

Abstract
As a highly efficient delivery system, lentiviral vectors (LVs) have become a powerful tool to assess the antiviral efficacy of RNA drugs such as short hairpin RNA (shRNA) and decoys. Furthermore, recent advanced systems allow controlled expression of the effector RNA via coexpression of a tetracycline/doxycycline (DOX) responsive repressor (tTR-KRAB). Herein, this system was utilized to assess the antiviral effects of LV-encoded shRNAs targeting three conserved regions on the pregenomic RNA of hepatitis B virus (HBV), namely the region coding for the reverse transcriptase (RT) domain of the viral polymerase (LV-HBV-shRNA1), the core promoter (CP; LV-HBV-shRNA2), and the direct repeat 1 (DR1; LV-HBV-shRNA3). Transduction of just the LV-HBV-shRNA vectors into the stably HBV expressing HepG2.2.15 cell line showed significant reductions in secreted HBsAg and HBeAg, intracellular HBcAg as well as HBV RNA and DNA replicative intermediates for all vectors, however, most pronouncedly for the DR1-targeting shRNA3. The corresponding vector was therefore applied in the DOX-controlled system. Notably, strong interference with HBV replication was found in the presence of the inducer DOX whereas the antiviral effect was essentially ablated in its absence; hence, the silencing effect of the shRNA and consequently HBV replication could be strictly regulated by DOX. This newly established system may therefore provide a valuable platform to study the antiviral efficacy of RNA drugs against HBV in a regulated manner, and even be applicable in vivo.
Electronic supplementary material
The online version of this article (doi:10.1007/s11262-013-0886-2) contains supplementary material, which is available to authorized users.
Keywords: Hepatitis B virus, HepG2.2.15 cell line, Lentiviral vector, RNAi, Doxycycline
Introduction
Chronic hepatitis B virus (HBV) infection is associated with a high risk for developing liver cirrhosis or hepatocellular carcinoma (HCC), and thus still is one of the major problems threatening human health globally. The major challenges for anti-HBV therapy are to overcome the low efficacy, high rate of drug resistance and adverse effects of currently available drugs [1, 2]. Hence, there is an urgent need for developing new therapeutic strategies.
One potential strategy is RNA interference (RNAi), a gene silencing regulatory mechanism present virtually in all eukaryotes, which employs double-stranded RNA molecules to degrade, or functionally inactivate, target RNAs in a sequence-specific manner [3–5]. Numerous studies have shown that replication of various viruses including HIV-1 [6–8], HCV [9–11], HSV-1 [12], SARS-associated coronavirus [13] as well as HBV [14–17], can be suppressed by RNAi, using synthetic siRNAs and plasmid-based shRNAs. However, due to their short half-life and low transfection efficiency, clinical application is limited. In this regard, non-replicating recombinant viral vectors with high delivery efficiency may provide a novel antiviral option [18–20].
HIV-1-derived LVs, intensively optimized during the last decade, are one of the most promising systems. They not only provide a non-pathogenic and latent infection by integrating into the host genome, but also demonstrate high transduction efficiency in both dividing and non-dividing cells with persistent transgene expression [21–23]. Currently LV-based vaccines have already been tested in preclinical trials for protection against or treatment of HIV-1 [9]. Moreover, several routes of LVs administration invoking sustained anti-HIV immune responses have been explored in mice [24, 25]. These findings suggest that the LVs may also be suitable delivery vectors for gene therapy in antiviral treatment. In addition, a novel tetracycline, or doxycycline (DOX), respectively, regulated RNAi system based on LVs has been developed to avoid the undesirable toxicity that may be induced by long-term shRNA expression [26, 27]. In this system, a fusion protein (tTR-KRAB) of the DNA binding domain of the Escherichia coli tetracycline repressor (tTR) and the approximately 75 amino acid KRAB repressor is likewise delivered by an LV, LV-tTR-KRAB. KRAB suppresses transcription from both RNA polymerase II and RNA polymerase III promoters within a distance of up to 3 kb from its binding site [28, 29]. The mechanism is not entirely known but seems to involve local heterochromatin formation and histone deacetylation, and/or an indirect influence of the arrangement of the basal transcription machinery [30–32]. When a promoter is juxtaposed with tet operator (tetO) sequences, as in the LV-HBV-shRNA vectors used in this study, the tTR-KRAB protein is specifically targeted to this promoter by the tTR domain and represses shRNA transcription in the absence of DOX. Conversely, addition of DOX releases tTR-KRAB from tetO, thus permitting shRNA expression [26] (Fig. 1b).
Fig. 1.
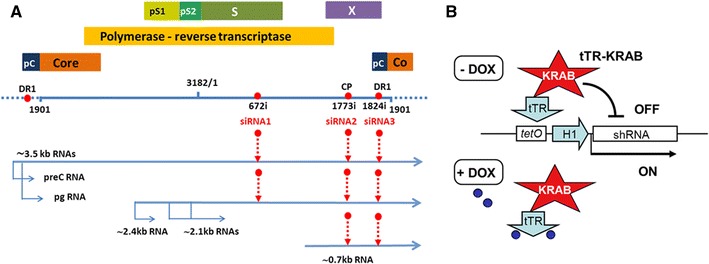
a Schematic diagram of the HBV genome and location of siRNA target sites. A linearized version of the 3,182 bp HBV genome (Genbank accession no.: J02203) is shown; one unit-length genome comprises the sequence from position 1,901 to 1,900; in HepG2.2.15 cells the genome is integrated as head-to-tail dimer, symbolized by the dashed extensions. The first nt of the siRNA target sites is indicated by a number followed by “i”. All target sites have a ≥95 % conservation rate in HBV genotypes A-H [43]. Individual genes are depicted as bars; pC, precore; pS1, preS1; pS2, preS2. Viral transcripts comprise the 3.5 kb precore RNA which includes the precore start codon for HBeAg production, and the slightly shorter pgRNA which serves as mRNA for core protein and polymerase, and as template for new DNA genomes. Note that the DR1 region is present twice on these terminally redundant RNAs. The 2.4 kb RNA encodes the large envelope protein L (PreS1/PreS2/S), the 2.1 kb RNA encode the M (PreS2/S) and the S protein. The 0.8 kb RNA is the mRNA for HBx. b Principle of DOX-controlled shRNA expression. On the LV-HBV-shRNA expression vectors, the H1 promoter driving shRNA transcription is juxtaposed with tetO sequences. In the absence of DOX, the tTR-KRAB fusion protein, co-transduced into the same cell by the LV-tTR-KRAB vector, binds to tetO and suppresses shRNA transcription (“OFF”). Addition of DOX (small blue spheres) causes release of tTR-KRAB and shRNA is transcribed (“ON”)
In this study, we constructed LV-HBV-shRNA vectors targeting three functionally important and therefore highly conserved regions on the HBV genome (Fig. 1a), namely DR1 (target site nt positions 1,824–1,842), the core promoter (CP; target site nt positions 1,773–1,793), and the RT domain encoding region of the polymerase gene (target site nt positions 672–690). DR1 is an essential cis-element for replication; the 3′ DR1 copy on the pregenomic (pg) RNA contains the acceptor site for the DNA primer generated during initiation of reverse transcription at 5′ ε [33–35]. CP controls transcription [36, 37] of the pgRNA which serves as template for HBV DNA synthesis and also as mRNA for polymerase and core protein [35, 38], and of the precore RNA from which the precore protein precursor of secreted HBeAg [39] is translated. Moreover, the CP region overlaps with the ϕ cis-element which facilitates first-strand template switch and initiation of minus-strand DNA synthesis [40, 41]. The RT region of the HBV polymerase is essential for reverse transcriptase activity [42]; on top, the same nucleotide sequence overlappingly encodes part of the S gene. Not the least, the suitability as RNAi targets of DR1 and the specific RT region chosen was already demonstrated using simple shRNA expression plasmids [43].
