1 activators such as Hm1a. However, Hj1a and Hj2a should be useful pharmacological tools for probing NaV channel function, and their dual mode of action on Nav1.1 suggests an alternative pharmacological route to developing drugs for treatment of DS. Future SAR studies will facilitate development of analogues with greater subtype selectivity for development as therapeutics for NaV1.1-related epilepsies such as DS.
Acknowledgments
We acknowledge funding support from The University of Queensland (International Postgraduate Scholarship to C.Y.C. and Early Career Researcher Grant UQECR1945637 to L.V.B.), the Australian National Health & Medical Research Council (Program Grant APP1072113 and Principal Research Fellowship APP1136889 to G.F.K.), the U.S. National Science Foundation (Grant DEB-1638902 to D.R.R. and Graduate Research Fellowship to M.J.W.), and the Florida State University Council on Research and Creativity. We thank Alexander Ullrich, Kevin Wyss, and Felix Otto for providing H. jayakari scorpions for venom extraction, Dr Sean Millard (School of Biomedical Sciences, The University of Queensland) for fruit flies, and Dr Geoff Brown (Department of Agriculture and Fisheries, Queensland, Australia) for sheep blowflies.
Glossary
Abbreviations
- AED
antiepileptic drug
- CNS
central nervous system
- CSαα
cystine-stabilized α-helix–loop–helix
- CSαβ
cystine-stabilized α-helix/β-sheet
- DS
Dravet syndrome
- FA
formic acid
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- HEK
human embryonic kidney
- ICK
inhibitor cystine knot
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- LC-MS/MS
liquid chromatography coupled tandem mass spectrometry
- MBP
maltose binding protein
- NaV channel
voltage-gated sodium channel
- PVDF
polyvinylidene fluoride
- RP-HPLC
reversed-phase high performance liquid chromatography
- SAR
structure–activity relationship
- TEV
tobacco etch virus
- TFA
trifluoroacetic acid
- VSD
voltage-sensing domain
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.9b00079.
Mass spectra; SDS-PAGE gel analysis; insecticidal toxicity for Hj1a, rHj1a, and rHj2a (PDF)
The authors declare no competing financial interest.
This article is made available for a limited time sponsored by ACS under the ACS Free to Read License, which permits copying and redistribution of the article for non-commercial scholarly purposes.
Supplementary Material
References
- Catterall W. A. (2012) Voltage-gated sodium channels at 60: structure, function and pathophysiology. J. Physiol. 590, 2577–2589. 10.1113/jphysiol.2011.224204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W. A. (2010) Ion channel voltage sensors: structure, function, and pathophysiology. Neuron 67, 915–928. 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein M.; Westenbroek R. E.; Yu F. H.; Jones C. J.; Scheuer T.; Catterall W. A. (2015) Genetic background modulates impaired excitability of inhibitory neurons in a mouse model of Dravet syndrome. Neurobiol. Dis. 73, 106–117. 10.1016/j.nbd.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai C.; Abe Y.; Westenbroek R. E.; Scheuer T.; Catterall W. A. (2014) Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. U. S. A. 111, e3139 10.1073/pnas.1411131111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L.; Mann E. O.; Hang G. B.; Barth A. M.; Cobos I.; Ho K.; Devidze N.; Masliah E.; Kreitzer A. C.; Mody I.; Mucke L.; Palop J. J. (2012) Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721. 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depienne C.; Trouillard O.; Saint-Martin C.; Gourfinkel-An I.; Bouteiller D.; Carpentier W.; Keren B.; Abert B.; Gautier A.; Baulac S.; Arzimanoglou A.; Cazeneuve C.; Nabbout R.; LeGuern E. (2008) Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J. Med. Genet. 46, 183–191. 10.1136/jmg.2008.062323. [DOI] [PubMed] [Google Scholar]
