

Abstract
During 2008–2009, fifteen field infectious bronchitis viruses (IBVs) were isolated from commercial chicken farms in Thailand. After sequencing of the complete S1 gene, phylogenetic analysis was performed and this found that the Thai IBV isolates were divided into three distinct groups, unique to Thailand (group I), QX-like IBV (group II), and Massachusetts type (group III). This finding indicated that the recent Thai IBVs evolved separately and that at least three groups of viruses are circulating in Thailand. The recombination analysis of the S1 gene demonstrated that the 5′-terminus of the group I was similar to isolate THA001 which was unique to Thailand, isolated in 1998 whereas the 3′-terminus was similar to the group II. Moreover, the analysis of the S1 gene of the group II showed that the 5′-terminus was similar to QXIBV, isolated in China whereas the remaining region at the 3′-terminus was similar to the Chinese strain JX/99/01. The results indicated that the recombination events occurred in the S1 gene between the field strains. Based on these facts, the field IBV in Thailand has undergone genetic recombination.
Keywords: Infectious bronchitis virus, S1 gene, Recombination
Introduction
Infectious bronchitis (IB) is an acute viral respiratory disease of chickens and results in a significant economic loss to commercial chicken industries in many countries of the world. The disease is characterized by respiratory signs including gasping, coughing, sneezing, tracheal rales, and nasal discharge [1]. All ages of chickens are susceptible to IBV infection, but the clinical signs are more severe in young chickens [2]. In hens, respiratory distress and a decrease in egg production have been reported [3]. Some strains of IBV can cause acute nephritis and urolithiasis associated with a high mortality of infected chickens [4, 5]. In addition, IBV has also been reported to cause proventriculitis [6]. Furthermore, the disease is a risk factor for secondary bacterial infections resulting in an even higher morbidity and mortality rate [5].
Infectious bronchitis virus (IBV), the causative agent of IB, is a coronavirus. The genome of IBV consists of positive sense single stranded RNA, approximately 27.6 kilobases in length [7], that is encoded for four structural proteins: nucleocapsid (N) protein, envelope (E) protein, membrane (M) glycoprotein, and spike (S) glycoprotein [8]. The S glycoprotein is post-translationally cleaved into the S1 and S2 subunits [9]. The S1 subunit, located on the outside of virion, is responsible for the fusion between the virus envelope and the cell membrane of the host [7]. It contains virus neutralization and serotype-specific epitopes that are formed by amino acid within the defined hypervariable region (HVR); therefore, the molecular characterization of IBV is based on an analysis of the S1 gene [10].
The continuous emergence of new serotypes or variant strains of IBV has been reported world-wide [3, 4, 11–13]. The events are thought to be generated by mutation processes including deletion and insertion of the nucleotides within IBV genome; moreover, the evolution by genetic recombination has also been reported [12]. The new serotypes or variant strains of IBV can cause disease in vaccinated chickens [3, 4, 11]. Therefore, these emergences are of great concern to poultry producers.
In Thailand, the outbreak of IB was initially reported during 1953–1954 [14]. Since then, IB has continued to be an economically important disease in the Thai poultry industry and can be found all over the country [15, 16]. Recently, we characterized IBV isolated in Thailand from January to June 2008 by analysis of the HVR of S1 genes and found that the Thai IBV isolates were divided into two groups, QX-like IBV and that unique to the Thai strain [17]. In this study, the objective was to determine the genetic variation exhibited within Thai IBV isolates in 2008–2009 by analysis of the complete S1 genes and comparing them with previously published strains.
Materials and methods
Viruses
Fifteen Thai IBV isolates used in this study are listed in Table 1. All of them were isolated from commercial poultry farms in Thailand which had been experiencing of respiratory disease between January 2008 and October 2009. All flocks had been vaccinated against IB with commercial live attenuated H120.
Table 1.
