

Abstract
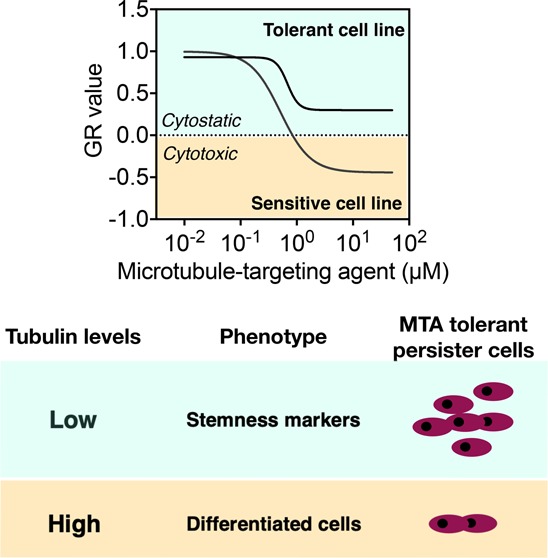
Sensitivity to microtubule-targeting agents (MTAs) varies among cancers and predicting the response of individual cancer patients to MTAs remains challenging. As microtubules possess vast molecular heterogeneity generated by tubulin isotypes and their post-translational modifications, we questioned whether this heterogeneity can impact MTA sensitivity. We investigated microtubule heterogeneity in 15 glioblastoma cell lines and measured sensitivity of orthogonal MTAs using a per-division growth rate inhibition method that corrects for the confounding effects of variable cell proliferation rates. We found that the tubulin profile is unique for each glioblastoma cell line and that the total α- and β-tubulin levels impact on MTA sensitivity. The baseline levels of α- and β-tubulin were up to 20% lower in cells that were not effectively killed by MTAs. We report that lower α/β-tubulin expression is associated with lack of cell differentiation and increased expression of stemness markers. The dedifferentiated stem-like cells with low α/β-tubulin levels survive MTAs treatment via reversible nonmutational dormancy. Our findings provide novel insights into the relationships between microtubules and MTAs and lay a foundation for better understanding of the sensitivity of cancer cells to MTAs.
Keywords: glioblastoma, microtubule-targeting agents, tubulin code, drug sensitivity, drug-tolerant persister cells
Introduction
Microtubules are cytoskeletal polymers composed of α/β-tubulin heterodimers and are involved in mitosis, cell motility, intracellular transport, and maintenance of cell shape. Because of their multiple cellular functions, microtubules are the direct target of microtubule-targeting agents (MTAs) that are clinically used to kill cancer cells.1 Although microtubules are highly conserved in their 3D structures, there is a significant diversity at the molecular level.2 Microtubule diversity stems from genes encoding for eight α- and nine β-tubulin isotypes, and from their post-translational modifications.3 These modifications range from the well-known phosphorylation to tubulin-specific modifications such as detyrosination and associated removal of penultimate glutamate yielding Δ2 α-tubulin. Together, tubulin isotypes and their post-translational modifications form the tubulin code, which fine-tunes specific functions of microtubules.2
Tubulin heterogeneity has been associated with tumor resistance to MTAs.4 For example, the overexpression of βIII-, βIV-, and βV-tubulins caused acquired resistance to taxanes and vinca alkaloids.5 Post-translational modifications have been also proposed to affect the efficacy of MTAs;1,4 however, this remains unsubstantiated by experimental data. In addition to the acquired resistance, drug-tolerant states contribute to the reduction of cancer drug efficacy. Initial exposure of cells to cancer drugs does not kill all cells, giving rise to a subpopulation of surviving cells that has been named drug-tolerant persister cells. Drug-tolerant persister cells usually use epigenetic reprogramming to survive the treatment and when drugs are removed, they regrow into a new population that is as equally drug-sensitive as the parental population.6−9 Two studies to date have shown that exposure of cancer cells to taxanes generates drug-tolerant persister cells,10,11 suggesting that the drug tolerance plays a role in MTA efficacy.
Glioblastoma is the most aggressive primary brain tumor with a median patient survival of 15 months. Given the extensive intratumoral molecular heterogeneity of glioblastomas12 and the lack of molecularly targeted drugs that can permeate the blood–brain barrier,13 glioblastoma remains a major unmet medical need. Although MTAs showed promising efficacy against glioblastoma cells, clinically approved MTAs are ineffective for the treatment of these tumors as their large molecular weight (>800 g/mol) and polarity renders them unable to cross the blood–brain barrier. Hence, there has been increasing research interest toward the development of effective MTA delivery methods14−16 or identification of small-molecule MTAs able to cross the blood–brain barrier.17−20
To advance the development of MTAs as pharmacological agents against glioblastoma, we characterized microtubules in three standard (U87, U251, A172) and 12 glioblastoma stem cell (GSC) lines.21 In parallel, we determined efficacy of clinical MTAs (paclitaxel, vinblastine, ixabepilone) and colchicine-site binders (colchicine, nocodazole, tivantinib, and CMPD1).19,22,23 Colchicine-site binders are generally smaller compared to taxanes, epothilones and vinca alkaloids and as the molecular weight is a critical parameter for blood–brain barrier permeability,24 this subclass of MTAs is more relevant to the development of glioblastoma therapeutics. We calculated conventional drug sensitivity metrics IC50, Hill coefficient h (marker of cell-to-cell variability), Emax (maximum efficacy), and area under the curve (AUC) which combines IC50 with Emax.(25) However, because the drug sensitivity is often confounded by unequal division rates across cell lines, we also performed growth rate (GR) inhibition analysis which yielded per-division drug potency (GR50), efficacy (GRmax), Hill coefficient (hGR), and area-over the curve (GRAOC).26−28 Finally, we investigated if some components of the tubulin code mediate MTA sensitivity.
Results
Tubulin Isotypes and Post-translational Modifications
As the tubulin expression is predominantly regulated post-transcriptionally such that protein levels poorly reflect the transcript levels,29,30 we investigated microtubule modifications by immunoblotting lysates from unsynchronized cells at three different passages. The absolute values of immunoblot signals were not normalized to the house-keeping proteins GAPDH, Hsp90, or β-actin as their levels varied greatly between cell lines (Figure 1). Equal loading was monitored with Coomassie staining (Figure S1) and immunoblot signals were normalized to the corresponding signals in A172 (Figure S2) and RN1 (Figure S3) cells. Both normalizations revealed comparable variability in the expression of tubulin isotypes and their modifications. The coefficients of variation (CV) show 20%, 25%, and 69% variability in the total expression of α-, β-, and γ-tubulin, respectively. The variability of the individual β-tubulin isotypes levels ranged from 26% (βI- and βIII-) to 54% (βII-tubulin, Figure 1a).