- Ogiwara I.; Miyamoto H.; Morita N.; Atapour N.; Mazaki E.; Inoue I.; Takeuchi T.; Itohara S.; Yanagawa Y.; Obata K.; Furuichi T.; Hensch T.; Yamakawa K. (2007) NaV1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. 27, 5903–5914. 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F. H.; Mantegazza M.; Westenbroek R. E.; Robbins C. A.; Kalume F.; Burton K. A.; Spain W. J.; McKnight G. S.; Scheuer T.; Catterall W. A. (2006) Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149. 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- Richards K. L.; Milligan C. J.; Richardson R. J.; Jancovski N.; Grunnet M.; Jacobson L. H.; Undheim E. A. B.; Mobli M.; Chow C. Y.; Herzig V.; Csoti A.; Panyi G.; Reid C. A.; King G. F.; Petrou S. (2018) Selective NaV1.1 activation rescues Dravet syndrome mice from seizures and premature death. Proc. Natl. Acad. Sci. U. S. A. 115, 8077–8085. 10.1073/pnas.1804764115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King G. F. (2011) Venoms as a platform for human drugs: translating toxins into therapeutics. Expert Opin. Biol. Ther. 11, 1469–1484. 10.1517/14712598.2011.621940. [DOI] [PubMed] [Google Scholar]
- Tarcha E. J.; Olsen C. M.; Probst P.; Peckham D.; Munoz-Elias E. J.; Kruger J. G.; Iadonato S. P. (2017) Safety and pharmacodynamics of dalazatide, a Kv1.3 channel inhibitor, in the treatment of plaque psoriasis: a randomized phase 1b trial. PLoS One 12, e0180762 10.1371/journal.pone.0180762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil C. G.; Walker D. G.; Miller D. M.; Butte P.; Morrison B.; Kittle D. S.; Hansen S. J.; Nufer K. L.; Byrnes-Blake K. A.; Yamada M.; Lin L. L.; Pham K.; Perry J.; Parrish-Novak J.; Ishak L.; Prow T.; Black K.; Mamelak A. N. (2019) Phase 1 safety, pharmacokinetics, and fluorescence imaging study of tozuleristide (BLZ-100) in adults with newly diagnosed or recurrent gliomas. Neurosurgery 85, E641–E649. 10.1093/neuros/nyz125. [DOI] [PubMed] [Google Scholar]
- Bosmans F.; Tytgat J. (2007) Voltage-gated sodium channel modulation by scorpion α-toxins. Toxicon 49, 142–158. 10.1016/j.toxicon.2006.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedraza Escalona M.; Possani L. D. (2013) Scorpion beta-toxins and voltage-gated sodium channels: interactions and effects. Front. Biosci., Landmark Ed. 18, 572–587. 10.2741/4121. [DOI] [PubMed] [Google Scholar]
- Smith J. J.; Hill J. M.; Little M. J.; Nicholson G. M.; King G. F.; Alewood P. F. (2011) Unique scorpion toxin with a putative ancestral fold provides insight into evolution of the inhibitor cystine knot motif. Proc. Natl. Acad. Sci. U. S. A. 108, 10478–10483. 10.1073/pnas.1103501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouhat S.; Jouirou B.; Mosbah A.; De Waard M.; Sabatier J. M. (2004) Diversity of folds in animal toxins acting on ion channels. Biochem. J. 378, 717–726. 10.1042/bj20031860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S.; Gao B.; Tytgat J. (2005) Phylogenetic distribution, functional epitopes and evolution of the CSαβ superfamily. Cell. Mol. Life Sci. 62, 2257–2269. 10.1007/s00018-005-5200-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestele S.; Catterall W. A. (2000) Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 82, 883–892. 10.1016/S0300-9084(00)01174-3. [DOI] [PubMed] [Google Scholar]