Spike glycoprotein cleavage recognition sites of Thai IBV isolates
| IBV isolates | Group of isolate | Years of isolation | Cleavage sites | Accession number |
|---|---|---|---|---|
| THA40151 | I | 2008 | Arg–Arg–His–Arg–Arg | GQ503612 |
| THA50151 | I | 2008 | Arg–Arg–His–Arg–Arg | GQ503613 |
| THA60151 | I | 2008 | Arg–Arg–His–Arg–Arg | GQ503614 |
| THA80151 | II | 2008 | Arg–Arg–His–Arg–Arg | GQ503616 |
| THA90151 | I | 2008 | Arg–Arg–His–Arg–Arg | GQ503617 |
| THA171051 | II | 2008 | Arg–Arg–His–Arg–Arg | GQ885126 |
| THA211051 | II | 2008 | Arg–Arg–His–Arg–Arg | GQ885128 |
| THA241251 | III | 2008 | Arg–Arg–Phe–Arg–Arg | GQ885131 |
| THA260152 | II | 2009 | Arg–Arg–His–Arg–Arg | GQ885133 |
| THA280252 | III | 2009 | Arg–Arg–Phe–Arg–Arg | GQ885134 |
| THA290252 | III | 2009 | Arg–Arg–Phe–Arg–Arg | GQ885135 |
| THA310252 | II | 2009 | Arg–Arg–His–Arg–Arg | GQ885137 |
| THA320352 | III | 2009 | Arg–Arg–Phe–Arg–Arg | GQ885138 |
| THA351052 | II | 2009 | Arg–Arg–His–Arg–Arg | GU111581 |
| THA361052 | II | 2009 | Arg–Arg–His–Arg–Arg | GU111582 |
Arg arginine, Phe phenylalanine, His histidine
Virus isolation and RNA extraction
Before the virus isolation, all of the samples were screened and had positive results with nested-PCR for the presence of IBV described by Pohuang et al. [17]. In brief, the trachea and lung samples were taken from pools of chickens from the same farm. The samples were prepared as 10% w/v suspensions in phosphate-buffered saline (pH 7.4) and centrifuged at 1,800×g for 10 min. The supernatants were then collected for RNA extraction using Viral Nucleic Acid Extraction Kit (Real Biotech, Taiwan) following the manufacturer’s instructions. The extracted RNA was subjected to nested-PCR using the primer sets and reaction conditions described previously [17]. The supernatants of IBV-positive samples were inoculated into 9–11-day-old embryonated chicken eggs. Each egg received 0.2 ml of the supernatant. The inoculated eggs were incubated at 37°C and candled daily. After 96 h post-inoculation, allantoic fluids were harvested. A further blind serial passage was performed in a similar way. All of the allantoic fluids were harvested and stored at –70°C. Then, the allantoic fluids were used for RNA extraction as described above.
Primers and RT-PCR amplification
The primer sets used in this study were newly designed to amplify the full length of the S1 gene. Two sets of primers were used and the primer sequences were as follows: the primers used for amplification of the 5′ half of the S1 gene were 5′ GCCAGTTGTTAATTTGAAAAC 3′ and 5′ TAATAACCACTCTGAGCTGT 3′, and the primers used for amplification of the 3′ half of S1 gene were 5′ ACTGGCAATTTTTCAGATGG 3′ and 5′ AACTGTTAGGTATGAGCACA 3′. The reverse transcription-polymerase chain reaction (RT-PCR) was accomplished in one step by using the AccessQuick RT-PCR System (Promega, Madison, WI, USA). RT was performed at 48°C for 45 min and heating at 94°C for 5 min. PCR was then performed by 35 cycles of denaturation at 94°C for 60 s, annealing at 54°C for 30 s, extension at 72°C for 60 s, and final extension at 72°C for 10 min. The PCR product was analyzed by electrophoresis on 1.2% agarose gel, followed by staining with ethidium bromide (0.5 μg/ml) and then was visualized by using an ultraviolet transilluminator.
Product purification and sequencing
The RT-PCR products were cut from the gel and purified using the Wizard SV Gel and PCR Clean-Up system (Promega, Madison, WI, USA) as the protocols suggested by the manufacturer. The purified RT-PCR products were sequenced in both of the forward and the reverse direction by commercial service (First Base, Selangor, Malaysia).
