

Abstract
Hepatitis C virus (HCV) infects more than 2 % of the world population with highest prevalence in parts of Africa and Asia. Past standard of care using interferon α and ribavirin had adverse effects and showed modest efficacy for some HCV genotypes spurring the development of direct acting antivirals (DAAs). Such DAAs target viral proteins and are thus better tolerated but they suffer from emergence of vial resistance. Furthermore, DAAs are often HCV genotype specific. Novel drug candidates targeting host factors required for HCV propagation, so called host-targeting antivirals (HTAs), promise to overcome both caveats. The genetic barrier to resistance is usually considered to be high for HTAs and all HCV genotypes presumably use the same host factors. Recent data, however, challenge these assumptions, at least for some HTAs. Here, we highlight the most important host-targeting strategies against hepatitis C and critically discuss their opportunities and risks.
Keywords: Hepatitis C, Host-targeting antivirals, Scavenger receptor class B type I, Phosphatidylinositol 4-kinase III alpha, Cyclophilin A, MicroRNA-122
Introduction
Hepatitis C virus (HCV) chronically infects more than 2 % of the world’s population with highest incidents in Africa and Asia [1]. Out of these patients 20 % will develop severe liver disease 10 to 25 years after contraction [2]. As a result, chronic hepatitis C is the number one indication for liver transplantation in many countries. As a member of the family of Flaviviridae HCV particles bear a plus strand RNA genome and a glycoprotein decorated envelope. HCV can be classified into six epidemiologically relevant genotypes and many subtypes with intergenotypic sequence variability at the nucleotide level of greater than 30 % [3]. This high diversity of HCV is caused by a fast replication rate combined with an error prone replication machinery. Owing to its pronounced genomic variation, HCV can evade immune recognition and become resistant to antiviral drugs.
After the discovery of HCV as the etiological agent causing non-A non-B hepatitis in 1989 [4], chronic hepatitis C patients were initially treated with interferon-α (IFNα). This original treatment was effective in only a fraction of treated patients, was poorly tolerated and required medication for 48 weeks. During the 1990s, IFN based therapy regiments were refined by addition of the nucleoside analogue ribavirin, and usage of pegylated IFNα (PEG-IFNα) derivatives increasing viral response rates [5]. After construction of the first infectious clone in 1997 [6] and the creation of the HCV replicon system [7] development of novel improved therapeutic strategies significantly gained momentum. A search for better drugs concentrated on direct acting antivirals (DAA), i.e., agents that specifically interfere with viral proteins required for propagation. New cell culture models for a genotype 2a isolate that permit analysis of the complete viral replication cycle in cell culture aided these efforts [8–10]. Since 2011, the first two DAAs, which are inhibitors of the viral protease NS3/4A, boceprevir and telaprevir, are on the market and achieve response rates of ~ 80 % in genotype 1 patients, when combined with ribavirin and PEG-IFNα. However, although this triple therapy has clearly improved response rates and shortened treatment duration, side effects, costs and exclusive licensing for genotype 1-infected patients limit its application. Fortunately, other promising virus-targeting drugs are in clinical trials, e.g., inhibitors of the NS5A phosphoprotein and the NS5B viral RNA-dependent RNA polymerase. Two major shortcomings of virus-targeting DAAs are the emergence of resistance mutations and -at least for several classes of DAAs (e.g., protease inhibitors)- a pronounced genotype-dependent efficacy. Numerous in vitro studies in combination with a growing number of HCV sequencing data from patients undergoing DAA treatment underline that the virus can develop drug-resistance and fitness restoring compensatory mutations [11]. Thus, DAAs will typically be used in combination therapy as in the current standard of care, which includes one of the two available protease inhibitors combined with PEG-IFNα and ribavirin. Again, as ribavirin and IFN are contraindicated in many patients and moreover cause harsh side effects that limit compliance, current drug development aims for future IFN-sparing and possibly also ribavirin-free combination-therapy regimens. In fact, numerous combination therapies involving DAAs with different targets on the virus are in clinical development (compare www.clinicaltrials.gov). It is expected that these trials will identify optimal drug combinations. Given the variability of HCV combined with the heterogeneity of patients with regards to co-morbidities, degree of liver disease and genetic background, it is likely that several combination therapy regimen will evolve, which are tailored toward specific patient and virus groups.
An emerging third group of antivirals, so called host-targeting antivirals (HTA), may be part of such future combination therapies, in particular as HTAs hold the promise of overcoming some of the caveats of DAAs. HTAs are antibodies, RNAs or small molecules, which interfere with host factors needed for HCV propagation. Intense research in the past decade revealed many molecular details of the HCV life cycle, including the usage of human proteins and a microRNA during entry, genome replication, particle assembly and/or release [12, 13•, 14]. This knowledge allowed targeted design of numerous HTAs, some of which with promising clinical trial results (Table 1). Host-targeting antiviral strategies are assumed to have two major advantages: First, the resistance barrier to host-targeting therapies is supposed to be high since host factors are genetically stable. Exceptions to this will be critically discussed in the specific chapters below. Second, usage of host factors is thought to be independent of the HCV genotype and thus HTAs should have pan-genotypic activity. Importantly, this assumption has not been fully supported by experimental evidence yet, as culture systems for genotypes other than genotype 1 and 2 were not available until recently. To date, the development of novel tissue culture models including chimeric viruses for all six major HCV genotypes [15] allows a detailed mechanistic and preclinical analysis of HTAs and a careful evaluation of their clinical use. With these tools we are beginning to understand that host factor usage might be HCV genotype dependent or at least blockage of host factors could have varying efficacies for different genotypes. Lastly, it is largely unknown, if genetic diversity of host molecules required by the virus influences efficacy of HTA-based antiviral strategies. With regards to PEG-IFNα/ribavirin therapy, work from the past four years highlights that host variability, i.e., polymorphisms in the vicinity of the IL28B gene locus, can affect the natural course and treatment outcome of hepatitis C [16•, 17–21]. In the future, stem cell technologies, like generation of induced pluripotent stem cells and their differentiation into hepatocytes, might allow addressing the effect of host genetic diversity on HCV infection [22–24]. Unquestionably, HTAs are emerging antivirals, which could complete the toolbox of HCV interfering strategies in the future. While DAA therapies have extensively been discussed elsewhere [25–27], we will here discuss the most advanced HTAs against HCV, mention early stage HTAs and highlight opportunities and risks of HTA therapy.
Table 1.
Host-targeting antivirals for hepatitis C therapy
| Inhibitor | Life cycle step targeted | Host factor targeted | Adverse effects | Resistance mutations (found in vitro) | Genotype specificity | Clinical phase |
|---|---|---|---|---|---|---|
| ITX 5061 | entry | SCARB1 | increased serum HDL | N415D (E2) | 1 - 6 a; 1 | 1b |
| anti-SRBI | entry | SCARB1 | N.A. | N.A. | 1, 2, 4, 6 | preclinical |
| anti-CD81 | entry | CD81 | N.A. | N.A. | 1, 2, 4 | preclinical |
| Anti-CLDN1 | entry | CLDN1 | N.A. | N.A. | 1 - 6 a | preclinical |
| erlotinib | entry | EGFR | rashes, diarrhea, lung, liver and kidney problems | N.A. | 1 - 6 a | preclinical |
| dansatinib | entry | ehprin A2 | rashes, diarrhea, lung, liver and kidney problems | N.A. | 1 - 6 a | preclinical |
| ezitimibe | entry | NPC1L1 | nausea, stomach pain, fever, loss of appetite, jaundice | N.A. | 1 - 6 a | preclinical |
| alisporivir | RNA replication | CypA | abdominal pain, fatigue | D320E/Y321N (NS5A) | 1, 2, 3 | 3 |
| SCY-635 | RNA replication | CypA | none reported | D320E/Y321N (NS5A), T77K/I432V (NS5B) | 1, 2, 3 | 2a |
| NIM811 | RNA replication | CypA | none reported | N.A. | 1 | 1 |
| AL-9 | RNA replication | PI4KIIIa | N.A. (PI4KIIIa −/− mice: lethal gastrointestinal disorders) | NS5A domain 1, NS4B C-terminus | 1, 2 | preclinical |
| Compound A and B | RNA replication | PI4KIIIa | N.A. (PI4KIIIa −/− mice: lethal gastrointestinal disorders) | NS5A domain 1, NS4B C-terminus | 1, | preclinical |
| Miravirsen | RNA replication | miR-122 | reduced serum cholesterol | N.A. | 1 - 6 a; 1 | 2a |
| Celgosivir | assembly and release | α-glucosidase I | mild to moderate gastrointestinal disorders | N.A. | 1 - 6 a; 1 | 2b, halted |
| DGAT1 inhibitor | assembly and release | DGAT1 | reduced plasma triglycerides | N.A. | 2 | preclinical |
| Py-2 | assembly and release | PLA2GA4 | N.A. | N.A. | 1, 2, 3, 5 a | preclinical |
N.A. data not available
aIn vitro data
Targeting HCV Cell Entry: Bona Fide Entry Factors
HCV host cell invasion is a complex multi-step process that requires numerous host cell factors and cell surface proteins. Among these, four so-called entry factors are indispensable for productive HCV uptake: scavenger receptor class B type I (SCARB1), the tetraspanin CD81 and the two tight junction molecules claudin-1 (CLDN1) and occludin (OCLN) [28–32]. While SCARB1 and CD81 bind the E2 glycoprotein on HCV particles [28, 29], it is unknown whether E2 directly interacts with CLDN1 or OCLN. However, lack of any one of the four proteins renders cells non-permissive to HCV [31]. Elegant kinetic studies using blocking antibodies against individual entry factors suggest their stepwise usage [30, 33–35]. SCARB1 initially binds HCV particles and may prime them for efficient CD81 binding [33, 36]. Interaction with CD81 in turn is thought to induce a conformational change in the E2 glycoprotein required for low pH-dependent membrane fusion in the endosome [37]. In contrast, CLDN1 and OCLN play a late role in entry shortly before particle uptake through clathrin-mediated endocytosis [30, 35]. Apart from the tight temporal control of HCV entry, the process is likely to be spatially regulated since liver-resident hepatocytes are polarized and the entry factors are differentially localized at the basolateral cell pole or the cellular tight junction. SCARB1 and CD81 reside on the basolateral side, facing the liver sinusoids, while CDLN1 and OCLN localize to the tight junctions between the apical and basolateral compartment. Circumstantial evidence suggests that CD81-bound HCV laterally translocates along the plasma membrane toward tight junctions, where uptake occurs [38, 39]. Lack of good polarized cell culture systems, inefficient HCV labeling and imaging techniques and the low specific infectivity of HCV derived from cell culture hamper, however, studies to provide direct evidence for this model. Unquestionably, CD81, SCARB1, CLDN1 and OCLN are required for HCV entry in vivo as demonstrated in human entry factor transgenic mice and human liver chimeric mouse models [40–43].
