

Abstract
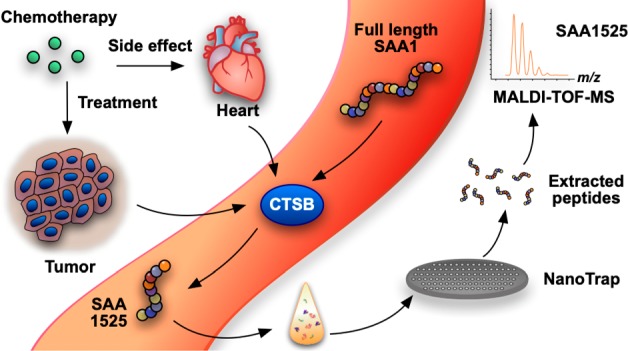
Improvements in long-term cancer survival rates have resulted in an increase in the prevalence of chemotherapy-linked cardiac failure, but treatment-induced cardiac injuries may not be detected until long after therapy. Monitoring cardiac function is recommended; however, cardiovascular injury in cancer patients differs from those with primary cardiac dysfunction, which limits the utility of traditional cardiac biomarkers. Here we examined plasma levels of peptides produced by cathepsin B, which is released during chemotherapy-induced cardiac injury. We applied nanotrap fractionation to enrich plasma peptides from cancer patients treated with or without chemotherapy. Peptides associated with chemotherapy-induced cardiotoxicity, but not other cardiac injury, were identified by mass spectrometry, and their dependence on cathepsin B activity was determined using enzyme inhibition experiments. We found that a peptide (SAA-1525) derived from serum amyloid A1 was significantly increased in cardiotoxicity patients, and its production was inhibited when plasma samples were pretreated with cathepsin B specific inhibitors. Plasma SAA-1525 also correlated with other markers of cardiac injury. Analysis of plasma SAA-1525 levels may hold potential as a rapid and minimally invasive method to monitor subclinical injury, thereby allowing timely intervention to mitigate further cardiac damage and avoid more severe clinical presentation.
Keywords: peptide, SAA-1525, cathepsin B, chemotherapy, cardiotoxicity
Introduction
Cancer treatment advances have markedly increased cancer survival rates but have also increased the risk that patients will develop chronic treatment-related multisystemic diseases.1 Cardiovascular injury has emerged as a major inadvertent consequence of cancer therapy,2−4 and chemotherapy-linked congestive heart failure has been linked to a 3.5-fold increase in mortality versus idiopathic cardiomyopathy.5 Cardiac injury events may occur during chemotherapy or shortly after its completion, but symptoms of cardiac injury may not be apparent for months or sometimes even years.6
Monitoring of cardiac function is recommended to detect subclinical myocardial damage that may occur during or after chemotherapy but is expensive and time-intensive.7,8 Most approaches used in clinical practice to assess cardiac function (e.g., echocardiographic evaluation of left ventricular ejection fraction (LVEF) or radionuclide ventriculography) have poor predictive power due to their low sensitivity to detect subclinical myocardial injury.9 Biomarkers of cardiac injury, including both troponin I (TnI) and the N-terminal pro-B-type natriuretic peptide (NT-proBNP), are promising diagnostic tools for early identification, assessment, and monitoring of cardiotoxicity. However, despite the proposed utility of these biomarkers, concern over their lack of sensitivity or specificity and their untraceable release kinetics during chemotherapy limit their diagnostic utility.10 Creatine kinase (CK) level or creatine kinase-muscle/brain (CK-MB) activity is an acceptable alternative for cardiac injury evaluation when troponin or NT-proBNP mass assays are not available or feasible,11 but strenuous exercise may also increase both CK and CK-MB, although with a lower relative index. CK-MB level can also be increased by kidney failure and, rarely, by chronic muscle disease, low thyroid hormone (T3, T4) levels, and alcohol abuse. Better diagnostic or prognostic biomarkers are needed to monitor cardiotoxicity and to facilitate preventive trials to potentially improve cardiovascular and oncologic morbidity and mortality.
Recent studies have revealed that treatment with the chemotherapeutics Gemcitabine and 5-fluorouracil can trigger the release of cathepsin B (CTSB) from lysosomes to mediate inflammasome activation.12 CTSB is also required for doxorubicin-induced cell death,13 and cell viability following doxorubicin treatment is significantly enhanced when CTSB expression is inhibited by treatment with a CTSB siRNA or specific inhibitor.13 Studies focusing on the expression and activity of cathepsins in failing cardiac tissue also suggest that cathepsins can trigger and promote cardiac remodeling14,15 and that attenuating the expression and activity of cathepsins by blocking the angiotensin II type 1 receptor should attenuate cardiac remodeling and dysfunction. However, there are no assays that profile cathepsin activity in blood as a biomarker of cardiac injury.
Circulating levels of specific peptides can impart important information about disease states and may yield greater diagnostic sensitivity than their parent proteins as they can also reflect the activity of disease-associated protease activities. Therefore, peptides are considered to have great potential as biomarkers for screening, diagnosis, and therapy monitoring.16−18 We therefore reasoned that measurement of CTSB-catalyzed peptides may offer more information about the cardiotoxicity and measured the level of peptides differentially expressed in the circulation of cancer patients that develop cardiotoxicity in response to chemotherapy to identify candidate peptide biomarkers that might reflect increased cytosolic or circulating CTSB activity in response to chemotherapy-induced cardiotoxicity. Thus, we analyzed the plasma proteome for CTSB-generated peptides associated with cardiotoxicity to identify suitable biomarker candidates.