Sequence and phylogenetic analysis
The nucleotide sequences of the S1 gene from the ATG start site to the cleavage recognition site of fifteen Thai IBV isolates were assembled, aligned, and compared with published IBV sequences deposited in the GenBank database. The first time they were compared with published IBV sequences deposited in the GenBank database using a BLAST search via the National Center of Biotechnology Information (http://www.ncbi.nlm.nih.gov/BLAST/). Sequence identities by BLAST analysis were included in the alignment and phylogenetic construction. The multiple sequence alignments and determination of the nucleotide and amino acid identities were performed using BioEdit version 7.0.5.2 [18]. A phylogenetic tree of the nucleotide sequences was constructed with the neighbor-joining method using MEGA version 4 [19]. The bootstrap values were determined from 1000 replicates of the original data. The S1 gene sequences of the fifteen IBV isolates were submitted to the GenBank database (Table 1). The other S1 gene sequences from the GenBank database which were used for comparison or phylogenetic analysis in this study included M41 (AY561711), Ma5 (AY561713), H120 (M21970), H52 (AF352315), Spain/98/308 (DQ064807), Beuadette (DQ001334), JMK (L14070), QXIBV (AF193423), LX4 (AY338732), LS2 (AY278246), BJ (AY319651), CK/CH/LTJ/95I (DQ167151), CK/CH/LAH/99I (DQ167129), JX/99/01 (AF210735), MH5365/95 (EU086600), A1211 (AF250006), A1171 (AF250005), 3385/06 (GQ229247), 3376/06 (GQ229244), CK/CH/Guangxi/Luchan2/0910 (GU938404), Spain/98/315 (DQ386095) CK/CH/Fujian/Putian3/0910 (GU938410), 4/91 (AF093794), UK2/91 (Z83976), Ark DPI (AF006624), Australian T (AY775779), N1/62 (AIU29522), Armidale (DQ490205), THA001 (GQ906705).
Recombination detection
Putative recombinant sequence and its parental strains were identified with Simplot version 3.5.1 [20]. The nucleotide identity was performed by using Kimura (2-parameter) method with a transition-transversion ratio of 2. The window width and the step size were 200 and 20 bp, respectively. Bootscan analysis was also carried out employing subprogram embedded in SimPlot, using the signals of 70% or more of the observed permuted trees for indication of the potential recombination events [21]. Recombination breakpoints were analyzed by maximization of χ2 using the program Findsites included in the SimPlot [22].
Results
Phylogenetic analysis
A phylogenetic tree was constructed using the nucleotide sequences of the S1 genes (position from ATG start site to the cleavage recognition site) of the field Thai IBV isolates and the GenBank deposited sequences. Fifteen Thai IBV isolates were separated into three distinct groups (Fig. 1). Group I consisted of five isolates including THA40151, THA50151, THA60151, and THA90151. The isolates in group I showed evolutionary distances from each other and this group was unique to Thailand. Group II consisted of twenty isolates including THA80151, THA171051, THA211051, THA260152, THA310252, THA351052, and THA361052 and had a close relationship with Chinese QXIBV. Group III consisted of seven isolates including THA241251, THA 280252, THA290252, and THA320352 and had a close relationship with the Massachusetts type.
Fig. 1.
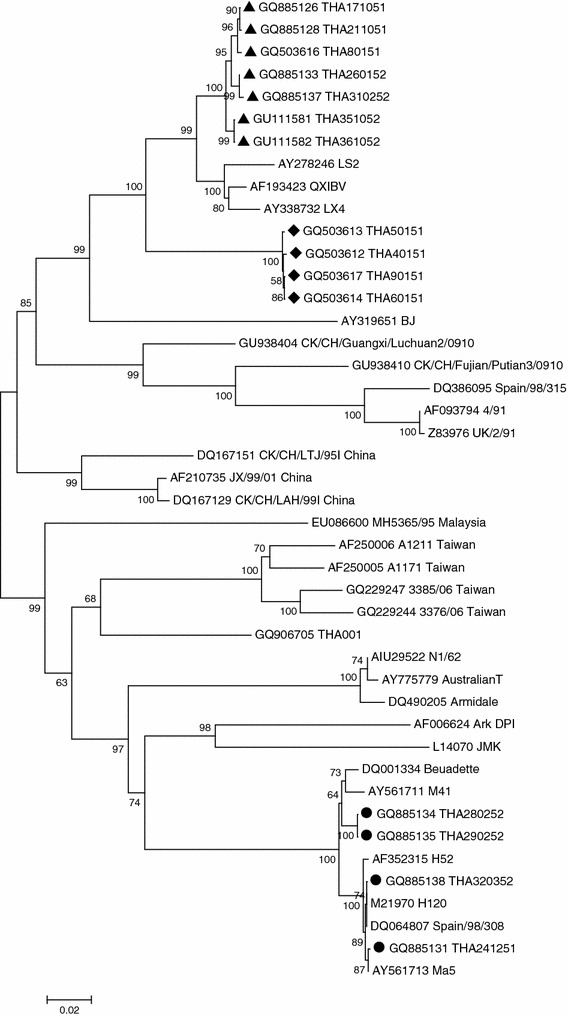
Phylogenetic tree based on the complete nucleotide sequences of the S1 gene between Thai IBV isolates, group I (filled diamond), group II (filled triangle), and group III (filled circle) and published sequences
Analysis of the spike glycoprotein cleavage recognition site
There were two different cleavage recognition site sequences observed among the Thai IBV isolates (Table 1). The most common cleavage recognition site sequence was Arg–Arg–His–Arg–Arg observed in both the group I and the group II Thai IBV. Another cleavage recognition site sequence was Arg–Arg–Phe–Arg–Arg observed in the group III Thai IBV.
