

Abstract
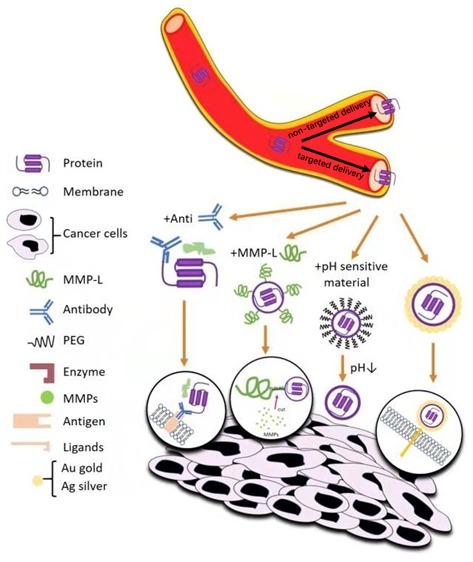
Great attention has been paid to cytotoxic proteins (e.g., ribosome-inactivating proteins, RIPs) possessing high anticancer activities; unlike small drugs, cytotoxic proteins can effectively retain inside the cells and avoid drug efflux mediated by multidrug resistance transporters due to the large-size effect. However, the clinical translation of these proteins is severely limited because of various biobarriers that hamper their effective delivery to tumor cells. Hence, in order to overcome these barriers, many smart drug delivery systems (DDS) have been developed. In this review, we will introduce two representative type I RIPs, trichosanthin (TCS) and gelonin (Gel), and overview the major biobarriers for protein-based cancer therapy. Finally, we outline advances on the development of smart DDS for effective delivery of these cytotoxic proteins for various applications in cancer treatment.
Keywords: trichosanthin, gelonin, ribosome-inactivating protein, tumor-targeting drug delivery, cell-penetrating peptide
Although molecularly targeted therapeutics (e.g., monoclonal antibodies and protein kinase inhibitors) have been successfully applied in clinics for cancer therapy, many challenges such as drug resistance and low response rate still remain to be resolved. To meet clinical needs, continuing efforts have been made to discover novel therapeutic agents. In this regard, great attention has been paid to cytotoxic proteins possessing high anticancer activities. Ribosome-inactivating proteins (RIPs) are attractive drug candidates for the treatment of cancer (Table 1). These RIPs possess unparalleled efficiency for inhibiting protein synthesis that could lead to tumor cell death at extremely low concentration (at picomolar) unmatched by other anticancer small drugs, once they could reach their target cytosolic ribosomes.1 Specifically, recent advances in genetic recombination technology allowed mass production of recombinant RIPs at industrial scale, and various fusion proteins could be produced with relative ease. On the basis of this advancement, RIPs with improved druggability could be engineered. However, clinical translation of these RIPs for cancer therapy is still far from reaching the goal. The main difficulty lies in drug delivery challenges, owing to the biobarriers in the body such as the vascular barriers, intratumoral barriers, and intracellular barriers. In order to overcome these obstacles, various smart drug delivery systems (DDS) based on prodrug-type strategies have been developed.2 The main idea for the prodrug type strategies is (1) to create a cell-permeable protein drug that can efficiently penetrate into the cells to exert their anticancer activity and (2) to adopt a carrier system that could curb the activity of the drugs during blood circulation but be activated as they reach the targeted cells/tissues. To accomplish the first goal, RIPs have been modified with cell-penetrating peptides (CPPs) and tumor-targeting ligands. Characterized by the capacity for delivering the attached cargoes into nearly any type of cells, CPPs have been widely applied for intracellular drug delivery. However, their nonspecific cell uptake could potentially cause side toxicity when coupled with cytotoxic proteins.3 To address the second issue, the protein drugs can be linked/immobilized to different nanostructures such as nanoparticles, as well as to antibodies for enhancing the tumor-targeting delivery efficiency. The necessity for delivery systems also lies in protecting the toxic proteins from proteolytic degradation and improving their pharmacokinetic profiles. In this article, we will focus on the properties and mechanisms of actions of two representative type I RIPs: trichosanthin (TCS) and gelonin (Gel). We will further summarize the development of smart DDS for tumor-targeting delivery of TCS and Gel.
Table 1. Types of Ribosome-Inactivating Proteins and Their Molecular Targets.
| type of RIP | samples | target | ref |
|---|---|---|---|
| type I RIP | tricosanthin | depurinates A4324 positioned in the conserved α-sarcin/ricin loop | (4) |
| gelonin | depurinates A4324 positioned in the conserved α-sarcin/ricin loop | (5) | |
| depurinates poly(ADP-ribosyl)ated poly(ADP-ribose) polymerase | (6) | ||
| saporin | depurinates A4324 positioned in the conserved α-sarcin/ricin loop | (7) | |
| depurinates poly(ADP-ribosyl)ated poly(ADP-ribose) polymerase | (6) | ||
| momordin | depurinates A4324 positioned in the conserved α-sarcin/ricin loop | (8) | |
| depurinates poly(ADP-ribosyl)ated poly(ADP-ribose) polymerase | (6) | ||
| PAP | removes A4324, A4321, and G4323 from the eukaryotic large rRNA | (9) | |
| type II RIP | ricin | modifies both or either G4323 and A4324 nucleoside residues | (10) |
| depurinates poly(ADP-ribosyl)ated poly(ADP-ribose) polymerase | (6) | ||
| agglutinin | depurinates A4324 nucleoside residue | (11) | |
| abrin | modifies both or either G4323 and A4324 nucleoside residues | (10) | |
| modeccin | modifies both or either G4323 and A4324 nucleoside residues | (10) |
Trichosanthin
Trichosanthin (TCS) is an active protein component (27 kDa) isolated from a Chinese herb, Tian Hua Fen, the root of Trichosanthes kirilowii Maximis, and is a well-studied type I RIP for its antitumor and antivirus activities (Table 2). Its precursor consists of 289 amino acids, and the active form of TCS is obtained from its precursor, by cleavage of 23 and 19 amino acids from its N- and C-terminal ends, respectively. The amino acid sequence of TCS reveals homology to ricin A chain.23,24 TCS, as a typical type I RIP, depurinates adenine-4324 positioned in the conserved α-sarcin/ricin loop of rat 28S rRNA. Structural analysis of TCS revealed two domains and five conserved catalytic residues.4 A modified form of TCS with deletion of 7 amino acids from the C-terminal (C7-TCS) caused a reduction in its pH-dependent membrane insertion ability; deletion of these amino acid residues led to conformational changes and oligomerization, resulting in a reduced level of ribosome-inactivating property (in vitro) and cytotoxicity (in vivo).25 Notably, TCS is an approved gynecological drug in China for ectopic pregnancies, hydatidiform moles, chorionic epithelioma, and abortion.
Table 2. Structure of TCS and List of Cancer Types and Virus Tested for the Activity of TCSa 2,12−22.

Protein structure was modeled online (https://swissmodel.expasy.org/interactive).