Interference with HCV entry factors is a strategy to block de novo infection of naïve cells (Fig. 1). Therefore, they are promising drugs for preventive therapy in chronic hepatitis C patients, who undergo liver transplantation. Indeed, re-infection of liver graft tissue is universal and unfortunately disease progression after transplantation is commonly accelerated. For hepatitis C treatment, virus-targeting entry blockers, i.e., neutralizing antibodies against the HCV glycoprotein E2, are unfavorable, as E2 is the most variable among the HCV proteins. Due to the error prone viral replication, an infected individual carries a swarm of related viral variants (quasispecies), which may comprise viruses that escape neutralizing antibodies. Consequently, there is a strong need for the development of HTAs interfering with virus entry. The most advanced entry-blocking compound is a small molecule inhibitor of SCARB1 termed ITX 5061 [44]. This orally bioavailable drug showed a good safety profile in clinical trials with 280 subjects [44]. In a clinical phase 1b study in treatment naive chronic genotype 1 patients only 1 of 7 patients showed a virological response [45]. However, in a post-transplant setting a better efficacy profile is possible. In preclinical tests using HCV pseudotypes, ITX 5061 had broad specificity for genotypes 1 through 6 confirming that SCARB1-dependence is conserved among viral genotypes and raising the hope that SCARB1-targeting molecules like ITX 5061 could be pan-genotypic HCV entry inhibitors [44]. Binding studies involving soluble truncated E2 protein indicate that ITX 5061 interferes with the interaction of E2 with SCARB1, which is likely the reason for its antiviral activity [44]. In vitro resistance selection revealed that a single nucleotide change leads to an amino acid substitution in HCV E2 (N415D) which renders HCV insensitive to ITX 5061 [46]. On the one hand, this demonstrates that HCV can in theory evade HTA therapy by mutating the viral binding partner of the targeted host factor and in fact suggests a low genetic barrier to resistance. On the other hand, the N415 residue is highly conserved among sequenced HCV isolates and only one in 1300 reported sequences in the Los Alamos National Laboratories HCV database shows aspartic acid at this position [46]. Notably, the same mutation was reported upon long term passage of the JFH1 virus in tissue culture [47] and further characterization revealed that it rendered the virus highly susceptible to neutralization by serum IgG from chronically HCV-infected patients. Therefore, immune-mediated constraints may prevent the virus from developing this type of ITX 5061 resistance in vivo. Moreover, the N415D ITX 5061 resistant virus was still sensitive to protease inhibitors, arguing that ITX 5061 could be used in combination with virus-targeting DAAs. Finally, as patients will receive ITX 5061 only for a comparably short duration in a post-transplant setting, the risk for resistance emergence may be low. Nevertheless, careful monitoring of viral variants emerging during in vivo application of this compound will be critical. Apart from drug resistance, an expected caveat of targeting host molecules and possibly their endogenous function are side effects. Those appear minimal for ITX 5061. The only reported but not adverse side effect of ITX 5061 is the inhibition of SCARB1 mediated high density lipoprotein (HDL) uptake leading to increased serum HDL levels [48]. In fact, ITX 5061 was originally tested as an anti-atherogenic compound. Lastly, it remains to be determined whether host genetic variance of SCARB1 could affect ITX 5061 inhibition. A recent study suggests that several lab-generated variants of SCARB1 all efficiently support HCV cell entry [49]. Given that ITX 5061 competes with HCV for SCARB1 binding, it is likely that the compound inhibits all SCARB1 variants, but future work, in particular on naturally occurring SCARB1 variants, will clarify this assumption. In summary, ITX 5061 is a promising small molecule, pan-genotypic HTA for preventive combination therapy of post-transplant hepatitis C patients.
Fig. 1.
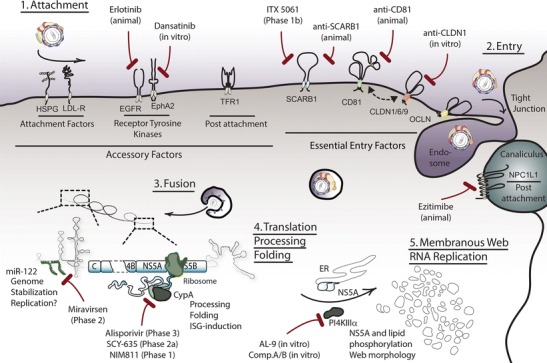
HCV life cycle and critical host factors for intervention. HCV requires host proteins and RNAs during its life cycle offering several points of intervention using HTAs. Initially, HCV-lipoviroparticles attach to LDL-R and heparan sulfate proteoglycans (HSPG) on the surface of hepatocytes, followed by a coordinated uptake requiring the essential entry factors SCARB1, CD81, CLDNs and OCLN. The latter four entry factors can be targeted by specific antibodies or small molecules, like ITX 5061, which blocks HCV-SCARB1 interactions and is in clinical phase 1b development. Accessory factors including receptor tyrosine kinases, transferrin receptor 1 (TFR1) and the cholesterol receptor NPC1L1 provide alternative targets for entry blockage. After internalization and particle uncoating, the HCV RNA genome replicates in specialized cytosolic compartments, termed membraneous webs. Formation of these replication complexes is aided by host and viral proteins. Inhibition of the host chaperone CypA by cyclosporine and its derivatives (alisporivir, SCY-635, NIM811) affects function of the viral NS5A protein and thereby inhibits HCV replication. Alisporivir, SCY-635, NIM811 are currently tested in clinical phases 3, 2 and 1, respectively. PI4KIIIα is a host lipid kinase required for membraneous web formation and its inhibition by small molecules, like AL-9, abolishes HCV replication. A third class of replication HTAs is comprised by antagomirs of the host miR-122. This microRNA binds to and stabilizes the HCV RNA genome and facilitates its translation. Moreover, an involvement of miR-122 in HCV RNA replication is being discussed. Sequestration of miR-122 by miravirsen, a clinical phase 2 HTA, strongly suppresses viral titers
Apart from small molecules, entry factor blocking antibodies efficiently inhibit HCV entry and are in preclinical development [41–43, 50]. The most advanced antibodies target SCARB1 and CD81. In a recent report, two neutralizing antibodies against SCARB1 (mAb8 and mAb151) blocked HCV infection and direct cell-to-cell spread in vitro and in vivo [43]. Notably, in a human liver xenotransplant mouse model (uPA-SCID) both anti-SCARB1 antibodies prevented HCV infection in a prophylactic setting, when five antibody dosages were given over a period of two weeks starting one day prior to infection. However, viral rebound occurred at least one week after termination of antibody treatment, suggesting that a two-week treatment is insufficient to eliminate virus from peripheral reservoirs. In a three-day post-infection setting, the antibodies prevented infection in three out of five mice and reduced viral dissemination in the remaining two mice. Similar to ITX 5061, SCARB1 antibodies are pan-genotypic and show no adverse side effects, thus presenting an alternative option for preventive therapy. Future studies will have to show whether anti-SCARB1 therapy is efficient in a broad range of orthotropic liver transplant patients.
In contrast to SCARB1 antibodies, CD81 antibodies only block HCV infection in a prophylactic setting in the xenotransplant uPA-SCID mouse model [41], suggesting that targeting SCARB1 is superior to blockage of CD81. Although the reason for this is not clear, this discrepancy may point to a differential role of these entry factors during virus transmission. In vitro data indicate that HCV is transmitted by cell-free, secreted virus particles and also by direct cell-to-cell transmission between neighboring cells [51–53]. This latter mode of infection may be particularly relevant in vivo and could facilitate viral escape from neutralizing antibodies. Notably, while CD81 is absolutely essential for infection by cell-free virus, its importance for direct cell-to-cell transmission has been disputed [52, 53]. On the one hand, Fofana et al. identified a CD81 antibody, which –at least in vitro– also interferes with cell-to-cell spread [54]. The epitope for this antibody currently remains elusive, but this finding raises the hope that development of CD81 antibodies for post-infection treatment could be possible. On the other hand, recent evidence confirmed that CD81 facilitates, but is not absolutely essential for, cell-to-cell transmission in vitro [55]. If that holds true in vivo, the absence of an essential role for CD81 during cell-to-cell spread may limit efficacy of CD81 antibodies compared to antibodies targeting entry factors critical for both cell-free and cell-to-cell transmission. As mentioned above, SCARB1 neutralizing antibodies potently repress cell-to-cell transmission in vitro [53] and in humanized mice [42, 43]. Taken together, these results suggest that SCARB1 is more important for direct cell-to-cell spread than CD81, which in turn explains why targeting SCARB1 blocks HCV infection in vivo more effectively than CD81.