Comparison of complete S1 gene
When the BLAST search was performed, we found that groups I, II, and III had a nucleotide identity of about 89, 97, and 95–100% with other IBV strains deposited in GenBank database, respectively. The complete nucleotide and deduced amino acid sequences of the S1 gene of the fifteen Thai IBV isolates were determined and compared with each other. Group I Thai IBV isolates had nucleotide identities of 99.8–100% and amino acid identities of 99.3–100% with each other. Group II Thai IBV isolates had nucleotide identities of 98.8–100% and amino acid identities of 96.3–100% with each other. Group III Thai IBV isolates had nucleotide identities of 97.9–100% and amino acid identities of 95.2–100% with each other. When comparison of the nucleotide sequences among the groups was made, less than 90.1, 74.5, and 73.5% identity was found between groups I and II, between groups I and III, and between groups II and III, respectively. For the amino acid identity among the groups, less than 80.4, 52.2, and 53.1% identity was found between groups I and II, between groups I and III, and between groups II and III, respectively.
Recombination in the S1 gene
The recombination events in the S1 gene sequences of Thai IBV were analyzed by using the Simplot analysis. In the similarity plot, the strains were considered as recombinants if any crossover event took place between two putative parental strains. By this analysis, the recombination events were found in groups I and II, but not in group III Thai IBV. Overall, groups I and II were clustered into a different group based on phylogenetic analysis of the S1 gene (Fig. 1). However, when the similarity plot of group I (represented by isolate THA90151) was performed, the 3′-terminus of the S1 gene was found to be similar with each other (Fig. 2). Interestingly, The 5′-terminus of the group I was similar to isolate THA001 which was unique to Thailand, isolated in 1998 (Fig. 2). The positions of recombination breakpoints were estimated at nt 679–699 (the maximization of χ2 = 146.4). The similarity plot of group II (represented by isolate THA80151) showed that the 5′-terminus of the S1 gene was found to be similar to Chinese QXIBV but a short region at the 3′-terminus was similar to the Chinese strain JX/99/01 (Fig. 3). The positions of recombination breakpoints were estimated at nt 1531–1537 (the maximization of χ2 = 71.0).
Fig. 2.
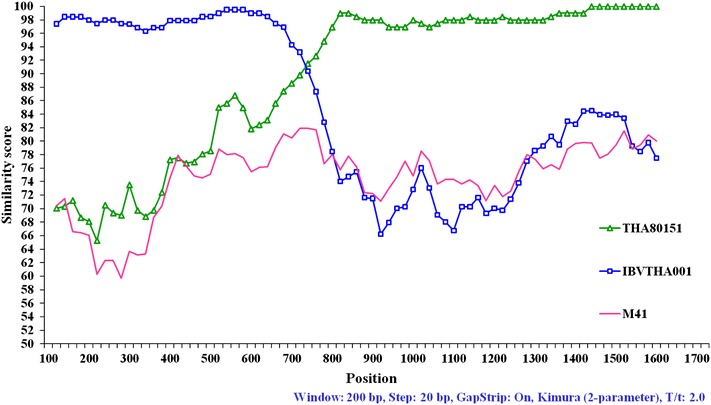
Similarity plot of the S1 gene of the group I Thai IBV (represented by THA90151). Isolate THA80151 (filled triangle) and isolate THA001 (filled square) were used as putative parental strains when isolate THA90151 was queried. M41 (no fill) was used as an outlier sequence
Fig. 3.