Figure 1.
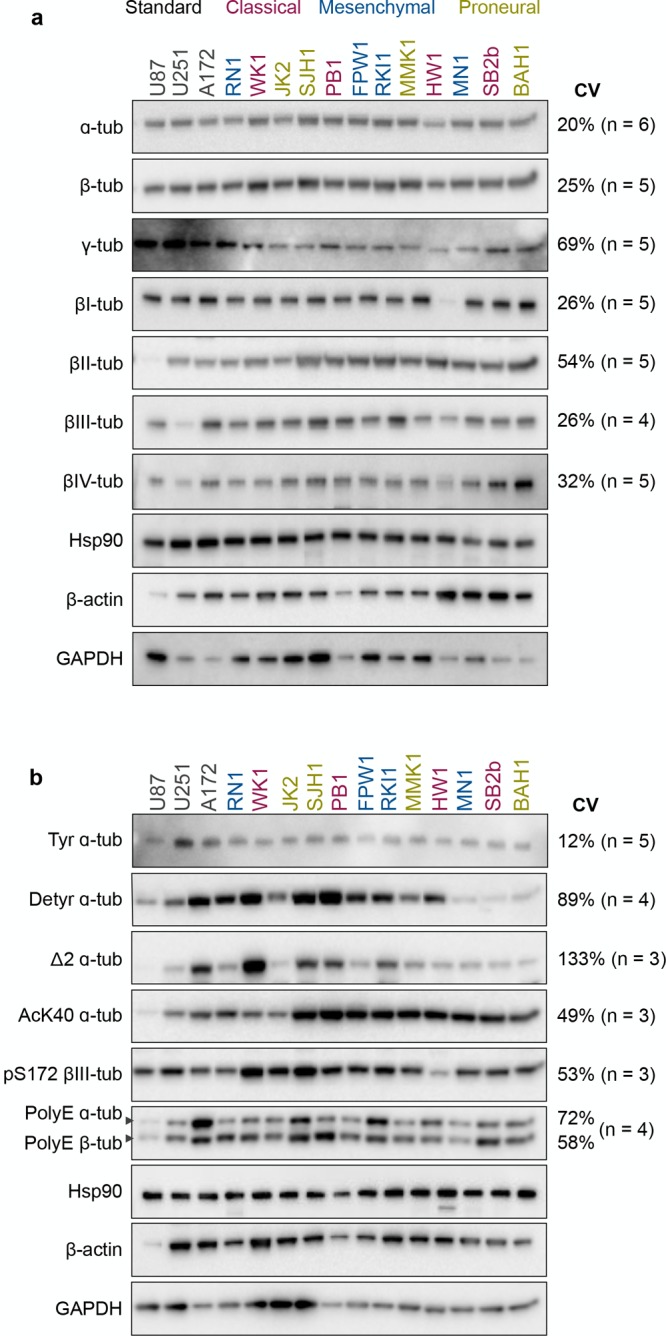
Tubulin isotypes and post-translational modifications in glioblastoma cells. Western blot (5 μg of total protein) analysis of tubulin isotypes (a) and their post-translational modifications (b) in glioblastoma cell lines. Colors of the cell lines indicated the glioblastoma subtype: classical, mesenchymal, and proneural. Representative immunoblots and coefficient of variation (CV) of at least three independent experiments are shown. Quantification and normalization are presented in Figures S2 and S3.
When assessing the post-translational modifications, we found 12%, 89%, and 133% variability across the levels of tyrosinated, detyrosinated, and Δ2 α-tubulin, respectively (Figure 1b). The acetylation of α-tubulin, with the exception of WK1 and JK2 lines, was high in the GSC lines when compared to the standard cell lines (CV = 49%). There was a 53% variability in the phosphorylation of βIII-tubulin and 58–72% variability in the polyglutamylation of α- and β-tubulin (Figure 1b). In summary, tubulin isotypes and their post-translational modifications varied greatly (up to 133% variability) across 15 glioblastoma cell lines.
MTAs Sensitivity
Multiparametric analysis of the dose–response curves fitted to the relative cell count (Figure 2a and Figure S4) yielded values for IC50, Emax, AUC, and Hill coefficient h (Tables S3–S6). Live-cell imaging revealed that standard A172, U87, and U251 cells had overall shorter doubling times (35–55 h, Figure 3b). The doubling time of GSC lines ranged from 49 h (proneural MMK1) to 140 h (proneural SJH1), with no clear trend for cell lines representing the same glioblastoma subtype. We then used the GRcalculator tool and the population doubling times to generate GR curves (Figure 2c and Figure S4) and calculated GR metrics (Tables S3–S6) for colchicine (Figure 2d), nocodazole (Figure 2e), tivantinib (Figure 2f) and CMPD1 (Figure 2g). We used GR metrics to analyze glioblastoma cell sensitivity to MTAs, as they incorporate cell proliferation rates and are therefore considered more accurate descriptors of drug efficacy.26
Figure 2.

MTA sensitivity in glioblastoma cells. (a) Schematic of relative drug sensitivity metrics calculated from a dose–response curve fitted to a relative cell count. (b) Proliferation rates of glioblastoma cell lines were determined with the IncuCyte platform and CellCounter + + online tool. (c) Schematic of grow inhibition (GR) metrics from a dose–response curve fitted to GR values. Relative and GR metrics for colchicine (d), nocodazole (e), tivantinib (f) and CMPD1 (g) were calculated from dose responses determined with CellTiter-Blue viability assay after 5 days of drug treatment and proliferation rates using the GRcalculator tool. Data are mean ± SEM (n = 3). All values are listed in Tables S3–S6.
Figure 3.

Spearman’s correlations between tubulin expression and MTA efficacy. (a,b) Pairwise distribution and correlation of GRmax and the expression of α-tubulin and β-tubulinin glioblastoma stem cell lines. (c,d) Pairwise distribution and correlation of GRAOC and the expression of α-tubulin and β-tubulinin glioblastoma stem cell lines.