Antibodies binding CLDN1 efficiently block HCV entry in vitro [50] and may thus be another promising avenue for clinical development. Notably, CLDN6 and CLDN9 can substitute for lack of CLDN1 in human non-liver cells [56, 57]. Although it was originally assumed that this broad tropism toward different members of the CLDN protein family is common to all HCV isolates, this notion was recently challenged: Using cell lines expressing either only CLDN1, CLDN6 or both molecules, Haid et al. showed that all tested viral strains efficiently used CLDN1 while only some strains also used CLDN6 [58]. Importantly, viruses capable of using both CLDN1 and CLDN6 were not fully neutralized by CLDN1-specific antibodies, if the same cell co-expressed a modest level of CLDN6. These findings point toward a possible viral escape from CLDN1-targeting agents for viral strains with broad CLDN-tropism. However, Fofana et al. were unable to observe an additive inhibitory effect when combining CLDN1 and CLDN6-specific antibodies on Huh-7.5.1 cells challenged with HCV [54]. This discrepancy might result from the use of host cell lines with different CLDN1 and six expression levels. Notably, primary human hepatocytes of some donors express low but detectable CLDN6 at the cell surface and CLDN6 transcript expression is highly variable in HCV patient derived liver biopsies [54, 58]. Thus, the potential risk of viral escape from CLDN1-targeting strategies via use of CLDN6 may not only depend on the viral strain but also on host determinants, i.e., differential abundance of CLDN6. Clearly, the relevance of viral CLDN tropism and its potential implication for development of CLDN1-targeting strategies requires further investigation, e.g., in xenotransplanted mice with human hepatocytes from donors with distinct CLDN6 expression. Aside from these aspects related to efficacy and potential viral escape, possible side effects of anti-CLDN treatments should be carefully considered. Currently, in vitro data suggests that CLDN1 endogenous functions remain unaltered upon antibody treatment [54]. Consequently, side effects of anti-CLDN1 treatment are expected to be low. Confirmation of this assumption in preclinical models will be critical before finally evaluating CLDN1-targeting therapy.
For the fourth essential entry factor, OCLN, no neutralizing antibodies have been reported to date. However, as OCLN deficient mice have a severe phenotype including growth retardation, infertility and bone thinning, preclinical tests need to carefully investigate possible side effects of OCLN-targeting in humans [59]. In conclusion, small molecule inhibitors and antibodies targeting SCARB1 are the most advanced HCV entry-targeting agents. Given the essential and pan-genotypic role of SCARB1 for both cell-free and cell-to-cell transmission, SCARB1 blockage holds the promise for a well-tolerated therapy to prevent allograft infection of chronic hepatitis C transplant patients.
Targeting HCV Cell Entry: Entry Co-factors and Modulators
Apart from the four essential entry factors, additional molecules have been shown to be involved in HCV cell entry. These include attachment factors like glucosaminoglycans [34, 60] and the low density lipoprotein receptor (LDL-R) [61, 62], receptor tyrosine kinases [63•], and Nieman-Pick C1-like 1 and transferrin receptor 1 [64, 65]. This multitude of host factors offers numerous levels for interference (Fig. 1). Among the most prominent entry co-factors are the two receptor tyrosine kinases epidermal growth factor receptor (EGFR) and ephrin A2 (EphA2) [63•]. Inhibition of EGFR and EphA2 using the small molecules erlotinib and dansatinib blocks HCV entry in vitro with an IC50 of 500 nM for both compounds. In a human liver chimeric mouse model daily preventive erlotinib treatment reduces serum titers of HCV approximately ten-fold compared to placebo treated animals. Viral titers rebound to serum titers of control animals shortly after treatment termination. Together, these findings provide evidence that erlotinib at least partially represses HCV infection in vivo. Moreover, in vitro erlotinib inhibits both cell-free and cell-to-cell transmission of HCV and silencing of EGFR suggest that the usage of EGFR is HCV genotype independent [63•]. If, however, erlotinib is able to fully ablate HCV cell entry in vivo and whether the virus can escape this type of treatment by resistance mutations is currently not clear. Adverse effects of erlotinib, which is an approved anti-cancer drug, include rashes, diarrhea, lung, liver and kidney problems [66]. Occurrence of side effects is in line with the fact that EGFR is ubiquitously expressed and has important physiological functions including cell proliferation, migration and adhesion. While the intensity of side effects associated with erlotinib and many other anti-cancer agents may be acceptable in malignant disease, it may preclude their use for a not quite as immediately life threatening condition such as chronic HCV infection. Lastly, polymorphisms in EGFR seem to correlate with the outcome of erlotinib treatment in non-small cell lung cancer, suggesting that host genetic variance could also influence antiviral therapy outcome [67]. In summary, targeting of conserved host factors with less critical endogenous functions seems favorable over interference with receptor tyrosine kinases.
Novel mechanistic studies on the role of EGFR revealed that it signals through the GTPase HRas and the kinase BRaf for HCV receptor complex assembly [68]. Small molecule inhibitors of both signaling molecules exist and are licensed for anti-cancer therapy. We currently lack preclinical in vivo data for their usefulness in anti-HCV therapy. In principle, targeting HRas and BRaf offers an additional mode of intervention with HCV cell entry, possibly with superior efficacy and/or reduced side effects when compared with erlotinib.
A second and less frequently discussed HCV entry modulator is the cholesterol receptor Niemann-Pick C1-like 1 (NPC1L1) [64]. This protein is a 13 transmembrane molecule, which is expressed on the apical side of hepatocytes and mediates cholesterol absorption. In vitro the small molecule NPC1L1 inhibitor ezetimibe decreased HCV infection at least five-fold at a 30 μM dose. In a human liver chimeric mouse model ezitimibe prevented infection of two out of seven mice when a three-week dosing was started two weeks prior to infection. Treatment onset two days prior to infection surprisingly did not prevent HCV infection of mice. Although NPC1L1 seems to reduce entry of all HCV genotypes and although ezitimibe is a licensed cholesterol-lowering drug with little side effects [69], the overall modest efficacy in mice does not favor application of NPC1L1 inhibitors in monotherapy. However, in conjunction with other HTAs or DAAs ezitimibe may be useful to improve treatment efficacy.
Taken together, targeting HCV entry modulators seems less efficient than targeting of bona fide entry factors, likely due to their accessory rather than essential role during HCV cell entry. Still, compounds with high tolerability could be a valuable addition in combination therapy, in particular after orthotropic liver transplantation.
Targeting HCV RNA Replication: Cyclophilin A
While blockage of HCV entry into hepatocytes is a promising strategy to prevent de novo infection of naïve cells, targeting HCV replication, i.e., amplification of the viral genome, holds the promise of efficiently eradicating HCV from already infected tissue. HCV replication takes place in the cytoplasm of the host cell, where virus encoded RNA-dependent RNA polymerase (NS5B) amplifies the plus strand RNA genome. To this end, HCV induces with the aid of host factors specialized membranous compartments [70–73], at which multiple viral proteins including non-structural proteins NS3, NS4A, NS4B, NS5A and NS5B and host factors assemble the HCV replication complex [74]. Hence, induction, assembly and function of HCV replication complexes involve numerous host factors offering multiple targets for intervention. Cyclophilin B (CypB) was one of the first host factors reported to be crucial for HCV replication [75]. Cyclophilins are highly conserved peptidyl-prolyl isomerases, which catalyze the isomerization of peptide bonds at proline residues from trans to cis. Such transformations either aid folding of newly synthesized proteins or change the structure of already folded proteins. Interestingly, cyclophilins are essential replication factors for a number of viruses, including HIV, herpes simplex virus, vaccinia virus, vesicular stomatitis virus and coronavirus. In the context of HCV replication, more recent evidence indicates that cyclphilin A (CypA) rather than CypB is used by HCV as co-factor and that the isomerase activity of CypA is essential for this function [76–79]. Moreover, biochemical and genetic analyses revealed an interaction between subdomains of NS5A and CypA [80, 81], which could promote viral protein folding, regulate polyprotein processing and thereby facilitate RNA replication. Targeting of CypA by the cyclic polypeptide immunosuppressant agent cyclosporine A (CsA) prevents the interaction of CypA and NS5A across all viral genotypes and has strong anti-HCV activity in vitro (Fig. 1) [82, 83].