Results
Study Subject Characteristics
As described in Table 1, this study enrolled a total of 40 patients (50% women; median age 68 years; IQR 62–74 years); 13 consecutive patients who received chemotherapy (Chemo) and 27 consecutive patients who declined chemotherapy (Non-Chemo). In the Chemo group, 8 patients exhibited plasma TnI or NT-proBNP values consistent with cardiotoxicity (Ctox), while 19 Non-Chemo patients were found to have cardiac damage (Cdam) using the same criteria. The goal of this study was to identify a potential universal biomarker for prediction of chemotherapy-induced cardiotoxicity, without reference to a specific cancer type or chemotherapy drug. Study subjects with various tumor types were consecutively enrolled and segregated into chemotherapy and nonchemotherapy groups based on their decision to accept or refuse chemotherapy, without restriction on their cancer type or drug regimen for those who elected to receive chemotherapy. Patients enrolled in this study received a variety of chemotherapeutics, including imatinib, anastrozole, exemestane, BYL719, ABT-199, pracinostat, bosutinib, nilotinib, and abiraterone. This study was designed to identify candidate plasma biomarkers of cardiotoxicity differentially expressed in the Chemo group (Ctox vs nCtox), while excluding factors that were also similarly regulated in the Non-Chemo group (Cdam vs nCdam). Since previous cardiac injury was present for subjects in both the treatment and nontreatment groups, it was necessary to distinguish biomarkers that were solely related to cardiac damage following patient exposure to chemotherapy. As a result, both the nCtox and Non-Chemo groups were considered to be negative controls and were used to exclude markers not specific for chemotherapy-induced cardiac injury.
Table 1. Clinical Characteristics of Patients Who Received Chemotherapy or Declined Chemotherapya.
| chemotherapy (n = 13) |
nonchemotherapy (n = 27) |
|||||
|---|---|---|---|---|---|---|
| variable | noncardiotoxicity (nCtox) | cardiotoxicity (Ctox) | noncardiac damage (nCdam) | cardiac damage (Cdam) | Pa | Pb |
| number, n (female) | 5 (4) | 8 (1) | 8 (5) | 19 (10) | 0.0149* | >0.6375 |
| age, y, median (IQR) | 70 (63–73) | 55 (46–71) | 65 (55–72) | 68 (59–75) | >0.99 | >0.99 |
| LVEF, %, median (IQR) | 59.5 (55.5–64.25) | 58 (58–62.75) | 62 (50–62.5) | 57.5 (53.25–60.5) | >0.99 | >0.99 |
| oncological disease, n (%) | ||||||
| leukemia | 6 (75) | 7 (37) | ||||
| breast cancer | 3 (60) | 2 (25) | 1 (5) | |||
| lymphoma | 1 (13) | 1 (5) | ||||
| prostate cancer | 2 (25) | |||||
| stomach cancer | 1 (20) | 1 (13) | 1 (5) | |||
| lung cancer | 1 (13) | 1 (5) | ||||
| others | 1 (20) | 3 (38) | 8 (42) | |||
| Pro-BNP, pg/mL, median (IQR) | 372.9 (251.3–726.6) | 10346 (8068–17650) | 235.1 (104.6–610.2) | 3035 (1409–7081) | 0.0047 | 0.0055 |
| TnI, ng/mL, median (IQR) | 0.03 (0.03–0.03) | 0.54 (0.0475–0.775) | 0.03 (0.03–0.03) | 0.15 (0.05–0.75) | 0.0267 | 0.0019 |
| CK, U/L, median (IQR) | 38 (20–121) | 173 (99.25–228) | 30 (24–46) | 35.5 (20–204.5) | >0.99 | >0.99 |
| CK-MB, μg/L, median (IQR) | 2 (1.65–2.2) | 5.15 (1.425–9.025) | 1.8 (1.15–2.55) | 2.9 (2.5–5.3) | 0.6125 | >0.99 |
Pa, P-value between nCtox and Ctox groups. Pb, P-value between nCdam and Cdam groups. *, P-value = 0.031 after data correction by binary logistic regression.
Ctox incidence was lower for women (20%) than for men (88%), but the Cdam rate was similar for women (67%) and men (75%). The Ctox gender difference remained significant after correction by binary logistic regression, implying that men are more susceptible to cardiotoxicity, in agreement with results previously reported for patients treated with doxorubicin,19 although these results remain controversial. The median age was lower in Ctox versus nCtox patients (55 vs 70 years), but it was similar in Cdam and nCdam patients (68 and 65 years). Cancer types were not equally distributed among the Chemo and Non-Chemo treatment groups or their subgroups. Leukemia cases were highly represented in the Ctox and Cdam groups (75 and 37%, respectively) with remaining Ctox and Cdam cases being associated with a range of other cancer types.
Ctox and Cdam groups were defined by elevated TnI or NT-proBNP plasma level. Both markers were elevated in the Ctox versus nCtox and Cdam versus nCdam subjects, but no differences in CK and CK-MB were detected between the groups electing to undergo or forsake chemotherapy or the Ctox and Cdam groups (not shown).
Plasma SAA-1525 Levels Are Elevated in Subjects with Ctox
We employed our previously described nanotrap platform20 to isolate and enrich small plasma peptides which were quantified and identified by MALDI-TOF MS. This approach identified more than 400 peptide peaks, including 27 that were ≥2-fold enriched in Ctox versus nCtox samples (Figure 1, Table S1), and only 4 of these peaks (m/z, 1525.8, 1030.52, 1115.66, and 1177.66) distinguished (p < 0.01) Ctox and nCtox cases (Table S1) but not Cdam and nCdam cases. Because three of the peptides (m/z, 1030.52, 1115.66, and 1177.66) could not be successfully sequenced, further analysis was only performed on the 1525.8 m/z peptide (Figure 2A,B). A 2-way ANOVA analysis was performed to evaluate the statistical significance of 1525.8 m/z peptide differences between all groups (Table S2). 1525.8 m/z peptide levels were determined to be significantly higher in the Ctox group versus the nCtox (p = 0.0011), nCdam (p = 0.0002), or Cdam (p = 0.0004) groups. LC-MS/MS analysis determined the sequence of peptide 1525.8 m/z to be PNHFRPAGLPEKY (Figure 2C), which matched the C-terminus of SAA1 (Figure 2D), a major acute-phase protein that is primarily produced by liver cells during cases of infection, tissue injury, malignancy, and other sources of inflammation.21 Circulating levels of acute-phase protein that serve as biomarkers of inflammation are frequently elevated in cancer and with inflammation-associated cardiac damage. We therefore retrieved the results of high-sensitivity C-reactive protein (hs-CRP) assays performed to assess systemic inflammation from records of the study subjects. We found that hs-CRP levels were significantly increased in both the Ctox and Cdam groups (Figure S1), implying that acute inflammation, as reflected by elevated hs-CRP levels, is associated with both cardiotoxicity and cardiac damage in the absence of chemotherapy in our study cohort. This data also strongly suggests that elevated SAA-1525 does not directly reflect acute inflammation, since the Ctox and Cdam groups exhibit similar hs-CRP levels but SAA-1525 is elevated only in the Ctox group. A MEROPS database analysis of SAA1 identified 37 candidate CTSB cleavage sites (Figure S2) as well as multiple potential cleavage sites for cathepsin L (11 sites), matrix metalloprotease 1 (MMP1; 2 sites) and MMP3 (7 sites), and the merpin A alpha and beta subunits (1 and 2 sites, respectively), although none of these cleavage sites directly matched the ends of the SAA-1525 peptide.