- Cestèle S.; Scheuer T.; Mantegazza M.; Rochat H.; Catterall W. A. (2001) Neutralization of gating charges in domain II of the sodium channel α subunit enhances voltage-sensor trapping by a β-scorpion toxin. J. Gen. Physiol. 118, 291–302. 10.1085/jgp.118.3.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S.; Herzig V.; King G. F. (2018) Dipteran toxicity assays for determining the oral insecticidal activity of venoms and toxins. Toxicon 150, 297–303. 10.1016/j.toxicon.2018.06.077. [DOI] [PubMed] [Google Scholar]
- Ward M. J.; Ellsworth S. A.; Rokyta D. R. (2018) Venom-gland transcriptomics and venom proteomics of the Hentz striped scorpion (Centruroides hentzi; Buthidae) reveal high toxin diversity in a harmless member of a lethal family. Toxicon 142, 14–29. 10.1016/j.toxicon.2017.12.042. [DOI] [PubMed] [Google Scholar]
- Ward M. J.; Rokyta D. R. (2018) Venom-gland transcriptomics and venom proteomics of the giant Florida blue centipede, Scolopendra viridis. Toxicon 152, 121–136. 10.1016/j.toxicon.2018.07.030. [DOI] [PubMed] [Google Scholar]
- Krueger F. (2017) Babraham Bioinformatics Trim Galore, version 0.4.4; www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
- Zhang J.; Kobert K.; Flouri T.; Stamatakis A. (2014) PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620. 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokyta D. R.; Lemmon A. R.; Margres M. J.; Aronow K. (2012) The venom-gland transcriptome of the eastern diamondback rattlesnake (Crotalus adamanteus). BMC Genomics 13, 312. 10.1186/1471-2164-13-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr M. G.; Haas B. J.; Yassour M.; Levin J. Z.; Thompson D. A.; Amit I.; Adiconis X.; Fan L.; Raychowdhury R.; Zeng Q.; Chen Z.; Mauceli E.; Hacohen N.; Gnirke A.; Rhind N.; di Palma F.; Birren B. W.; Nusbaum C.; Lindblad-Toh K.; Friedman N.; Regev A. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y.; Wu G.; Tang J.; Luo R.; Patterson J.; Liu S.; Huang W.; He G.; Gu S.; Li S.; Zhou X.; Lam T. W.; Li Y.; Xu X.; Wong G. K.; Wang J. (2014) SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30, 1660–1666. 10.1093/bioinformatics/btu077. [DOI] [PubMed] [Google Scholar]
- Bankevich A.; Nurk S.; Antipov D.; Gurevich A. A.; Dvorkin M.; Kulikov A. S.; Lesin V. M.; Nikolenko S. I.; Pham S.; Prjibelski A. D.; Pyshkin A. V.; Sirotkin A. V.; Vyahhi N.; Tesler G.; Alekseyev M. A.; Pevzner P. A. (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P.; Longden I.; Bleasby A. (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277. 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Li W.; Jaroszewski L.; Godzik A. (2001) Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics 17, 282–283. 10.1093/bioinformatics/17.3.282. [DOI] [PubMed] [Google Scholar]
- Chow C. Y.; Cristofori-Armstrong B.; Undheim E. A.; King G. F.; Rash L. D. (2015) Three peptide modulators of the human voltage-gated sodium channel 1.7, an important analgesic target, from the venom of an Australian tarantula. Toxins 7, 2494–2513. 10.3390/toxins7072494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker A. A.; Madio B.; Jin J.; Undheim E. A.; Fry B. G.; King G. F. (2017) Melt with this kiss: paralyzing and liquefying venom of the assassin bug Pristhesancus plagipennis (Hemiptera: Reduviidae). Mol. Cell. Proteomics 16, 552–566. 10.1074/mcp.M116.063321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilov I. V.; Seymour S. L.; Patel A. A.; Loboda A.; Tang W. H.; Keating S. P.; Hunter C. L.; Nuwaysir L. M.; Schaeffer D. A. (2007) The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteomics 6, 1638–1655. 10.1074/mcp.T600050-MCP200. [DOI] [PubMed] [Google Scholar]