Within the MTAs binding to the colchicine site on microtubules, colchicine was the most potent MTA (GR50 ∼ 10 nM, Figure 3d) with fully cytostatic effect in the classical HW1, proneural JK2, and MMK1 cell lines (GRmax close to 0). Positive GRmax in the mesenchymal RKI1 line indicates a partially cytostatic effect. In the remaining cell lines, GRmax centered around −0.4, which corresponds to partial cytotoxicity. However, the GRmax values for colchicine did not reach the value of −1, which corresponds to the killing of all cells. This incomplete cell killing efficacy implicates that each glioblastoma cell line contains a subpopulation of cells able to survive colchicine treatment. This is supported by the shallow slope (low hGR values) of the dose–response curves (Figure 3d), an indicator of high cell-to-cell variability. GRAOC values were also low (GRAOC < 1) for the majority of cell lines, further confirming weak colchicine efficacy. Cell lines HW1, RKI1, JK2, and MMK1 were identified as the least sensitive to colchicine (GRmax close to 0 or positive; low GRAOC) and presenting high cell-to-cell variability (low hGR). Although proneural SJH1 cell line reached the lowest negative GRmax value (GRmax = −0.75), we do not consider this cell line as the most sensitive one. The population doubling time for SJH1 cells was 140 h, corresponding to a 0.8 division over the course of a 120 h viability assay, and GR values obtained from slow growing cell lines were considered to be ambiguous.31 Similar drug sensitivity metrics were obtained for nocodazole (Figure 3e), tivantinib (Figure 3f) and CMPD1 (Figure 3g).
In summary, the per-division metrics unanimously identified HW1, RKI1, JK2, and MMK1 as the least MTA sensitive (GRmax close to 0 or positive, low GRAOC, low hGR). The most sensitive cells to MTAs were WK1 and MN1 with the lowest negative GRmax and the highest positive GRAOC values. Intriguingly, glioblastoma sensitivity to MTAs is not subtype specific and within each subtype there were cell lines with high (mesenchymal MN1) and low (mesenchymal RKI1) sensitivity. Another important observation was that despite a 20-fold difference in potency, colchicine (GR50 = 13 nM) and CMPD1 (GR50 = 273 nM) displayed the same efficacy in the most sensitive WK1 cells (GRmax = −0.48 and −0.47, respectively).
Correlations of the MTAs Sensitivity with the Tubulin Code
Given the observed tubulin heterogeneity (Figure 1) and MTAs sensitivity (Figure 2) in the GSC lines, we questioned whether the tubulin code has an effect on MTAs efficacy. We calculated Spearman’s correlation coefficients between the tubulin code metrics normalized to A172 and MTAs parameters for each agent (Tables S7–S10). Data obtained with standard A172, U87, and U251 cell lines were excluded, as these cells were grown under different conditions (serum-grown vs serum-free media for GSC) and were found to possess nearly identical tubulin code profiles and MTA sensitivities.
While there were numerous correlations between the tubulin features and the relative (IC50, Emax, h, AUC) metrics for individual MTAs, these correlations were not consistent across the four MTAs (Tables S7–S10). Importantly, consistent negative (ρ < −0.6; P < 0.05) correlations were found between the total levels of α-tubulin and the GRmax values of all four drugs (Figure 3a). Negative correlations were also found between GRmax values of colchicine, nocodazole, CMPD1, and expression of β-tubulin (ρ < −0.5, P < 0.06). Furthermore, positive correlations were found between the GRAOC and total α-tubulin (Figure 3c) as well as β-tubulin (Figure 3d). Except for tivantinib (P < 0.08), the remaining GRAOC correlations were statistically significant (ρ > 0.58, P < 0.05). Since data obtained with BAH1 cells were identified as outliers, we reanalyzed data sets excluding BAH1 data and confirmed correlations between the MTA efficacy metrics and expression of α- and β-tubulin (Figure S5). Together, these correlations suggest that the lower expression of α/β-tubulin is linked to weak MTA efficacy (i.e., higher GRmax and lower GRAOC).
Total Tubulin Levels Impact MTAs Efficacy
To validate the quantity of α/β-tubulins as determinants of cellular response to MTAs, we performed additional experiments with WK1, RN1 (higher α/β-tubulin expression), JK2, and RKI1 cells (lower α/β-tubulin expression). Quantitative capillary-based immunoassay analysis confirmed decreasing order of α- and β-tubulin expression in WK1 > RN1 > JK2 > RKI1 cells (Figure 4a). Colchicine treatment was cytotoxic to WK1 and RN1 but cytostatic to JK2 and RKI1 (Figure 4b). The GR dose–response curves for paclitaxel, vinblastine, and ixabepilone exhibited the same efficacy trend (Figure 4c). These clinical MTAs were cytotoxic to WK1 and RN1 (higher α/β-tubulin expression) but cytostatic to JK2 and RKI1 cells (lower α/β-tubulin expression). To further confirm that overall tubulin levels may be linked to the efficacy of MTAs, we performed an orthogonal assay that quantifies surviving drug-tolerant cells.8,10 On the basis of the colchicine GR curves (Figure 4d), we hypothesized that surviving subpopulations will be larger in JK2 and RKI1 cells. As expected, significantly more colchicine-tolerant persister cells were detected in RKI1 cells when compared to WK1 and RN1 (Figure 4d,e). Similarly, the percentage of drug-tolerant cells was larger in the vinblastine-treated RKI1 cells when compared to vinblastine-treated WK1 cells (Figure 4f).
Figure 4.

Cells with lower levels of α/β-tubulin generate more drug-tolerant cells. (a) Capillary-based immunoassays of total α- and β-tubulin. Representative images and mean value of three independent experiments are shown. (b) GRmax values for colchicine in four glioblastoma cell lines. (n = 3–5, one-way ANOVA). (c) RKI1 cells were treated with paclitaxel, vinblastine, and ixabepilone for 5 days and CellTiter-Blue viability assay was performed. GR values and dose–response curves were generated with the GRcalculator tool. Each curve is a mean of three independent cell viability assays, where each data point was done in triplicate. (d,e) GR curves for colchicine were extracted from Figure S4. Cells were treated with colchicine (500 nM) and stained with Nuclear-ID Red on day 0 and day 14. Representative images (d) and quantification (e) of five independent experiments are shown (mean ± SEM). (f) Cells were treated with vinblastine (25 nM) and stained with Nuclear-ID Red on day 0 and day 14. Representative images and quantification (mean ± SEM) of three independent experiments are shown.