TCS as an Anticancer Agent
TCS causes apoptosis in cancer cells. Reactive oxygen species (ROS), mitochondrial and endoplasmic reticulum changes, and changes in the expression of apoptosis-related genes showed involvement in this process.26 However, a possible correlation between N-glycosidase activity and TCS-induced apoptosis remains yet to be explored.27 The anticancer effects of TCS have been established against hepatoma and lung cancer both in vitro and in vivo, and its effects as an immunolesioning agent against melanoma were confirmed by in vitro experiments.13 In addition, the antitumor effects of TCS on choriocarcinoma, cervical cancer, breast cancer, leukemia and lymphoma, colon carcinoma, stomach adenocarcinoma, and prostatic cancer were reported.12 In most cases, these antitumor effects were linked with apoptosis, resulting from the inactivation of ribosomes. TCS was also found to have antiproliferative and apoptosis-inducing effects in human MCF-7 (estrogen-dependent) and MDA-MB-231 (estrogen-independent) cells; induction of cell apoptosis by TCS was linked with the activation of both caspase-8 and -9 regulatory pathways with subsequent processes, such as caspase-3 activation and enhanced PARP cleavage.28 Furthermore, this work revealed DNA fragmentation in the nude mice bearing MDA-MB-231. Caspase pathways including those of caspase-8, -9, and -3 were activated by TCS in leukemia HL60 cells, and TCS-induced apoptosis was suggested to be mediated by mitochondrial and endoplasmic reticulum stress signaling through caspase-3.15 Furthermore, in HeLa cells, the suppression of cell proliferation by TCS was reported to be linked to the inhibition of protein kinase C (PKC) and protein kinase A (PKA), which suggested that the TCS-induced antitumor proliferation was induced by suppression of PKA/MAPK signaling pathway.16 Involvement of adenylyl cyclase activity and cyclic adenosine monophosphate in TCS-induced apoptosis was explained by their suppression initiated by the influx of extracellular Ca2+, but this suppression was demolished with the activation of PKC and PKA.29 In nude mouse models implanted with CNE1 (well-differentiated) and CNE2 (poorly differentiated) nasopharyngeal cancer cells, it was revealed that the antitumor effects of TCS were associated with apoptosis and partially with the suppression of telomerase activity.17 Low-density lipoproteins (LDL) were the major receptors for phagocytosis in JAR and BeWo choriocarcinoma cell lines, and LDL receptor-related protein 1 (LRP1) was responsible for TCS binding and endocytosis. This was the possible basis of abortifacient and antichoriocarcinoma activity of TCS.30 TCS was also reported to act on activation of the methylation-silenced tumor suppressor genes, induction of demethylation, and downregulation of DNA methyl transferase 1 (DNMT1).31 Furthermore, the cDNA array analysis approach was applied to establish TCS-induced gene expression profile changes in human CaSki cells, and the results revealed that TCS increases the expression of Smac (a mitochondrial protein that promotes cytochrome C dependent caspase activation) in CaSki cells and supports Smac demethylation.32 However, prolonged treatment resulted in resistance to TCS in the CaSki tumor cells
TCS as an Antiviral Agent
TCS also exhibits antiviral activity against various types of viral infections. For instance, TCS was demonstrated its protective role against infectious brain injury induced by herpex simplex virus (HSV-1) in mice.20 TCS has also been considered a candidate for treatment of HIV/AIDS.21 The anti-HIV activity of TCS was found to correlate with its ribosome-inactivating property; however, there may be other mechanisms involved in its anti-HIV effects.22 Antiviral activity of TCS was also related with its capacity to reduce the elevated levels of mitogen-activated protein kinase p38 (p38MAPK) and B-cell lymphoma 2 (Bcl-2) induced by HSV-1 infection, thereby lowering viral replication.33
Gelonin
Gelonin (Gel), which consists of 258 amino acids with an approximate molecular weight of 28 kDa, is a type I RIP derived from the seeds of Gelonium multiflorum(43) (Table 3). It shares a 33% sequence homology with TCS and ricin.44 Similar to TCS, Gel possesses N-glycosidase activity and depurinates adenine-4324 positioned in the conserved α-sarcin/ricin loop of 28S rRNA.43,45 Gel has also been extensively studied for its applicability as an anticancer and antiviral agent.46 However, as a typical type I RIP, without the help of intracellular drug carriers, Gel is poorly internalized into cells and could exert little therapeutic effect.47,48 Since the first discovery of Gel by Stirpe in 1980,43 the development of various Gel fusion proteins has widely been attempted to enhance its delivery efficacy. Specifically, thanks to the advance in genetic engineering, recombinant Gel could be produced from E. coli and was found to possess protein synthesis inhibition activity equipotent to that of the native plant-derived protein.49 To date, there have been reported various Gel fusion proteins with anticancer activity superior to that of the unmodified Gel.50,37,51,41,42
Table 3. Structure of Gel and the Cell Lines Tested for Gel Activitya 5,34−42.

Protein structure was modeled online (https://swissmodel.expasy.org/interactive).
Gel Fusion Proteins as Anticancer Agents
A variety of modified Gel proteins have been obtained by either chemical conjugation or using genetic recombination methods. One of the earliest studies was the chemical conjugation of rGel to a humanized anti-CD33 M195 monoclonal antibody via a disulfide bond for tumor-targeting delivery.36 The obtained HuM195-Gel conjugate showed high cytotoxicity on the CD33 positive cell lines (HL6O, OCIJAML2, and OCI/AML5), while unconjugated antibody and Gel alone had less effect at equimolar concentrations.36 Another example is the VEGF121-rGel which was composed of an isoform of vascular endothelial growth factor (VEGF121; 121 amino acids) and recombinant Gel (rGel) coupled via a G4S (GGGGGS) linker.38 VEGF121-rGel was selectively cytotoxic to endothelial cells that overexpress KDR/Flk-1 receptor (IC50: 0.5–1 nM; up to 600-fold lower than rGel); the endothelial cells with overexpression of KDR were more sensitive (60-fold) to VEGF121-rGel than were nondividing cells. VEGF121/rGel treatment resulted in significant inhibition in tumor growth in an animal model with human melanoma (A-375) or prostate (PC-3) xenografts.38
The Gel fused with B-lymphocyte stimulator (rGel/BLyS) showed selective binding and internalization to the cells expressing BLyS receptors. Among the tested B cell tumor cell lines, three mantle cell lymphoma (MCL) (JeKo-1, Mino, and SP53) and 2 diffuse large B cell lymphoma (DLBCL) cell lines (SUDHL-6 and OCI-Ly3) were most sensitive to rGel/BLyS (IC50: 2–5 pM and 0.001–5 nM for MCL and DLBCL).34 The therapeutic mechanism for rGel/BLyS was associated with the binding to the cell-expressing BLyS receptors and constitutively active NF-κB; after treatment, NF-κB targets (e.g., Bcl-Xl, Mcl-1 and survivin) were downregulated.35 rGel/BLyS treatment could also downregulate IL-6R and STAT3 activity, while upregulating Bax and apoptosis.52 rGel/BLyS could specifically enter CD19+ B-CLL lymphocytes via selective binding with BAFF-R and induced apoptosis, with a potent protein synthesis inhibition 1800-fold more effective than that of rGel in patient-derived leukemic lymphocyte samples.39 In subsequent studies, this rGel/BLyS was found to be also effective for various B cell tumors.34
Cao et al. prepared rGel fusion proteins with anti-HER2 human single-chain antibody (C6.5) using different peptide linkers as follows: (1) L linker (GGGGS), (2) Fdt (AGNRVRRSVG; contains furin cleavage site), and (3) Fpe (TRHRQPRGWEQL; contains furin cleavage site).53 The results revealed that Fdt enabled precise cleavage at the designated position by recombinant furin with 100% efficiency, while only 18.5% cleavage for Fpe at pH 7.2 was observed. In animal studies, C6.5/rGel with the Fdt linker (C6.5-Fdt-Gel) showed the least anticancer activity.53 The results of a binding study demonstrated that among the three C6.5/rGel samples when incubated at 37 °C in the presence of human plasma the C6.5-Fdt-Gel construct showed the highest reduction (20% reduction) in cell binding.54
Recently, Shin and co-workers genetically engineered a variety of fusion proteins coupling rGel with different types of peptide carriers (e.g., melittin, chlorotoxin, F3 peptide, and anti-IGF-1R affibody).37,51,41,42 Melittin, a typical pore-forming toxin, possesses the ability to create pores in the cell membrane, which in turn could destabilize the cell membranes. The pore formation can facilitate intracellular delivery of attached cargoes as well as directly cause death of tumor cells. However, to kill tumor cells by melittin alone, a high concentration (above micromolar) is required. The fused Gel-melittin was significantly more cytotoxic than either the native Gel or melittin in different cancer cell lines (e.g., HeLa, 9L, U87 MG, CT26, and LS174T).37 This could be explained by the enhanced uptake of Gel by the help of melittin. Chlorotoxin, a 36-mer peptide derived from scorpion venom, has attracted great interest as an effective carrier to target brain tumor cells because of its selective binding ability to the tumor cell-expressed MMP-2. Recombinant Gel-chlorotoxin fusion (Gel-CLTX) showed a 4.1-fold higher cell uptake than that of rGel in U87 MG human glioblastoma cells and a 25-fold higher cytotoxicity against U87 MG cells as compared to that in nontumorous 293 HEK cells.5 F3 peptide is a 31-mer peptide that binds to nucleolins selectively exposed on the surface of tumor and angiogenic endothelial cells. By genetically fusing F3 to rGel, selective and enhanced cellular uptake and cytotoxicity were observed in cancer cells such as U87 MG, HeLa, LnCaP, and 9L.51,5 Furthermore, in a following study, Gel fusion proteins coupled with multiple (2 or 3) F3 peptides showed an even higher cell uptake and augmented cytotoxicity as compared to those of Gel fusion with one F3. Consistently, in a LNCaP s.c. xenograft tumor mouse model, a greater tumor growth inhibition rate was observed from the group treated with tandem-multimeric Gel-F3.41 The Gel fusion with anti-IGF-1R affibody also showed selective and augmented (22-fold) cell uptake and cytotoxicity against IGF-1R overexpressed U87 MG brain cancer cells, compared with those of rGel.5
Biobarriers against Therapeutic Protein Delivery
Biobarriers that normally protect our body could paradoxically serve as formidable obstacles for potential anticancer protein drugs such as RIPs to successfully reach the tumor site. These barriers include endothelial/epithelial cell membranes, the reticuloendothelial system (RES), a complex networks of blood vessels, abnormal blood flow, and interstitial pressure gradients in tumors. Along with these biobarriers, the immune surveillance system clears any hazardous substances that invade the body,55 and moreover, even after successfully reaching the tumor cells, the tumor cell membranes function as a critical barrier that controls the final destiny of the protein drugs.56 Hence, developing strategies to effectively overcome the biobarriers is an imminent task for the success of protein-based cancer therapy.