Figure 1.

Rational progression of the discovery of cardiotoxicity biomarkers from sample preparation to identification of cardiotoxicity specific peptide in human biological fluids.
Figure 2.

Peptide SAA-1525 identification. (A) SAA-1525 LC-MS signal normalized to spiked-in ACTH fragment (Mean ± SE; **, p < 0.01 by two-way ANOVA). (B) ROC analysis for SAA-1525 differentiation of Ctox and nCtox patients. (C) SAA-1525 peptide peak LC-MS/MS sequence. (D) CTSB cleavage sites in SAA1. Blue arrows indicate CTSB cleavage sites. * indicates a cleavage site not assigned to CTSB activity.
Cleavage of SAA-1525 from SAA1 Requires CTSB Activity
SAA1 reportedly exists as a hexamer, with its subunits displaying a unique four-helix bundle fold in which its C-terminal tail (Figure 2D) forms extensive salt bridges and hydrogen bonds that are critical to the stability of the helix bundle.22 Notably, a CTSB cleavage site in the C-terminus of SAA1 (aa 95) is required for its activation. Furthermore, a recent study indicated that SAA-1525 can be released from a SAA-2126 (aa 104–122) peptide produced by CTSB activity, through hydrolysis of the Asp–Pro bond at the N-terminus of SAA-1525 under neutral and slightly acidic conditions.23 Given the relationship between CTSB and SAA-2126 on SAA-1525 formation, we hypothesized that CTSB activity was responsible for the observed increase in circulating SAA-1525 in Ctox patient samples. We therefore spiked recombinant SAA1 into plasma samples with high (159.94–703.51 ng/mL) and low (25.66–47.00 ng/mL) CTSB concentrations, in marked excess of the native SAA1 concentration, to determine the effect of plasma CTSB concentration on the production of SAA-2126 and SAA-1525 peptides. Both SAA-1525 and SAA-2126 levels were increased in samples with high versus low concentrations of CTSB (Figure 3A,B). However, no difference in SAA-1525 level was detected in samples spiked with a synthetic SAA-2126 peptide (Figure 3C), indicating that SAA-2126 was not the direct precursor to SAA-1525. To confirm that CTSB activity was required for SAA-1525 production, plasma samples with varying levels of CTSB were treated with and without the CTSB specific inhibitors E6424 and ALLM,25 spiked with an excess of recombinant SAA1 protein and analyzed for SAA-1525 production. We found that SAA-1525 release was consistently reduced in all samples treated with either CTSB inhibitor (Figure 3D), indicating that SAA-1525 production was CTSB-dependent.
Figure 3.

SAA-1525 production is CSTB-dependent. ACTH-normalized LC-MS signal for cleavage of recombinant SAA1 to (A) SAA-1525 and (B) SAA-2126 and (C) cleavage of synthetic SAA-2126 peptide to SAA-1525 in human plasma with high and low CTSB levels. Mean ± SE; ****, p < 0.0001 by Student’s t test. (D) Cleavage of SAA1 to SAA-1525 in human plasma treated with or without the CTSB-specific inhibitors E64 and ALLM.
SAA-1525 Correlates More Strongly with Ctox than Does CTSB
To better understand the relationship between SAA-1525 and traditional cardiac injury biomarkers, we analyzed correlations between plasma SAA-1525 and TnI, NT-proBNP, CK, and CK-MB levels, and compared these values to the matching correlations obtained with plasma CTSB (Figure 4, Table S3), despite not having all of these tests performed for each patient’s evaluation. We found that in Chemo patients, the plasma SAA-1525 levels exhibited strong correlation (Figure 4A) with TnI (a heart attack marker), followed by NT-proBNP (a marker of heart failure), CK (a marker of cardiac and skeletal muscle injury) and CK-MB (a more specific marker of cardiac injury). SAA-1525 correlations with all these markers were reduced in the Non-Chemo group. For the plasma CTSB levels, Chemo patients exhibited strong correlations (Figure 4B) with plasma levels of NT-proBNP, CK, and TnI but had less of a correlation with CK-MB, while correlations with all these markers, except CK-MB, were decreased in the Non-Chemo group. NT-proBNP still retained a strong CTSB correlation in the Non-Chemo group, but the TnI correlation was only half that found in the Chemo group, and there was no longer any correlation with CK.
Figure 4.

Radar chart representing the correlation between laboratory indicators and (A) SAA-1525 or (B) CTSB. Each axis displays the laboratory indicators and the values are Spearman r.
Discussion
Early identification of patients at risk for cardiotoxicity remains an elusive but critical goal for cardiologists and oncologists who need definitive tests to help mitigate or prevent long-term cardiac damage. New biomarkers are needed to better identify patients at risk for cardiac damage during Chemo to reduce treatment-related Ctox and to manage long-term cardiac risk in cancer survivors. Monitoring of such biomarker levels may also allow for midtreatment intervention to further improve the personalization of medicine and patient outcomes.