Fractional killing has been previously attributed to insufficient target engagement in surviving cells.25 However, immunofluorescence of WK1 and RKI1 cells treated with colchicine revealed dose-dependent disruption of the microtubules in all cells, suggesting target engagement in the entire cell populations (Figure 5a). To ensure that cell survival was not due to the overexpression of drug efflux proteins, we performed control experiments with inhibitors of drug efflux pumps. The GR curves for colchicine (Figure 5b), CMPD1, and paclitaxel (Figure S6a,b) were identical between naïve RKI1 cells and RKI1 cells cotreated with efflux inhibitors verapamil, MK571, and CP-100356. Furthermore, the size of the RKI1 subpopulation surviving a 14-day treatment with colchicine did not decrease when cells were cotreated with verapamil (MDR1/BCRP inhibitor32), elacridar, and zosuquidar (MDR1 inhibitors33) or MK571 (MRP1 inhibitor34) (Figure 5c). Finally, flow cytometry analysis revealed no changes in MDR1 levels between colchicine-naïve (day 0) and colchicine-surviving cells (day 14) (Figure S6). Together, these data suggest that cell lines with lower α/β-tubulin expression are less sensitive to MTAs and generate larger populations of drug-tolerant cells. This phenomenon appears to be independent of drug efflux proteins and insufficient target engagement.
Figure 5.

Target engagement and expression of stem-cell markers. (a) WK1 and RKI1 cells were treated with colchicine for 5 days and stained with Alexa488-labeled anti-β-tubulin antibody (green) and DAPI (blue). Representative images of two independent experiments are shown. (b) RKI1 cells were treated with colchicine ± efflux inhibitors for 5 days and CellTiter-Blue viability assay was performed. GR values and dose–response curves were generated with the GRcalculator tool. (c) RKI1 cells were treated with 500 nM colchicine (Col) ± verapamil (25 μM), elacridar (1 μM), zosiquidar (1 μM), and MK571 (10 μM) for 14 days and stained with Nuclear-ID Red. Representative images and quantification (mean ± SEM) of 3–6 independent experiments are shown. (d) Heat map representation of the RNA-seq expression (red, high; blue, low) pattern of the transcription factors specific for glioblastoma stem cells in WK1, RN1, JK2, and RKI1 cells. (e) Western blot analysis of SOX2 (30 μg of total protein), α- and β-tubulin (1 μg total protein). Representative immunoblots and quantification presented as fold change relative to WK1 (set as 1) are shown. Data are mean ± SEM (n = 4–7; unpaired t test WK1 vs RKI1, ***P < 0.001, ****P < 0.0001). (f) Cells were fixed and stained with AlexaFluor594-labeled anti-β-tubulin (green), AlexaFluor488-labeled anti-nestin (yellow) antibodies and DAPI (blue). Representative images and quantification of three independent experiments are shown.
MTA Tolerant Cells Overexpress Stemness Markers and Convert to Dormant Cells
We observed that MTA tolerant RKI1 cells were proliferating faster than MTA-sensitive WK1 cells (Figure 2b). As fast proliferation is a functional criterion of glioblastoma stem cells,35 we hypothesized that the degree of stemness might be related to MTA sensitivity. Data from RNA sequencing analysis revealed that 13 out of 20 transcription factors specific for glioblastoma stem cells35−37 are upregulated in RKI1 cells (low MTA sensitivity) when compared to WK1 (high MTA sensitivity) cells (Figure 5d). At the protein level, SOX2 expression increases as the expression of α/β-tubulin decreases (Figure 5e). Having also confirmed lower β-tubulin expression in the JK2 and RKI1 cells by immunofluorescence, we further show that the expression of nestin is higher in the MTA-tolerant JK2 and RKI1 cells compared to the MTA-sensitive WK1 and RN1 (Figure 5f). Thus, MTA tolerant cell lines express higher levels of stemness markers and lower baseline levels of α/β-tubulin.
To further understand how glioblastoma cells survive MTA treatment, we performed flow cytometry analysis of colchicine-surviving RKI1 cells and found that drug-tolerant cells expressed features of dormant cells:38−40 large granular morphology (Figure 6a) and polyploidy (Figure 6b), high p-p38:p-ERK1/2 ratio (Figure 6c) and upregulation of the key dormancy markers DEC2, NR2F1, and p27 (Figure 6d). On the contrary, the levels of mRNA coding for p21, which is a marker of senescence (irreversible cell cycle arrest), decreased (Figure 6d). As dormancy is reversible, colchicine-surviving RKI1 and WK1 cells were allowed to recover in the absence of the drug and then expanded. The fraction of drug-tolerant cells was smaller in the WK1 cell line compared to that in the RKI1, and WK1 surviving cells required longer recovery time. However, both RKI1 and WK1 drug-tolerant subpopulations eventually regained proliferation (Figure 6e). The MTA dose–response curves for the parental (drug-naïve) and expanded drug-tolerant persister cells were indistinguishable (Figure 6f,g), implicating that drug-sensitive cells arise from drug-tolerant cells. Thus, the incomplete killing of glioblastoma stem cells by MTAs is not caused by a stable subpopulation of drug-resistant cells but rather by a fraction of cells that is able to resonate between proliferative (i.e., drug-sensitive) and dormant (i.e., drug-tolerant) states. These drug-tolerant fractions are larger and more aggressive in cell lines expressing lower baseline levels of α- and β-tubulin.
Figure 6.

Prolonged colchicine treatment induces dormancy. (a) RKI1 cells were treated with colchicine (500 nM), stained, and analyzed by flow cytometry for size (FSC-A) and granularity (SSC-A). Geometric mean of FSC-A (size) and SSC-A (granularity) was graphed using Prism v7.0 (GraphPad). Data are mean ± SEM (n = 3). (b) RKI1 cells were treated with colchicine (500 nM), stained for DNA with Vybrant DyeCycle Violet Stain and analyzed by flow cytometry. Representative FACS plots of three independent experiments are shown. (c) RKI1 cells were treated with colchicine (500 nM), stained with PE-eFluor610 conjugated p-ERK1/2 and APC conjugated p-p38 antibodies. The geometric mean of p-p38:p-ERK1/2 was graphed using Prism v7.0 (GraphPad). Data are mean ± SEM (n = 3). (d) Lysates of untreated (day 0) and colchicine (500 nM) treated (day 14) RKI1 cells were analyzed by RT-PCR. mRNA levels were normalized to GAPDH mRNA. Data are mean ± SEM (n = 3). (e) RKI1 and WK1 cells (day 0) were treated with colchicine (500 nM) for 14 days (day 14). Cells were allowed to recover until they regained their normal morphology (expanded DTPs). (f) RKI1 and (g) WK1 parental and expanded DTPs were treated with colchicine and CMPD1. GR curves were generated from dose responses determined with CellTiter-Blue viability assay after 5 days of drug treatment using the GRcalculator tool. Each curve is a mean of three independent cell viability assays, where each data point was done in triplicate.