Vascular Barriers
Compared to oral administration, intravenous administration of higher molecular compounds, especially proteins, can be a more reliable route for ensuring sufficient bioavailability. However, despite full access to blood circulation via intravenous administration, there are still obstacles for the protein drugs to reach the pathological site. The vascular barriers, primarily the endothelial cell tight junctions, block the drugs from getting out of the circulation and only allow selective transport.57 The typical case for the vascular barrier is the blood–brain barrier (BBB).58 The BBB is selectively permeable to nutrients and oxygen that are essential for the brain. The transport of vital substances for maintaining normal brain functions is generally mediated by the receptor-mediated transcytosis (RMT) system through vesicular trafficking machinery of the endothelium, and therefore, it is useful to exploit the RMT system to deliver therapeutics into the brain.59
Another approach to deliver anticancer drugs into the central nervous system (CNS) is to reversibly open the tight junction of the endothelial cells of BBB. In this respect, special features, such as bioavailability and biodegradability, are required for the drug carriers.60 Previously, it was found that endothelial sphingosine 1-phosphate receptor-1 (S1P1) promotes the barrier function against compounds with a size smaller than 10 kDa. Its knockout in mice caused a breach of BBB for small-molecular substances. Therefore, temporarily inhibiting endothelial S1P1 might provide an effective way to deliver cytotoxic proteins through the BBB.59 Apart from the physical barriers in the vascular system, when the blood arrives at certain organs like the kidney and liver, a significant part of the drugs may also be eliminated, which could diminish the portion of drugs that could reach the tumor tissue. In this process, the blood flow rate may take an effect.61
Also, there are many immunocytes and plasma proteins circulating in the blood. When a drug is transported inside the vessel, some of the drug molecules may lose activity by binding with plasma proteins or being caught by the mononuclear phagocytes. In certain cases, this may cause further immune responses and side effects, leading to discontinuation of the treatment. For example, during blood circulation, TCS protein drugs could be captured by macrophages and induce various immunomodulatory effects including upregulation of IL-4 and -13 and inhibition of INF-γ. TCS was reported to increase the expression of macrophage IL-10 and monocyte chemoattractant protein-1, and decrease IL-12 amd TNF-α levels.62 The extent of cell uptake appeared dependent upon the underlying mechanism (receptor-mediated vs nonspecific internalization). For instance, the peritoneal exudate cells (PEC) were found to be more sensitive to the cytotoxic effects of Gel than nonphagocytic cells, possibly due to mannose-receptor-mediated internalization.63 Overall, the protein drugs should be protected or masked from various interactions with blood components for improving their in vivo pharmacokinetic profiles.
Intratumoral Barriers
Once the anticancer drugs reach the targeted tumor tissue, they are confronted by intratumoral/endothelial barriers including dense extracellular matrix and high interstitial fluid pressure (IFP). The endothelial barriers play a crucial role in strictly maintaining tissue and vascular homeostasis.64 The integrity of endothelial barriers relies on a number of factors existing in the microenvironment. In tumor tissue, this barrier often gets impaired, and the blood vessel networks become leaky and disorganized. Subsequently, this enhanced vascular permeability can drive infiltration of inflammatory cells, but angiogenesis and extravasation of tumor cells cause high IFP.65 Specifically, this high IFP was responsible for poor perfusion and limited delivery of therapeutic agents. The high IFP is associated with a high resistance to mass transfer, and the condition could even aggravate the vascular abnormalities, such as high vessel tortuosity or generation of a large number of narrow tumor capillaries.66 Eventually, these overall events impact drug delivery into the tumor tissue, as well as affect cancer cell proliferation, survival, and invasion.67 Extracellular matrix (ECM) serves as a major intratumoral barrier. Previously, the ECM had been considered merely an intercellular filling, but recently, it has been discovered as a physiologically active component responsible for maintaining tissue homeostasis.68 The composition and organization of the ECM are tissue-specific and dynamically regulated. However, when this regulation is abnormal, the barrier function could be impaired, and this could lead to the development and progression of diseases such as cancer.68 As cancer progresses, the tumor size increases rapidly, and the tumor becomes densely packed with ECM among the cancer cells, tumor-associated fibroblasts/macrophages (TAFs/TAMs), and other cells. This series of events not only hampers the diffusion of drugs through the extracellular space but also exacerbates the tumor condition by facilitating cancer cell proliferation and metastasis.69 The growth factors released from immunocytes could facilitate remodeling of the ECM of primary tumors and facilitate the engraftment of metastasizing cancer cells in distant organs.69 Overall, these intratumoral barriers heavily affect the degree of drug penetration and distribution inside the tumor tissue. RIPs have a much larger size than that of conventional small-molecule drugs; they encounter these significant obstacles that prevent them from reaching the tumor cells.
Intracellular Barriers
Even when the anticancer drugs could finally reach the tumor cells, there yet remains the intracellular barrier to be overcome (Figure 1). The first challenge is to bind to the membrane of the tumor cells. In this regard, nonspecific electrostatic interaction with the cellular membrane components or receptor-mediated binding could mediate further internalization processes based on the interaction strength.70 Positively charged nanoparticles were found to more readily bind to the cancer cell membranes, possibly due to the highly negative surface of cancer cells compared to the normal cells.71 Uptake of large molecules, such as proteins or nanoparticles, occurs mainly by endocytosis. Depending on the size of the substance, endocytosis proceeds in two common pathways: (1) clathrin-mediated endocytosis (e.g., nonpermeable particles up to 200 and 500 nm) and (2) caveolae-mediated endocytosis (e.g., up to 5 μm particles).70 Endocytic sorting, as another barrier for drug delivery, could recycle the endosomal drugs back to extracellular space either by clathrin-dependent or -independent pathways.72 Therefore, the challenge remains at the last stage of intracellular drug delivery: endosomal escape. Specifically for proteins such as RIPs, the macromolecules are likely to be degraded in the harsh lysosomal environment with high acidity and abundant enzymes.45 As the targets of the RIPs are located in the cytosol compartment, RIPs must escape from the endosomes and eventually take action for killing the tumor cells.
Figure 1.

Intracellular fate of cytotoxic proteins.
Delivery Strategies for CPP-Modified Protein Drugs
There are many biobarriers in the “journey” of drugs from the administration site to the action site. Among these, the intracellular barrier has remained a formidable obstacle for cytotoxic protein delivery. In this regard, various approaches have been attempted, and the discovery of CPPs has suggested an effective way to resolve this problem.73 For example, the TAT peptide which is the most widely used CPP in drug delivery has shown capacity for carrying cargoes into almost all types of cells. The cargoes varied from small molecules to proteins, genes, and even nanoparticles.73 Nevertheless, because of the nonselectivity in their cell internalization, there yet remains the challenge of how to keep the drug inactive (and safe) during blood circulation but selectively activate the drug in the tumor. In order to overcome such challenges, various types of smart strategies have been developed that include the following: (1) Antibody Targeted Triggered Electrically Modified Prodrug Type Strategy (ATTEMPTS), (2) enzyme-triggered systems, (3) pH-triggered and charge–charge-sensitive systems, (4) nanobased strategy, and (5) light-induced internalization systems. In this section, we will specifically introduce a novel prodrug-type smart DDS developed upon TCS and Gel.
ATTEMPTS
The ATTEMPTS system is a Smart DDS developed by Yang’s lab for delivering protein drugs to treat different cancers (Figure 2).2 The architecture of the ATTEMPTS system consists of (1) a targeting component, a tumor-targeting ligand conjugated to heparin that possesses highly dense negative charge, and (2) a drug component, a protein drug modified with a cationic CPP. The two components can form a stable complex via charge interaction. The antibody mediates tumor-targeting delivery, and the CPP can facilitate internalization of the coupled protein drugs into tumor cells with high efficiency. A short, cationic CPP such as low-molecular-weight protamine (LMWP) can provide a stable yet reversible binding with heparin, allowing timely dissociation triggered by protamine via competitive substitution of LMWP. By binding to heparin, the activity of the protein drugs in the complex could be effectively curbed during blood circulation due to steric inhindrance.74 Overall, the ATTEMPTS approach could provide an effective means for tumor-targeting of protein drugs and, furthermore, once arrived at the tumor site, an efficient and safe way to exploit the strong cell-internalizing ability of CPPs.75 A proof-of-concept study of ATTEMPTS was carried out by Yang’s research group utilizing T84.66 as the tumor targeting ligand.76 T84.66 is a well-studied monoclonal antibody that selectively binds to carcinoembryonic antigen (CEA) which is a widely accepted colon cancer cell biomarker.77 In that study, T84.66-heparin conjugate was prepared by chemical conjugation, and TAT peptide (YGRKKRRQRRR)-modified Gel (TAT-gelonin) was genetically engineered. Similar to the Gel fusion protein modified with LMWP (VSRRRRRRGGRRRR) that exerted significantly increased cytotoxic activity against cancer cells (20–120-fold lower IC50 than that of Gel),48 TAT-gelonin showed augmented cytotoxic effects on the tested cancer cell lines. By simply mixing the two components of the ATTEMPTS system, the T84.66-heparin/TAT-gelonin complex could be easily prepared. Through in vitro and in vivo characterization, this complex displayed selective and enhanced anticancer activity against CEA-overexpressing colon cancer cells. These results demonstrated the feasibility of applying the ATTEMPTS system for protein-toxin-based cancer therapy.76
Figure 2.