Therefore, we explored the feasibility of exploiting LVs for efficient delivery of shRNAs against the DR1, CP, and RT regions, and assessed their antiviral efficacies in the stably HBV-transfected human hepatoma cell line HepG2.2.15. Subsequently, the anti-DR1 shRNA, exerting the strongest interference, was selected to further evaluate its antiviral efficacy in the DOX-controllable system. As shown below, antiviral activity was strictly DOX-dependent, allowing to profoundly affect HBV gene expression and replication in a regulated fashion.
Materials and methods
Construction of vectors expressing shRNAs
The vector pSuper [44], containing the human H1 promoter, was used as a backbone to generate a series of shRNA expression plasmids by inserting the following annealed oligonucleotides (the actual target sequences in the sense oligonucleotides are underlined) between the HindIII and BglII sites: pSuper-HBV-shRNA1 targeting the RT region (nt positions 672–690) 5′-GAT CCC CGC TCA GTT TAC TAG TGC CTT TCA AGA GAT GGC ACT AGT AAA CTG AGC TTT TTA-3′ (sense) and 3′-GGG CGA GTC AAA TGA TCA CGG AAA GTT CTC TAC CGT GAT CAT TTG ACT CGA AAA ATT CGA-5′ (antisense); pSuper-HBV-shRNA2 targeting the CP region (nt positions 1,773–1,793) 5′-GAT CCC CCT AGG AGG CTG TAG GCA TAA ATT CAA GAG ATT TAT GCC TAC AGC CTC CTA GTT TTT A-3′ (sense) and 3′-GGG GAT CCT CCG ACA TCC GTA TTT AAG TTC TCT AAA TAC GGA TGT CGG AGG ATC AAA AAT TCG A-5′ (antisense); pSuper-HBV-shRNA3 targeting DR1 (nt 1,824–1,842) 5′-GAT CCC CTT CAC CTC TGC CTA ATC ATT TCA AGA GAA TGA TTA GGC AGA GGT GAA TTT TTA-3′ (sense) and 3′-GGG AAG TGG AGA CGG ATT AGT AAA GTT CTC TTA CTA ATC CGT CTC CAC TTA AAA ATT CGA-5′ (antisense). shRNA2 and shRNA3 target all viral transcripts, shRNA1 targets all transcripts except the HBx mRNA (Fig. 1a). Each shRNA targets the pgRNA serving as the template for HBV genomic replication as well as the mRNA for the core antigen and the polymerase. Moreover, shRNA-1 also targets the HBV HBsAg mRNAs, while shRNA-2 and shRNA-3 target both the HBsAg mRNAs and HBx mRNAs (Fig. 1a). As a negative control, oligonucleotides 5′-CCT AGT GAA GGG CGT ATG ATT ATT CAA GAG ATA ATC ATA CGC CCT TCA CTA GTT TTTA-3′ (sense) and 3′-GGG GAT CAC TTC CCG CAT ACT AAT AAG TTC TCT ATT AGT ATG CGG GAA GTG ATC AAA AAT TCG A-5′ (antisense) were employed to construct an analogous pSuper vector, pSuper-shRNA-NC, which encodes an shRNA lacking any perfect match to HBV or cellular sequences. All shRNA target sequences were BLAST searched against the human genomic plus transcript database (NCBI) to exclude excessive similarity with human genes or mRNAs. The sequences of the shRNA expression cassettes including the H1 promoter in all plasmids were verified by DNA sequencing.
The lentiviral pLV-HBV-shRNA expression vectors were constructed by inserting the 300 bp EcoRI and ClaI fragments containing the H1 promoter plus shRNA sequences from the respective pSuper-HBV-shRNA plasmids into pLVTH [26] which also encodes green fluorescent protein (GFP) to facilitate determination of viral titers. All constructs were confirmed by restriction endonuclease analysis.
Virus production, concentration and titration
Recombinant lentiviruses (LV-HBV-shRNA and LV-tTR-KRAB) were generated from 293T cells (6 × 106 cells/100 mm plate) by transient transfection of three plasmids [45] with LipofectamineTM 2,000 (Invitrogen, Carlsbad, US). In brief, to produce LV-HBV-shRNAs, 293T cells were co-transfected with 15 μg of pLV-HBV-shRNA, 10 μg of pCMVΔR8.91 [46] expressing the HIV-1 gag/pol, tet, and rev genes required for efficient lentivirus production, and 5 μg of pMD2.G [46] expressing the VSV envelope glycoprotein G; LV-tTR-KRAB was produced analogously from pLV-tTR-KRAB [26]. LV particles were harvested 48 h later, centrifuged to remove cell debris at 3,000×g for 5 min, filtered through 0.45 μm cellulose acetate filters (Millipore), and concentrated by ultra-high speed centrifugation at 70,000 g for 2 h at 4 °C. The virus stocks were stored at −80 °C until use. Infectious titers were determined by transduction of 293T cells with known amounts of the stock solutions (tenfold dilutions corresponding to 1, 0.1, 0.01, 0.001 μl and 0.0001 μl per 1.5 ml medium in one well of a 6-well plate, seeded with 50,000 cells) and counting of GFP-positive cells. Typical titers were around 108 transducing units/ml.
Cell culture and transduction
293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco Life Technologies) supplemented with 10 % fetal bovine serum (FBS; Gibco). The human hepatoblastoma cell line HepG2.2.15, stably transfected by head-to-tail dimers of HBV DNA, was cultured in DMEM supplemented with 10 % FBS and 200 ng/ml G418 (Gibco). The cell cultures were maintained at 37 °C in a moist atmosphere containing 5 % CO2.
Transduction of LV-HBV-shRNA vectors into HepG2.2.15 cells was carried out using Polybrene (Sigma-Aldrich). In brief, HepG2.2.15 cells were seeded at a density of 1 × 105 cells per well into 6-well plates. After 24 h, cells were inoculated with LV-HBV-shRNA vector particles at a multiplicity of infection (MOI) of 10 in the presence of 6 μg/ml polybrene. After overnight incubation, the culture medium was replaced with fresh complete medium. Transduction efficiency was routinely >95 %, as indicated by the fraction of GFP-positive cells.