Discussion
The analysis of the tubulin heterogeneity and MTA efficacy presented in this paper provides novel insights into the relationships between microtubules and MTAs. Furthermore, our data showing that the tubulin expression and post-translational modifications within a tissue-specific cancer type are unique for each cell line expand the concept of cancer heterogeneity. Genetic and epigenetic profiling have revealed the existence of various molecular subtypes within a tissue-specific tumor, but these subclassifications have been largely limited to alteration in signaling pathways and not to the house-keeping proteins such as tubulin.
Prior literature has reported variability in the expression of β-tubulin isotypes.30 However, other components of the tubulin code, such as α- and γ-tubulin and the post-translational modifications were not known. We observed a considerable variability (19–133%) in the levels of α-, β-, and γ-tubulin and their post-translational modifications. Thus, in addition to cell signaling abnormalities found in glioblastoma (Table S1), such as EGFR amplification and PTEN deletion,12 microtubule modifications appear unique for each tumor. The cell line-specific tubulin code is likely a connection to different phenotypes of these cell lines, and we anticipate that deeper understanding of these differences will improve our effort to effectively target glioblastoma cells. Importantly, we also show that the tubulin characteristics found in the standard glioblastoma cell lines routinely used in research are not representative of those found in the clinically relevant glioblastoma stem cell lines. As such, future studies into the roles of the tubulin code in glioblastoma should not be based on data obtained from the standard serum-grown cell lines.
MTAs sensitivity was variable across glioblastoma cell lines, ranging from partially cytotoxic to partially cytostatic responses. We found that the glioblastoma cell sensitivity to MTAs is independent of tubulin isotypes and the post-translational modifications investigated in this project. Surprisingly, the baseline levels of α- and β-tubulin correlated with the sensitivity to MTAs. The most sensitive cells contained the highest levels of α- and β-tubulin. This finding is in agreement with the concept that high expression of the targeted protein is usually a good predictor of responsiveness to drugs targeting that protein.41 However, while this hypothesis has been validated with receptor ligands and molecularly targeted cancer drugs, MTAs were not expected to fit into this concept, as microtubules are abundantly expressed in every cell. Our data showing that MTAs sensitivity declines with the decreasing levels of α- and β-tubulins points toward similarities in the mechanism of action of molecularly targeted drugs and MTAs. Moreover, our work confirms that drug efficacy parameters on a per-division basis have higher information content than conventional metrics.25,26,42 While the conventional dose–response curves fitted to relative cell count were overlapping, the growth rate corrected curves revealed significant differences in the maximum efficacy (Figure S4). Furthermore, when correlating MTA sensitivity with tubulin features, the per-division GRmax and GRAOC but not the conventional Emax and AUC metrics enabled us to identify the relationship between the α/β-tubulin levels and sensitivity to MTAs.
The most prevalent model of MTAs efficacy is based on the extensive clinical and in vitro data indicating that β-tubulin isotypes overexpression is involved in resistance to taxanes and vinca alkaloids (reviewed in ref (30)). Our somewhat contrasting data show that the high baseline levels of α/β-tubulin are determinants of good MTAs sensitivity in glioblastoma models. Nevertheless, it is important to highlight the difference in the experimental approaches. The majority of MTAs efficacy studies analyzed the expression of tubulin isotypes in drug-resistant cancer cells grown in media containing MTAs, thus describing mechanisms of the acquired resistance. Here, we have taken a different approach and analyzed how basal tubulin levels affect the first exposure of cells to MTAs. Furthermore, we identified that low α/β-tubulin levels mark MTAs tolerance but not resistance. Although not strictly defined, the difference between drug tolerance and resistance lies in the ability of a cell to proliferate in the presence of the drug.43 Unlike resistant cells which survive and proliferate in the presence of a drug, tolerant cells are able to survive but do not proliferate while exposed to the drug. As we show in the follow-up experiments, MTA treatment led to survival of drug-tolerant persister cells which resumed proliferation only upon the MTA removal.
Finally, we show that decreased α/β-tubulin expression is associated with lack of cell differentiation. The higher is the dedifferentiation state, the lower is the α/β-tubulin expression. Intriguingly, dedifferentiated cells survive the first exposure to MTA chemotherapy by activating a dormant state. Similar observations have been reported with molecularly targeted kinase inhibitor dasatinib.8 Whether the MTA-induced dormancy in glioblastoma cells is a primary step preceding acquired drug resistance remains to be investigated.
Small molecule MTAs able to penetrate the blood–brain barrier are in the preclinical and clinical development. In cancer treatments, the personalized medicine approach to genetically diverse tumors is now well-established with molecularly targeted drugs. We show in this study that chemotherapy with MTAs might face the same challenge, as tubulin profile and sensitivity to MTAs vary greatly across glioblastoma cell lines. We discovered that low α/β-tubulin expression limits the efficacy of MTAs, arguing the one-drug-fits-all concept for MTAs.
Methods
Cell Lines
Standard A172 (cat. no. 88062428), U251 (cat. no. 09063001) and U87 (cat. no. 89081402) glioblastoma cell lines were obtained from the European Collection of Authenticated Cell Cultures (ECACC, Salisbury, UK) through Cell Bank Australia in 2014. Cells were cultured in DMEM supplemented with 10% FBS (InterPath) and antibiotic–antimycotic solution (Life Technologies) at 37 °C and 5% CO2. Glioblastoma stem cell lines were derived from glioblastoma specimens. The protocols were approved by the Human Ethics Committee of the Royal Brisbane & Women’s Hospital (RBWH 2004/161). The RNA sequencing, mutational profile (Table S1), and subtype assignment for these cells have been published.21 GSC lines were cultured in KnockOut DMEM/F-12 basal medium supplemented with StemPro NSC SFM supplement, 2 mM GlutaMAX-ICTS, 20 ng/mL EGF, 10 ng/mL FGF-β, and antibiotic–antimycotic solution (all Life Technologies) as adherent cells on flasks coated with MatriGel Matrix (Corning Life Sciences). All cell cultures were routinely tested for mycoplasma infection, and the cumulative length of culturing did not exceed 15 passages.
Western Blotting
Cell lysates were prepared by adding 1 mL RIPA buffer to a T175 flask of unsynchronized cells at 80–90% confluency. Protein concentrations were determined with Pierce BCA assay kit (ThermoFisher Scientific), following manufacturer’s instructions. Lysates were resolved (2 h, 95 V) on 4–12% SDS-PAGE gels and transferred onto PVDF membranes using iBlot 2, P3 for 7 min (all Life Technologies). Membranes were blocked with 5% BSA in TBST, incubated with primary antibodies (Table S2) in 5% BSA in TBST overnight at 4 °C and with secondary antibody for 1 h at room temperature. All secondary antibody preparations were in 5% skim milk in TBST, with the exception of antityrosinated α tubulin and antiphospho βIII-tubulin, which were prepared in 5% BSA. Detection was performed with Immobilon Western HRP Substrate Luminol-Peroxidase reagent (MerckMillipore) and the ChemiDoc MP Imaging System (Bio-Rad). Densitometry quantification was done with ImageLab software (BioRad).