Many pathological processes are associated with aberrant protease expression or activity, which can be detected as altered peptide profiles. Multiple studies have reported that high levels of circulating CTSB are associated with heart disease.26−28 CTSB expression is induced in cardiac myoblasts after doxorubicin treatment29 and is reported to have roles in pressure-overload myocardial remodeling,28 dilated cardiomyopathy,30 and doxorubicin-induced cell death.13 However, CTSB must be activated by post-translational modification; thus, CTSB protein concentrations alone do not necessarily reflect CTSB activity associated with cardiac pathogenesis. Measurement of circulating levels of CTSB-produced peptides offers an attractive means of gauging in vivo protease activity, since these peptides are likely to transit through their tissue of origin and enter the circulation. However, such peptides often circulate at low concentration and may be masked by highly abundant proteins, such as albumin and immunoglobulin.18,31 Serum or plasma peptide isolation thus is a major challenge for biomarker discovery, since no standard technique offers efficient recovery from the complex mixtures found in liquid biopsies. In this study, we used a nanopore-based approach to enrich circulating peptides for MS analysis and identified those that were enriched in Ctox subject samples and could be produced by Ctox-associated proteases to increase the accuracy and sensitivity of biomarker selection.32,33
Our results revealed that a SAA1 peptide was significantly increased in plasma samples of Ctox cases but not in other negative control groups and that production of this peptide from spiked-in SAA1 could be blocked by pretreatment of Ctox plasma samples with CTSB inhibitors. We combined the cardiomyocytes-damage-associated CTSB with enzymatic hydrolysis peptide together and found the novel cancer-drug-induced cardiotoxicity biomarker SAA-1525. Our preliminary results demonstrate, to our knowledge for the first time, that the proteolytic activity of CTSB is clearly linked to the cleavage patterns of its catalytic substrate SAA1 in the blood and was determined by means of a rapid, reproducible, sensitive, precise, and minimally invasive approach.
As previously described, SAA1 belongs to the class of acute-phase proteins, those that are induced by inflammation-related conditions such as infection and malignancy.21 SAA1 expression is not specific, however, and did not significantly differ among the study cohorts, unlike its cleavage product, SAA-1525. Plasma levels of both CTSB and SAA-1525 are distinct between Ctox and nCtox patients but not Cdam and nCdam patients. Further studies are needed to analyze the relative merits of measuring CTSB and SAA-1525 for Ctox prediction. However, we hypothesize that SAA-1525 will more accurately reflect CTSB-mediated activity associated with cardiac injury than will the concentration of circulating CTSB, since CTSB inhibitors in plasma may reduce the correlation between CTSB abundance and activity and thus diminish its effectiveness as a biomarker. In addition to providing more accurate patient information, SAA-1525 measurement may also offer advantages from a workflow perspective, since the current approach requires only a 30 min nanotrap isolation step prior to analysis by LC-MS/MS, which may compare favorably to an immunoassay-based analysis of total plasma CTSB abundance. It does not appear that the enhanced correlation of SAA-1525 with cTnI detected in the Chemo group reflects inflammation-related cardiac injury that is indirectly related to chemotherapy, since similar levels of inflammation were observed in both the Chemo and Non-Chemo groups. We therefore propose that proteolysis of SAA1 by CTSB is the critical factor that regulates the differential SAA-1525 levels detected in these groups.
Chemotherapy drugs, including those of the anthracycline class, are the agents most frequently implicated in associated cardiotoxic side effects.34 A wide range of chemotherapy drugs have reported associations with cardiotoxicity,35 including antimetabolite agents,36 microtubule-targeting drugs,37 and protein kinase inhibitors.38 There is thus an urgent clinical demand for a marker that can be serially monitored to predict the development of cardiotoxicity in response to a variety of different chemotherapeutics. Our results suggest that circulating SAA 1525 has the potential to address this unmet clinical requirement. Our results indicate that SAA 1525 is produced by proteolysis of circulating SAA1 by CTSB. We speculate chemotherapy resulting in cardiotoxicity may increase the activity, expression, or release of CTSB from injured cardiomyocytes to increase the hydrolysis of circulating SAA1 and thereby increase circulating SAA-1525 levels. Several studies support the potential for such a mechanism. The chemotherapeutic nilotinib is reported to induce cardiotoxicity by increasing CTSB activity,39 while bosutinib40 has been reported to induce permeabilization of the lysosomal membrane, leading to lysosomal dysfunction and the release of CTSB. Both drugs were administered to individuals in the Ctox group. Other commonly used chemotherapeutics, including gemcitabine and 5-fluorouracil, can also trigger CTSB release by lysosomal permeabilization.12 In summary, accumulated evidence suggests that cardiomyocyte expression of CTSB can be induced by various chemotherapeutics. Further studies may thus be useful to determine if altered CTSB expression, activation, or release represents a common mechanism associated with chemotherapeutics that are associated with enhanced levels of chemotherapy. A better understanding of the responsible mechanism might allow for improved screening of the cardiotoxic potential of future chemotherapeutics. Knowledge of which drugs can induce cardiotoxicity through CTSB dysregulation would also provide evidence for the utility of screening these patients for altered SAA-1525 to detect the development of cardiotoxicity.
Our results imply that plasma SAA-1525 is specifically elevated in the chemotherapy-induced cardiotoxicity group independent of potential inflammation-induced cardiac damage as assessed by the level of circulating hs-CRP, through the proteolytic activity of CTSB. This suggests that plasma SAA-1525 may have great potential as a rapid and minimally invasive predictive marker for chemotherapy-related cardiotoxicity. However, further studies are required to reproduce our results and assess the predictive utility of SAA1 for cardiotoxicity in studies using specific therapeutic regimens in different patient populations. Multiple treatment regimens (chemotherapy and/or chemoradiation) were included in this pilot study; however, due to the small group sizes, we were unable to stratify blood SAA-1525 levels as a predictive marker for response to individual treatment regimens or to generalize these levels for the response to all cardiotoxicity agents temporarily. Larger prospective cohorts, with well-defined treatment regimens, should be analyzed to address these questions in the future work, which may have major clinical significance.