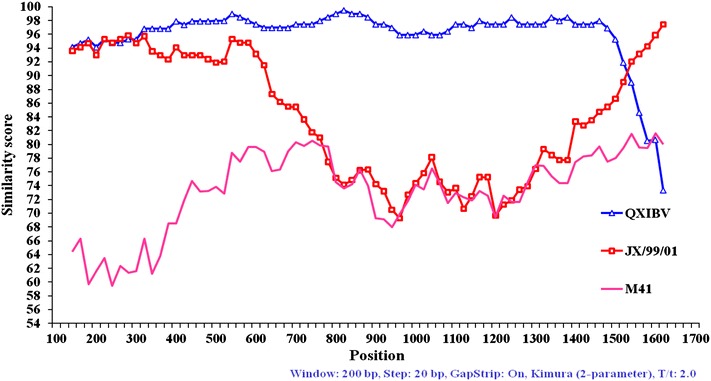
Similarity plot of the S1 gene of the group II Thai IBV (represented by THA80151). QXIBV (filled triangle) and JX/99/01 (filled square) were used as putative parental strains when isolate THA80151 was queried. M41 (no fill) was used as an outlier sequence
Discussion
Previously, we demonstrated that IBVs isolated in Thailand between January and June 2008 were divided into two distinct genotypic groups based on an analysis of the HVR of the S1 genes [17]. After that, we continued to collect the IBV samples until October 2009. Furthermore, the complete S1 gene sequences were determined in order to obtain more information about the evolution of Thai IBV. Herein, we found that fifteen Thai IBV isolates between 2008 and 2009 were divided into three distinct genotypic groups. When compared with our previous report [17], the group I Thai IBV was clustered into a different group but still unique to Thailand, the group II was clustered in the group of QX-like IBV and the group III reported only in this study, was clustered into the Massachusetts type. The result suggested that at least three groups of IBV are circulating at the present time in Thailand.
Although the basic amino acid residues of the cleavage recognition site of IBV did not correlate with cleavability, host cell range and virulence as orthomyxoviruses and paramyxoviruses, these sequences correlated with the geographic distribution of IBV [23]. Two spike glycoprotein cleavage recognition sites were found in the Thai IBV isolates. The cleavage recognition site sequence, Arg–Arg–His–Arg–Arg, was found in the groups I and II Thai IBV. This cleavage recognition site sequence had been previously reported in Chinese IBV isolates [24]. Another cleavage recognition site sequence, Arg–Arg–Phe–Arg–Arg, was found in the group III Thai IBV. This cleavage recognition site sequence has been found in many countries [23, 24]. Based on these facts, the groups I and II Thai IBV isolates have a close relationship with Chinese IBV isolates.
Overall, the group I Thai IBV appeared to be different from previous published strains by phylogenetic analysis. Interestingly, when the potential of recombination event was analyzed, the 5′-terminus of the S1 gene were similar to isolate THA001 which was isolated in Thailand in 1998 [18] but the remaining sequences were similar to the group II Thai IBV. Surprisingly, when the S1 genes of the group II Thai IBV were analyzed, recombination event was also observed in the nucleotide sequences near the cleavage recognition site. Although the group II Thai IBV appeared to be similar to QXIBV, a region near the cleavage recognition site was found to be similar to JX/99/01 isolated China in 1999 [24]. Our primary concern was that the possible viral recombination resulting from the co-infection of heterologous field strains in the chicken flocks. The recombination may be subsequently occurring with co-infection with QXIBV and JX/99/01 resulting for the occurrence of the group II Thai IBV. After that, the co-infection and exchange of genetic information between the group II Thai IBV and isolate THA001 resulted in the occurrence of the group I Thai IBV. As reported by Wang et al. [25], the evidence of natural recombination could occur within the S1 gene of IBV field isolates. The natural recombination events observed here indicated that the S1 gene of IBV was the potential site for recombination and that the exchange of genetic information could occur in more than one region of the gene.
The recombination event is though to occur by the switching of the polymerase from one template to another during the genomic synthesis [26]. Specifically, an intergenic (IG) consensus sequences (CTGAACAA or CTTAACAA) serve as recombination “hot spots” [27]. Sometime, the presence of homologous nucleotide sequence regions between the strains may serve as a potential recombination junction or cross-over site [25]. Although the IG consensus sequences were not observed in this study we found the highly conserved nucleotide sequences present around the recombination breakpoint regions in the putative parental strains (data not shown). The result suggested that the homologous nucleotide sequence regions between the strains may play a role as a cross-over site of our isolates.