CsA is not only antiviral, but also immunosuppressive, since the CsA-CypA complex inhibits calcineurin and thereby suppresses T helper cells [84, 85]. This dual role stimulated the development of derivatives, which retain antiviral activity without being immunosuppressive. Currently, numerous CypA inhibitors with exclusive antiviral effect are in preclinical and clinical trials [86]. Among these, alisporivir (Debio 025), NIM811 and SCY-635 are the most extensively studied drug candidates. Alisporivir showed efficient reduction of viral load (2-log to 4-log depending on the study) for genotype 1, 2, 3 and 4 during monotherapy [87–89]. Moreover, combination with INFα/ribavirin resulted in additive effects. In a phase 2 clinical trial with such combined triple therapy the incidence of adverse effects was low. Thereafter, a phase 3 trial in treatment-naïve genotype 1 patients was initiated, but soon halted by the FDA due to occurrence of pancreatitis with one fatal outcome upon treatment with IFN and alisporivir. Therefore IFN/alisporivir combinations are precluded from future clinical development. However, trials with IFN-free alisporivir treatment regimens with improved safety profile will resume. Apart from its broad HCV genotype specificity alisporivir seems to act independently of the host genetic background. A recent study investigated host variability of CypA and found that rare nonsynonymous SNPs in CypA not only rendered cells largely resistant to HCV infection, but also residual replication was still sensitive to CypA inhibition [90]. Lastly, CypA inhibitors appear to have a high genetic barrier to development of viral resistance in vivo. However, in vitro resistance toward anti-cyclophilin compounds can be selected either by long term viral passage in the presence of the drugs or by selecting viruses in host cells with reduced cyclophilin abundance [78, 91, 92]. Currently described resistance mutations map to domain 2 and 3 of NS5A supporting the concept that CypA facilitates HCV replication via modification of NS5A function. Interestingly, some of these resistance mutations are located at the C-terminus of NS5A close to the cleavage site between NS5A and NS5B. Moreover, these alterations were shown to modulate polyprotein cleavage at the NS5A-NS5B site. This supports the notion that cyclophilin A fine tunes protein folding processes that are required for optimal polyprotein cleavage and in turn replication complex assembly [78]. Although some of the mutations described in vitro confer a high degree of viral resistance (in part shifting the EC50 value by more than 40-fold [91]), so far no resistance mutations have been described during treatment of HCV patients. In fact, during alisporivir monotherapy no viral breakthrough was observed suggesting that in vivo the barrier to viral resistance is high [93]. Notably, in the same study one alisporivir null responder was identified. If, however, absence of viral response in this case was due to viral or host resistance is not clear and merits further investigation.
A possible explanation for this rare treatment failure could be a recently reported second antiviral mechanism of cyclophilin derivatives, which is independent from CypA-NS5A complex disruption. The alternative non-immunosupressive CypA inhibitor, SCY-635, which is currently in clinical phase 2a trials, seems to reconstitute IFN signaling and in turn increase innate antiviral defenses. First evidence for this stems from a clinical trial with SCY-635 monotherapy in chronic HCV genotype 1 infected patients. In this context, SCY-635 not only dose dependently repressed viral load but also caused increased plasma levels of IFNα, IFN λ 1 and 3 as well as 2´5´oligoadenylate synthase 1 (2´5´OAS-1), a key IFN stimulated gene (ISG) [94••]. Meanwhile, two possible molecular links between SCY-635, IFN and ISG induction were disclosed: Bobardt and colleagues reported that CypA binds IFN regulatory factor 9 (IRF9) and that NS5A competes with IRF9 for CypA binding [95]. As IRF9 is the DNA binding part of the transcription factor IFN-stimulated gene factor 3 (ISGF3), it is critical to transmit IFN-induced JAK/STAT signaling to the nucleus for expression of ISGs [96]. Importantly, inhibition of CypA by cyclosporine prevents IRF9-CypA complex formation and thereby enhances IFN-induced expression of ISGs [95]. On the other hand, Watashi et al. noted that in IFN-treated HCV infected cells, SCY-635 decreases phosphorylation of protein kinase R (PKR) [97]. Since phosphorylated PKR downregulates expression of ISG at the level of translation, it is possible that SCY-635-dependent repression of PKR phosphorylation enhances translation of ISGs [98]. Therefore, blockade of CypA by SCY-635 may increase expression of ISGs and antiviral activity of IFN by both transcriptional and post-transcriptional mechanisms. It will be interesting to dissect to what extent these mechanisms contribute to the antiviral activity of CypA-targeting strategies and if both antiviral mechanisms are shared by the different compounds targeting CypA. Provided concerns regarding the safety of CypA-targeting HTAs can be eliminated, these agents could be attractive pan-genotypic therapeutics.
Targeting HCV RNA Replication: Phosphatidylinositol 4-kinase III alpha (PI4KIIIα)
Genome wide RNA interference screens and in depth cell culture replication assays with HCV replicons and full length infectious virus have revealed numerous additional host dependency factors, that could in principle serve as antiviral targets [99–107]. One of the most prominent and most consistently identified host factors for HCV replication is PI4KIIIα [101–106]. This protein belongs to a family of enzymes that catalyze phosphorylation of lipids at position four of their inositol moiety. The resulting phosphoinositides (PIs) reside at the cytosolic leaflet of vesicle and organelle membranes, where they play a critical role in the recruitment and activity of signaling proteins [108, 109]. To date, four mammalian phosphatidylinositol 4-kinases are known (PI4KII α and β and PI4KIII α and β), which differ in their subcellular localization and create distinct PI pools, thus contributing to vesicle trafficking and lipid transport [108]. PI4KIIIα is located at the endoplasmic reticulum and the plasma membrane and was found to be the primary mammalian PI4K that influences HCV replication [102]. Notably, silencing of PI4KIIIα dramatically reduces HCV RNA replication and concomitantly results in a clustered distribution of viral non-structural proteins and aberrant ultrastructure of the membranous web [105], which is an accumulation of membrane vesicles and the site of HCV RNA replication [70, 71]. Interestingly, HCV NS5A directly interacts with PI4KIIIα and this interaction stimulates kinase activity of the enzyme [102, 105]. More recently, the binding site of PI4KIIIα was mapped to a highly conserved region within domain 1 of NS5A. Moreover, PI4KIIIα, although being a lipid kinase, was reported in the same study to modulate the phosphorylation status of NS5A [110]. Although it is currently unclear if this effect is direct or indirect, likely both the PI4KIIIα-dependent regulation of NS5A phosphorylation and local accumulation of phosphatidylinositol 4-phosphate pools are important for HCV replication [110]. Since reduced levels of PI4KIIIα and viral NS5A mutations ablating the interaction with PI4KIIIα both cause aberrant ultrastructure of HCV replication complexes, it is reasonable to assume that the PI4KIIIα-NS5A interplay is essential for proper assembly and function of membrane bound HCV replication complexes.
To date, two studies have addressed if targeting the interaction between NS5A and PI4KIIIα or the enzymatic activity of PI4KIIIα itself would be a useful strategy to control HCV RNA replication. Bianco et al. reported that AL-9, a 4-anilino quinazoline, targets the kinase activity of PI4KIIIα in vitro and within liver cells [111]. Interestingly, 4-anilino quinazoline compounds had previously been reported as HCV inhibitors [111–114], and based on putative resistance mutations within NS5A, presumed to target NS5A. Until now, none of these mutations was however shown to confer resistance to quinazolines [111, 112]. Nevertheless, given the interaction between PI4KIIIα and NS5A and the observation that aniline quinazoline moieyties are frequently present in kinase inhibitors, it seems likely that AL-9 targets PI4KIIIα and thereby inhibits HCV replication. Additional compelling evidence that PI4KIIIα-inhibitors interfere with HCV replication was reported by Vaillancourt [115•]. Using a PI4KIIIα kinase assay to screen more than 500,000 compounds various specific inhibitors of this enzyme were identified. Molecules belonging to three different chemotypes – all of which are unrelated to the 4-anilino quinazoline compounds described above – potently repressed kinase activity in vitro and HCV replication in replicon assays. Importantly, inhibition of other kinases was excluded by in vitro assays involving various kinases and lipid kinases. These studies also revealed 15 to 20-fold selectivity toward PI4KIIIα compared to PI4KIIIβ. Besides this, the antiviral activity of the compounds correlated with the degree of PI4KIIIα inhibition further supporting the notion that the compounds are antiviral due to blockade of PI4KIIIα. Finally, viral resistance was selected and revealed that specific mutations within the C-terminus of NS4B and domain 1 of NS5A reduce sensitivity of HCV to these molecules by up to 20-fold. Interestingly, the resistance mutations permitted efficient HCV replication in cells with silenced PI4KIIIα highlighting that they confer reduced dependence of HCV on functional PI4KIIIα and indeed that the compounds target PI4KIIIα. Combining these observations with the ultrastructural analyses of replication complexes from cells with PI4KIIIα silencing or mutant viruses with reduced PI4KIIIα-binding, it is reasonable to assume that recruitment of PI4KIIIα by NS5A and the enzymatic activity of PI4KIIIα are crucial for proper assembly/morphology and function of HCV replication complexes (Fig. 1). Inhibitors of PI4KIIIα disturb NS5A binding and viral resistance to these molecules is attained by reduced dependence on PI4KIIIα due to altered function of NS5A and/or NS4B. Of note, kinase inhibitor resistance mutations decreased HCV RNA replication in Huh-7.5 with endogenous levels of PI4KIIIα. Thus, resistance is likely linked to a decrease in viral fitness, which may contain emergence of these mutations in vivo. To explore the physiological role of PI4KIIIα in vivo, Villaincourt et al. created knockout mice with conditional lesion of the PI4KIIIα gene locus [115•]. Unfortunately, induction of the gene defect in homozygous animals caused lethal gastrointestinal disorders. Therefore, the critical physiological role of PI4KIIIα and possible side effects of therapies targeting this enzyme will probably limit further development of this class of inhibitors for future HCV therapy.