Cell Viability
Cells (2 × 103 cells/well) were plated in 96-well plates, with the exception of PB1 cells (8 × 103 cells/well). Cells were treated with DMSO (vehicle), colchicine (Tocris, cat. no. 1364), nocodazole (Tocris, cat. no. 1228), tivantinib (Selleckchem, cat. no. S2753), CMPD1 (Santa Cruz, cat. no. sc-203138), paclitaxel (Tocris, cat. no. 1097), vinblastine (Tocris, cat. no. 1256) and ixabepilone (AdooQ Bioscience, cat. no. A11449), at log 3 8-point dilution row for 5 days, with or without cotreatment with efflux pump inhibitors verapamil (Sigma-Aldrich, cat. no. V4629), MK571 (Sigma-Aldrich, cat. no. M7571) or CP-100356 (Sigma-Aldrich, cat. no. PZ0171). CellTiter-Blue dye (Promega) was added at 37 °C for 2–4 h, and fluorescence was measured with a Tecan M200 PRO+ microplate reader (Tecan) at 585 nm. Data were normalized to DMSO-treated controls (set as 1). Relative and per-division metrics were calculated from the viability data and proliferation rates using the GRcalculator online tool.26,42 Graphs were recreated from the GRcalculator online tool using Prism 7.0 (GraphPad).
Proliferation Rate
Cells were seeded at 500, 1 × 103, 2 × 103, and 4 × 103 cells/well in the 96-well plates. Proliferation was monitored by analyzing the occupied area (% confluence) of cell images over 7 days using the IncuCyte IC S3 2018A software (Essen Bioscience). Population doubling times were computed with several time points, from different initial cell densities using the “cell calculator++” tool (doubling-time.com; Roth V., 2006).
DTP Generation and Expansion
WK1 and RKI1 cells (1 × 106) were treated with 500 nM colchicine for 14 days. Fresh media with drug was added every 3 days. At day 14, drug-tolerant persisters were allowed to recover in drug-free media. Cells were monitored every 3 days until they regained their morphology and expansion started when cells resembled their parental cell lines. Images were taken using Zeiss Axio Vert.A1 microscope and ZEN 2–blue edition software (Zeiss). Expanded DTPs were seeded at 2 × 103 cells/well density for the viability assays in Figure 7.
Flow Cytometry
RKI1 cells (1.2 × 106) were treated with colchicine (500 nM) and samples were collected at day 0 (untreated), day 7, and day 14. Floating and adherent cells were washed with ice-cold PBS. For cell viability, samples were stained using LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (ThermoFisher Scientific) for 30 min at 4 °C in the dark as per manufacturer’s instructions. Samples were centrifuged at 300g for 5 min at 4 °C, washed with ice-cold PBS with 2% FBS (blocking buffer), and incubated in FITC conjugated human-CD243 (MDR-1) antibody/106 cells (ThermoFisher Scientific, 1:10 in blocking buffer) in 50 μL reaction volume on ice for 45 min. Samples were centrifuged, washed twice with the blocking buffer, and incubated in fixation buffer (BioLegend) for 10 min at room temperature (RT) as per manufacturer’s instructions, followed by permeabilization by adding 350 μL of ice-cold 100% methanol with vortexing. For dormancy analysis, samples were washed twice with blocking buffer before being incubated with p-ERK1/2 (PE-eFluor610 conjugate, ThermoFisher Scientific) and p-p38 (APC conjugate, ThermoFisher Scientific) antibodies/106 cells in 1:10 dilution with blocking buffer in 50 μL reaction vol for 45 min on ice. For DNA analysis, samples were washed twice with blocking buffer and stained with Vybrant DyeCycle Violet Stain (ThermoFisher Scientific) at 37 °C for 30 min as per manufacturer’s instructions. Samples were analyzed on LSRFortessa X-20 running FACSDiVa v6 software (BD Biosciences). Live and dead cells were discriminated by a live cell gating, and expression levels of CD243, p-ERK1/2, p-p38, and DNA analyses were performed on the live cell gate; all data were analyzed using FlowJo v10.3. Unstained and single stained samples were used as compensation controls.
Nuclear-ID Red Staining
WK1, RN1, JK2, and RKI1 (1 × 105) were treated with 500 nM colchicine ±25 μM verapamil, 1 μM elacridar (Tocris, cat no. 4646), 1 μM zosuquidar (Tocris, cat no. 5456) or 10 μM MK571 (Sigma-Aldrich, cat no. M7571) for 14 days. Fresh media containing drugs was added every 3 days. Untreated (day 0) and treated (day 14) cells were stained with Nuclear-ID red stain (Enzo Lifesciences) at 1:1,000 dilution in StemPro media. Cells were incubated with the stain for 30 min prior to mounting onto microscope slides using Dako Fluorescence Mounting Medium (Agilent). Images were taken using Zeiss Axio Scope.A1 and ZEN 2–blue edition software (Zeiss) and processed using Fiji.
Capillary Immunoassay
Total cell lysates at 1 μg/μL (α-tubulin) and 0.25 μg/μL (β-tubulin) were analyzed using an automated capillary electrophoresis system Wes (ProteinSimple), as per manufacturer’s instructions. Wes Separation Capillary Cartridges for 12–230 kDa (ProteinSimple) were used. The primary antibodies used in Western blotting were also used for the capillary immunoassay at 1:50 dilution (α-tubulin, GAPDH) or 1:1,000 dilution (β-tubulin). Signals were detected with Anti-Rabbit and Anti-Mouse Detection Modules for Jess, Wes, Peggy Sue, or Sally Sue and analyzed using Compass software (all ProteinSimple).
qRT-PCR
qRT-PCR was carried out according to standard protocols. RKI1 cells (1.2 × 106) were treated with colchicine (500 nM) for 14 days. Fresh media containing colchicine was added every 3 days. Cells were lysed at day 0 (untreated) and day 14. RNeasy mini kit (Qiagen) was used to isolate RNA from cell lysates as per manufacturer’s instructions. cDNA was generated using Applied Biosystems High-Capacity cDNA Reverse Transcription kit (Life Technologies) as per manufacturer’s instructions. qRT-PCR was performed using DEC2, p27 (both Integrated DNA Technologies), N2RF1 (Qiagen, cat no. QT00089355), p21 (Qiagen, cat no. QT00005803), and GAPDH primers (Qiagen, cat no. QT00079247) with KAPA SYBR FAST Universal 2X qPCR Master Mix (Kapa Biosystems). RT-PCR was run on LightCycler 480 (Roche). The cycling conditions were as follows: 10 min at 95 °C followed by 40 cycles, each consisting of 10 s at 95 °C and 30 s at 60 °C. Samples were run in triplicate. Threshold cycles (Ct) were calculated using the LightCycler 480 software. Relative quantification using the comparative Ct method was used to analyze the data output. Values were expressed as fold change over corresponding values for the control by the 2-ΔΔCt method.