Methods
Study Population
Plasma samples were collected at M. D. Anderson Center Cancer in Houston, TX, from July 2014 to August 2014 from cancer patients who received or declined to receive chemotherapy and who were provided written informed consent to participate in this study, using an Institutional Review Board approved study participant enrollment form. Patients were excluded from study enrollment if they were <18 years of age or if they had an LVEF of <50%, valvular heart disease, or severe hypertension. Study subjects with various tumor types were consecutively enrolled at first diagnosis and segregated into chemotherapy (Chemo) and nonchemotherapy (Non-Chemo) groups based on their decision to accept or refuse chemotherapy treatment. Chemo subjects were assigned to the cardiotoxicity (Ctox) cohort if their plasma TnI levels were >0.04 ng/mL41 or if their plasma NT-proBNP levels were >450, >900, or >1800 pg/mL in patients of ages <50, 50–75, and >75 years, respectively.42,43 All other Chemo patients were classified into the noncardiotoxicity (nCtox) cohort. Non-Chemo group subjects were similarly divided into cohorts exhibiting cardiac damage (Cdam) or no cardiac damage (nCdam) cohorts using the same TnI and NT-proBNP criteria.
Nanotrap Fractionation of Low-Molecular Weight Plasma Peptides
Plasma samples of study participants were fractionated on mesoporous silica nanotraps44 to isolate low-molecular-weight (<10 kDa) peptides (Figure 1), which were then identified and quantified by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). Briefly, individual wells on nanotrap-coated, 4 in. silicon wafers were loaded with 6 μL of human plasma. After 30 min of incubation at 25 °C in a humidified chamber, each well was aspirated and washed with 10 μL of deionized water. The peptides retained in the nanotrap were then released with 90 s of incubation with 6 μL of a 0.1% trifluoroacetic acid/50% acetonitrile solution. MALDI-TOF MS data were analyzed with FlexAnalysis software v1.33 (Bruker Daltonic, Billerica, MA). The mean area of selected monoisotopic peaks was determined from duplicate samples after normalization against a spiked-in calibration standard peptide consisting of ACTH fragment with a length of 18–39 amino acids (aa).45
Peptide Identification by LC-MS/MS
Nanotrap-enriched plasma peptide samples were dried by vacuum centrifugation, suspended in 0.1% formic acid, and then subjected to LC-MS/MS using an Orbitrap Velos Pro MS instrument coupled with an UltiMate 3000 NanoLC system (Thermo Scientific). Loaded peptides were fractionated with a linear gradient of 2–37% acetonitrile with 0.1% formic acid at a flow rate of 300 nL/min on an EASY-Spray C18 LC column (15 cm × 75 μm I.D. and 3 μm particle size; Thermo Scientific), and the resulting MS/MS spectra were analyzed against the UniProt (www.uniprot.org) human proteome database using Mascot Server 2.3.0 (Matrix Science, London, UK) and a mass tolerance of 0.5 Da.
We identified peptides that were selectively enriched (>2-fold) in the Ctox versus nCtox plasma samples (Table S1) and employed one-way ANOVA to select only those peptides where this difference reached statistical significance (p < 0.01). To identify peptides that might arise from CTSB activity we used the MEROPS database (https://www.ebi.ac.uk/merops/cgi-bin/substrates?id=C01.060) to analyze proteins that matched the enriched peptides to determine if CTSB was known or predicted to produce the target peptides.
Measurement of Plasma CTSB Concentration and Activity
Plasma CTSB concentrations were analyzed with commercial ELISA kits (R&D Systems, Inc.) using 10 μL of patient plasma. CTSB serum amyloid A1 (SAA1) substrates24 were identified by a search of the MEROPS database. An MS-identified SAA1 peptide (SAA-2126) that was produced in response to CTSB activity was synthesized by GL Biochem. MALDI-TOF MS was used to measure CTSB activity on SAA1 protein and SAA-2126 peptide substrates spiked into plasma samples with high (159.94–703.51 ng/mL) and low (25.66–47.00 ng/mL) CTSB protein concentrations. Samples were spiked with 8.5 uM SAA1 or 10 uM SAA-2126 and analyzed for their SAA-1525 and SAA-2126 levels. Plasma SAA-1525 concentrations were also measured in samples incubated with or without the CTSB inhibitors E64 or ALLM (25 mM, Sigma-Aldrich).24,25
Statistical Analysis
Mass spectrometry data was analyzed with MarkerView software version 1.2.1 (AB SCIEX, Concord, Canada), and the spiked-in peptide calibration standard (ACTH aa 18–39; 2465 m/z) was used to normalize peptide signal intensity to identify peptides that exhibited a ≥2-fold increase in Ctox than nCtox group. The abundance of these target peptides was then analyzed by one-way ANOVA to identify peptides that significantly differed (p-value < 0.01) between the Ctox and nCtox groups but not between the Cdam and nCdam groups. Statistical analyses (1-way ANOVA, 2-way ANOVA, Chi-square tests, receiver operating characteristic (ROC), and correlation curve analyses) were performed using GraphPad Prism 7 software (GraphPad Software, Inc.). Age, NT-proBNP, TnI, CK, CK-MB, and peptide differences between subgroups were analyzed by 1-way ANOVA, with p-values < 0.05 considered to be statistically significant. Gender differences between groups were analyzed by χ2 tests (GraphPad Prism 7 software) and verified by binary logistic regression (SPSS 17.0 software, USA). Data in Table 1 were expressed as medians with the interquartile range (IQR).
Acknowledgments
This work was partially supported by research funding to T.Y.H. from the NIH (U01CA214254, R01HD090927, R01AI122932, R01AI113725, and R21Al126361-01) and the Arizona Biomedical Research Commission (ABRC) young investigator award and by research funding to J.F. from the Fred Hutchinson Cancer Research Center (0000917241) and the NIH (R03AI140977). Dr. Qing He Meng at The University of Texas M. D. Anderson Cancer Center’s Department of Laboratory Medicine generously provided all clinical samples analyzed in this study.