- Klint J. K.; Senff S.; Saez N. J.; Seshadri R.; Lau H. Y.; Bende N. S.; Undheim E. A. B.; Rash L. D.; Mobli M.; King G. F. (2013) Production of recombinant disulfide-rich venom peptides for structural and functional analysis via expression in the periplasm of E. coli. PLoS One 8, e63865 10.1371/journal.pone.0063865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez N. J.; Cristofori-Armstrong B.; Anangi R.; King G. F. (2017) A strategy for production of correctly folded disulfide-rich peptides in the periplasm of E. Methods Mol. Biol. 1586, 155–180. 10.1007/978-1-4939-6887-9_10. [DOI] [PubMed] [Google Scholar]
- Marley J.; Lu M.; Bracken C. (2001) A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 20, 71–75. 10.1023/A:1011254402785. [DOI] [PubMed] [Google Scholar]
- Mobli M.; Maciejewski M. W.; Gryk M. R.; Hoch J. C. (2007) An automated tool for maximum entropy reconstruction of biomolecular NMR spectra. Nat. Methods 4, 467–468. 10.1038/nmeth0607-467. [DOI] [PubMed] [Google Scholar]
- Vranken W. F.; Boucher W.; Stevens T. J.; Fogh R. H.; Pajon A.; Llinas M.; Ulrich E. L.; Markley J. L.; Ionides J.; Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins: Struct., Funct., Genet. 59, 687–696. 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Delaglio F.; Cornilescu G.; Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223. 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378. 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bende N. S.; Dziemborowicz S.; Mobli M.; Herzig V.; Gilchrist J.; Wagner J.; Nicholson G. M.; King G. F.; Bosmans F. (2014) A distinct sodium channel voltage-sensor locus determines insect selectivity of the spider toxin Dc1a. Nat. Commun. 5, 4350. 10.1038/ncomms5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunagar K.; Undheim E. A.; Chan A. H.; Koludarov I.; Munoz-Gomez S. A.; Antunes A.; Fry B. G. (2013) Evolution stings: the origin and diversification of scorpion toxin peptide scaffolds. Toxins 5, 2456–2487. 10.3390/toxins5122456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S.; Bosmans F.; Tytgat J. (2004) Adaptive evolution of scorpion sodium channel toxins. J. Mol. Evol. 58, 145–153. 10.1007/s00239-003-2534-2. [DOI] [PubMed] [Google Scholar]
- Shafee T. M.; Lay F. T.; Phan T. K.; Anderson M. A.; Hulett M. D. (2017) Convergent evolution of defensin sequence, structure and function. Cell. Mol. Life Sci. 74, 663–682. 10.1007/s00018-016-2344-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada G.; Garcia B. I.; Schiavon E.; Ortiz E.; Cestele S.; Wanke E.; Possani L. D.; Corzo G. (2007) Four disulfide-bridged scorpion beta neurotoxin CssII: heterologous expression and proper folding in vitro. Biochim. Biophys. Acta, Gen. Subj. 1770, 1161–1168. 10.1016/j.bbagen.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Dash T. S.; Shafee T.; Harvey P. J.; Zhang C.; Peigneur S.; Deuis J. R.; Vetter I.; Tytgat J.; Anderson M. A.; Craik D. J.; Durek T.; Undheim E. A. B. (2019) A centipede toxin family defines an ancient class of CSαβ defensins. Structure 27, 315–326. 10.1016/j.str.2018.10.022. [DOI] [PubMed] [Google Scholar]
- Krezel A. M.; Kasibhatla C.; Hidalgo P.; MacKinnon R.; Wagner G. (1995) Solution structure of the potassium channel inhibitor agitoxin 2: caliper for probing channel geometry. Protein Sci. 4, 1478–1489. 10.1002/pro.5560040805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochnicka-Chalufour A.; Corzo G.; Satake H.; Martin-Eauclaire M. F.; Murgia A. R.; Prestipino G.; D’Suze G.; Possani L. D.; Delepierre M. (2006) Solution structure of discrepin, a new K+-channel blocking peptide from the α-KTx15 subfamily. Biochemistry 45, 1795–1804. 10.1021/bi0519248. [DOI] [PubMed] [Google Scholar]