Schematic illustration of the ATTEMPTS system.
In a previous report, Gel was genetically modified with TAT peptide. When compared with Gel, the TAT-gelonin fusion protein displayed greater cell internalization and enhanced cytotoxicity on various cell lines (229-, 391-, 93-, and 108-fold higher activities against LS174T, HCT116, MDCK and 293 HEK cells, respectively).40 With the addition of polyanionic heparin and formation of complex of heparin/TAT-gelonin, the cell internalization of TAT-gelonin was dramatically reduced. Charge–charge interaction is a key mechanism for controlled release of protein drugs in the ATTEMPTS system. Yang’s lab also developed a simplified version of the ATTEMPTS system based on a passive-targeting strategy without modification of antibody.78 The complex of heparin/TAT-gelonin exhibited a prodrug-type feature due to the inhibition of the CPPs-mediated cell entry, but the exposure of the CPPs can be achieved by adding the trigger protamine which is an antagonist of heparin for dissociation of TAT-gelonin from the heparin counterpart.78
Enzyme-Triggered Systems
CPP-modified protein toxins can efficiently kill almost all types of tumor cells. Nevertheless, because of their rapid clearance and nonselective action, highly effective drug delivery strategies are in need. For glioma treatment, we developed a DDS for TCS-based therapy (Figure 3). The structure of the DDS was a fusion protein composed of four components: (1) TCS as a therapeutic agent, (2) lactoferrin as the carrier, (3) a peptide sequence linker that is specifically cleaved by glioma-associated matrix metalloproteinase 2 (MMP-2), and (4) a CPP.79 Once the DDS enters the glioma tissues led by the targeting ligand lactoferrin, TCS-CPP would be dissociated from the carrier lactoferrin, mediated by MMP-2 cleavage. TCS-CPP was thus internalized to the tumor cells. The study showed that this DDS enhanced the antiglioma activity.79 In another study, intein-based site-selective PEGylation was used to develop a PEG-based DDS for the TCS.80 A fusion protein consisting of TCS, LMWP (cell-penetrating sequence), an MMP-2 substrate peptide (PLGLAG), and a terminal intein motif was genetically engineered. By using the intein-based site-selective PEGylation technique, a PEG was conjugated to the MMP-2 cleavable peptide at C-terminal. This system showed obvious prodrug-like properties. The PEGylated TCS was stable in the bloodstream, but with cleavage by MMP-2 in the tumors, PEG was detached. The deshielded TCS-LMWP showed efficient cell uptake and potent antitumor activity. In the animal studies using the MMP-2-overexpressing HT1080 nude mice, the PEGylated TCS-based DDS showed longer plasma circulation, reduced immunogenicity, lower systemic toxicity, and improved tumoricidal effects.80 In a further study, when the PEGylated TCS-based DDS was combined with paclitaxel, it showed synergistic antitumor activity both in vitro and in vivo and reversed paclitaxel resistance in the A549/T lung tumor model.81 The antiresistance mechanism was related to inhibition of the paclitaxel-induced elevated caspase-9 phosphorylation as well as activation of caspase-3 and induction of apoptosis.
Figure 3.

Enzyme-responsive prodrug-type DDS for targeting CPP-modified toxin to brain tumor cells. Protein structures were modeled online on https://swissmodel.expasy.org/interactive.
We also developed a novel DDS termed the Smart Hitchlike via Endogenous Albumin-Trichosanthin Hinge (SHEATH) system. The SHEATH system is composed of (1) TCS, (2) TAT peptide, (3) a protease substrate peptide (consisting of proline, threonine, and asparagine; PTN), and (4) an albumin-binding domain (ABD) (Figure 4).82 The TCS-TAT-PTN-ABD fusion protein, constructed by genetic recombination, could automatically form conjugates with albumin mediated by the binding of ABD. The results showed a higher cell uptake efficiency of TCS-TAT-PTN-ABD, suggesting the effective dissociation of TCS-TAT from the conjugate of TCS-TAT-PTN-ABD/albumin, due to proteolytic cleavage of the PTN peptides and the subsequent TAT-mediated cell internalization of protein drugs.
Figure 4.

SHEATH system based on the noncovalent nanoconjugate between a recombinant TCS protein and the endogenous albumin. After injection, such a multifunctional fusion protein would bind with the endogenous serum albumin in the bloodstream and hitchhike to the tumor site for enhanced accumulation, where the protease-substrate peptide would be cleaved and the rTCS fragment unhitched from the carrier for taking pharmacological action. Reprinted with permission from ref (82). Copyright 2019 Royal Society of Chemistry.
pH-Triggered Systems
An acidic extracellular environment inside the tumor tissue, caused by lactate secretion from glycolysis, is a driving factor for tumor progression and metastasis.83 A pH-triggered smart DDS is commonly built to undergo structural alteration based on the pH change between the normal tissues and tumors as well as between the inside and outside of tumor cells.84 A common design of a pH-triggered DDS is to modify the protein-drugs-loaded nanoparticles with a pH-sensitive polymer. For instance, a pH-responsive copolymer (stearoyl-PEG-polySDM) with an apparent pKa of 7.2 was used to modify the protein-loaded liposomes, thus rendering liposomes bearing a negative surface charge in neutral pH and stable due to the charge repulsion.85 However, the charge density would decrease in acidic condition (e.g., pH 6.5), which lead to aggregation of the liposomes due to the colloidal instability and facilitated the accumulation of liposomal drug in the tumor sites. Another interesting example is a pH-responsive toxin protein constructed by genetic engineering.86 Listeriolysin O (LLO) is a cytolysin with a pore-forming function in cell membranes, which is characterized with a pH sensor identified in its structure. In this work, LLO was engineered for developing a pH-triggering pore-forming mutant, based on detailed structural analysis coupled with molecular dynamics and mutational analysis.
Nanobased Strategy
To date, many types of nanoparticles have been developed, and their application for drug delivery has revolutionized the pharmaceutical field. The use of nanoparticles could generally provide (1) increased plasma stability of drugs, (2) selective and augmented drug accumulation in tumor tissue, (3) codelivery of different types of drugs, as well as (4) effective ways to overcome multidrug resistance.87 Specifically, for proteins, the nanoparticles could also help to preserve their molecular conformation and biological activity. Furthermore, nanoparticles can also facilitate the cellular entry of poorly cell-permeable protein drugs and, moreover, help their endosomal escape after internalization. For example, Gel barely enters cells and has a limited access to cytosol. To overcome this issue, Provoda et al. encapsulated Gel in a pH-sensitive liposome with a pore-forming protein LLO,88 which possesses translocation ability to escape from the endosome. The cellular results showed that this liposomal Gel had rapid cytocidal effects on B16 cell with a high efficiency (IC50: 100 pM). These results were further supported by in vivo studies using the B16 melanoma mice model.88
The nanoparticles can also serve as an effective platform for the codelivery of different drugs enabling combination therapies. For instance, a nanoparticle-based strategy has been developed to codeliver TCS and albendazole (ABZ, an antihelmintic drug that can inhibit tubulin polymerization) to tumor cells (Figure 5).89 This nanoparticle system was composed of a self-assembling complex of ABZ-encapsulated negatively charged albumin silver nanoparticles and cationic cell penetrating peptide-modified TCS. These nanocomplexes were effective for the treatment of drug-resistant A549/T lung tumor and prevention of metastasis in vivo.
Figure 5.

Schematic illustration of tumor delivery and synergistic effect via codelivery of TCS and ABZ by silver nanoparticles. Reprinted with permission from ref (89). Copyright 2017 American Chemical Society.