Glossary
Abbreviations
- SAA-1525
serum amyloid A1 peptide, 1525 m/z
- LVEF
left ventricular ejection fraction
- TnI
troponin I
- NT-proBNP
N-terminal pro-B-type natriuretic peptide
- CK
creatine kinase
- CK-MB
creatine kinase-muscle/brain
- CTSB
cathepsin B
- Chemo
chemotherapy
- Non-Chemo
nonchemotherapy
- Ctox
cardiotoxicity
- nCtox
noncardiotoxicity
- Cdam
cardiac damage
- nCdam
no cardiac damage
- MALDI-TOF
MS matrix-assisted laser desorption/ionization time-of-flight mass spectrometry
- SAA1
serum amyloid A1
- ROC
receiver operating characteristic
- IQR
interquartile range
- MMP1
matrix metalloprotease 1
- MMP3
matrix metalloprotease 3
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsptsci.9b00035.
Author Contributions
T.Y.H. and J.F. designed the study. J.F. and F.Z. performed the experiment. F.Z., R.J.W., T.Y.H., J.F., C.J.L., and B.N. analyzed data. B.N. created the TOC. F.Z., C.J.L., B.N., T.Y.H., and J.F. wrote the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Xu J.; Murphy S. L.; Kochanek K. D.; Bastian B. A. (2016) Deaths: Final Data for 2013. National vital statistics reports: from the Centers for Disease Control and Prevention, National Center for Health Statistics. National Vital Statistics System 64 (2), 1–119. [PubMed] [Google Scholar]
- Chen J.; Long J. B.; Hurria A.; Owusu C.; Steingart R. M.; Gross C. P. (2012) Incidence of heart failure or cardiomyopathy after adjuvant trastuzumab therapy for breast cancer. J. Am. Coll. Cardiol. 60 (24), 2504–12. 10.1016/j.jacc.2012.07.068. [DOI] [PubMed] [Google Scholar]
- Minichillo S.; Gallelli I.; Barbieri E.; Cubelli M.; Rubino D.; Quercia S.; Dall’Olio M.; Rapezzi C.; Zamagni C. (2017) Trastuzumab resumption after extremely severe cardiotoxicity in metastatic breast cancer patient: a case report. BMC Cancer 17 (1), 722. 10.1186/s12885-017-3712-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. Y.; Yin B. B.; Jia D. Y.; Hou Y. L. (2017) Association between obesity and trastuzumab-related cardiac toxicity in elderly patients with breast cancer. Oncotarget 8 (45), 79289–79297. 10.18632/oncotarget.17808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felker G. M.; Thompson R. E.; Hare J. M.; Hruban R. H.; Clemetson D. E.; Howard D. L.; Baughman K. L.; Kasper E. K. (2000) Underlying Causes and Long-Term Survival in Patients with Initially Unexplained Cardiomyopathy. N. Engl. J. Med. 342 (15), 1077–1084. 10.1056/NEJM200004133421502. [DOI] [PubMed] [Google Scholar]
- Bovelli D.; Plataniotis G.; Roila F. (2010) Cardiotoxicity of chemotherapeutic agents and radiotherapy-related heart disease: ESMO Clinical Practice Guidelines. Annals of Oncology 21, v277–v282. 10.1093/annonc/mdq200. [DOI] [PubMed] [Google Scholar]
- Wouters K. A.; Kremer L. C. M.; Miller T. L.; Herman E. H.; Lipshultz S. E. (2005) Protecting against anthracycline-induced myocardial damage: a review of the most promising strategies. Br. J. Haematol. 131 (5), 561–578. 10.1111/j.1365-2141.2005.05759.x. [DOI] [PubMed] [Google Scholar]
- Ganz W. I.; Sridhar K. S.; Ganz S. S.; Gonzalez R.; Chakko S.; Serafini A. (2004) Review of Tests for Monitoring Doxorubicin-lnduced Cardiomyopathy. Oncology 53 (6), 461–470. 10.1159/000227621. [DOI] [PubMed] [Google Scholar]
- Steinherz L. J.; Graham T.; Hurwitz R.; Sondheimer H. M.; Schwartz R. G.; Shaffer E. M.; Sandor G.; Benson L.; Williams R. (1992) Guidelines for Cardiac Monitoring of Children During and After Anthracycline Therapy: Report of the Cardiology Committee of the Childrens Cancer Study Group. Pediatrics 89 (5), 942–949. [PubMed] [Google Scholar]
- Mueller M.; Biener M.; Vafaie M.; Doerr S.; Keller T.; Blankenberg S.; Katus H. A.; Giannitsis E. (2012) Absolute and Relative Kinetic Changes of High-Sensitivity Cardiac Troponin T in Acute Coronary Syndrome and in Patients with Increased Troponin in the Absence of Acute Coronary Syndrome. Clin. Chem. 58 (1), 209–218. 10.1373/clinchem.2011.171827. [DOI] [PubMed] [Google Scholar]
- Apple F. S.; Jesse R. L.; Newby L. K.; Wu A. H.B.; Christenson R. H.; Christenson R. H.; Apple F. S.; Cannon C. P.; Francis G.; Jesse R.; et al. (2007) National Academy of Clinical Biochemistry and IFCC Committee for Standardization of Markers of Cardiac Damage Laboratory Medicine Practice Guidelines: analytical issues for biochemical markers of acute coronary syndromes. Clin. Chem. 53 (4), 547–551. 10.1373/clinchem.2006.084715. [DOI] [PubMed] [Google Scholar]
- Bruchard M.; Mignot G.; Derangere V.; Chalmin F.; Chevriaux A.; Vegran F.; Boireau W.; Simon B.; Ryffel B.; Connat J. L.; Kanellopoulos J.; Martin F.; Rebe C.; Apetoh L.; Ghiringhelli F. (2013) Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 19 (1), 57–64. 10.1038/nm.2999. [DOI] [PubMed] [Google Scholar]