QXIBV was first described and identified in China [28], after that this IBV genotype spread and became one of the most prominent genotypes in many countries [29–31]. Although the complete S1 gene of the group II Thai IBV was of 95.5–96.0% nucleotide identity to Chinese QXIBV, the nucleotide sequences at 3′-terminus near the cleavage recognition site were different from the Chinese QXIBV. We found that this region was closely related to the Chinese isolate JX/99/01. Interestingly, this change was not found when the comparisons of the S1 gene among QXIBV reported in others countries were analyzed (data not shown). These findings suggest that the group II Thai IBV had gone through evident evolution change in Thailand.
The isolates in the group III were clustered into the Massachusetts type. The Massachusetts type was also isolated in many countries world-wide including Asian countries such as China, Japan, and South Korea [24]. Some of the Massachusetts type isolated here may be a field challenge which comes from the point mutation of vaccine strains because they had 97.5–99.9% nucleotide and 93.2–99.7% amino acid identity to the vaccine strains (data not shown). It has been shown that the S1 gene of the 4/91 pathogenic virus differs only 0.6% from the vaccine strain but it cannot conclude that the small number of sequence differences are responsible for the attenuation of pathogenicity [32]. However, the possibility that some of them were re-isolation of vaccine strains could not be excluded due to the 100% identity with the Massachusetts type, the vaccine strain used in Thailand. As indicated in the previous report, we could not conclusively distinguish between the vaccine strain and field challenge of the same genotype, especially when a sequence identity of between 99 and 100% was found [33].
The data obtained from this study indicated that the IBV in Thailand undergoes genetic recombination. The natural recombination is contributing to the emergence of a new genotypes or IBV variants in the field. This event leads to difficulty in the prevention and control of IB. Thus, the development of the novel measures in prevention and control of IB must be required.
Acknowledgments
This study was financially supported by grants from the 90th Anniversary of Chulalongkorn University fund (Ratchadaphiseksomphot Endowment Fund), Chulalongkorn University.
References
- 1.Parsons D, Ellis MM, Cavannagh D, Cook AKJ. Vet. Rec. 1992;131:408–411. doi: 10.1136/vr.131.18.408. [DOI] [PubMed] [Google Scholar]
- 2.Animas BS, Otsuki K, Hanayama M, Sanekata T, Tsubokura M. J. Vet. Med. Sci. 1994;56:443–447. doi: 10.1292/jvms.56.443. [DOI] [PubMed] [Google Scholar]
- 3.Gough RE, Randall CJ, Dagless M, Alexander DJ, Cox WJ, Pearson D. Vet. Rec. 1992;130:493–494. doi: 10.1136/vr.130.22.493. [DOI] [PubMed] [Google Scholar]
- 4.Liu SW, Kong XG. Avian Path. 2004;33:321–327. doi: 10.1080/0307945042000220697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ziegler AF, Ladman BS, Dunn PA, Schneider A, Davison S, Miller PG, Lu H, Weinstock D, Slem M, Eckroade RJ, Gelb J., Jr Avian Dis. 2002;46:847–858. doi: 10.1637/0005-2086(2002)046[0847:NIBIPC]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 6.Yu L, Jiang Y, Low S, Wang Z, Nam SJ, Liu W, Kwang J. Avian Dis. 2001;45:416–424. doi: 10.2307/1592981. [DOI] [PubMed] [Google Scholar]
- 7.Boursnell MEG, Brown TDK, Foulds IJ, Green PF, Tomley FM, Binns MM. J. Gen. Virol. 1987;68:57–77. doi: 10.1099/0022-1317-68-1-57. [DOI] [PubMed] [Google Scholar]