- Landon C.; Cornet B.; Bonmatin J. M.; Kopeyan C.; Rochat H.; Vovelle F.; Ptak M. (1996) 1H-NMR-derived secondary structure and the overall fold of the potent anti-mammal and anti-insect toxin III from the scorpion Leiurus quinquestriatus quinquestriatus. Eur. J. Biochem. 236, 395–404. 10.1111/j.1432-1033.1996.00395.x. [DOI] [PubMed] [Google Scholar]
- Tugarinov V.; Kustanovich I.; Zilberberg N.; Gurevitz M.; Anglister J. (1997) Solution structures of a highly insecticidal recombinant scorpion α-toxin and a mutant with increased activity. Biochemistry 36, 2414–2424. 10.1021/bi961497l. [DOI] [PubMed] [Google Scholar]
- Zhu L.; Peigneur S.; Gao B.; Tytgat J.; Zhu S. (2013) Two recombinant α-like scorpion toxins from Mesobuthus eupeus with differential affinity toward insect and mammalian Na+ channels. Biochimie 95, 1732–1740. 10.1016/j.biochi.2013.05.009. [DOI] [PubMed] [Google Scholar]
- Durek T.; Vetter I.; Wang C. I.; Motin L.; Knapp O.; Adams D. J.; Lewis R. J.; Alewood P. F. (2013) Chemical engineering and structural and pharmacological characterization of the α-scorpion toxin OD1. ACS Chem. Biol. 8, 1215–1222. 10.1021/cb400012k. [DOI] [PubMed] [Google Scholar]
- Cologna C. T.; Peigneur S.; Rustiguel J. K.; Nonato M. C.; Tytgat J.; Arantes E. C. (2012) Investigation of the relationship between the structure and function of Ts2, a neurotoxin from Tityus serrulatus venom. FEBS J. 279, 1495–1504. 10.1111/j.1742-4658.2012.08545.x. [DOI] [PubMed] [Google Scholar]
- Bosmans F.; Martin-Eauclaire M.-F.; Swartz K. J. (2008) Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 456, 202–208. 10.1038/nature07473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H.; Liu D.; Wu K.; Lei J.; Yan N. (2019) Structures of human NaV1.7 channel in complex with auxiliary subunits and animal toxins. Science 363, 1303–1308. 10.1126/science.aaw2493. [DOI] [PubMed] [Google Scholar]
- Santos R.; Ursu O.; Gaulton A.; Bento A. P.; Donadi R. S.; Bologa C. G.; Karlsson A.; Al-Lazikani B.; Hersey A.; Oprea T. I.; Overington J. P. (2017) A comprehensive map of molecular drug targets. Nat. Rev. Drug Discovery 16, 19–34. 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revell J. D.; Lund P. E.; Linley J. E.; Metcalfe J.; Burmeister N.; Sridharan S.; Jones C.; Jermutus L.; Bednarek M. A. (2013) Potency optimization of Huwentoxin-IV on hNav1.7: a neurotoxin TTX-S sodium-channel antagonist from the venom of the Chinese bird-eating spider Selenocosmia huwena. Peptides 44, 40–46. 10.1016/j.peptides.2013.03.011. [DOI] [PubMed] [Google Scholar]
- Rashid M. H.; Huq R.; Tanner M. R.; Chhabra S.; Khoo K. K.; Estrada R.; Dhawan V.; Chauhan S.; Pennington M. W.; Beeton C.; Kuyucak S.; Norton R. S. (2015) A potent and Kv1.3-selective analogue of the scorpion toxin HsTX1 as a potential therapeutic for autoimmune diseases. Sci. Rep. 4, 4509. 10.1038/srep04509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassagnon I. R.; McCarthy C. A.; Chin Y. K.-Y.; Pineda S. S.; Keramidas A.; Mobli M.; Pham V.; De Silva T. M.; Lynch J. W.; Widdop R. E.; Rash L. D.; King G. F. (2017) Potent neuroprotection after stroke afforded by a double-knot spider-venom peptide that inhibits acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. U. S. A. 114, 3750–3755. 