- Bien S.; Rimmbach C.; Neumann H.; Niessen J.; Reimer E.; Ritter C. A.; Rosskopf D.; Cinatl J.; Michaelis M.; Schroeder H. W.; Kroemer H. K. (2010) Doxorubicin-induced cell death requires cathepsin B in HeLa cells. Biochem. Pharmacol. 80 (10), 1466–77. 10.1016/j.bcp.2010.07.036. [DOI] [PubMed] [Google Scholar]
- Cheng X. W.; Obata K.; Kuzuya M.; Izawa H.; Nakamura K.; Asai E.; Nagasaka T.; Saka M.; Kimata T.; Noda A.; Nagata K.; Jin H.; Shi G. P.; Iguchi A.; Murohara T.; Yokota M. (2006) Elastolytic cathepsin induction/activation system exists in myocardium and is upregulated in hypertensive heart failure. Hypertension 48 (5), 979–87. 10.1161/01.HYP.0000242331.99369.2f. [DOI] [PubMed] [Google Scholar]
- Cheng X. W.; Murohara T.; Kuzuya M.; Izawa H.; Sasaki T.; Obata K.; Nagata K.; Nishizawa T.; Kobayashi M.; Yamada T.; Kim W.; Sato K.; Shi G. P.; Okumura K.; Yokota M. (2008) Superoxide-dependent cathepsin activation is associated with hypertensive myocardial remodeling and represents a target for angiotensin II type 1 receptor blocker treatment. Am. J. Pathol. 173 (2), 358–69. 10.2353/ajpath.2008.071126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann M.; Streb C. (2006) Selective oxidation of indole by chloroperoxidase immobilized on the mesoporous molecular sieve SBA-15. J. Porous Mater. 13 (3–4), 347–352. 10.1007/s10934-006-8029-y. [DOI] [Google Scholar]
- Liotta L. A.; Ferrari M.; Petricoin E. (2003) Clinical proteomics: written in blood. Nature 425 (6961), 905. 10.1038/425905a. [DOI] [PubMed] [Google Scholar]
- Li Y.; Li Y.; Chen T.; Kuklina A. S.; Bernard P.; Esteva F. J.; Shen H.; Ferrari M.; Hu Y. (2014) Circulating proteolytic products of carboxypeptidase N for early detection of breast cancer. Clin. Chem. 60 (1), 233–42. 10.1373/clinchem.2013.211953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myrehaug S.; Pintilie M.; Yun L.; Crump M.; Tsang R. W.; Meyer R. M.; Sussman J.; Yu E.; Hodgson D. C. (2010) A population-based study of cardiac morbidity among Hodgkin lymphoma patients with preexisting heart disease. Blood 116 (13), 2237–40. 10.1182/blood-2010-01-263764. [DOI] [PubMed] [Google Scholar]
- Wu H. J.; Li Y.; Fan J.; Deng Z.; Hu Z.; Liu X.; Graviss E. A.; Ferrari M.; Ma X.; Hu Y. (2014) Antibody-free detection of Mycobacterium tuberculosis antigen using customized nanotraps. Anal. Chem. 86 (4), 1988–96. 10.1021/ac4027669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabay C.; Kushner I. (1999) Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 340 (6), 448–54. 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- Lu J.; Yu Y.; Zhu I.; Cheng Y.; Sun P. D. (2014) Structural mechanism of serum amyloid A-mediated inflammatory amyloidosis. Proc. Natl. Acad. Sci. U. S. A. 111 (14), 5189–94. 10.1073/pnas.1322357111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spodzieja M.; Rafalik M.; Szymańska A.; Kołodziejczyk A. S.; Czaplewska P. (2013) Interaction of serum amyloid A with human cystatin C—assessment of amino acid residues crucial for hCC–SAA formation (part II). J. Mol. Recognit. 26 (9), 415–425. 10.1002/jmr.2283. [DOI] [PubMed] [Google Scholar]
- Rocken C.; Menard R.; Buhling F.; Vockler S.; Raynes J.; Stix B.; Kruger S.; Roessner A.; Kahne T. (2005) Proteolysis of serum amyloid A and AA amyloid proteins by cysteine proteases: cathepsin B generates AA amyloid proteins and cathepsin L may prevent their formation. Ann. Rheum. Dis. 64 (6), 808–15. 10.1136/ard.2004.030429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T.; Kishi M.; Saito M.; Tanaka T.; Higuchi N.; Kominami E.; Katunuma N.; Murachi T. (1990) Inhibitory effect of di- and tripeptidyl aldehydes on calpains and cathepsins. J. Enzyme Inhib. 3 (3), 195–201. 10.3109/14756369009035837. [DOI] [PubMed] [Google Scholar]
- Blondelle J.; Lange S.; Greenberg B. H.; Cowling R. T. (2015) Cathepsins in heart disease-chewing on the heartache?. American journal of physiology. Heart and circulatory physiology 308 (9), H974–6. 10.1152/ajpheart.00125.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra S.; Kumar M.; Manchanda M.; Singh R.; Thakur B.; Rani N.; Arava S.; Narang R.; Arya D. S.; Chauhan S. S. (2017) Clinical significance of cathepsin L and cathepsin B in dilated cardiomyopathy. Mol. Cell. Biochem. 428 (1–2), 139–147. 10.1007/s11010-016-2924-6. [DOI] [PubMed] [Google Scholar]
- Wu Q. Q.; Xu M.; Yuan Y.; Li F. F.; Yang Z.; Liu Y.; Zhou M. Q.; Bian Z. Y.; Deng W.; Gao L.; Li H.; Tang Q. Z. (2015) Cathepsin B deficiency attenuates cardiac remodeling in response to pressure overload via TNF-alpha/ASK1/JNK pathway. American journal of physiology. Heart and circulatory physiology 308 (9), H1143–54. 10.1152/ajpheart.00601.2014. [DOI] [PubMed] [Google Scholar]
- Bao G. Y.; Wang H. Z.; Shang Y. J.; Fan H. J.; Gu M. L.; Xia R.; Qin Q.; Deng A. M. (2012) Quantitative proteomic study identified cathepsin B associated with doxorubicin-induced damage in H9c2 cardiomyocytes. BioSci. Trends 6 (6), 283–287. 10.5582/bst.2012.v6.6.283. [DOI] [PubMed] [Google Scholar]