Light-Induced Internalization Systems
A major bottleneck challenge of many cytotoxic proteins (e.g., TCS and Gel) for successful application to cancer therapy lies in their nearly cell-impermeant property. This obstacle includes not only their difficulty in initially entering the cell membrane but also escaping from the endosomes once internalized. For the CPP-modified proteins, it has been previously reported that facilitating their endosomal escape could result in greater translocation to the cytosol.90 To this regard, photochemical internalization (PCI) has widely been studied.91,92 PCI is a technique that uses light to activate the photosensitizers (e.g., porphyrin, phthalocyanine or chlorin dyes etc.), taken up by the target cells. As a product of the photosensitizer activation, reactive oxygen species (ROS) are generated. ROS can interact with the endosomal membranes and lead to the disruption, eventually causing a subsequent release of the endosomal substances (Figure 6).90 Theoretically, this strategy could be applied to any therapeutic drug, including protein toxins. Therefore, highly potent but cell-impermeant protein toxins are an ideal drug model for studying the PCI-based drug delivery strategy. For instance, Berg et al. reported that PCI treatment with Gel and photosensitizer in cervical carcinoma cells led to a 200-fold greater cytotoxicity than single treatment.93 Furthermore, there was eradication of sarcoma in 50% of the Gel-PCI treated mice, while a significant inhibition of tumor growth was observed in the remaining 50% mice.94 There are also reports of PCI studies with Gel fusion proteins. For instance, Bull-Hansen et al. performed PCI studies with HER2-targeting MH3-B1/Gel fusion proteins for treatment of ovarian cancer.91 With this PCI treatment, both SKOV-3 cells (resistant to trastuzumab and MH3-B1/Gel monotherapies) and SK-BR-3 cells (sensitive to trastuzumab and MH3-B1/Gel monotherapies) showed great cell killing effects to similar extent. Furthermore, Berstad et al. engineered Gel/EGF fusion protein and tested the PCI efficacy in vitro and invivo.92 The results showed that PCI treatment induced an efficient cytosolic release of Gel/EGF selectively in EGFR-expressing A431 tumor cells, in which apoptosis, necrosis, and autophagy. The animal study further demonstrated a high treatment efficacy. Weyergang et al. also studied the PCI treatment effect with VEGFR-targeting VEGF121/Gel fusion proteins.95 The VEGF121/Gel efficiently entered VEGFR1- and VEGFR2-expressing cells and caused cytocydal effects in combination of PCI treatment. In animal studies, synergistic effects on inhibition of tumor growth and perfusion were observed, showing a 50% complete remission (CR) of allografts; interestingly, however, CR was not achieved in athymic nude mice, suggesting potent antitumor immunity was induced by VEGF121/Gel-PCI treatment.
Figure 6.

Photochemical internalization (PCI) of cytotoxic proteins. Once the photosensitizers and the gelonin fusion proteins associate to the cancer cell membrane, they become (A) internalized by endocytosis. (B) While both the photosensitizers and gelonin fusion proteins colocalized in the endosomes, with laser irradiation, reactive oxygen species (ROS) are generated and ruptures the endosomal membrane. (C) The released gelonin fusion proteins inhibit the substrate ribosomes in the cytosol and induce cell death.
Conclusion
This review discusses the major biobarriers against delivering protein toxins to the tumor site and outlines the advances in smart DDS developed for tumor-targeting delivery of RIPs. The RIPs possess extraordinary potency for inhibiting protein synthesis and have long been considered as the promising anticancer agents. Nevertheless, their inability to internalize tumor cells (in case of the type 1 RIP) and nonselectively toxic nature severely halted the clinical translation of these proteins for cancer therapy. Owing to the discovery and application of various CPPs, efficient intracellular delivery of proteins is feasible. However, the CPP-mediated protein delivery methods have a disadvantage of nonselectivity in cell uptake. Therefore, it is long-sought goal to exploit the high efficiency and selectivity of CPP-mediated delivery, and many efforts have been made. However, clinical translation of CPP-modified proteins is still far from reach and further exploration is needed. Currently, the prodrug-type strategies have been mostly investigated for developing effective DDS for keeping the CPP-modified protein drugs inactive in blood circulation but becoming fully activated in the tumors. CPP-based protein therapies will emerge as promising modes of tumor treatment in the future. In addition, the nanotechnology-based delivery systems of protein toxins could also be a potential solution because of the ability of nanomediated tumor-targeting and intratumoral/intracellular delivery.
Acknowledgments
We are thankful for the support of NFSC (81925035, 81673382, and 81521005), the Strategic Priority Research Program of CAS (XDA12050307), the National Special Project for Significant New Drugs Development (2018ZX09711002-010-002), Shanghai Sci-Tech innovation Action Plan (19431903100), the Fudan-SIMM Joint Research Fund (FU-SIMM20174009), and the project of constructing biomaterials with controllable release of biologically active proteins and polyphenols by Uzbek Academy of Sciences (TA-FA-F6-002). Dr. A. M. Asrorov is a recipient of CAS PIFI Fellowship (2019PB0076) and Belt & Road Young Scientist Award (Shanghai, 18430740800).
The authors declare no competing financial interest.
This article is made available for a limited time sponsored by ACS under the ACS Free to Read License, which permits copying and redistribution of the article for non-commercial scholarly purposes.
References
- Shapira A.; Benhar I. (2010) Toxin-based therapeutic approaches. Toxins 2 (11), 2519–83. 10.3390/toxins2112519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Zhang M.; Min K. A.; Wang H.; Shin M. C.; Li F.; Yang V. C.; Huang Y. (2018) Improved Protein Toxin Delivery Based on ATTEMPTS Systems. Curr. Drug Targets 19 (4), 380–392. 10.2174/1389450118666170302094758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze S. R.; Ho A.; Vocero-Akbani A.; Dowdy S. F. (1999) In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285 (5433), 1569–72. 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Shi W. W.; Wong K. B.; Shaw P. C. (2018) Structural and Functional Investigation and Pharmacological Mechanism of Trichosanthin, a Type 1 Ribosome-Inactivating Protein. Toxins 10 (8), 335. 10.3390/toxins10080335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham S. H.; Min K. A.; Shin M. C. (2017) Molecular tumor targeting of gelonin by fusion with F3 peptide. Acta Pharmacol. Sin. 38 (6), 897–906. 10.1038/aps.2017.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri L.; Brigotti M.; Perocco P.; Carnicelli D.; Ciani M.; Mercatali L.; Stirpe F. (2003) Ribosome-inactivating proteins depurinate poly(ADP-ribosyl)ated poly(ADP-ribose) polymerase and have transforming activity for 3T3 fibroblasts. FEBS Lett. 538 (1–3), 178–82. 10.1016/S0014-5793(03)00176-5. [DOI] [PubMed] [Google Scholar]
- Giansanti F.; Flavell D. J.; Angelucci F.; Fabbrini M. S.; Ippoliti R. (2018) Strategies to Improve the Clinical Utility of Saporin-Based Targeted Toxins. Toxins 10 (2), 82. 10.3390/toxins10020082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo Y.; Tsurugi K.; Lambert J. M. (1988) The site of action of six different ribosome-inactivating proteins from plants on eukaryotic ribosomes: the RNA N-glycosidase activity of the proteins. Biochem. Biophys. Res. Commun. 150 (3), 1032–6. 10.1016/0006-291X(88)90733-4. [DOI] [PubMed] [Google Scholar]