Immunofluorescence
For β-tubulin immunofluorescence in DTPs, cells (8 × 103) were treated with colchicine (5–500 nM) for 5 days. For nestin and β-tubulin immunofluorescence, untreated cells (1 × 105) were fixed with ice-cold 4% PFA for 20 min at RT and blocked in 5% BSA/PBS for 20 min. Cells were incubated with anti-nestin (1:25, R&D Systems) and anti-β-tubulin antibody (1:200, Abcam). Secondary antibodies were Alexa488-conjugated antimouse IgG (against nestin) and Alexa594-conjugated antirabbit IgG (against β-tubulin) (Life Technologies). Cell nuclei were counterstained using Prolong Gold mounting media with DAPI (Life Technologies). Images were acquired under 40× objectives on a Zeiss Axio Scope.A1 microscope using ZEN 2–blue edition software (Zeiss). Images were processed using Fiji software.
Data and Statistical Analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology. All immunoblots and immunofluorescence images are representatives of at least three independent experiments. All cellular assays were repeated at least three times, and each was run in duplicate or triplicate. All results are expressed as mean ± SEM. All statistical analyses were performed using Prism v7.0 (GraphPad). Correlation analysis between tubulin isotypes/post-translational modifications and MTA efficacy was performed using Spearman’s correlation method as not all data sets followed a normal distribution. Two-tailed P values are listed for these correlations. An independent t test was performed for comparison of two data sets, and 1-way ANOVA followed by Tukey’s post hoc test was used when there were multiple groups to compare. In all cases, P < 0.05 was considered significant.
Acknowledgments
This work was supported by funding from The University of Sydney, Brain Foundation Australia, Cancer Institute New South Wales and the National Health & Medical Research Council of Australia. R.H.A. is supported by The University of Sydney RTP scholarship, and L.M. has been supported by Cancer Institute NSW Career Development Fellowship (15/CDF/107).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsptsci.9b00045.
Genotypes of glioblastoma stem cell lines; details of primary antibodies; MTAs sensitivity metric values; Spearman’s correlation coefficients between MTAs sensitivity metrics with tubulin code; representative image of Coomassie stained gels; expression of tubulin isotypes and post-translational modifications normalized to A172 and RN1 signals; dose–response curves for MTAs in glioblastoma cell lines; Spearman’s correlations between tubulin expression and MTAs efficacy excluding BAH1 data; data obtained with inhibitors of efflux pumps (PDF)
Author Contributions
R.H.A. and L.M. designed the study. R.H.A., A.R. and D.I.V. performed experiments and data analysis under L.M. supervision. T.G.J., B.W.S. and B.W.D. generated, characterized, and provided GSC lines.
The authors declare no competing financial interest.
Supplementary Material
References
- Steinmetz M. O.; Prota A. E. (2018) Microtubule-targeting agents: Strategies to hijack the cytoskeleton. Trends Cell Biol. 28, 776–792. 10.1016/j.tcb.2018.05.001. [DOI] [PubMed] [Google Scholar]
- Roll-Mecak A. (2019) How cells exploit tubulin diversity to build functional cellular microtubule mosaics. Curr. Opin. Cell Biol. 56, 102–108. 10.1016/j.ceb.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadadhar S.; Bodakuntla S.; Natarajan K.; Janke C. (2017) The tubulin code at a glance. J. Cell Sci. 130, 1347. 10.1242/jcs.199471. [DOI] [PubMed] [Google Scholar]
- Kavallaris M. (2010) Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 10, 194–204. 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- Parker A. L.; Teo W. S.; McCarroll J. A.; Kavallaris M. (2017) An emerging role for tubulin isotypes in modulating cancer biology and chemotherapy resistance. Int. J. Mol. Sci. 18, 1434. 10.3390/ijms18071434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangauer M. J.; et al. (2017) Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. 10.1038/nature24297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S. V.; et al. (2010) A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80. 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liau B. B.; et al. (2017) Adaptive chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance. Cell Stem Cell 20, 233–246. 10.1016/j.stem.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch A.; et al. (2013) Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 23, 811–825. 10.1016/j.ccr.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvi M. P.; et al. (2017) Taxane-platin-resistant lung cancers co-develop hypersensitivity to JumonjiC Demethylase inhibitors. Cell Rep. 19, 1669–1684. 10.1016/j.celrep.2017.04.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman A.; et al. (2015) Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat. Commun. 6, 6139. 10.1038/ncomms7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan C. W.; et al. (2013) The somatic genomic landscape of glioblastoma. Cell 155, 462–477. 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S.; Louis D. N.; Curry W. T.; Batchelor T. T.; Dietrich J. (2013) Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end?. Nat. Rev. Clin. Oncol. 10, 14–26. 10.1038/nrclinonc.2012.204. [DOI] [PubMed] [Google Scholar]
- Liu Y.; et al. (2014) Paclitaxel loaded liposomes decorated with a multifunctional tandem peptide for glioma targeting. Biomaterials 35, 4835–4847. 10.1016/j.biomaterials.2014.02.031. [DOI] [PubMed] [Google Scholar]
- Kang T.; et al. (2014) iNGR-modified PEG-PLGA nanoparticles that recognize tumor vasculature and penetrate gliomas. Biomaterials 35, 4319–4332. 10.1016/j.biomaterials.2014.01.082. [DOI] [PubMed] [Google Scholar]
- Zhang B.; et al. (2015) UPA-sensitive ACPP-conjugated nanoparticles for multi-targeting therapy of brain glioma. Biomaterials 36, 98–109. 10.1016/j.biomaterials.2014.09.008. [DOI] [PubMed] [Google Scholar]