10.1073/pnas.1614728114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuis J. R.; Dekan Z.; Wingerd J. S.; Smith J. J.; Munasinghe N. R.; Bhola R. F.; Imlach W. L.; Herzig V.; Armstrong D. A.; Rosengren K. J.; Bosmans F.; Waxman S. G.; Dib-Hajj S. D.; Escoubas P.; Minett M. S.; Christie M. J.; King G. F.; Alewood P. F.; Lewis R. J.; Wood J. N.; Vetter I. (2017) Pharmacological characterisation of the highly NaV1.7 selective spider venom peptide Pn3a. Sci. Rep. 7, 40883. 10.1038/srep40883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B.; Murray J. K.; Andrews K. L.; Sham K.; Long J.; Aral J.; Ligutti J.; Amagasu S.; Liu D.; Zou A.; Min X.; Wang Z.; Ilch C. P.; Kornecook T. J.; Lin M. J.; Be X.; Miranda L. P.; Moyer B. D.; Biswas K. (2018) Discovery of tarantula venom-derived NaV1.7-inhibitory JzTx-V peptide 5-Br-Trp24 analogue AM-6120 with systemic block of histamine-induced pruritis. J. Med. Chem. 61, 9500–9512. 10.1021/acs.jmedchem.8b00736. [DOI] [PubMed] [Google Scholar]
- Pennington M. W.; Czerwinski A.; Norton R. S. (2018) Peptide therapeutics from venom: current status and potential. Bioorg. Med. Chem. 26, 2738–2758. 10.1016/j.bmc.2017.09.029. [DOI] [PubMed] [Google Scholar]
- Holford M.; Daly M.; King G. F.; Norton R. S. (2018) Venoms to the rescue. Science 361, 842–844. 10.1126/science.aau7761. [DOI] [PubMed] [Google Scholar]
- Osteen J. D.; Herzig V.; Gilchrist J.; Emrick J. J.; Zhang C.; Wang X.; Castro J.; Garcia-Caraballo S.; Grundy L.; Rychkov G. Y.; Weyer A. D.; Dekan Z.; Undheim E. A. B.; Alewood P.; Stucky C. L.; Brierley S. M.; Basbaum A. I.; Bosmans F.; King G. F.; Julius D. (2016) Selective spider toxins reveal a role for NaV1.1 channel in mechanical pain. Nature 534, 494–499. 10.1038/nature17976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez N. J.; Senff S.; Jensen J. E.; Er S. Y.; Herzig V.; Rash L. D.; King G. F. (2010) Spider-venom peptides as therapeutics. Toxins 2, 2851–2871. 10.3390/toxins2122851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undheim E. A.; Mobli M.; King G. F. (2016) Toxin structures as evolutionary tools: using conserved 3D folds to study the evolution of rapidly evolving peptides. BioEssays 38, 539–548. 10.1002/bies.201500165. [DOI] [PubMed] [Google Scholar]
- Deuis J. R.; Mueller A.; Israel M. R.; Vetter I. (2017) The pharmacology of voltage-gated sodium channel activators. Neuropharmacology 127, 87–108. 10.1016/j.neuropharm.2017.04.014. [DOI] [PubMed] [Google Scholar]
- King G. F.; Vetter I. (2014) No gain, no pain: NaV1.7 as an analgesic target. ACS Chem. Neurosci. 5, 749–751. 10.1021/cn500171p. [DOI] [PubMed] [Google Scholar]
- Ogiwara I.; Miyamoto H.; Tatsukawa T.; Yamagata T.; Nakayama T.; Atapour N.; Miura E.; Mazaki E.; Ernst S. J.; Cao D.; Ohtani H.; Itohara S.; Yanagawa Y.; Montal M.; Yuzaki M.; Inoue Y.; Hensch T. K.; Noebels J. L.; Yamakawa K. (2018) NaV1.2 haplodeficiency in excitatory neurons causes absence-like seizures in mice. Commun. Biol. 1, 1–16. 10.1038/s42003-018-0099-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders S. J.; Campbell A. J.; Cottrell J. R.; Moller R. S.; Wagner F. F.; Auldridge A. L.; Bernier R. A.; Catterall W. A.; Chung W. K.; Empfield J. R.; George A. L. Jr.; Hipp J. F.; Khwaja O.; Kiskinis E.; Lal D.; Malhotra D.; Millichap J. J.; Otis T. S.; Petrou S.; Pitt G.; Schust L. F.; Taylor C. M.; Tjernagel J.; Spiro J. E.; Bender K. J. (2018) Progress in understanding and treating SCN2A-mediated disorders. Trends Neurosci. 41, 442–456. 10.1016/j.tins.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.; Li Z.; Huang X.; Huang G.; Gao S.; Shen H.; Liu L.; Lei J.; Yan N. (2019) Molecular basis for pore blockade of human Na+ channel NaV1.2 by the mu-conotoxin KIIIA. Science 363, 1309–1313. 10.1126/science.aaw2999. [DOI] [PubMed] [Google Scholar]