HBsAg and HBeAg ELISA
The effects of LV-HBV-shRNAs on the secretion of HBsAg and HBeAg were assessed at 24, 48, 72, 96 h, and 1 week after transduction with the LV-HBV-shRNA vectors using enzyme-linked immunosorbent assay (ELISA) kits following the manufacturer’s instructions (Kehua Co. Shanghai, China).
RNA preparation and assay of HBV RNA
To quantify HBV mRNA after introducing LV-HBV-shRNAs into HepG2.2.15 cell line, total RNA was extracted from the transduced cells at 96 h post-transduction using Trizol reagent (Invitrogen, USA) and reverse-transcribed (RT) using oligo dT priming. Quantitative real-time PCR was carried out using SYBRGreen PCR Master Mix chemistry (Toyobo, Japan) following the manufacturer’s instructions, and the values were normalized to β-actin mRNA as the internal control. The primer sequences were 5′-TCG TGT TAC AGG CGG GGT TT-3′ (forward) and 5′-GAC TGC GAA TTT TGG CCA AG-3′ (reverse) for the HBV S region, and 5′-GAA CGG TGA AGG TGA CAG-3′ (forward) and 5′-TAG AGA GAA GTG GGG TGG-3′ (reverse) for β-actin. All reactions were run in duplicate using the Stratagene Mx3000P QPCR System (Agilent Technologies).
Quantitation of HBV replicative intermediates by Southern blotting and real-time PCR
The levels of encapsidated HBV replicative DNA intermediates were assessed by Southern blotting. Encapsidated HBV DNA was purified using the method described by Guo et al. [47]. In brief, the cells were washed once with ice-cold phosphate-buffered saline (PBS) and lysed in 300 μl lysis buffer (50 mM Tris-HCl (pH7.4), 1 mM EDTA, 1 % NP-40) per well, then the cell lysates were placed on ice for 30 min and centrifuged for 5 min at 16,000×g. The supernatants were adjusted to 10 mM MgCl2 and treated with 100 μg/ml of DNase I and 100 μg/ml RNase for 30 min at 37 °C. The reactions were stopped by addition of EDTA. Proteins were digested with 0.5 mg/ml of proteinase K in the presence of 1 % sodium dodecyl sulfate for 2 h at 37 °C. Nucleic acids were purified by phenol–chloroform (1:1) extraction and ethanol precipitation and dissolved in ddH2O. HBV DNA isolated from the intracellular core particles was separated on a 1.0 % agarose gel, blotted onto a nylon membrane and hybridized with a 32P-labeled unit-length HBV DNA fragment. Bands were visualized by phosphorimaging (Cyclone Plus, Perkin Elmer).
To exam viral load in the culture supernatant, HBV DNA was extracted using the TIANamp Virus DNA/RNA Kit (Tiangen Biotech Co. Beijing, China) and quantified by real-time polymerase chain reaction using SybrGreen PCR Master Mix chemistry (Toyobo, Japan).
Primer sequences were as follows: 5’-TGC CAA CTG GAT CCT GCG CG-3′(forward) and 5′-TTC ACG GTG GTC TCC ATG CG-3′ (reverse). Amplification was performed at 95 °C for 5 min and then 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 20 min for a total of 40 cycles, followed by 72 °C for 10 min for total HBV DNA, then followed by 95 °C for 1 min, 55 °C for 30 s and 95 °C for 30 s for signals collection. The level of expression of HBV genes was measured using threshold cycle C t (C t threshold cycle).
Western blotting
HBV core protein was detected by immunoblotting using an anti-HBcAg mouse monoclonal antibody. In brief, the treated cells were washed twice with ice-cold PBS and lysed in 100 μl of lysis buffer (50 mM Tris-HCl(pH 8.0); 150 mM NaCl; 0.1 % SDS; 1 % NP40; 0.5 % deoxycholic acid; 0.5 % sodium azide; and 100 μg/ml PMSF) per well of a 6-well plate. The total protein from each sample was loaded on 12 % SDS-PAGE. Subsequently, gels were electroblotted to a PVDF membrane, which was then soaked for 30 min in blocking solution, and incubated for 1 h at room temperature with a mouse monoclonal anti-HBcAg antibody (1:1,000 diluted, kindly provided by Dr. M. Nassal, University Hospital Freiburg, Germany); as internal control the housekeeping protein GAPDH on the same blot was detected using a mouse anti-GAPDH monoclonal antibody (1:1,000 diluted, Invitrogen). After three washes, the blots were incubated with appropriate peroxidase conjugates secondary antibody and bands were visualized using the DAB Western Blot kit (Thermo).
DOX regulation of shRNA expression by co-transduction of LV-tTR-KRAB
HepG2.2.15 cells were seeded into a 6-well plate at a density of 2 × 105 cells per well and transduced 16 h later with LV-HBV-shRNA3 and the LV-tTR-KRAB vector particles at a ratio of 1:1 in the presence of 6 μg/ml polybrene. After 12-h incubation, the transduced cells were split into two equal aliquots. One aliquot was maintained in the absence, the other in the presence of 5 μg/ml DOX for 2 weeks. To monitor a potential direct impact of DOX on shRNA expression from LV-HBV-shRNA in the absence of tTR-KRAB or on the parental HepG.2.2.15 cells, cells transduced with only LV-HBV-shRNA3 or naive HepG2.2.15 cells were also cultured for 2 weeks in the presence or 5 μg/ml DOX and analyzed in parallel for HBV gene expression and replication as described above.
Statistical analysis
All experiments were performed three times independently. The data were expressed as 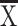 ± standard deviation (SD). Comparison between groups was performed by an independent-samples t test. The level of significance was set to p < 0.05.
± standard deviation (SD). Comparison between groups was performed by an independent-samples t test. The level of significance was set to p < 0.05.
Results
To study the effect of LV-mediated RNAi on HBV replication in a cell culture model, we used the HepG2.2.15 cell line which contains stable HBV head-to-tail dimer integrates [48] that support viral gene expression and replication, including formation of infectious HBV virions. It is therefore widely used as an in vitro model for HBV replication and appeared well suited for the current study. To this end, the cells were seeded at a density of 1 × 105 cells per well of a 6-well plate and inoculated 24 h later with the respective LVs at a multiplicity of infection (MOI) of 10; vector titers in the stocks had been determined using the LV built-in GFP to microscopically count the number of positive cells in dependence of the applied vector stock volume. After overnight incubation, the vector-containing media were replaced by fresh media, and data were collected at the time points indicated below. The GFP assay showed that routinely >95 % of the cells were successfully transduced under the above described conditions.
shRNA impact on secreted HBV antigens
As an initial test for the functionality of the shRNAs, HepG2.2.15 cells were transfected with the pSuper-HBV-shRNA plasmids and the negative control vector pSuper-shRNA-NC; untreated HepG2.2.15 cells served as reference. HBsAg and HBeAg in the supernatants after 24, 48 and 72 h were then determined by ELISA. As shown in Supplementary Fig. 1, shRNA3 achieved the strongest reductions, followed by shRNA2 and shRNA1; no significant differences between cells receiving the negative control plasmid and untransfected cells were seen. HBsAg levels were maximally reduced by 70–80 %, HBeAg levels by 20–30 %. These data confirmed that the HBV-specific shRNAs acted as intended. Higher levels of inhibition would not be expected because all cells contain integrated HBV but only a fraction of them are transfected with the shRNA plasmid.