Targeting HCV RNA Replication: MicroRNA-122
Apart from proteinaceous host factors, HCV requires a microRNA for efficient replication. MicroRNAs (miRNAs) are 20 to 22 nucleotides long non-coding RNA molecules, which typically bind to mRNA and arrest translation or induce mRNA cleavage and degradation, thus having emerged as powerful regulators of gene expression [116]. Not surprisingly, viruses have evolved to exploit this cellular machinery. In fact, several DNA viruses such as herpesviruses encode viral miRNAs and use these for tuning expression of both host and viral RNAs [117]. Although HCV, like most RNA viruses, does not encode miRNAs itself, it depends on these RNA molecules in a unique way, thus offering a potential target for antiviral intervention. Specifically, miRNA-122 (miR-122), a liver-specific miRNA, that regulates numerous genes involved in fatty acid and cholesterol metabolism, binds to the 5´non translated region of the HCV RNA genome. Two tandem binding sites for miR-122 have been characterized [118] and the site specific binding of miR-122 has been reported to facilitate translation of the viral RNA [119] and to stabilize the HCV RNA leading to an accumulation of vial genomes [120–123]. Although presence of miR-122 is not absolutely essential for HCV RNA replication, its high abundance is crucial for efficient replication [124]. Thus, the liver-specific expression of miR-122 likely contributes to the overt hepatotropism of HCV [125, 126].
Inactivation of miR-122 using a complementary locked nucleic acid-modified oligonucleotide (miravirsen or SPC3649) reduces HCV titers in vitro and in HCV infected chimpanzees by 2–3 logs (Fig. 1) [127••]. Moreover, broad HCV genotype specificity in vitro suggests a wide usage for a miravirsen-based therapy in patients [128]. Genetically engineered variants of HCV, which lack the miR-122 binding site, are resistant to miravirsen and still viable although showing fitness loss [128]. Notably, in monotherapy clinical trials and in cell culture so far no resistance mutations to miravirsen emerged [129] . Therefore, the barrier to viral resistance to this drug seems high. With regard to side effects, preclinical studies in chimpanzees suggest that miravirsen treatment reduces serum cholesterol levels, but neither induces toxicity nor histopathological changes. More importantly, in a recent phase 2a clinical trial five weekly injections of miravirsen reduced viral titers up to 3 logs without adverse side effects or resistance emergence [130••, 131••]. In fact, some treated patients cleared HCV RNA during miravirsen monotherapy. Furthermore, miravirsen treatment both in chimpanzees and humans elicited a continuous and prolonged antiviral effect that lasted for several weeks after cessation of therapy [127••]. While miravirsen administration is currently only possible through the less attractive parenteral route, an advantage of miravirsen therapy could be the longlasting effect. Pharmacokinetic patient studies reveal a 37 day plasma half-life and suggest that miravirsen may be administered only once per months [132]. Collectively, in vitro and in vivo data provide firm evidence that targeting miR-122 is an efficacious and –at least in these transient treatment regimens– well tolerated future therapeutic option.
In spite of the encouraging initial results, recent findings regarding the physiological role of miR-122 warrant caution: Mice lacking miR-122 are viable but develop steatohepatitis, fibrosis and hepatocellular carcinoma. Importantly, reconstitution of miR-122 reduces tumor incidence demonstrating that miR-122 acts as tumor suppressor in mice [133]. To date, the molecular details of miR-122´s anti-tumor activity are unclear. Certainly, more research is needed to exclude that transient sequestration of miR-122 could promote tumorigenesis. If severe adverse side effects occur, the long half-life of miravirsen will make its serum levels difficult to control, in particular since there is no readily available means of terminating the drug’s action. Apart from this note of caution regarding miR-122 as host target, it will be interesting to explore if the association of chronic HCV infection with hepatocellular carcinoma is connected with the virus usurping and sequestering miR-122. In conclusion, miravirsen is a pan-genotypic, effective HTA with low risk of resistance emergence. Critical evaluation of adverse effects will clarify if miravirsen will be part of future INF-free regimen against HCV.
Targeting HCV Assembly and Release
Cell based assays to dissect the pathways and steps of HCV particle assembly and virus release have been available only for a relatively short time [8–10]. Therefore, host-targeting antiviral strategies focusing on these late stages of the viral replication cycle are least advanced. The first reported assembly blockers were iminosugars, which target α-glucosidase I, an ER enzyme required for HCV glycoprotein folding and maturation [134–136]. However, comparably modest efficacy of the iminosugar celgosivir in genotype 1 patients led to termination of clinical trials [137]. Nevertheless, more recent reports have highlighted several cellular co-factors that assist virus production and that may be future targets for antiviral therapies.
It has been known for a long time that HCV travels through the blood stream in tight association with lipoproteins [8]. Moreover, careful proteomic analysis of serum-derived HCV revealed the presence of apolipoprotein E (ApoE), ApoC1, ApoB, and ApoA1 in the lipoviroparticle [8, 138, 139]. For cell culture derived HCV (HCVcc) ApoE, and ApoC1, have been reported to associate with particles and the lipid composition of HCVcc was found to resemble the one of very low density lipoprotein (VLDL) [140]. Given this interplay of HCV with lipoproteins, it was not surprising that host factors involved in assembly and release of VLDL seem to aid production of infectious viral progeny: Specifically, microsomal triglyceride transfer protein (MTTP), which is involved in loading of lipids onto nascent ApoB, as well as ApoB and ApoE, both components of VLDL, were reported to contribute to virus production. As a consequence, modulators of VLDL production and secretion emerged as potential antivirals. For some of these compounds including MTTP and ApoB inhibitors, preclinical results are available and show modest antiviral activities. Notably, recent evidence suggests that among the different factors of the VLDL pathway implicated in HCV virus production, only ApoE is absolutely essential, since production of infectious HCV can be reconstituted in an engineered human kidney derived cell line (293 T), which does not produce VLDL and lacks endogenous expression of MTTP and ApoB [141]. Although these HCV particles with minimal lipoprotein coat may not be as infectious as natural HCV it is therefore possible that HCV could escape MTTP and ApoB targeting strategies. On the other hand, these findings favor development of ApoE-targeting strategies. Moreover, ApoE associated with HCV particles plays a critical role during viral cell entry by facilitating attachment of virions to cellular heparin sulfate proteoglycans [142–144], so that ApoE-targeting compounds may arrest both assembly and entry of HCV particles.
Besides host factors of the VLDL pathway additional cellular proteins have recently been shown to contribute to virus production. For instance, cellular lipid modifying enzymes like diacylglycerol acyl transferase 1 (DGAT1) and the cytosolic phospholipase A2 (PLA2GA4) contribute to production of infectious HCV progeny [145, 146]. Specifically, DGAT1 catalyzes triglyceride biosynthesis and thereby promotes lipid droplet formation. HCV is thought to assemble on the surface of lipid droplets and in vitro DGAT1 inhibition or silencing was shown to reduce HCV genotype 2a assembly and release by limiting the trafficking of HCV core to these orgenelles [145]. Intriguingly, upon inhibition of DGAT1, the related enzyme DGAT2 seemed to compensate the endogenous function of DGAT1. This in vitro finding indicates that adverse effects of DGAT1 inhibition may be low. Moreover, DGAT1 inhibitors are in development for treatment of obesity and display little adverse effects [147]. The second lipid modifying enzyme reported to be involved in HCV assembly, PLA2GA4, cleaves glycerophospholipids with arachidonic acid at the sn2-position. Thereby PLA2GA4 alters membrane fluidity and curvature and releases arachidonic acid, which is a precursor for inflammatory mediators. Hence, PLA2GA4 inhibitors like pyrrolidine-2 (Py-2) are in preclinical development for treatment of inflammatory disorders. In a recent study by Menzel et al. Py-2 inhibited assembly and release of HCV genotypes 1, 2, 3 and 5 in vitro and treatment with arachidonic acid restored HCV infectivity [146]. Thus PLA2GA4 inhibitors present a possible avenue for assembly blockage and future preclinical tests could connect to the existing pipelines of pyrrolidines as anti-inflammatory agents.
In summary, the identification of host lipoproteins and enzymes required for HCV assembly and release provides novel HTA development options. As we currently lack knowledge of possible side effects and efficacy of such HTAs in vivo, future preclinical work needs to elucidate if compounds targeting the last step of the HCV life cycle merit further development as possible anti-HCV therapeutics.
Conclusion: Opportunities and Risks of Future HCV Therapies
After 15 years of IFNα/ribavirin based therapies against hepatitis C, novel treatment regimens developed rapidly since the approval of the first DAAs in 2011. An increased understanding of the molecular virology of HCV and of the usage of host factors for propagation led to the discovery of a multitude of antiviral targets. Initial efforts focused on the inhibition of viral enzymes or viral structural proteins. This is reflected by the overall distribution of FDA-approved antiviral drugs. In 2012, the FDA listed 47 virus-targeting drugs including those against HIV and influenza virus and only 10 HTAs [148]. The latter will, however, gain increasing attention as they often have a high genetic barrier for resistance emergence and display a broad specificity for various genotypes and subtypes of a given virus. The most prominent HTAs against HCV include entry inhibitors and replication inhibitors. Preclinical studies are, however, starting to reveal that some of the proposed advantages of HTAs do not apply. In rare cases genotype specificity is observed as different genotypes can engage different host factors as exemplified by the usage of CLDN1 and CLDN6 for HCV entry. In other cases the virus can acquire resistance mutations, which lead to usage of an altered binding site on the same host factor. For instance, NS5A mutations can confer resistance to CypA-targeting drugs. These findings suggest that even HTAs should be used in combination therapy with other drugs. An obvious caveat of HTAs is the possibility of side effects. Consequently, preclinical and early clinical studies need to carefully evaluate dosing and treatment duration of host-targeting therapies. It should be noted that the reported side effects for some HTAs in clinical phases like ITX 5061 and SCY-635 are low. However, caution is warranted as the termination of alisporivir/IFN clinical trials shows. In summary, at least some HTA therapies appear to have a superior side effect profile compared to past IFNα-based standard of care, which showed strong, sometimes intolerable adverse effects over a 24 to 48 week treatment period. Lastly, we are just beginning to understand the role of host genetics in HCV infection. The best example is a polymorphism upstream of the IL28B gene, which correlates with development of chronicity and IFNα-based treatment response [18–21]. Future research needs to address whether or not HCV host factors are genetically diverse in the human population and what impact polymorphisms have on HCV infection and response to HTA treatment. Nonetheless, the era of virus- and host-targeting antivirals against HCV holds a big promise to chronic HCV patients. Many previous non-responders can now be treated with the new protease inhibitors in combination with IFNα/ribavirin and patients undergoing treatment benefit from reduced side effects and shortened therapy duration. Future development of HTAs and novel INFα-free possibly all-oral combination therapy is expected to further increase quality of life of chronic HCV patients. Certain patient groups, e.g., HIV co-infected individuals and patients with late stage liver disease, might require individualized therapy. Thus, intense research on DAAs and HTAs is needed to find the most effective drug combinations with least adverse effects. Finally, a better understanding of host and virus genetic diversity and their influence on drug efficacy might allow personalized treatment in the future and thereby guarantee cost effective use of novel drugs and optimal therapy outcome for individual patients.