- Ge J.; Zhao G.; Chen R.; Li S.; Wang S.; Zhang X.; Zhuang Y.; Du J.; Yu X.; Li G.; Yang Y. (2006) Enhanced myocardial cathepsin B expression in patients with dilated cardiomyopathy. Eur. J. Heart Failure 8 (3), 284–9. 10.1016/j.ejheart.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Villanueva J.; Shaffer D. R.; Philip J.; Chaparro C. A.; Erdjument-Bromage H.; Olshen A. B.; Fleisher M.; Lilja H.; Brogi E.; Boyd J.; et al. (2005) Differential exoprotease activities confer tumor-specific serum peptidome patterns. J. Clin. Invest. 116 (1), 271–284. 10.1172/JCI26022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouamrani A.; Hu Y.; Tasciotti E.; Li L.; Chiappini C.; Liu X.; Ferrari M. (2010) Mesoporous silica chips for selective enrichment and stabilization of low molecular weight proteome. Proteomics 10 (3), 496–505. 10.1002/pmic.200900346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Bouamrani A.; Tasciotti E.; Li L.; Liu X.; Ferrari M. (2010) Tailoring of the nanotexture of mesoporous silica films and their functionalized derivatives for selectively harvesting low molecular weight protein. ACS Nano 4 (1), 439–51. 10.1021/nn901322d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen P. A. (2018) Anthracycline cardiotoxicity: an update on mechanisms, monitoring and prevention. Heart (London, U. K.) 104 (12), 971–977. 10.1136/heartjnl-2017-312103. [DOI] [PubMed] [Google Scholar]
- Rhea I. B.; Oliveira G. H. (2018) Cardiotoxicity of Novel Targeted Chemotherapeutic Agents. Current treatment options in cardiovascular medicine 20 (7), 53. 10.1007/s11936-018-0649-4. [DOI] [PubMed] [Google Scholar]
- Jensen S. A.; Hasbak P.; Mortensen J.; Sorensen J. B. (2010) Fluorouracil Induces Myocardial Ischemia With Increases of Plasma Brain Natriuretic Peptide and Lactic Acid but Without Dysfunction of Left Ventricle. J. Clin. Oncol. 28 (36), 5280–5286. 10.1200/JCO.2009.27.3953. [DOI] [PubMed] [Google Scholar]
- Cargill R. I.; Boyter A. C.; Lipworth B. J. (1994) Reversible myocardial ischaemia following vincristine containing chemotherapy. Respir Med. 88 (9), 709–10. 10.1016/S0954-6111(05)80074-5. [DOI] [PubMed] [Google Scholar]
- Kim D. W.; Tiseo M.; Ahn M. J.; Reckamp K. L.; Hansen K. H.; Kim S. W.; Huber R. M.; West H. L.; Groen H. J. M.; Hochmair M. J.; et al. (2017) Brigatinib in Patients With Crizotinib-Refractory Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer: A Randomized, Multicenter Phase II Trial. J. Clin. Oncol. 35 (22), 2490–2498. 10.1200/JCO.2016.71.5904. [DOI] [PubMed] [Google Scholar]
- Yang Q.; Zhang C.; Wei H.; Meng Z.; Li G.; Xu Y.; Chen Y. (2017) Caspase-Independent Pathway is Related to Nilotinib Cytotoxicity in Cultured Cardiomyocytes. Cell. Physiol. Biochem. 42 (6), 2182–2193. 10.1159/000479993. [DOI] [PubMed] [Google Scholar]
- Noguchi S.; Shibutani S.; Fukushima K.; Mori T.; Igase M.; Mizuno T. (2018) Bosutinib, an SRC inhibitor, induces caspase-independent cell death associated with permeabilization of lysosomal membranes in melanoma cells. Vet. Comp. Oncol. 16 (1), 69–76. 10.1111/vco.12312. [DOI] [PubMed] [Google Scholar]
- Melanson S. E.; Morrow D. A.; Jarolim P. (2007) Earlier detection of myocardial injury in a preliminary evaluation using a new troponin I assay with improved sensitivity. Am. J. Clin. Pathol. 128 (2), 282–6. 10.1309/Q9W5HJTT24GQCXXX. [DOI] [PubMed] [Google Scholar]
- Januzzi J. L.; van Kimmenade R.; Lainchbury J.; Bayes-Genis A.; Ordonez-Llanos J.; Santalo-Bel M.; Pinto Y. M.; Richards M. (2006) NT-proBNP testing for diagnosis and short-term prognosis in acute destabilized heart failure: an international pooled analysis of 1256 patients: the International Collaborative of NT-proBNP Study. Eur. Heart J. 27 (3), 330–7. 10.1093/eurheartj/ehi631. [DOI] [PubMed] [Google Scholar]
- Weber M.; Hamm C. (2005) Role of B-type natriuretic peptide (BNP) and NT-proBNP in clinical routine. Heart (London, U. K.) 92 (6), 843–849. 10.1136/hrt.2005.071233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Zhao Z.; Fan J.; Lyon C. J.; Wu H. J.; Nedelkov D.; Zelazny A. M.; Olivier K. N.; Cazares L. H.; Holland S. M.; Graviss E. A.; Hu Y. (2017) Quantification of circulating Mycobacterium tuberculosis antigen peptides allows rapid diagnosis of active disease and treatment monitoring. Proc. Natl. Acad. Sci. U. S. A. 114 (15), 3969–3974. 10.1073/pnas.1621360114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buskirk A. D.; Hettick J. M.; Chipinda I.; Law B. F.; Siegel P. D.; Slaven J. E.; Green B. J.; Beezhold D. H. (2011) Fungal pigments inhibit the matrix-assisted laser desorption/ionization time-of-flight mass spectrometry analysis of darkly pigmented fungi. Anal. Biochem. 411 (1), 122–8. 10.1016/j.ab.2010.11.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.