- Hudak K. A.; Wang P.; Tumer N. E. (2000) A novel mechanism for inhibition of translation by pokeweed antiviral protein: depurination of the capped RNA template. RNA 6 (3), 369–80. 10.1017/S1355838200991337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo Y.; Mitsui K.; Motizuki M.; Tsurugi K. (1987) The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J. Biol. Chem. 262 (12), 5908–5912. [PubMed] [Google Scholar]
- Cheng J.; Lu T. H.; Liu C. L.; Lin J. Y. (2010) A biophysical elucidation for less toxicity of agglutinin than abrin-a from the seeds of Abrus precatorius in consequence of crystal structure. J. Biomed. Sci. 17, 34. 10.1186/1423-0127-17-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha O.; Niu J.; Ng T. B.; Cho E. Y.; Fu X.; Jiang W. (2013) Anti-tumor action of trichosanthin, a type 1 ribosome-inactivating protein, employed in traditional Chinese medicine: a mini review. Cancer Chemother. Pharmacol. 71 (6), 1387–93. 10.1007/s00280-013-2096-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilabert-Oriol R.; Weng A.; Mallinckrodt B.; Melzig M. F.; Fuchs H.; Thakur M. (2014) Immunotoxins constructed with ribosome-inactivating proteins and their enhancers: a lethal cocktail with tumor specific efficacy. Curr. Pharm. Des. 20 (42), 6584–643. 10.2174/1381612820666140826153913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei B.; Huang Q.; Huang S.; Mai W.; Zhong X. (2016) Trichosanthin-induced autophagy in gastric cancer cell MKN-45 is dependent on reactive oxygen species (ROS) and NF-kappaB/p53 pathway. J. Pharmacol. Sci. 131 (2), 77–83. 10.1016/j.jphs.2016.03.001. [DOI] [PubMed] [Google Scholar]
- Li J.; Xia X.; Ke Y.; Nie H.; Smith M. A.; Zhu X. (2007) Trichosanthin induced apoptosis in HL-60 cells via mitochondrial and endoplasmic reticulum stress signaling pathways. Biochim. Biophys. Acta, Gen. Subj. 1770 (8), 1169–80. 10.1016/j.bbagen.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Wang P.; Chen L. L.; Yan H.; Li J. C. (2009) Trichosanthin suppresses HeLa cell proliferation through inhibition of the PKC/MAPK signaling pathway. Cell Biol. Toxicol. 25 (5), 479–88. 10.1007/s10565-008-9102-x. [DOI] [PubMed] [Google Scholar]
- Kang M.; Ou H.; Wang R.; Liu W.; Mao Y.; Tang A. (2013) Effect of trichosanthin on apoptosis and telomerase activity of nasopharyngeal carcinomas in nude mice. JBUON 18 (3), 675–682. [PubMed] [Google Scholar]
- Xu J.; Gao D. F.; Yan G. L.; Fan J. M. (2009) Induced apoptotic action of recombinant trichosanthin in human stomach adenocarcinoma MCG803 cells. Mol. Biol. Rep. 36 (6), 1559–64. 10.1007/s11033-008-9352-y. [DOI] [PubMed] [Google Scholar]
- Wen D.; Wang J.; Yan H.; Chen J.; Xia K.; Liu J.; Zhang A. (2015) Effect of Radix Trichosanthis and Trichosanthin on Hepatitis B Virus in HepG2.2.15 Cells. J. Nanosci. Nanotechnol. 15 (3), 2094–8. 10.1166/jnn.2015.9271. [DOI] [PubMed] [Google Scholar]
- Chen G. F.; Huang W. G.; Chen F. Y.; Shan J. L. (2006) Protective effects of trichosanthin in Herpes simplex virus-1 encephalitis in mice. Zhongguo Dang Dai Er Ke Za Zhi 8 (3), 239–241. (in Chinese). [PubMed] [Google Scholar]
- Fang E. F.; Ng T. B.; Shaw P. C.; Wong R. N. (2011) Recent progress in medicinal investigations on trichosanthin and other ribosome inactivating proteins from the plant genus Trichosanthes. Curr. Med. Chem. 18 (28), 4410–4417. 10.2174/092986711797200499. [DOI] [PubMed] [Google Scholar]
- Wang J. H.; Nie H. L.; Tam S. C.; Huang H.; Zheng Y. T. (2002) Anti-HIV-1 property of trichosanthin correlates with its ribosome inactivating activity. FEBS Lett. 531 (2), 295–8. 10.1016/S0014-5793(02)03539-1. [DOI] [PubMed] [Google Scholar]
- Maraganore J. M.; Joseph M.; Bailey M. C. (1987) Purification and characterization of trichosanthin. Homology to the ricin A chain and implications as to mechanism of abortifacient activity. J. Biol. Chem. 262 (24), 11628–11633. [PubMed] [Google Scholar]
- Collins E. J.; Robertus J. D.; LoPresti M.; Stone K. L.; Williams K. R.; Wu P.; Hwang K.; Piatak M. (1990) Primary amino acid sequence of alpha-trichosanthin and molecular models for abrin A-chain and alpha-trichosanthin. J. Biol. Chem. 265 (15), 8665–8669. [PubMed] [Google Scholar]
- Zhang F.; Lu Y. J.; Shaw P. C.; Sui S. F. (2003) Change in pH-dependent membrane insertion characteristics of trichosanthin caused by deletion of its last seven C-terminal amino acid residues. Biochemistry (Moscow) 68 (4), 436–45. 10.1023/A:1023656114906. [DOI] [PubMed] [Google Scholar]
- Zhang C.-y.; Gong Y.-x.; Ma H.; An C.-c.; Chen D.-y. (2001) Reactive oxygen species involved in trichosanthin-induced apoptosis of human choriocarcinoma cells. Biochem. J. 355 (3), 653–661. 10.1042/bj3550653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Li X.; Li J. C. (2010) Possible mechanisms of trichosanthin-induced apoptosis of tumor cells. Anat. Rec. 293 (6), 986–92. 10.1002/ar.21142. [DOI] [PubMed] [Google Scholar]
- Fang E. F.; Zhang C. Z.; Zhang L.; Wong J. H.; Chan Y. S.; Pan W. L.; Dan X. L.; Yin C. M.; Cho C. H.; Ng T. B. (2012) Trichosanthin inhibits breast cancer cell proliferation in both cell lines and nude mice by promotion of apoptosis. PLoS One 7 (9), e41592 10.1371/journal.pone.0041592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q.; Bai T.; Shen S.; Li L.; Ding H.; Wang P. (2011) Increase of cytosolic calcium induced by trichosanthin suppresses cAMP/PKC levels through the inhibition of adenylyl cyclase activity in HeLa cells. Mol. Biol. Rep. 38 (4), 2863–8. 10.1007/s11033-010-0432-4. [DOI] [PubMed] [Google Scholar]
- Jiao Y.; Liu W. (2010) Low-density lipoprotein receptor-related protein 1 is an essential receptor for trichosanthin in 2 choriocarcinoma cell lines. Biochem. Biophys. Res. Commun. 391 (4), 1579–84. 10.1016/j.bbrc.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Song H.; Hu H.; Cui L.; You C.; Huang L. (2012) Trichosanthin inhibits DNA methyltransferase and restores methylation-silenced gene expression in human cervical cancer cells. Mol. Med. Rep. 6 (4), 872–8. 10.3892/mmr.2012.994. [DOI] [PubMed] [Google Scholar]
- Cui L.; Song J.; Wu L.; Huang L.; Wang Y.; Huang Y.; Yu H.; Huang Y.; You C. C.; Ye J. (2015) Smac is another pathway in the anti-tumour activity of Trichosanthin and reverses Trichosanthin resistance in CaSki cervical cancer cells. Biomed. Pharmacother. 69, 119–24. 10.1016/j.biopha.2014.10.027. [DOI] [PubMed] [Google Scholar]
- Huang H.; Chan H.; Wang Y. Y.; Ouyang D. Y.; Zheng Y. T.; Tam S. C. (2006) Trichosanthin suppresses the elevation of p38 MAPK, and Bcl-2 induced by HSV-1 infection in Vero cells. Life Sci. 79 (13), 1287–92. 10.1016/j.lfs.2006.03.047. [DOI] [PubMed] [Google Scholar]
- Lyu M. A.; Cheung L. H.; Hittelman W. N.; Marks J. W.; Aguiar R. C.; Rosenblum M. G. (2007) The rGel/BLyS fusion toxin specifically targets malignant B cells expressing the BLyS receptors BAFF-R, TACI, and BCMA. Mol. Cancer Ther. 6 (2), 460–70. 10.1158/1535-7163.MCT-06-0254. [DOI] [PubMed] [Google Scholar]
- Lyu M. A.; Rai D.; Ahn K. S.; Sung B.; Cheung L. H.; Marks J. W.; Aggarwal B. B.; Aguiar R. C.; Gandhi V.; Rosenblum M. G. (2010) The rGel/BLyS fusion toxin inhibits diffuse large B-cell lymphoma growth in vitro and in vivo. Neoplasia 12 (5), 366–75. 10.1593/neo.91960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliaro L. C.; Liu B.; Munker R.; Andreeff M.; Freireich E. J.; Scheinberg D. A.; Rosenblum M. G. (1998) Humanized M195 monoclonal antibody conjugated to recombinant gelonin: an anti-CD33 immunotoxin with antileukemic activity. Clin. Cancer Res. 4 (8), 1971–1976. [PubMed] [Google Scholar]