Next, analogous experiments were performed with the LV-HBV-shRNA vector transduced cells, except that the observation period was extended to 1 week. All HBV-shRNA expressing LVs led to significant (p < 0.01) reductions in HBsAg (Fig. 2a) and HBeAg (Fig. 2b) compared to the untreated and the control shRNA vector transduced cells. The antiviral effects slightly increased from 24 h to 72 h post-transduction and then remained at similar levels for the entire observation period. As in the pSuper-HBV-shRNA transfected cells, shRNA3 exerted the strongest effects, however, the level of inhibition was much more pronounced, especially for HBsAg. shRNA3 achieved around 90–98 % inhibition, shRNA2 around 80–90 %, and shRNA1 around 60–70 %. HBeAg suppression was generally less efficient, with maximum inhibition rates of 60–70 % for shRNA3. The reasons for the lower impact on HBeAg are not clear but similar observations had also been reported by others [49, 50]. The negative control shRNA vector exhibited no suppressive effect on HBsAg or HBeAg levels compared to the untreated cells, corroborating that neither the shRNA nor the LV itself had a negative impact on the cells, and demonstrating the HBV specificity of the other shRNAs.
Fig. 2.
Impact of LV-delivered shRNAs on secreted HBV antigens. HepG2.2.15 cells were transduced with LV-HBV-shRNA1 (targeting the RT region), LV-HBV-shRNA2 (targeting the CP region), or LV-HBV-shRNA3 (targeting the DR1 region). At the indicated timepoints post-transduction the levels of HBsAg (a) and HBeAg (b) were determined by ELISA. Untreated HepG2.2.15 cells served as reference, cells transduced with LV-HBV-shRNA-NC expressing an irrelevant shRNA as control for nonspecific effects. Values obtained for the transduced cells were normalized to those obtained for untreated HepG2.2.15 cells which were set as 100 %. All data are shown as mean ± SD from three independent experiments
Impact of LV-expressed shRNAs on HBV RNA
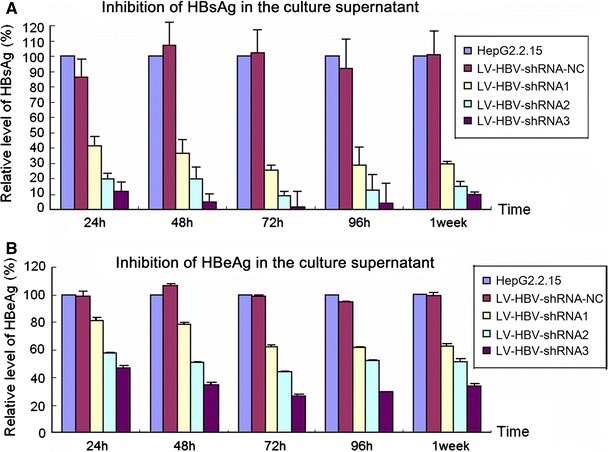
One week post-transduction, total RNA was extracted from the cells and analyzed by real-time RT-PCR using HBV-specific and β-actin specific primers; the relative ratios of HBV to β-actin specific signals compared to that in untreated HepG2.2.15 cells are shown in Fig. 3. Consistent with the ELISA data, shRNA3 caused a massive (close to 95 %) reduction in HBV RNA, followed by shRNA2 (around 77 %) and shRNA1 (30 %). Also in this assay, HBV RNA remained unaffected by the nonspecific shRNA encoding negative control vector.
Fig. 3.
Impact of LV-delivered shRNAs on HBV RNA levels. HepG2.2.15 cells were transduced with the indicated LV-HBV-shRNA vectors; untreated HepG2.2.15 cells served as reference. At 96 h post-transduction, total RNA was isolated and HBV-specific transcripts were quantitated by real-time RT-PCR using primers specific for the HBV S gene. Quantitation of β-actin mRNA in the same samples served for normalization. Values are given as the ratio of HBV-specific versus β-actin specific RNAs; the ratio obtained for untreated HepG2.2.15 cells was set as 100 %. Values shown are the mean of three independent experiments; bars indicate SD. Numerical mean values were 97.6 ± 7.4 % for LV-HBV-NC (negative control); 70.0 ± 3.6 % for LV-HBV-shRNA1; 22.4 ± 1.2 % for LV-HBV-shRNA2; and 5.3 ± 1.9 % forLV-HBV-shRNA3
Impact of LV-expressed shRNAs on HBV DNA replication and core expression
The level of HBV DNA in extracellular particles was assessed by real-time PCR and that in intracellular nucleocapsids by Southern blotting. As shown in Fig. 4, LV-HBV-shRNA3 targeting DR1 decreased extracellular viral load by about 17-fold at 1 week post-transduction compared to untreated and negative control shRNA vector transduced cells. Inhibition by LV-HBV-shRNA2 was comparable, and that by LV-HBV-shRNA1 about half as strong. More pronounced inhibition of HBV replication than of HBV transcription or protein expression has also previously been observed for some anti-HBV shRNAs [43] and might reflect effects beyond RNA degradation.
Fig. 4.
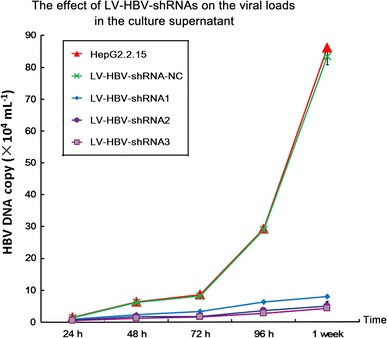
Impact of LV-delivered shRNAs on HBV viral load in the culture supernatant. HepG2.2.15 cells were transduced with the indicated LV-HBV-shRNA vectors. At the indicated timepoints, HBV DNA in the culture supernatants was determined by real-time PCR. Values are expressed as mean from three independent determinations; error bars indicate SD
Overall consistent data were obtained by Southern blot analysis of HBV DNAs associated with intracellular nucleocapsid (Fig. 5). Almost no replicative DNA intermediates were found in the LV-HBV-shRNA2 and LV-HBV-shRNA3 transduced cells; higher levels were seen in LV-HBV-shRNA1 transduced cells but they were still reduced compared to untreated or control LV-shRNA transduced cells.