Compliance with Ethics Guidelines
Conflict of Interest
Gisa Gerold has no conflicts of interest to declare. She received grants from the German Academy of Science Leopoldina, the Human Frontier Science Program and the German Liver Foundation. Thomas Pietschmann is a paid board member for Janssen and Biotest, and his institution receives grants from ERC, DFG, and Helmholtz Association.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the authors.
Contributor Information
Gisa Gerold, Email: gisa.gerold@twincore.de.
Thomas Pietschmann, Email: thomas.pietschmann@twincore.de.
References
Papers of particular interest, published recently, have been highlighted as: •Of importance ••Of major importance
- 1.Gravitz L. Introduction: a smouldering public-health crisis. Nature. 2011;474:S2. doi: 10.1038/474S2a. [DOI] [PubMed] [Google Scholar]
- 2.Seeff LB. Natural history of chronic hepatitis C. Hepatology. 2002;36:S35. doi: 10.1053/jhep.2002.36806. [DOI] [PubMed] [Google Scholar]
- 3.Simmonds P. The origin of hepatitis C virus. Curr Top Microbiol Immunol. 2013;369:1. doi: 10.1007/978-3-642-27340-7_1. [DOI] [PubMed] [Google Scholar]
- 4.Choo QL, et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 5.Manns MP, Wedemeyer H, Cornberg M. Treating viral hepatitis C: efficacy, side effects, and complications. Gut. 2006;55:1350. doi: 10.1136/gut.2005.076646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kolykhalov AA, et al. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science. 1997;277:570. doi: 10.1126/science.277.5325.570. [DOI] [PubMed] [Google Scholar]
- 7.Lohmann V, et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 8.Zhong J, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102:9294. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindenbach BD, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 10.Wakita T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat med. 2005;11:791. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vermehren J, Sarrazin C. The role of resistance in HCV treatment. Best Pract Res Clin Gastroenterol. 2012;26:487. doi: 10.1016/j.bpg.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 12.Zeisel MB, Felmlee DJ, Baumert TF. Hepatitis C virus entry. Curr Top Microbiol Immunol. 2013;369:87. doi: 10.1007/978-3-642-27340-7_4. [DOI] [PubMed] [Google Scholar]
- 13.Lohmann V. Hepatitis C virus RNA replication. Curr Top Microbiol Immunol. 2013;369:167. This is the first study describing a small molecule inhibitor for HCV entry. [DOI] [PMC free article] [PubMed]
- 14.Lindenbach BD. Virion assembly and release. Curr top microbiol immunol. 2013;369:199. doi: 10.1007/978-3-642-27340-7_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gottwein JM, et al. Development and characterization of hepatitis C virus genotype 1–7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology. 2009;49:364. doi: 10.1002/hep.22673. [DOI] [PubMed] [Google Scholar]
- 16.Hayes CN, Imamura M, Aikata H, Chayama K. Genetics of IL28B and HCV--response to infection and treatment. Nat rev Gastroenterol hepatol. 2012;9:406. doi: 10.1038/nrgastro.2012.101. [DOI] [PubMed] [Google Scholar]
- 17.Prokunina-Olsson L, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat genet. 2013;45:164. doi: 10.1038/ng.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doehring A, et al. Screening for IL28B gene variants identifies predictors of hepatitis C therapy success. Antivir Ther. 2010;15:1099. doi: 10.3851/IMP1689. [DOI] [PubMed] [Google Scholar]
- 19.Lange CM, et al. Impact of donor and recipient IL28B rs12979860 genotypes on hepatitis C virus liver graft reinfection. J hepatol. 2010. [DOI] [PubMed]
- 20.Thomas DL, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ge D, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 22.Wu X, et al. Productive hepatitis C virus infection of stem cell-derived hepatocytes reveals a critical transition to viral permissiveness during differentiation. PLoS Pathog. 2012;8:e1002617. doi: 10.1371/journal.ppat.1002617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwartz RE, et al. Modeling hepatitis C virus infection using human induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2012;109:2544. doi: 10.1073/pnas.1121400109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roelandt P, et al. Human pluripotent stem cell-derived hepatocytes support complete replication of hepatitis C virus. J Hepatol. 2012;57:246. doi: 10.1016/j.jhep.2012.03.030. [DOI] [PubMed] [Google Scholar]
- 25.Buhler S, Bartenschlager R. New targets for antiviral therapy of chronic hepatitis C. Liver int off j Int Assoc Stud Liver. 2012;32(Suppl 1):9. doi: 10.1111/j.1478-3231.2011.02701.x. [DOI] [PubMed] [Google Scholar]
- 26.Scheel TK, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat med. 2013;19:837. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manns MP, von Hahn T. Novel therapies for hepatitis C - one pill fits all? Nat rev Drug discov. 2013;12:595. doi: 10.1038/nrd4050. [DOI] [PubMed] [Google Scholar]
- 28.Scarselli E, et al. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO j. 2002;21:5017. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pileri P, et al. Binding of hepatitis C virus to CD81. Science. 1998;282:938. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 30.Evans MJ, et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 31.Ploss A, et al. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature. 2009;457:882. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu S, et al. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J virol. 2009;83:2011. doi: 10.1128/JVI.01888-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeisel MB, et al. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology. 2007;46:1722. doi: 10.1002/hep.21994. [DOI] [PubMed] [Google Scholar]
- 34.Koutsoudakis G, et al. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J virol. 2006;80:5308. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sourisseau M, et al. Temporal analysis of hepatitis C virus cell entry with occludin directed blocking antibodies. PLoS pathog. 2013;9:e1003244. doi: 10.1371/journal.ppat.1003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bankwitz D, et al. Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J virol. 2010;84:5751. doi: 10.1128/JVI.02200-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma NR, et al. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J Biol Chem. 2011;286:30361. doi: 10.1074/jbc.M111.263350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brazzoli M, et al. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J virol. 2008;82:8316. doi: 10.1128/JVI.00665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farquhar MJ, et al. Hepatitis C virus induces CD81 and claudin-1 endocytosis. J virol. 2012;86:4305. doi: 10.1128/JVI.06996-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dorner M, et al. A genetically humanized mouse model for hepatitis C virus infection. Nature. 2011;474:208. doi: 10.1038/nature10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.• Meuleman P et al. Anti-CD81 antibodies can prevent a hepatitis C virus infection in vivo. Hepatology. 2008;48:1761. This paper demonstrates for the first time that blocking antibodies targeting HCV entry factors can prevent HCV infection in a mouse model. It also provides the first in vivo evidence for CD81 as critical HCV entry factor. [DOI] [PubMed]
- 42.Meuleman P, et al. A human monoclonal antibody targeting scavenger receptor class B type I precludes hepatitis C virus infection and viral spread in vitro and in vivo. Hepatology. 2012;55:364. doi: 10.1002/hep.24692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.• Lacek K et al. Novel human SR-BI antibodies prevent infection and dissemination of HCV in vitro and in humanized mice. J hepatol. 2012;57:17. This preclinical study using a humanized mouse model highlights the potential use of anti-SCARB1 antibodies in preventive therapy of HCV infection. [DOI] [PubMed]
- 44.• Syder AJ et al. Small molecule scavenger receptor BI antagonists are potent HCV entry inhibitors. J hepatol. 2011;54:48. This is the first study describing a small molecule inhibitor for HCV entry. The compound is currently the most advanced entry blocker and passed clinical phase 1a. [DOI] [PubMed]
- 45.Sulkowski MS et al. Safety and antiviral activity of the HCV entry inhibitor ITX5061 in treatment-naive HCV- infected adults: a randomized, double-blind, Phase 1b Study. J infect dis. 2013. [DOI] [PMC free article] [PubMed]
- 46.Zhu H, et al. Evaluation of ITX 5061, a scavenger receptor B1 antagonist: resistance selection and activity in combination with other hepatitis C virus antivirals. J infect dis. 2012;205:656. doi: 10.1093/infdis/jir802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dhillon S, et al. Mutations within a conserved region of the hepatitis C virus E2 glycoprotein that influence virus-receptor interactions and sensitivity to neutralizing antibodies. J virol. 2010;84:5494. doi: 10.1128/JVI.02153-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masson D, et al. Increased HDL cholesterol and apoA-I in humans and mice treated with a novel SR-BI inhibitor. Arterioscler thromb vasc biol. 2009;29:2054. doi: 10.1161/ATVBAHA.109.191320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Westhaus S, et al. Characterization of the inhibition of hepatitis C virus entry by in vitro-generated and patient-derived oxidized low-density lipoprotein. Hepatology. 2013;57:1716. doi: 10.1002/hep.26190. [DOI] [PubMed] [Google Scholar]
- 50.Fofana I, et al. Monoclonal anti-claudin 1 antibodies prevent hepatitis C virus infection of primary human hepatocytes. Gastroenterology. 2010;139:953. doi: 10.1053/j.gastro.2010.05.073. [DOI] [PubMed] [Google Scholar]
- 51.Timpe JM, et al. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology. 2008;47:17. doi: 10.1002/hep.21959. [DOI] [PubMed] [Google Scholar]
- 52.Witteveldt J, et al. CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J gen virol. 2009;90:48. doi: 10.1099/vir.0.006700-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brimacombe CL, et al. Neutralizing antibody-resistant hepatitis C virus cell-to-cell transmission. J virol. 2011;85:596. doi: 10.1128/JVI.01592-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fofana I, et al. A Novel Monoclonal Anti-CD81 Antibody Produced by Genetic Immunization Efficiently Inhibits Hepatitis C Virus Cell-Cell Transmission. PloS one. 2013;8:e64221. doi: 10.1371/journal.pone.0064221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Catanese MT, et al. Different Requirements for Scavenger Receptor Class B Type I in Hepatitis C Virus Cell-Free versus Cell-to-Cell Transmission. J virol. 2013;87:8282. doi: 10.1128/JVI.01102-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng A, et al. Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J virol. 2007;81:12465. doi: 10.1128/JVI.01457-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meertens L, et al. The tight junction proteins claudin-1, -6, and −9 are entry cofactors for hepatitis C virus. J virol. 2008;82:3555. doi: 10.1128/JVI.01977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haid S et al. Isolate-dependent use of Claudins for cell entry by hepatitis C virus. Hepatology. 2013. [DOI] [PubMed]
