

Abstract
Porcine epidemic diarrhea virus (PEDV) has caused devastating impact on pig-rearing industry in China and current vaccine is not effective against the circulating PEDV variants. In the present study, the full-length genome sequence from a PEDV isolate (CH/HNQX-3/14) was determined. The complete genome sequence analysis showed that the CH/HNQX-3/14 possessed unique deletion regions in the S and ORF3 genes. It was identified as a recombinant strain using phylogenetic analysis and recombination detection program. Further analyses of the full-length sequence suggest that CH/HNQX-3/14 is a natural recombinant between the attenuated vaccine strains (CV777 and DR13) and circulating wild-type strain (CH/ZMDZY/11). The recombination occurred not only in structural protein-coding region (S1 and N genes) but also in non-structural protein-coding region (replicases 1a and ORF3 genes). These results provided new evidence that PEDV strains circulating in China underwent recombination between vaccine and field strains, suggesting that recombination contributes to the genetic diversity of PEDV. Our findings provide valuable information on PEDV evolution and underscore the need for ongoing surveillance of this economically important swine disease.
Keywords: Porcine epidemic diarrhea virus, Recombination, Genome sequencing and analysis
Introduction
PEDV is an enveloped virus possessing an approximately 28 kb, positive-sense, single-stranded RNA genome, belonging to the genus Alphacoronavirus of the family Coronaviridae [1, 2]. The virus is the causative agent of porcine epidemic diarrhea (PED), an acute and highly contagious enteric disease characterized by watery diarrhea, vomiting, and which produces high mortality in piglets [3]. The PEDV genome contains at least seven open reading frames (ORFs), encoding four structural proteins (spike [S], envelope [E], membrane [M], and nucleocapsid [N]), and three non-structural proteins (replicases 1a and 1b and ORF3) [3]. Of these, the S glycoprotein is the major structural protein and is essential for understanding the epidemiological status of PEDV in the field, diversity of PEDV isolates, and the association between genetic mutations and viral antigenicity [4, 5]. The ORF3 gene has been demonstrated to be associated with the virulence of PEDV [6]. The N protein provides a structural basis for the helical nucleocapsid, and is important for the induction of cell-mediated immunity [7]. Two long ORFs (ORF1a and ORF1b) genes, which occupy two thirds of the genome, encode the non-structural replicase polyproteins (replicase 1a and 1b) [8]. These dates increase our insight into PEDV genomic characteristics.
PED was first recognized in England in 1971 [1], and since then, outbreaks have been reported in Europe and Asia [9, 10], and more recently in the United States [11]. In 2014, outbreaks of PED reemerged in Belgium and France and pose more threat on swine industry in Europe [12, 13]. PEDV has been reported in many provinces of China since it was first documented in 1984 [14]. In particular, the devastating outbreak of PEDV since 2010 has caused a high number of deaths among suckling pigs and, as a consequence, substantial economic losses [15]. Although commercial vaccines against PEDV are available, traditional control strategies and the conventional vaccines have failed to control the disease. Currently, PEDV is still evolving within the swine population. To investigate the evolutionary process of PEDV strains, we have been conducting a systematic surveillance program on molecular epidemiology of PEDV circulating in China since 2010.
In our previous work, molecular epidemiology based on the ORF3 gene and partial S gene of PEDV revealed that the strains prevailing in central China were highly virulent PEDV variants, and some amino acid mutations within the epitope regions of S protein were found, which might be associated with vaccination failure [16]. In this study, we determined the whole genome sequence for a novel PEDV isolate (CH/HNQX-3/14) identified from Henan in central China. In addition, we used the whole genome sequence plus available genome sequences for other PEDV isolates to conduct phylogenetic and recombination analysis.
Materials and methods
Viruses and sequences
PEDV strain CH/HNQX-3/14 was isolated from small intestine sample of a naturally PEDV-infected piglet in Henan in 2014, and used for genome sequencing and genetic analysis. To investigate the full genetic characterization, 39 complete genomes of PEDV strains available from GenBank were retrieved and aligned with CH/HNQX-3/14 full-length genome determined in the present study. All strains used in this study are presented in Table 1.
Table 1.