Fig. 5.
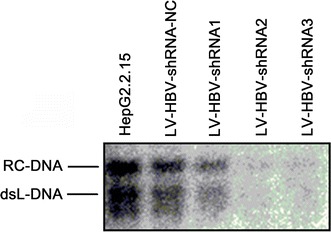
Impact of LV-delivered shRNAs on HBV replicative DNA intermediates in intracellular nucleocapsids. HepG2.2.15 cells were transduced with the indicated LV-HBV-shRNA vectors as described in Fig. 4. At 96 h post-transduction, nucleocapsid-associated viral DNAs were isolated and analyzed by Southern blotting, using a 32P-labeled full-length HBV DNA probe. The positions of relaxed-circular (RC) and double-stranded linear (dsL) DNA are indicated
As another parameter of antiviral activity, we monitored HBV core protein expression. Western blotting (Fig. 6) showed again a significant reduction in core protein levels compared to GAPDH in LV-HBV-shRNA2 and LV-HBV-shRNA3 transduced cells. Together, these data showed that transduction with LV-HBV-shRNA3 induced the strongest antiviral effects.
Fig. 6.
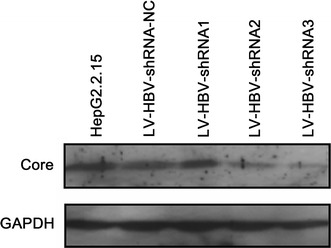
Impact of LV-delivered shRNAs on HBV core protein levels. HepG2.2.15 cells were transduced with the indicated LV-HBV-shRNA vectors as described in Fig. 4. Untreated HepG2.2.15 cells served as reference. Core protein in cellular lysates was detected by immunoblotting with a HBV core specific monoclonal antibody. Detection of cellular GAPDH present in the same samples served as loading control
Impact of DOX-inducible RNAi on HBV replication and expression in HepG2.2.15 cells
Based on the data reported above, we chose to employ vector LV-HBV-shRNA3 to investigate the feasibility of using the DOX-inducible RNAi system to control expression of the anti-HBV shRNA, and thus also HBV gene expression and replication, in a regulated manner.
To this end, HepG2.2.15 cells were subjected to various treatments. One group was transduced with just LV-HBV-shRNA3 as a reference; the second with LV-HBV-shRNA3 plus LV-tTR-KRAB which should inhibit expression of the anti-HBV shRNA; the third group received in addition DOX to relieve tTR-KRAB mediated suppression of RNAi. To monitor a potential direct (not tTR-KRAB) mediated effect of DOX on the shRNA vector, the fourth group was transduced with just LV-HBV-shRNA3 and cultured with DOX. Lastly, a potential impact of DOX on the HepG2.2.15 cells or HBV replication in the cells was addressed by culturing naive HepG2.2.15 cells in DOX containing medium. We then tested as before for secreted HBsAg and HBeAg, core protein and viral replicative DNA intermediates in intracellular nucleocapsids .
Fully consistent with the previous data, LV-HBV-shRNA3 on its own caused a strong reduction in HBsAg and less pronounced reduction in HBeAg (Fig. 7). This was not affected by the presence of DOX, which had also no impact on naive HepG2.2.15 cells. Co-transduction of LV-HBV-shRNA3 and LV-tTR-KRAB in the absence of DOX completely blocked the antiviral effect, giving very similar ELISA values as the untreated cells. The presence of DOX, however, completely restored HBsAg and HBeAg suppression to levels indistinguishable from those achieved with the shRNA3 vector alone. Suppression of secreted antigens in the presence of DOX, and inhibition of suppression in its absence, were largely maintained over 2 weeks. The trend toward slightly less efficient HBsAg suppression at the late time points was consistent with the results obtained by transduction of only the LV-HBV-shRNA vectors (Fig. 2a) and may reflect a slow selection of cells that produce less shRNA.
Fig. 7.
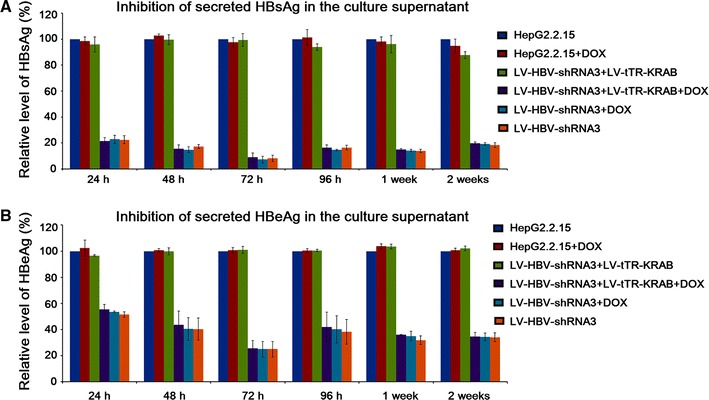
Regulation of HBsAg and HBeAg secretion by DOX-inducible shRNA targeting the DR1 region of HBV. HepG2.2.15 cells were transduced with LV-HBV-shRNA3 alone, or co-transduced with LV-tTR-KRAB; cells were maintained in the absence DOX or in the presence of 5 μg/ml DOX as indicated. Non-transduced HepG2.2.15 cells served as reference. Cell culture supernatants were analyzed at the indicated timepoints for HBsAg (a) and HBeAg (b) by ELISA. Note the absence of suppression in LV-tTR-KRAB co-transduced cells in the absence of DOX, and full restauration of suppression in the presence of DOX. All data shown are the mean from three independent experiments. Error bars indicate SD
Comparable results were obtained for core protein expression (Fig. 8). In the presence of DOX, cells co-transduced with the shRNA3 and the tTR-KRAB vector showed similarly low core protein levels as cells transduced with only the shRNA3 vector, whereas without DOX the tTR-KRAB vector co-transduced cells still contained high levels of core protein.
Fig. 8.

Regulation of HBV core protein expression by DOX-inducible shRNA targeting the DR1 region of HBV. HepG2.2.15 cells were transduced with LV-HBV-shRNA3 alone or co-transduced with LV-tTR-KRAB, and maintained in the presence or absence of DOX as indicated. Core protein and GAPDH present in cell lysates prepared 7 days post-transduction were analyzed by Western blotting as described in the legend to Fig. 6
Finally, these results were confirmed for nucleocapsid-associated viral DNAs by Southern blotting (Fig. 9). LV-HBV-shRNA3 alone, regardless of the presence of DOX, and LV-HBV-shRNA3 plus LV-tTR-KRAB in the presence of DOX all caused a similarly strong reduction, whereas omitting DOX in the co-transduction experiments completely blocked the antiviral effect.
Fig. 9.