- 8.D. Cavanagh, S.A. Naqi, in Diseases of poultry, 11th edn., ed. by A.M. Saif, Y.M. Fadly, L.R. McDougald, D.E. Swayne (Iowa State University Press, Ames, IA, 2003) pp. 101–119
- 9.Cavanagh D, Davis PJ, Pappin D, Binns MM, Boursnell M, Brown T. Virus Res. 1986;4:133–143. doi: 10.1016/0168-1702(86)90037-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kingham BF, Keeler CL, Jr, Nix WA, Ladman BS, Gelb J., Jr Avian Dis. 2000;44:25–335. doi: 10.2307/1592547. [DOI] [PubMed] [Google Scholar]
- 11.Gelb J, Jr, Wolff JB, Moran CA. Avian Dis. 1991;35:82–87. doi: 10.2307/1591298. [DOI] [PubMed] [Google Scholar]
- 12.Jia W, Karaca K, Parrish CR, Naqi SA. Arch. Virol. 1995;140:259–271. doi: 10.1007/BF01309861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pohuang T, Chansiripornchai N, Tawatsin A, Sasipreeyajan J. Indian Vet. J. 2009;86:1110–1112. [Google Scholar]
- 14.Chindavanig P. J. Thai Vet. Med. Assoc. 1962;12:1–7. [Google Scholar]
- 15.Antarasena C, Sahapong S, Aowcharoen B, Choe-ngern N, Kongkanunt R. Songklanakarin J. Sci. Technol. 1990;12:273–279. [Google Scholar]
- 16.Upatoom N, Jirathanawat V, Srihakim S, Leesirikul N, Chirawatanapong W, Bunyahotra R, Siriwan P, Likitdecharoj B. Thai J. Vet. Med. 1983;13:36–43. [Google Scholar]
- 17.Pohuang T, Chansiripornchai N, Tawatsin A, Sasipreeyajan J. J. Vet. Sci. 2009;10:219–223. doi: 10.4142/jvs.2009.10.3.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall TA. Nulc. Acids Symp. Ser. 1999;41:95–98. [Google Scholar]
- 19.Tamura K, Dudley J, Nei M, Kumar S. Mol. Biol. Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 20.Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. J. Virol. 1999;73:152–160. doi: 10.1128/jvi.73.1.152-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salminen MO, Carr JK, Burke DS, McCutchan FE. AIDS Res. Hum. Retrovir. 1995;11:1423–1425. doi: 10.1089/aid.1995.11.1423. [DOI] [PubMed] [Google Scholar]
- 22.Robertson DL, Hahn BH, Sharp PM. J. Mol. Evol. 1995;40:249–259. doi: 10.1007/BF00163230. [DOI] [PubMed] [Google Scholar]
- 23.Jackwood MW, Hilt DA, Callison SA, Lee CW, Plaza H, Wade E. Avian Dis. 2001;45:366–372. doi: 10.2307/1592976. [DOI] [PubMed] [Google Scholar]
- 24.Liu SW, Zhang QX, Chen JD, Han ZX, Liu X, Feng L, Shao YH, Rong JG, Kong XG, Tong GZ. Arch. Virol. 2006;151:1133–1148. doi: 10.1007/s00705-005-0695-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, Junker D, Collisson EW. Virology. 1993;192:710–716. doi: 10.1006/viro.1993.1093. [DOI] [PubMed] [Google Scholar]
- 26.Tolskaya EA, Romanova LA, Blinow VM, Virtorova EG, Sinyakov AN, Kolesnikova MS, Agol VI. Virology. 1987;161:54–61. doi: 10.1016/0042-6822(87)90170-X. [DOI] [PubMed] [Google Scholar]
- 27.Lee CW, Jackwood MW. Arch. Virol. 2000;145:2135–2148. doi: 10.1007/s007050070044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang YD, Wang YL, Zhang Z, Fan G, Jiang Y, Liu X, Ding J, Wang S. Chin. J. Anim. Quar. 1998;15:1–3. [Google Scholar]
- 29.Beato MS, De Battisti C, Terregino C, Drago A, Capua I, Ortali G. Vet. Rec. 2005;156:720. doi: 10.1136/vr.156.22.720. [DOI] [PubMed] [Google Scholar]
- 30.Bochkov YA, Batchenko GV, Shcherbakova LO, Borisov AV, Drygin VV. Avian Path. 2006;35:379–393. doi: 10.1080/03079450600921008. [DOI] [PubMed] [Google Scholar]
- 31.Gough RE, Cox WJ, Welchman D, Worthington KJ, Jones RC. Vet. Rec. 2008;162:99–100. doi: 10.1136/vr.162.3.99. [DOI] [PubMed] [Google Scholar]
- 32.Callison SA, Jackwood MW, Hilt DA. Avian Dis. 2001;45:492–499. doi: 10.2307/1592994. [DOI] [PubMed] [Google Scholar]
- 33.Worthington KJ, Currie RJW, Jones RC. Avian Path. 2008;37:247–257. doi: 10.1080/03079450801986529. [DOI] [PubMed] [Google Scholar]