- Senese S.; et al. (2014) Chemical dissection of the cell cycle: probes for cell biology and anti-cancer drug development. Cell Death Dis. 5, e1462 10.1038/cddis.2014.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhu S.; Harris F.; Lea R.; Snape T. J. (2014) Small-molecule clinical trial candidates for the treatment of glioma. Drug Discovery Today 19, 1298–1308. 10.1016/j.drudis.2014.02.007. [DOI] [PubMed] [Google Scholar]
- Gurgis F.; Akerfeldt M.; Heng B; Wong C; Adams S; Guillemin G.; Johns T.; Chircop M; Munoz L (2015) Cytotoxic activity of the MK2 inhibitor CMPD1 in glioblastoma cells is independent of MK2. Cell Death Discovery 1, 15028. 10.1038/cddiscovery.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phoa A. F.; et al. (2015) Pharmacology of novel small-molecule tubulin inhibitors in glioblastoma cells with enhanced EGFR signalling. Biochem. Pharmacol. 98, 587–601. 10.1016/j.bcp.2015.10.014. [DOI] [PubMed] [Google Scholar]
- Stringer B. W.; et al. (2019) A reference collection of patient-derived cell line and xenograft models of proneural, classical and mesenchymal glioblastoma. Sci. Rep. 9, 4902. 10.1038/s41598-019-41277-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama R.; et al. (2013) Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res. 73, 3087–3096. 10.1158/0008-5472.CAN-12-3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; et al. (2016) Structures of a diverse set of colchicine binding site inhibitors in complex with tubulin provide a rationale for drug discovery. FEBS J. 283, 102–111. 10.1111/febs.13555. [DOI] [PubMed] [Google Scholar]
- Rankovic Z. (2017) CNS Physicochemical property space shaped by a diverse set of molecules with experimentally determined exposure in the mouse brain. J. Med. Chem. 60, 5943–5954. 10.1021/acs.jmedchem.6b01469. [DOI] [PubMed] [Google Scholar]
- Fallahi-Sichani M.; Honarnejad S.; Heiser L. M.; Gray J. W.; Sorger P. K. (2013) Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat. Chem. Biol. 9, 708. 10.1038/nchembio.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M.; Niepel M.; Chung M.; Sorger P. K. (2016) Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 13, 521. 10.1038/nmeth.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M.; Niepel M.; Subramanian K.; Sorger P. K. (2017) Designing drug-response experiments and quantifying their results. Curr. Protocols Chem. Biol. 9, 96–116. 10.1002/cpch.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niepel M.; Hafner M.; Chung M.; Sorger P. K. (2017) Measuring Cancer Drug Sensitivity and Resistance in Cultured Cells. Curr. Protocols Chem. Biol. 9, 55–74. 10.1002/cpch.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilmar A.; Garcia-Foncillas J.; Huarriz M.; Santoni-Rugiu E.; Sorensen J. B. (2012) RT-PCR versus immunohistochemistry for correlation and quantification of ERCC1, BRCA1, TUBB3 and RRM1 in NSCLC. Lung Cancer 75, 306–312. 10.1016/j.lungcan.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Parker A. L.; Teo W. S.; McCarroll J. A.; Kavallaris M. (2017) An emerging role for tubulin isotypes in modulating cancer biology and chemotherapy resistance. Int. J. Mol. Sci. 18, 1434. 10.3390/ijms18071434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M.; et al. (2017) Quantification of sensitivity and resistance of breast cancer cell lines to anti-cancer drugs using GR metrics. Sci. Data 4, 170166. 10.1038/sdata.2017.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittapalli R. K.; et al. (2016) ABCG2 and ABCB1 Limit the Efficacy of Dasatinib in a PDGF-B–Driven Brainstem Glioma Model. Mol. Cancer Ther. 15, 819. 10.1158/1535-7163.MCT-15-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman S.; et al. (2017) Eradication of tumors through simultaneous ablation of CD276/B7-H3-positive tumor cells and tumor vasculature. Cancer Cell 31, 501–515. 10.1016/j.ccell.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang X.; et al. (2018) A Promising Microtubule Inhibitor Deoxypodophyllotoxin Exhibits Better Efficacy to Multidrug-Resistant Breast Cancer than Paclitaxel via Avoiding Efflux Transport. Drug Metab. Dispos. 46, 542. 10.1124/dmd.117.079442. [DOI] [PubMed] [Google Scholar]
- Lathia J. D.; Mack S. C.; Mulkearns-Hubert E. E.; Valentim C. L. L.; Rich J. N. (2015) Cancer stem cells in glioblastoma. Genes Dev. 29, 1203–1217. 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suva M. L.; Rheinbay E.; Gillespie S. M.; Patel A. P.; Wakimoto H.; Rabkin S. D.; Riggi N.; Chi A. S.; Cahill D. P.; Nahed B. V.; Curry W. T.; Martuza R. L.; Rivera M. N.; Rossetti N.; Kasif S.; Beik S.; Kadri S.; Tirosh I.; Wortman I.; Shalek A. K.; Rozenblatt-Rosen O.; Regev A.; Louis D. N.; Bernstein B. E. (2014) Reconstructing and Reprogramming the Tumor-Propagating Potential of Glioblastoma Stem-like Cells. Cell 157, 580–594. 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard S. M.; et al. (2009) Glioma Stem Cell Lines Expanded in Adherent Culture Have Tumor-Specific Phenotypes and Are Suitable for Chemical and Genetic Screens. Cell Stem Cell 4, 568–580. 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- Mirzayans R.; Andrais B.; Murray D. (2018) Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers 10, E118. 10.3390/cancers10040118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa M. S.; Avivar-Valderas A.; Bragado P.; Wen H.-C.; Aguirre-Ghiso J. A. (2011) ERK1/2 and p38α/β Signaling in Tumor Cell Quiescence: Opportunities to Control Dormant Residual Disease. Clin. Cancer Res. 17, 5850. 10.1158/1078-0432.CCR-10-2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluegen G.; et al. (2017) Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 19, 120. 10.1038/ncb3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niepel M.; et al. (2013) Profiles of Basal and Stimulated Receptor Signaling Networks Predict Drug Response in Breast Cancer Lines. Sci. Signaling 6, ra84 10.1126/scisignal.2004379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M.; Niepel M.; Sorger P. K. (2017) Alternative drug sensitivity metrics improve preclinical cancer pharmacogenomics. Nat. Biotechnol. 35, 500–502. 10.1038/nbt.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recasens A.; Munoz L. (2019) Targeting Cancer Cell Dormancy. Trends Pharmacol. Sci. 40, 128–141. 10.1016/j.tips.2018.12.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.