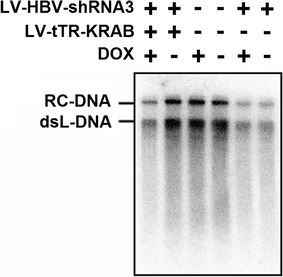
Regulation of HBV replication by DOX-inducible shRNA targeting the DR1 region of HBV. HepG2.2.15 cells were transduced with LV-HBV-shRNA3 alone or co-transduced with LV-tTR-KRAB, and maintained in the presence or absence of DOX as indicated. At 7 days post-transduction, intracellular nucleocapsid-associated viral DNA was isolated and analyzed by Southern blotting as described in the legend to Fig. 5
Together, these results showed first that DOX acted specifically on tTR-KRAB, as intended. More importantly, all assays confirmed complete reversibility of tTR-KRAB mediated RNAi suppression by DOX. Hence, the system allows to deliberately turn on and off HBV replication in HepG2.2.15 cells, without negative effects on the cells.
Discussion
Numerous studies exploring RNAi as a potential therapeutic option to inhibit viral replication have been reported, e.g., for HIV-1, HAV, HCV, HSV and others [12, 51, 52]. Although HBV is a DNA virus, RNA plays a key role in its replication as template for generation of new DNA genomes by reverse transcription. Therefore, RNAi can not only act on the viral mRNAs and prevent generation of viral proteins but directly interfere with HBV replication. This has been confirmed by several studies employing synthetic siRNAs and plasmid-based shRNAs targeting different regions of the HBV genome, both in vitro and in hydrodynamically transfected mice in vivo [53–55]. However, due to the short half-life of siRNAs and limited and variable transfection efficiency of shRNA plasmids, the inhibitory effects are transient, lasting only a few days, limiting their application for long-term gene silencing in mammalian systems [56]. This issue is partially overcome using adenovirus- or retrovirus-based vectors for shRNA expression which have successfully been applied to suppress HBV replication [49, 57]. However, adenoviral vectors do not integrate, are likely to be lost during cell division, and thus can achieve only transient shRNA expression. Furthermore, adenovirus is a very common human pathogen and in vivo delivery may be hampered by pre-existing host immune responses. Retroviruses, on the other hand, require cell division for integration, and hence are inefficient in transducing genes into non-dividing cells such as quiescent hepatocytes.
Recently developed drug-inducible lentiviral vectors as shRNA delivery vehicles [26] offer several advantages compared to the aforementioned systems. First, they integrate into the host genome, allowing for stable, persistent shRNA expression. Second, they transduce both dividing and non-dividing cells with high efficiency. Third, gene silencing can be regulated by addition of DOX, minimizing potential toxicity induced by long-term shRNA expression, such as induction of interferon responses [58] of “off-target” activities by or complementarity to non-target genes in only six or seven nucleotides of the guide strand [59]. So far, the inducible RNAi system has been applied in different areas including regulation of housekeeping gene expression and antiviral research on HIV-1 [9, 60]. Although lentivirus-mediated RNAi suppression of HBV replication has already been reported [50, 61], the current study, to our knowledge, is the first to establish the reversible effects of shRNAs on HBV via a drug-regulated lentiviral vector system.
First, our results demonstrate that LV-HBV-shRNA expressed anti-HBV shRNAs can induce efficient gene silencing in vitro. Consistent with the data from cells transfected with the parental pSuper-HBV-shRNA plasmids (Supplementary Fig. 1), LV-delivered shRNA2 targeting the CP region and especially shRNA3 targeting the DR1 region were more efficient than shRNA1 directed against the RT region in suppressing HBsAg and HBeAg (Fig. 2). However, both the level of inhibition and the duration of the silencing effect were much more pronounced for LV-delivered shRNA; for instance, LV-HBV-shRNA3 suppressed HBsAg production of HBsAg up to 98.5 % at 72 h post-transduction (Fig. 2), and highly efficient suppression was maintained for 2 weeks (Fig. 7). The most likely explanation is that transfection reaches only a fraction of the virus producing cells and the non-replicating plasmid is diluted out by cell division. In contrast, transduction by the LVs is highly efficient (around 95 % in our experiments) and the shRNA expression cassettes become stably integrated. The trend toward slightly lower suppression after 1 week may reflect a slow selection of cells with reduced shRNA expression. Overall similar results were observed for secreted HBeAg although suppression was less pronounced than for HBsAg, both in plasmid-transfected and in LV-transduced cell (Fig. 2 and Supplementary Fig. 1). There is no obvious explanation but similar results were reported by Uprichard et al. [49] and Deng et al. [50]. It was suggested that the relative RNAi resistance of the precore mRNA compared to the 2.4 and 2.1 kb mRNAs may be due to a more global protection within RNA-protein complexes on the rough endoplasmic reticulum [49]. An alternative explanation could be that expression of HBeAg in the cell system used is not limited by mRNA levels but by the efficacy of posttranslational steps of protein production.
At any rate, the RT-PCR assays (Fig. 3) demonstrated also a strong reduction in the levels of HBV RNA by the LV-HBV-shRNA2 vector (about 77 %) and particularly the LV-HBV-shRNA3 vector (around 95 %), without affecting the β-actin housekeeping gene mRNA. LV-HBV-shRNA1 caused only limited suppression of HBV RNA (30 %). This is probably due to poor target accessibility [62], which is affected by RNA structure and protein binding [63]. While these parameters are difficult to assess, the high activity of shRNA3 may relate to the fact that its DR1 target region is present in two copies on the pgRNA (see Fig. 1); RNAi targeting either the 5′ or the 3′ site would induce their degradation and, for the pgRNA, prevent it from acting as template for reverse transcription.
Results consistent with the secreted antigen and RNA data were also obtained for extracellular (Fig. 4) and intracellular (Fig. 5) viral DNA, as determined by qPCR and Southern blotting. Again, shRNA3 achieved a strong, sustained inhibition (>90 %). Somewhat surprising was the still strong suppression of extracellular HBV DNA by shRNA1; however, that overall replication is more vulnerable to RNAi than expression of individual gene products has previously been observed for some anti-HBV shRNAs [43]. This suggests that interference with replication may not exclusively occur by target RNA degradation but involve other mechanisms; for instance, sequence-specific binding of the guide RNA could block interaction sites on the pgRNA that are important for packaging or reverse transcription.
As LV-HBV-shRNA3 exerted the highest antiviral activity, we employed this vector to investigate the potential of the DOX-inducible tTR-KRAB system [26] to achieve regulated shRNA expression and consequently regulated HBV gene expression and replication.
Controls confirmed that DOX per se did neither affect HBV replication in naive HepG2.2.15 cells nor suppression of HBV replication in cells transduced with only LV-HBV-shRNA3. Importantly, however, co-transduced LV-tTR-KRAB completely abrogated the antiviral effect in the absence of DOX whereas addition of DOX fully restored antiviral activity, as shown for secreted viral antigens (Fig. 7), core protein expression (Fig. 8), and replication (Fig. 9).