- 59.Saitou M, et al. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol biol cell. 2000;11:4131. doi: 10.1091/mbc.11.12.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barth H, et al. Viral and cellular determinants of the hepatitis C virus envelope-heparan sulfate interaction. J virol. 2006;80:10579. doi: 10.1128/JVI.00941-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc Natl Acad Sci U S A. 1999;96:12766. doi: 10.1073/pnas.96.22.12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Owen DM, Huang H, Ye J, Gale M., Jr Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology. 2009;394:99. doi: 10.1016/j.virol.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lupberger J, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat med. 2011;17:589. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sainz B, Jr, et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat med. 2012;18:281. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martin DN, Uprichard SL. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc Natl Acad Sci U S A. 2013;110:10777. doi: 10.1073/pnas.1301764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hotta K, Kiura K. Safety profiles of erlotinib therapy in patients with advanced non-small-cell lung cancer. Expert rev anticancer ther. 2011;11:991. doi: 10.1586/era.11.74. [DOI] [PubMed] [Google Scholar]
- 67.Galvani E, Peters GJ, Giovannetti E. EGF receptor-targeted therapy in non-small-cell lung cancer: role of germline polymorphisms in outcome and toxicity. Futur oncol. 2012;8:1015. doi: 10.2217/fon.12.89. [DOI] [PubMed] [Google Scholar]
- 68.Zona L, et al. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell host microbe. 2013;13:302. doi: 10.1016/j.chom.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 69.Bays HE, Neff D, Tomassini JE, Tershakovec AM. Ezetimibe: cholesterol lowering and beyond. Expert rev cardiovasc ther. 2008;6:447. doi: 10.1586/14779072.6.4.447. [DOI] [PubMed] [Google Scholar]
- 70.Gosert R, et al. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J virol. 2003;77:5487. doi: 10.1128/JVI.77.9.5487-5492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Egger D, et al. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J virol. 2002;76:5974. doi: 10.1128/JVI.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Paul D, Hoppe S, Saher G, Krijnse-Locker J, Bartenschlager R. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J virol. 2013. [DOI] [PMC free article] [PubMed]
- 73.Romero-Brey I, et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS pathog. 2012;8:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Quinkert D, Bartenschlager R, Lohmann V. Quantitative analysis of the hepatitis C virus replication complex. J virol. 2005;79:13594. doi: 10.1128/JVI.79.21.13594-13605.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Watashi K, et al. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol cell. 2005;19:111. doi: 10.1016/j.molcel.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 76.Chatterji U, et al. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J biol chem. 2009;284:16998. doi: 10.1074/jbc.M109.007625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu Z, Yang F, Robotham JM, Tang H. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J virol. 2009;83:6554. doi: 10.1128/JVI.02550-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kaul A, et al. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS pathog. 2009;5:e1000546. doi: 10.1371/journal.ppat.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang F, et al. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J virol. 2008;82:5269. doi: 10.1128/JVI.02614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Foster TL, Gallay P, Stonehouse NJ, Harris M. Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J virol. 2011;85:7460. doi: 10.1128/JVI.00393-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Verdegem D, et al. Domain 3 of NS5A protein from the hepatitis C virus has intrinsic alpha-helical propensity and is a substrate of cyclophilin A. J biol chem. 2011;286:20441. doi: 10.1074/jbc.M110.182436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chatterji U, et al. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J hepatol. 2010;53:50. doi: 10.1016/j.jhep.2010.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Watashi K, Hijikata M, Hosaka M, Yamaji M, Shimotohno K. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology. 2003;38:1282. doi: 10.1053/jhep.2003.50449. [DOI] [PubMed] [Google Scholar]
- 84.Colgan J, Asmal M, Yu B, Luban J. Cyclophilin A-deficient mice are resistant to immunosuppression by cyclosporine. J immunol. 2005;174:6030. doi: 10.4049/jimmunol.174.10.6030. [DOI] [PubMed] [Google Scholar]
- 85.Borel JF, Feurer C, Gubler HU, Stahelin H. Biological effects of cyclosporin A: a new antilymphocytic agent. Agents actions. 1976;6:468. doi: 10.1007/BF01973261. [DOI] [PubMed] [Google Scholar]
- 86.Lin K, Gallay P. Curing a viral infection by targeting the host: the example of cyclophilin inhibitors. Antivir res. 2013;99:68. doi: 10.1016/j.antiviral.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Flisiak R, et al. The cyclophilin inhibitor Debio 025 combined with PEG IFNalpha2a significantly reduces viral load in treatment-naive hepatitis C patients. Hepatology. 2009;49:1460. doi: 10.1002/hep.22835. [DOI] [PubMed] [Google Scholar]
- 88.Nag A, Robotham JM, Tang H. Suppression of viral RNA binding and the assembly of infectious hepatitis C virus particles in vitro by cyclophilin inhibitors. J virol. 2012;86:12616. doi: 10.1128/JVI.01351-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pawlotsky J-M, et al. Alisporivir plus ribavirin is highly effective as interferon-free or interferon-add-on regimen in previously untreated HCV-G2 or G3 patients: SVR12 results from VITAL-1 Phase 2b study. J hepatol. 2012;56:S553. [Google Scholar]
- 90.von Hahn T, et al. Hepatocytes that express variants of cyclophilin A are resistant to HCV infection and replication. Gastroenterology. 2012;143:439. doi: 10.1053/j.gastro.2012.04.053. [DOI] [PubMed] [Google Scholar]
- 91.Yang F, et al. A major determinant of cyclophilin dependence and cyclosporine susceptibility of hepatitis C virus identified by a genetic approach. PLoS pathog. 2010;6:e1001118. doi: 10.1371/journal.ppat.1001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Coelmont L, et al. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PloS one. 2010;5:e13687. doi: 10.1371/journal.pone.0013687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.• Flisiak R et al. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology. 2008;47:817. This study describes a successful phase II study for the cyclophilin inhibitor alisporivir (Debio 025) in combination with PEG IFNalpha2a. In a randomized, placebo-controlled, multicenter trial alisporivir co-administration induced an additive RNA reduction in patients with genotypes 1 and 4 during a 4 week treatment window. Alisporivir is currently tested in phase 3 clinical trials and the most advanced host targeting antiviral against hepatitis C. [DOI] [PubMed]
- 94.Hopkins S, et al. The cyclophilin inhibitor SCY-635 suppresses viral replication and induces endogenous interferons in patients with chronic HCV genotype 1 infection. J hepatol. 2012;57:47. doi: 10.1016/j.jhep.2012.02.024. [DOI] [PubMed] [Google Scholar]
- 95.Bobardt M, et al. HCV NS5A and IRF9 compete for CypA binding. J hepatol. 2013;58:16. doi: 10.1016/j.jhep.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fu XY, Kessler DS, Veals SA, Levy DE, Darnell JE., Jr ISGF3, the transcriptional activator induced by interferon alpha, consists of multiple interacting polypeptide chains. Proc Natl Acad Sci U S A. 1990;87:8555. doi: 10.1073/pnas.87.21.8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Watashi K, Daito T, Sluder A, Borroto-Esoda K, Wakita T. Cyclophilin inhibitors potentiate interferon signaling through diminished PKR phosphorylation in HCV-infected cells. J hepatol. 2013;58:S5. [Google Scholar]
- 98.Garaigorta U, Chisari FV. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell host microbe. 2009;6:513. doi: 10.1016/j.chom.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Supekova L, et al. Identification of human kinases involved in hepatitis C virus replication by small interference RNA library screening. J biol chem. 2008;283:29. doi: 10.1074/jbc.M703988200. [DOI] [PubMed] [Google Scholar]
- 100.Tai AW, et al. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell host microbe. 2009;5:298. doi: 10.1016/j.chom.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vaillancourt FH, et al. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology. 2009;387:5. doi: 10.1016/j.virol.2009.02.039. [DOI] [PubMed] [Google Scholar]
- 102.Berger KL, et al. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc Natl Acad Sci U S A. 2009;106:7577. doi: 10.1073/pnas.0902693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Borawski J, et al. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J virol. 2009;83:10058. doi: 10.1128/JVI.02418-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Trotard M, et al. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB j off publ Fed Am Soc Exp Biol. 2009;23:3780. doi: 10.1096/fj.09-131920. [DOI] [PubMed] [Google Scholar]
- 105.Reiss S, et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell host microbe. 2011;9:32. doi: 10.1016/j.chom.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li Q, et al. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci U S A. 2009;106:16410. doi: 10.1073/pnas.0907439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ng TI, et al. Identification of host genes involved in hepatitis C virus replication by small interfering RNA technology. Hepatology. 2007;45:1413. doi: 10.1002/hep.21608. [DOI] [PubMed] [Google Scholar]
- 108.Balla A, Balla T. Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends cell biol. 2006;16:351. doi: 10.1016/j.tcb.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 109.D'Angelo G, Vicinanza M, Di Campli A, De Matteis MA. The multiple roles of PtdIns(4)P – not just the precursor of PtdIns(4,5)P2. J cell sci. 2008;121:1955. doi: 10.1242/jcs.023630. [DOI] [PubMed] [Google Scholar]
- 110.Reiss S, et al. The lipid kinase phosphatidylinositol-4 kinase III alpha regulates the phosphorylation status of hepatitis C virus NS5A. PLoS pathog. 2013;9:e1003359. doi: 10.1371/journal.ppat.1003359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bianco A, et al. Metabolism of phosphatidylinositol 4-kinase IIIalpha-dependent PI4P Is subverted by HCV and is targeted by a 4-anilino quinazoline with antiviral activity. PLoS pathog. 2012;8:e1002576. doi: 10.1371/journal.ppat.1002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Najarro P, Mathews N, Coclerill S, NS5A Inhibitors. In: Tan S-L, He Y, editors. Hepatitis C: Antiviral Drug Development; 2011. pp 271.