- Shin M. C.; Min K. A.; Cheong H.; Moon C.; Huang Y.; He H.; Yang V. C. (2016) Preparation and Characterization of Gelonin-Melittin Fusion Biotoxin for Synergistically Enhanced Anti-Tumor Activity. Pharm. Res. 33 (9), 2218–28. 10.1007/s11095-016-1959-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenendaal L. M.; Jin H.; Ran S.; Cheung L.; Navone N.; Marks J. W.; Waltenberger J.; Thorpe P.; Rosenblum M. G. (2002) In vitro and in vivo studies of a VEGF121/rGelonin chimeric fusion toxin targeting the neovasculature of solid tumors. Proc. Natl. Acad. Sci. U. S. A. 99 (12), 7866–71. 10.1073/pnas.122157899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmanapalli R.; Lyu M. A.; Du M.; Keating M. J.; Rosenblum M. G.; Gandhi V. (2007) The growth factor fusion construct containing B-lymphocyte stimulator (BLyS) and the toxin rGel induces apoptosis specifically in BAFF-R-positive CLL cells. Blood 109 (6), 2557–64. 10.1182/blood-2006-08-042424. [DOI] [PubMed] [Google Scholar]
- Shin M. C.; Zhang J.; Min K. A.; Lee K.; Moon C.; Balthasar J. P.; Yang V. C. (2014) Combination of antibody targeting and PTD-mediated intracellular toxin delivery for colorectal cancer therapy. J. Controlled Release 194, 197–210. 10.1016/j.jconrel.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M. C.; Min K. A.; Cheong H.; Moon C.; Huang Y. Z.; He H. N.; Yang V. C. (2017) Tandem-multimeric F3-gelonin fusion toxins for enhanced anti-cancer activity for prostate cancer treatment. Int. J. Pharm. 524 (1–2), 101–110. 10.1016/j.ijpharm.2017.03.072. [DOI] [PubMed] [Google Scholar]
- Ham S.; Min K. A.; Yang J. W.; Shin M. C. (2017) Fusion of gelonin and anti-insulin-like growth factor-1 receptor (IGF-1R) affibody for enhanced brain cancer therapy. Arch. Pharmacal Res. 40 (9), 1094–1104. 10.1007/s12272-017-0953-7. [DOI] [PubMed] [Google Scholar]
- Stirpe F.; Olsnes S.; Pihl A. (1980) Gelonin, a New Inhibitor of Protein-Synthesis, Nontoxic to Intact-Cells - Isolation, Characterization, and Preparation of Cyto-Toxic Complexes with Concanavalin-A. J. Biol. Chem. 255 (14), 6947–6953. [PubMed] [Google Scholar]
- Rosenblum M. G.; Kohr W. A.; Beattie K. L.; Beattie W. G.; Marks W.; Toman P. D.; Cheung L. (1995) Amino acid sequence analysis, gene construction, cloning, and expression of gelonin, a toxin derived from Gelonium multiflorum. J. Interferon Cytokine Res. 15 (6), 547–55. 10.1089/jir.1995.15.547. [DOI] [PubMed] [Google Scholar]
- Varkouhi A. K.; Scholte M.; Storm G.; Haisma H. J. (2011) Endosomal escape pathways for delivery of biologicals. J. Controlled Release 151 (3), 220–8. 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Nicolas E.; Beggs J. M.; Haltiwanger B. M.; Taraschi T. F. (1997) Direct evidence for the deoxyribonuclease activity of the plant ribosome inactivating protein gelonin. FEBS Lett. 406 (1–2), 162–4. 10.1016/S0014-5793(97)00267-6. [DOI] [PubMed] [Google Scholar]
- Li Z.; Qu Y.; Li H.; Yuan J. (2007) Truncations of gelonin lead to a reduction in its cytotoxicity. Toxicology 231 (2–3), 129–36. 10.1016/j.tox.2006.11.074. [DOI] [PubMed] [Google Scholar]
- Shin M. C.; Zhang J.; David A. E.; Trommer W. E.; Kwon Y. M.; Min K. A.; Kim J. H.; Yang V. C. (2013) Chemically and biologically synthesized CPP-modified gelonin for enhanced anti-tumor activity. J. Controlled Release 172 (1), 169–78. 10.1016/j.jconrel.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan P. A.; Garrison D. A.; Better M. (1993) Cloning and expression of a gene encoding gelonin, a ribosome-inactivating protein from Gelonium multiflorum. Gene 134 (2), 223–7. 10.1016/0378-1119(93)90097-M. [DOI] [PubMed] [Google Scholar]
- Stirpe F. (2004) Ribosome-inactivating proteins. Toxicon 44 (4), 371–83. 10.1016/j.toxicon.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Park T.; Min K. A.; Cheong H.; Moon C.; Shin M. C. (2019) Genetic engineering and characterisation of chlorotoxin-fused gelonin for enhanced glioblastoma therapy. J. Drug Target 27 (9), 950–958. 10.1080/1061186X.2018.1516221. [DOI] [PubMed] [Google Scholar]
- Lyu M. A.; Sung B.; Cheung L. H.; Marks J. W.; Aggarwal B. B.; Aguiar R. C.; Rosenblum M. G. (2010) The rGel/BLyS fusion toxin inhibits STAT3 signaling via down-regulation of interleukin-6 receptor in diffuse large B-cell lymphoma. Biochem. Pharmacol. 80 (9), 1335–42. 10.1016/j.bcp.2010.07.017. [DOI] [PubMed] [Google Scholar]
- Cao Y.; Marks J. D.; Marks J. W.; Cheung L. H.; Kim S.; Rosenblum M. G. (2009) Construction and characterization of novel, recombinant immunotoxins targeting the Her2/neu oncogene product: in vitro and in vivo studies. Cancer Res. 69 (23), 8987–95. 10.1158/0008-5472.CAN-09-2693. [DOI] [PubMed] [Google Scholar]
- Lyu M. A.; Cao Y. J.; Mohamedali K. A.; Rosenblum M. G. (2012) Cell-targeting fusion constructs containing recombinant gelonin. Methods Enzymol. 502, 167–214. 10.1016/B978-0-12-416039-2.00008-2. [DOI] [PubMed] [Google Scholar]
- Esser C.; Rannug A. (2015) The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 67 (2), 259–79. 10.1124/pr.114.009001. [DOI] [PubMed] [Google Scholar]
- Pohorille A.; Deamer D. (2009) Self-assembly and function of primitive cell membranes. Res. Microbiol. 160 (7), 449–56. 10.1016/j.resmic.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Su C.; Liu Y.; Li R.; Wu W.; Fawcett J. P.; Gu J. (2019) Absorption, distribution, metabolism and excretion of the biomaterials used in Nanocarrier drug delivery systems. Adv. Drug Delivery Rev. 143, 97. 10.1016/j.addr.2019.06.008. [DOI] [PubMed] [Google Scholar]
- Upadhyay R. K. (2014) Drug delivery systems, CNS protection, and the blood brain barrier. BioMed Res. Int. 2014, 869269. 10.1155/2014/869269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A. R.; Shusta E. V. (2007) Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. 24 (9), 1759–71. 10.1007/s11095-007-9379-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuter J. (2014) Drug delivery to the central nervous system by polymeric nanoparticles: what do we know?. Adv. Drug Delivery Rev. 71, 2–14. 10.1016/j.addr.2013.08.008. [DOI] [PubMed] [Google Scholar]
- High K. P. (2009) Overcoming barriers to adult immunization. J. Am. Osteopath Assoc 109, S25–S28. [PubMed] [Google Scholar]
- Xu W.; Hou W.; Yao G.; Ji Y.; Yeh M.; Sun B. (2001) Inhibition of Th1- and enhancement of Th2-initiating cytokines and chemokines in trichosanthin- treated macrophages. Biochem. Biophys. Res. Commun. 284 (1), 168–72. 10.1006/bbrc.2001.4940. [DOI] [PubMed] [Google Scholar]
- Madan S.; Ghosh P. C. (1992) Interaction of gelonin with macrophages: effect of lysosomotropic amines. Exp. Cell Res. 198 (1), 52–8. 10.1016/0014-4827(92)90148-2. [DOI] [PubMed] [Google Scholar]
- Zervantonakis I. K.; Hughes-Alford S. K.; Charest J. L.; Condeelis J. S.; Gertler F. B.; Kamm R. D. (2012) Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. U. S. A. 109 (34), 13515–20. 10.1073/pnas.1210182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzi S.; Hebda J. K.; Gavard J. (2013) Vascular permeability and drug delivery in cancers. Front. Oncol. 3, 211. 10.3389/fonc.2013.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen T. G.; Gaustad J. V.; Leinaas M. N.; Rofstad E. K. (2012) High interstitial fluid pressure is associated with tumor-line specific vascular abnormalities in human melanoma xenografts. PLoS One 7 (6), e40006 10.1371/journal.pone.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T.; Liu K.; Wu Y.; Fan J.; Chen J.; Li C.; Zhu G.; Wang Z.; Li L. (2013) High interstitial fluid pressure promotes tumor cell proliferation and invasion in oral squamous cell carcinoma. Int. J. Mol. Med. 32 (5), 1093–100. 10.3892/ijmm.2013.1496. [DOI] [PubMed] [Google Scholar]