In conclusion, our data confirm the DR1 region as a highly efficient target site for anti-HBV RNAi and the superior efficiency of shRNA delivery by lentiviral vectors, resulting in much stronger and sustained antiviral activity. Moreover, they demonstrate that the DOX-controlled system allows for fully reversible regulation of the antiviral shRNA effects and thus also of HBV replication in a stably HBV expressing human hepatoma cell line. Hence, this new system should provide a valuable platform to study the pharmacokinetics of RNA drugs in general, and against HBV in particular. The data also suggest the potential of this system for therapeutic purposes, possibly in combination with conventional anti-HBV drugs such as nucleos(t)ide analogs to a priori reduce viral loads. However, prior to any practical application for treatment of chronic HBV infection, further studies in animal models will be required to evaluate its efficiency and safety in vivo, including potential toxicity of the tTR-KRAB fusion protein.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 1 (TIFF 3,229 kb)
Acknowledgments
We thank Dr. Michael Nassal (University Hospital Freiburg, Germany) for helpful and constructive comments during preparation of the manuscript. This work was supported by grants from the National Major Science and Technology Special Projects for Infectious Diseases of China (2012ZX10004503-008, 2012ZX10001006-002, and 2012ZX10002006-002).
Abbreviations
- CP
Core promoter
- DOX
Doxycycline
- DR1
Direct repeat 1
- EN2
Enhancer II region
- HAV
Hepatitis A virus
- HBV
Hepatitis B virus
- HBcAg
Hepatitis core antigen
- HBsAg
Hepatitis B surface antigen
- HBeAg
Hepatitis B e antigen
- HCC
Hepatocellular carcinoma
- HCV
Hepatitis C virus
- HIV
Human immunodeficiency virus
- HSV
Herpes simplex virus
- KRAB
Krüppel associated box
- LV
Lentiviral vector
- pgRNA
Pregenomic RNA
- RC-DNA
Relax circular-DNA
- RNAi
RNA interference
- SARS
Severe Acute Respiratory Syndromes
- SIN
Self-inactivating
- shRNA
Short hairpin RNA
- siRNA
Small interfering RNA
- ssDNA
Single-stranded DNA
- tet O
Tet operator
- tTR
Tetracycline transrepressor
References
- 1.Kwon H, Lok AS. Nat. Rev. Gastroenterol. Hepatol. 2011;8:275–284. doi: 10.1038/nrgastro.2011.33. [DOI] [PubMed] [Google Scholar]
- 2.Xie YH, Hong R, Liu W, Liu J, Zhai JW. Virol. Sin. 2010;25:294–300. doi: 10.1007/s12250-010-3138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hammond SM, Bernstein E, Beach D, Hannon GJ. Nature. 2000;404:293–296. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- 4.Hannon GJ. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 5.Sharp PA. Genes Dev. 2001;15:485–490. doi: 10.1101/gad.880001. [DOI] [PubMed] [Google Scholar]
- 6.Wingard JB, Anderson B, Weissman D. Eur. J. Immunol. 2008;38:1310–1320. doi: 10.1002/eji.200738069. [DOI] [PubMed] [Google Scholar]
- 7.Morris KV, Rossi JJ. Gene Ther. 2006;13:553–558. doi: 10.1038/sj.gt.3302688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Novina CD, Murray MF, Dykxhoorn DM, Beresford PJ, Riess J, Lee SK, Collman RG, Lieberman J, Shankar P, Sharp PA. Nat. Med. 2002;8:681–686. doi: 10.1038/nm725. [DOI] [PubMed] [Google Scholar]
- 9.Westerhout EM, Vink M, Haasnoot PC, Das AT, Berkhout B. Mol. Ther. 2006;14:268–275. doi: 10.1016/j.ymthe.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 10.Zhao ZF, Yang H, Han DW, Zhao LF, Zhang GY, Zhang Y, Liu MS. World J. Gastroenterol. 2006;12:6046–6049. doi: 10.3748/wjg.v12.i37.6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson JA, Richardson CD. J. Virol. 2005;79:7050–7058. doi: 10.1128/JVI.79.11.7050-7058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang YQ, Lai W, Li H, Li G. Clin. Exp. Dermatol. 2008;33:56–61. doi: 10.1111/j.1365-2230.2007.02543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He ML, Zheng B, Peng Y, Peiris JS, Poon LL, Yuen KY, Lin MC, Kung HF, Guan Y. JAMA. 2003;290:2665–2666. doi: 10.1001/jama.290.20.2665. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y, Cheng G, Mahato RI. Pharm. Res. 2008;25:72–86. doi: 10.1007/s11095-007-9504-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pouessel G, Dieux-Coeslier A, Wacrenier A, Fabre M, Gottrand F. Am. J. Med. Genet. A. 2006;140:1028–1029. doi: 10.1002/ajmg.a.31192. [DOI] [PubMed] [Google Scholar]
- 16.Shlomai A, Shaul Y. Hepatology. 2003;37:764–770. doi: 10.1053/jhep.2003.50146. [DOI] [PubMed] [Google Scholar]
- 17.Wu KL, Zhang X, Zhang J, Yang Y, Mu YX, Liu M, Lu L, Li Y, Zhu Y, Wu J. Virus Res. 2005;112:100–107. doi: 10.1016/j.virusres.2005.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jia F, Zhang YZ, Liu CM. J. Biotechnol. 2007;128:32–40. doi: 10.1016/j.jbiotec.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 19.Li Z, He ML, Yao H, Dong QM, Chen YC, Chan CY, Zheng BJ, Yuen KY, Peng Y, Sun Q, Yang X, Lin MC, Sung JJ, Kung HF. BMB Rep. 2009;42:59–64. doi: 10.5483/BMBRep.2009.42.1.059. [DOI] [PubMed] [Google Scholar]
- 20.Seppen J, Rijnberg M, Cooreman MP, Oude Elferink RP. J. Hepatol. 2002;36:459–465. doi: 10.1016/S0168-8278(01)00308-7. [DOI] [PubMed] [Google Scholar]
- 21.Lei YC, Hao YH, Zhang ZM, Tian YJ, Wang BJ, Yang Y, Zhao XP, Lu MJ, Gong FL, Yang DL. World J. Gastroenterol. 2006;12:4492–4497. doi: 10.3748/wjg.v12.i28.4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naldini L, Blomer U, Gage FH, Trono D, Verma IM. Proc. Natl. Acad. Sci. USA. 1996;93:11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kafri T, Blomer U, Peterson DA, Gage FH, Verma IM. Nat. Genet. 1997;17:314–317. doi: 10.1038/ng1197-314. [DOI] [PubMed] [Google Scholar]
- 24.Buffa V, Negri DR, Leone P, Borghi M, Bona R, Michelini Z, Compagnoni D, Sgadari C, Ensoli B, Cara A. Viral Immunol. 2006;19:690–701. doi: 10.1089/vim.2006.19.690. [DOI] [PubMed] [Google Scholar]