- Pan X.; Li Z.; Zhou Q.; Shen H.; Wu K.; Huang X.; Chen J.; Zhang J.; Zhu X.; Lei J.; Xiong W.; Gong H.; Xiao B.; Yan N. (2018) Structure of the human voltage-gated sodium channel NaV1.4 in complex with β1. Science 362, eaau2486 10.1126/science.aau2486. [DOI] [PubMed] [Google Scholar]
- King G. F.; Escoubas P.; Nicholson G. M. (2008) Peptide toxins that selectively target insect NaV and CaV channels. Channels 2, 100–116. 10.4161/chan.2.2.6022. [DOI] [PubMed] [Google Scholar]
- Rodriguez de la Vega R. C.; Possani L. D. (2005) Overview of scorpion toxins specific for Na+ channels and related peptides: biodiversity, structure-function relationships and evolution. Toxicon 46, 831–844. 10.1016/j.toxicon.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Klint J. K.; Senff S.; Rupasinghe D. B.; Er S. Y.; Herzig V.; Nicholson G. M.; King G. F. (2012) Spider-venom peptides that target voltage-gated sodium channels: pharmacological tools and potential therapeutic leads. Toxicon 60, 478–491. 10.1016/j.toxicon.2012.04.337. [DOI] [PubMed] [Google Scholar]
- King G. F. (2019) Tying pest insects in knots: the deployment of spider-venom-derived knottins as bioinsecticides. Pest Manage. Sci. 75, 2437–2445. 10.1002/ps.5452. [DOI] [PubMed] [Google Scholar]
- Bohlen C. J.; Julius D. (2012) Receptor-targeting mechanisms of pain-causing toxins: How ow?. Toxicon 60, 254–264. 10.1016/j.toxicon.2012.04.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jami S.; Erickson A.; Brierley S. M.; Vetter I. (2018) Pain-causing venom peptides: insights into sensory neuron pharmacology. Toxins 10, 15. 10.3390/toxins10010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson S. D.; Mueller A.; Clayton D.; Starobova H.; Hamilton B. R.; Payne R. J.; Vetter I.; King G. F.; Undheim E. A. B. (2018) A comprehensive portrait of the venom of the giant red bull ant, Myrmecia gulosa, reveals a hyperdiverse hymenopteran toxin gene family. Sci. Adv. 4, eaau4640 10.1126/sciadv.aau4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.; Yang F.; Wei N.; Hong J.; Li B.; Luo L.; Rong M.; Yarov-Yarovoy V.; Zheng J.; Wang K.; Lai R. (2015) A pain-inducing centipede toxin targets the heat activation machinery of nociceptor TRPV1. Nat. Commun. 6, 8297. 10.1038/ncomms9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlen C. J.; Chesler A. T.; Sharif-Naeini R.; Medzihradszky K. F.; Zhou S.; King D.; Sanchez E. E.; Burlingame A. L.; Basbaum A. I.; Julius D. (2011) A heteromeric Texas coral snake toxin targets acid-sensing ion channels to produce pain. Nature 479, 410–414. 10.1038/nature10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin King J. V.; Emrick J. J.; Kelly M. J. S.; Herzig V.; King G. F.; Medzihradszky K. F.; Julius D. (2019) A cell-penetrating scorpion toxin enables mode-specific modulation of TRPA1 and pain. Cell 178, 1362–1374. 10.1016/j.cell.2019.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanaei-Zadeh H.; Marashi S. M.; Dehghani R. (2017) Epidemiological and clinical characteristics of scorpionism in Shiraz (2012–2016); development of a clinical severity grading for Iranian scorpion envenomation. Med. J. Islam Repub Iran 31, 27. 10.18869/mjiri.31.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward M. J.; Ellsworth S. A.; Nystrom G. S. (2018) A global accounting of medically significant scorpions: Epidemiology, major toxins, and comparative resources in harmless counterparts. Toxicon 151, 137–155. 10.1016/j.toxicon.2018.07.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.