- Walker C.; Mojares E.; Del Rio Hernandez A. (2018) Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 19 (10), 3028. 10.3390/ijms19103028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eble J. A.; Niland S. (2019) The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 36, 171. 10.1007/s10585-019-09966-1. [DOI] [PubMed] [Google Scholar]
- Au J. L.; Yeung B. Z.; Wientjes M. G.; Lu Z.; Wientjes M. G. (2016) Delivery of cancer therapeutics to extracellular and intracellular targets: Determinants, barriers, challenges and opportunities. Adv. Drug Delivery Rev. 97, 280–301. 10.1016/j.addr.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Le W.; Wang Y.; Li Z.; Wang D.; Lin L.; Cui S.; Hu J. J.; Hu Y.; Yang P.; Ewing R. C.; Shi D.; Cui Z. (2016) Targeting Negative Surface Charges of Cancer Cells by Multifunctional Nanoprobes. Theranostics 6 (11), 1887–98. 10.7150/thno.16358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B. D.; Donaldson J. G. (2009) Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 10 (9), 597–608. 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M. C.; Zhang J.; Min K. A.; Lee K.; Byun Y.; David A. E.; He H.; Yang V. C. (2014) Cell-penetrating peptides: achievements and challenges in application for cancer treatment. J. Biomed. Mater. Res., Part A 102 (2), 575–87. 10.1002/jbm.a.34859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell A. K.; Yates E. A.; Fernig D. G.; Turnbull J. E. (2004) Interactions of heparin/heparan sulfate with proteins: appraisal of structural factors and experimental approaches. Glycobiology 14 (4), 17R–30R. 10.1093/glycob/cwh051. [DOI] [PubMed] [Google Scholar]
- Wang H.; Moon C.; Shin M. C.; Wang Y.; He H.; Yang V. C.; Huang Y. (2017) Heparin-Regulated Prodrug-Type Macromolecular Theranostic Systems for Cancer Therapy. Nanotheranostics 1 (1), 114–130. 10.7150/ntno.18292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M. C.; Zhang J.; Min K. A.; He H.; David A. E.; Huang Y.; Yang V. C. (2015) PTD-Modified ATTEMPTS for Enhanced Toxin-based Cancer Therapy: An In Vivo Proof-of-Concept Study. Pharm. Res. 32 (8), 2690–2703. 10.1007/s11095-015-1653-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urva S. R.; Balthasar J. P. (2010) Target mediated disposition of T84.66, a monoclonal anti-CEA antibody: application in the detection of colorectal cancer xenografts. MAbs 2 (1), 67–72. 10.4161/mabs.2.1.10781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M. C.; Zhao J.; Zhang J.; Huang Y.; He H.; Wang M.; Min K. A.; Yang V. C. (2015) Recombinant TAT-gelonin fusion toxin: synthesis and characterization of heparin/protamine-regulated cell transduction. J. Biomed. Mater. Res., Part A 103 (1), 409–19. 10.1002/jbm.a.35188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Zhang M.; Jin H.; Li D.; Xu F.; Wu A.; Wang J.; Huang Y. (2017) Glioma Dual-Targeting Nanohybrid Protein Toxin Constructed by Intein-Mediated Site-Specific Ligation for Multistage Booster Delivery. Theranostics 7 (14), 3489–3503. 10.7150/thno.20578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. Z.; Zhang M.; Jin H. Y.; Tang Y. S.; Wang H. Y.; Xu Q.; Li Y. P.; Li F.; Huang Y. Z. (2017) Intein-mediated site-specific synthesis of tumor-targeting protein delivery system: Turning PEG dilemma into prodrug-like feature. Biomaterials 116, 57–68. 10.1016/j.biomaterials.2016.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Zhang M.; Jin H.; Tang Y.; Wu A.; Xu Q.; Huang Y. (2017) Prodrug-Like, PEGylated Protein Toxin Trichosanthin for Reversal of Chemoresistance. Mol. Pharmaceutics 14 (5), 1429–1438. 10.1021/acs.molpharmaceut.6b00987. [DOI] [PubMed] [Google Scholar]
- Chang Y.; Yao S.; Chen Y.; Huang J.; Wu A.; Zhang M.; Xu F.; Li F.; Huang Y. (2019) Genetically-engineered protein prodrug-like nanoconjugates for tumor-targeting biomimetic delivery via a SHEATH strategy. Nanoscale 11 (2), 611–621. 10.1039/C8NR08951E. [DOI] [PubMed] [Google Scholar]
- Kato Y.; Ozawa S.; Miyamoto C.; Maehata Y.; Suzuki A.; Maeda T.; Baba Y. (2013) Acidic extracellular microenvironment and cancer. Cancer Cell Int. 13 (1), 89. 10.1186/1475-2867-13-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reshetnyak Y. K. (2015) Imaging Tumor Acidity: pH-Low Insertion Peptide Probe for Optoacoustic Tomography. Clin. Cancer Res. 21 (20), 4502–4. 10.1158/1078-0432.CCR-15-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila-Caballer M.; Codolo G.; Munari F.; Malfanti A.; Fassan M.; Rugge M.; Balasso A.; de Bernard M.; Salmaso S. (2016) A pH-sensitive stearoyl-PEG-poly(methacryloyl sulfadimethoxine)-decorated liposome system for protein delivery: An application for bladder cancer treatment. J. Controlled Release 238, 31–42. 10.1016/j.jconrel.2016.07.024. [DOI] [PubMed] [Google Scholar]
- Kisovec M.; Rezelj S.; Knap P.; Cajnko M. M.; Caserman S.; Flasker A.; Znidarsic N.; Repic M.; Mavri J.; Ruan Y.; Scheuring S.; Podobnik M.; Anderluh G. (2017) Engineering a pH responsive pore forming protein. Sci. Rep. 7, 42231. 10.1038/srep42231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.; Liu E. G.; Cui Y. N.; Huang Y. Z. (2017) Nanotechnology-based combination therapy for overcoming multidrug-resistant cancer. Cancer Biol. Med. 14 (3), 212–227. 10.20892/j.issn.2095-3941.2017.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provoda C. J.; Stier E. M.; Lee K. D. (2003) Tumor cell killing enabled by listeriolysin O-liposome-mediated delivery of the protein toxin gelonin. J. Biol. Chem. 278 (37), 35102–8. 10.1074/jbc.M305411200. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Liang J.; Wu A.; Chen Y.; Zhao P.; Lin T.; Zhang M.; Xu Q.; Wang J.; Huang Y. (2017) Co-Delivery of Trichosanthin and Albendazole by Nano-Self-Assembly for Overcoming Tumor Multidrug-Resistance and Metastasis. ACS Appl. Mater. Interfaces 9 (32), 26648–26664. 10.1021/acsami.7b05292. [DOI] [PubMed] [Google Scholar]
- Martinez de Pinillos Bayona A.; Moore C. M.; Loizidou M.; MacRobert A. J.; Woodhams J. H. (2016) Enhancing the efficacy of cytotoxic agents for cancer therapy using photochemical internalisation. Int. J. Cancer 138 (5), 1049–57. 10.1002/ijc.29510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull-Hansen B.; Berstad M. B.; Berg K.; Cao Y.; Skarpen E.; Fremstedal A. S.; Rosenblum M. G.; Peng Q.; Weyergang A. (2015) Photochemical activation of MH3-B1/rGel: a HER2-targeted treatment approach for ovarian cancer. Oncotarget 6 (14), 12436–12451. 10.18632/oncotarget.3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berstad M. B.; Cheung L. H.; Berg K.; Peng Q.; Fremstedal A. S. V.; Patzke S.; Rosenblum M. G.; Weyergang A. (2015) Design of an EGFR-targeting toxin for photochemical delivery: in vitro and in vivo selectivity and efficacy. Oncogene 34 (44), 5582–5592. 10.1038/onc.2015.15. [DOI] [PubMed] [Google Scholar]
- Berg K.; Selbo P. K.; Prasmickaite L.; Tjelle T. E.; Sandvig K.; Moan J.; Gaudernack G.; Fodstad O.; Kjolsrud S.; Anholt H.; Rodal G. H.; Rodal S. K.; Hogset A. (1999) Photochemical internalization: a novel technology for delivery of macromolecules into cytosol. Cancer Res. 59 (6), 1180–1183. [PubMed] [Google Scholar]
- Dietze A.; Peng Q.; Selbo P. K.; Kaalhus O.; Muller C.; Bown S.; Berg K. (2005) Enhanced photodynamic destruction of a transplantable fibrosarcoma using photochemical internalisation of gelonin. Br. J. Cancer 92 (11), 2004–9. 10.1038/sj.bjc.6602600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyergang A.; Fremstedal A. S.; Skarpen E.; Peng Q.; Mohamedali K. A.; Eng M. S.; Cheung L. H.; Rosenblum M. G.; Waltenberger J.; Berg K. (2018) Light-enhanced VEGF121/rGel: A tumor targeted modality with vascular and immune-mediated efficacy. J. Controlled Release 288, 161–172. 10.1016/j.jconrel.2018.09.005. [DOI] [PubMed] [Google Scholar]