- 25.Iglesias MC, Mollier K, Beignon AS, Souque P, Adotevi O, Lemonnier F, Charneau P. Mol. Ther. 2007;15:1203–1210. doi: 10.1038/sj.mt.6300135. [DOI] [PubMed] [Google Scholar]
- 26.Wiznerowicz M, Trono D. J. Virol. 2003;77:8957–8961. doi: 10.1128/JVI.77.16.8957-8951.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amar L, Desclaux M, Faucon-Biguet N, Mallet J, Vogel R. Nucleic Acids Res. 2006;34:e37. doi: 10.1093/nar/gkl034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Margolin JF, Friedman JR, Meyer WK, Vissing H, Thiesen HJ, Rauscher FJ., III Proc. Natl. Acad. Sci. USA. 1994;91:4509–4513. doi: 10.1073/pnas.91.10.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Senatore B, Cafieri A, Di Marino I, Rosati M, Di Nocera PP, Grimaldi G. Gene. 1999;234:381–394. doi: 10.1016/S0378-1119(99)00182-1. [DOI] [PubMed] [Google Scholar]
- 30.Urrutia R. Genome Biol. 2003;4:231. doi: 10.1186/gb-2003-4-10-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuda E, Agata Y, Sugai M, Katakai T, Gonda H, Shimizu A. J. Biol. Chem. 2001;276:14222–14229. doi: 10.1074/jbc.M105316200. [DOI] [PubMed] [Google Scholar]
- 32.Schultz DC, Friedman JR, Rauscher FJ., III Genes Dev. 2001;15:428–443. doi: 10.1101/gad.869501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu N, Ji L, Maguire ML, Loeb DD. J. Virol. 2004;78:642–649. doi: 10.1128/JVI.78.2.642-649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nassal M. Virus Res. 2008;134:235–249. doi: 10.1016/j.virusres.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 35.Seeger C, Mason WS. Microbiol. Mol. Biol. Rev. 2000;64:51–68. doi: 10.1128/MMBR.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chisari FV. Am. J. Pathol. 2000;156:1117–1132. doi: 10.1016/S0002-9440(10)64980-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moolla N, Kew M, Arbuthnot P. J. Viral. Hepat. 2002;9:323–331. doi: 10.1046/j.1365-2893.2002.00381.x. [DOI] [PubMed] [Google Scholar]
- 38.Locarnini S. Semin. Liver Dis. 2004;24(Suppl 1):3–10. doi: 10.1055/s-2004-828672. [DOI] [PubMed] [Google Scholar]
- 39.Yu X, Mertz JE. J. Virol. 1996;70:8719–8726. doi: 10.1128/jvi.70.12.8719-8726.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abraham TM, Loeb DD. J. Virol. 2007;81:11577–11584. doi: 10.1128/JVI.01414-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abraham TM, Loeb DD. J. Virol. 2006;80:4380–4387. doi: 10.1128/JVI.80.9.4380-4387.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feng H, Hu K. Virol. Sin. 2009;24:509–517. doi: 10.1007/s12250-009-3076-6. [DOI] [Google Scholar]
- 43.Sun D, Rosler C, Kidd-Ljunggren K, Nassal M. J. Hepatol. 2010;52:817–826. doi: 10.1016/j.jhep.2009.10.038. [DOI] [PubMed] [Google Scholar]
- 44.Brummelkamp TR, Bernards R, Agami R. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 45.Li MJ, Rossi JJ. Methods Enzymol. 2005;392:218–226. doi: 10.1016/S0076-6879(04)92013-7. [DOI] [PubMed] [Google Scholar]
- 46.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. J. Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo H, Jiang D, Ma D, Chang J, Dougherty AM, Cuconati A, Block TM, Guo JT. J. Virol. 2009;83:847–858. doi: 10.1128/JVI.02008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Acs G, Sells MA, Purcell RH, Price P, Engle R, Shapiro M, Popper H. Proc. Natl .Acad. Sci. USA. 1987;84:4641–4644. doi: 10.1073/pnas.84.13.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uprichard SL, Boyd B, Althage A, Chisari FV. Proc. Natl. Acad. Sci. USA. 2005;102:773–778. doi: 10.1073/pnas.0409028102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deng L, Li G, Xi L, Yin A, Gao Y, You W, Wang X, Sun B. BMC Gastroenterol. 2009;9:73. doi: 10.1186/1471-230X-9-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanda T, Kusov Y, Yokosuka O, Gauss-Muller V. Biochem. Biophys. Res. Commun. 2004;318:341–345. doi: 10.1016/j.bbrc.2004.03.194. [DOI] [PubMed] [Google Scholar]
- 52.Kapadia SB, Brideau-Andersen A, Chisari FV. Proc. Natl. Acad. Sci. USA. 2003;100:2014–2018. doi: 10.1073/pnas.252783999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen Y, Du D, Wu J, Chan CP, Tan Y, Kung HF, He ML. Biochem. Biophys. Res. Commun. 2003;311:398–404. doi: 10.1016/j.bbrc.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 54.Ren XR, Zhou LJ, Luo GB, Lin B, Xu A. J. Viral. Hepat. 2005;12:236–242. doi: 10.1111/j.1365-2893.2005.00587.x. [DOI] [PubMed] [Google Scholar]
- 55.Ying C, De Clercq E, Neyts J. Biochem. Biophys. Res. Commun. 2003;309:482–484. doi: 10.1016/j.bbrc.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 56.McManus MT, Sharp PA. Nat. Rev. Genet. 2002;3:737–747. doi: 10.1038/nrg908. [DOI] [PubMed] [Google Scholar]
- 57.Jia F, Zhang YZ, Liu CM. Biotechnol. Lett. 2006;28:1679–1685. doi: 10.1007/s10529-006-9138-z. [DOI] [PubMed] [Google Scholar]
- 58.Judge A, MacLachlan I. Hum. Gene Ther. 2008;19:111–124. doi: 10.1089/hum.2007.179. [DOI] [PubMed] [Google Scholar]
- 59.Anderson EM, Birmingham A, Baskerville S, Reynolds A, Maksimova E, Leake D, Fedorov Y, Karpilow J, Khvorova A. RNA. 2008;14:853–861. doi: 10.1261/rna.704708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Puca R, Nardinocchi L, Bossi G, Sacchi A, Rechavi G, Givol D, D’Orazi G. Exp. Cell Res. 2009;315:67–75. doi: 10.1016/j.yexcr.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 61.Han J, Pan X, Gao Y, Weil L. Virus Res. 2010;150:129–134. doi: 10.1016/j.virusres.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 62.Tafer H, Ameres SL, Obernosterer G, Gebeshuber CA, Schroeder R, Martinez J, Hofacker IL. Nat. Biotechnol. 2008;26:578–583. doi: 10.1038/nbt1404. [DOI] [PubMed] [Google Scholar]
- 63.Pei Y, Tuschl T. Nat. Methods. 2006;3:670–676. doi: 10.1038/nmeth911. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 1 (TIFF 3,229 kb)