- 113.Schmitz U, Tan SL. NS5A–from obscurity to new target for HCV therapy. Recent patents anti-infect drug discov. 2008;3:77. doi: 10.2174/157489108784746597. [DOI] [PubMed] [Google Scholar]
- 114.Najarro P, Powell K, Budworth A, Hallott A, Harris R. A-831, a novel HCV inhibitor targeting NS5A. J hepatol. 2006;36:III. [Google Scholar]
- 115.Vaillancourt FH, et al. Evaluation of phosphatidylinositol-4-kinase IIIalpha as a hepatitis C virus drug target. J virol. 2012;86:11595. doi: 10.1128/JVI.01320-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wilson RC, Doudna JA. Molecular mechanisms of RNA interference. Annu rev biophys. 2013;42:217. doi: 10.1146/annurev-biophys-083012-130404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Grundhoff A, Sullivan CS. Virus-encoded microRNAs. Virology. 2011;411:325. doi: 10.1016/j.virol.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jopling CL, Schutz S, Sarnow P. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell host microbe. 2008;4:77. doi: 10.1016/j.chom.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Henke JI, et al. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO j. 2008;27:3300. doi: 10.1038/emboj.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li Y, Masaki T, Yamane D, McGivern DR, Lemon SM. Competing and noncompeting activities of miR-122 and the 5' exonuclease Xrn1 in regulation of hepatitis C virus replication. Proc Natl Acad Sci U S A. 2013;110:1881. doi: 10.1073/pnas.1213515110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Machlin ES, Sarnow P, Sagan SM. Masking the 5' terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc Natl Acad Sci U S A. 2011;108:3193. doi: 10.1073/pnas.1012464108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shimakami T, et al. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc Natl Acad Sci U S A. 2012;109:941. doi: 10.1073/pnas.1112263109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 124.Zhang C, Huys A, Thibault PA, Wilson JA. Requirements for human Dicer and TRBP in microRNA-122 regulation of HCV translation and RNA abundance. Virology. 2012;433:479. doi: 10.1016/j.virol.2012.08.039. [DOI] [PubMed] [Google Scholar]
- 125.Da Costa D, et al. Reconstitution of the entire hepatitis C virus life cycle in nonhepatic cells. J virol. 2012;86:11919. doi: 10.1128/JVI.01066-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vogt A et al. Recapitulation of the hepatitis C virus life-cycle in engineered murine cell lines. Virology. 2013. [DOI] [PMC free article] [PubMed]
- 127.Lanford RE, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li YP, Gottwein JM, Scheel TK, Jensen TB, Bukh J. MicroRNA-122 antagonism against hepatitis C virus genotypes 1–6 and reduced efficacy by host RNA insertion or mutations in the HCV 5' UTR. Proc Natl Acad Sci U S A. 2011;108:4991. doi: 10.1073/pnas.1016606108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Patick AK, et al. Sequence analysis of HCV variants from a phase IIa trial of miravirsen (MIR), an oligonucleotide targeting miR-122, in treatment naive patients with chronic HCV infection. J Hepatol. 2012;56(56):S476. [Google Scholar]
- 130.Reesink HW, et al. Final results - randomized, double-blind, placebo-controlled safety, anti-viral proof-of-concept study of miravirsen, an oligonucleotide targeting miR-122, in treatment-naive patients with genotype 1 chronic HCV infection. J hepatol. 2012;56:S26. [Google Scholar]
- 131.•• Janssen HL et al. Treatment of HCV infection by targeting microRNA. N Engl J Med 2013;368:1685. Clinical study demonstrating the efficacy of miravirsen in chronic genotype 1 patients without viral resistance emergence. [DOI] [PubMed]
- 132.Persson R, et al. Pharmacokinetics of miravirsen, a miR-122 inhibitor, predict the prolonged viral load reduction in treatment naive genotype 1 HCV infected patients. J hepatol. 2012;56:S477. [Google Scholar]
- 133.Hsu SH, et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J clin investig. 2012;122:2871. doi: 10.1172/JCI63539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chapel C, et al. Reduction of the infectivity of hepatitis C virus pseudoparticles by incorporation of misfolded glycoproteins induced by glucosidase inhibitors. J gen virol. 2007;88:1133. doi: 10.1099/vir.0.82465-0. [DOI] [PubMed] [Google Scholar]
- 135.Chapel C, et al. Antiviral effect of alpha-glucosidase inhibitors on viral morphogenesis and binding properties of hepatitis C virus-like particles. J gen virol. 2006;87:861. doi: 10.1099/vir.0.81503-0. [DOI] [PubMed] [Google Scholar]
- 136.Steinmann E, et al. Antiviral effects of amantadine and iminosugar derivatives against hepatitis C virus. Hepatology. 2007;46:330. doi: 10.1002/hep.21686. [DOI] [PubMed] [Google Scholar]
- 137.Durantel D. Celgosivir, an alpha-glucosidase I inhibitor for the potential treatment of HCV infection. Curr opin investig drugs. 2009;10:860. [PubMed] [Google Scholar]
- 138.Kono Y, Hayashida K, Tanaka H, Ishibashi H, Harada M. High-density lipoprotein binding rate differs greatly between genotypes 1b and 2a/2b of hepatitis C virus. J med virol. 2003;70:42. doi: 10.1002/jmv.10372. [DOI] [PubMed] [Google Scholar]
- 139.Nielsen SU, et al. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J virol. 2006;80:2418. doi: 10.1128/JVI.80.5.2418-2428.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Merz A, et al. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J biol chem. 2011;286:3018. doi: 10.1074/jbc.M110.175018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mancone C, et al. Ferritin heavy chain is the host factor responsible for HCV-induced inhibition of apoB-100 production and is required for efficient viral infection. J proteome res. 2012;11:2786. doi: 10.1021/pr201128s. [DOI] [PubMed] [Google Scholar]
- 142.Jiang J, et al. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J Virol. 2012;86:7256. doi: 10.1128/JVI.07222-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shi Q, Jiang J, Luo G. Syndecan-1 serves as the major receptor for attachment of hepatitis C virus to the surfaces of hepatocytes. J virol. 2013;87:6866. doi: 10.1128/JVI.03475-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jiang J, Wu X, Tang H, Luo G. Apolipoprotein e mediates attachment of clinical hepatitis C virus to hepatocytes by binding to cell surface heparan sulfate proteoglycan receptors. PloS one. 2013;8:e67982. doi: 10.1371/journal.pone.0067982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Herker E, et al. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat med. 2010;16:1295. doi: 10.1038/nm.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Menzel N, et al. MAP-kinase regulated cytosolic phospholipase A2 activity is essential for production of infectious hepatitis C virus particles. PLoS pathog. 2012;8:e1002829. doi: 10.1371/journal.ppat.1002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhao G, et al. Validation of diacyl glycerolacyltransferase I as a novel target for the treatment of obesity and dyslipidemia using a potent and selective small molecule inhibitor. J med chem. 2008;51:380. doi: 10.1021/jm7013887. [DOI] [PubMed] [Google Scholar]
- 148.de Chassey B, Meyniel-Schicklin L, Aublin-Gex A, Andre P, Lotteau V. New horizons for antiviral drug discovery from virus-host protein interaction networks. Curr opin virol. 2012;2:606. doi: 10.1016/j.coviro.2012.09.001. [DOI] [PubMed] [Google Scholar]