Origin and GenBank accession numbers of PEDV isolates used in this study
| Reference strains | Accession No | Origin | Reference strains | Accession No | Origin |
|---|---|---|---|---|---|
| CH/S | JN547228.1 | China, 1986 | PEDV-LYG | KM609212.1 | China, 2014 |
| LZC | EF185992.1 | China, 2007 | CH/GDZQ/2014 | KM242131.1 | China, 2014 |
| DR13 | JQ023161.1 | Korea, 2008 | AH-M | KJ158152.1 | China, 2014 |
| ZJCZ4 | JX524137.1 | China, 2011 | CH/HNQX-3/14 | KR095279 | China, 2014 |
| CH/FJND-3/2011 | JQ282909.1 | China, 2011 | USA/Indiana12.83/2013 | KJ645635.1 | USA, 2013 |
| BJ-2011-1 | JN825712.1 | China, 2011 | USA/Illinois63/2013 | KJ645659.1 | USA, 2013 |
| CH/ZMDZY/11 | KC196276.1 | China, 2011 | USA/Tennesse56/2013 | KJ645654.1 | USA, 2013 |
| CV777 | AF353511.1 | Belgium, 2011 | USA/Minnesota52/2013 | KJ645704.1 | USA, 2013 |
| AJ1102 | JX188454.1 | China, 2012 | USA/Colorado/2013 | KF272920.1 | USA, 2013 |
| CHGD-01 | JX261936.1 | China, 2012 | USA/Ohio69/2013 | KJ645665.1 | USA, 2013 |
| LC | JX489155.1 | China, 2012 | USA/Ohio60/2013 | KJ645657.1 | USA, 2013 |
| CH/ZJCX-1/2012 | KF840537.1 | China, 2012 | MN | KF468752.1 | USA, 2013 |
| JS-HZ2012 | KC210147.1 | China, 2012 | IA2 | KF468754.1 | USA, 2013 |
| AH2012 | KC210145.1 | China, 2012 | USA/Iowa96/2013 | KJ645688.1 | USA, 2013 |
| SHQP/YM/2013 | KJ196348.1 | China, 2013 | ON-018 | KM189367.2 | USA, 2014 |
| CH/JX-2/2013 | KJ526096.1 | China, 2013 | OH1414 | KJ408801.1 | USA, 2014 |
| CH/YNKM-8/2013 | KF761675.1 | China, 2013 | OH851 | KJ399978.1 | USA, 2014 |
| LZW | KJ777677.1 | China, 2014 | MEX/104/2013 | KJ645708.1 | Mexico, 2013 |
| PEDV-14 | KM609207.1 | China, 2014 | L00721/GER/2014 | LM645057.1 | Germany, 2014 |
| PEDV-15F | KM609208.1 | China, 2014 | GER/L00719/2014 | LM645058.1 | Germany, 2014 |
Determination of the full-length genome sequences of the CH/HNQX-3/14 strain
Viral RNA was extracted using the TRIzol reagent (Invitrogen, USA) following the manufacture’s instruction. Synthesis of the first-strand cDNA was carried out by reverse transcription using TAKARA Reverse Transcription Kit (Code No. RR014A). Full-length PEDV genome amplification was performed with the primers described previously [17]. For each amplicon, three to ten independent clones were sequenced to determine the consensus sequence of given genomic region. The full-length genome sequence of CH/HNQX-3/14 was assembled using ContigExpress program in the Vector NTI software package (Invitrogen). The validated genome sequence of CH/HNQX-3/14 strain was submitted to GenBank databases and assigned accession No. KR095279.
Phylogenetic and recombination analysis
Clustal X (Ver.1.81) program was used to align the full-length genome sequences of CH/HNQX-3/14 and reference sequences from the GenBank database [18]. Phylogenetic trees were constructed using the neighbor-joining (NJ) method implemented in Molecular Evolutionary Genetics Analysis (MEGA) software (version 5.2.1) [19]. The reliability of the NJ tree topologies was assessed using 1000 bootstrap replicates. Six recombination detection methods (RDP, GENECONV, BOOTSCAN, MaxChi, Chimaera, and SISCAN) implemented in the Recombination Detection Program v.4.16 (RDP4) [20] were used for identification of recombinant sequences and breakpoints. The default settings were used for all methods, and the highest acceptable P value cut-off was set to 0.01. Only those recombination events supported by more than 4 programs were considered to avoid dependence on a single methodology. Base-by-base comparison of the possible recombinants with parental strains was performed by BioEdit package (Ver.7.0.5) to further confirm the recombination events and elucidate the differences between amino acid sequences in neutralizing epitope regions.
Results
CH/HNQX-3/14 genome characterization
The complete genome of CH/HNQX-3/14 is 27, 997 nucleotides (nt) in length, excluding the poly(A) tail, and the genome organization is as follows: a 5′ untranslated region (UTR) of 296 nt (nt 1 to 296), an open reading frame 1a (ORF1a) and ORF1b encoding a replicase of 20, 345 nt (nt 297 to 20, 641, 6, 781 aa), a spike (S) gene of 4, 143 nt (nt 20, 638 to 24, 780, 1, 381 aa), an ORF3 of 648 nt (nt 24, 780 to 25, 427, 216 aa), an envelope (E) gene of 230 nt (nt 25, 408 to 25, 638, 76 aa), a membrane (M) gene of 681 nt (nt 25, 646 to 26, 326, 227 aa), a nucleocapsid (N) gene of 1, 326 nt (nt 26, 338 to 27, 663, 442 aa), and a 3′ UTR (nt 27, 664 to 27, 997).
Of note, compared with the vaccine strain CV777, the genome of CH/HNQX-3/14 possessed four remarkable deletion regions (nt 20, 813 to 20, 824, nt 21, 058 to 21, 063, nt 21, 127 to 21, 132 and 22, 252 to 22, 257) in the S gene, resulting in a continuous 4-amino acid deletion (59QGVN62) and three continuous 2-amino acid deletions (141ND142, 164DI165, and 539AS540) in the S protein, respectively. These deletion sites were mainly in the S1 domain (1–781 aa) of PEDV S protein-coding sequences. Besides, 11 single-nucleotide variants were located in the aforementioned variable region, 8 of which are non-synonymous. These results suggested the high variability in the N terminal part of the S protein. In addition, the deletion 48T and the insertion 83CCTT86 that occurred in the ORF1a gene were found same to the vaccine strain CV777. Further, the sequence analysis showed that there was a unique deletion (nt 25, 027 to 25, 053) in the ORF3 gene, which was similar to CV777 that 51-bp deletion in the ORF3 region.
Sequence comparison
Comprehensive comparison of full-length PEDV genomes from different years and locations revealed that CH/HNQX-3/14 had a nucleotide identity of 96.9–99.7 % with other complete PEDV genomes available in GenBank. Of note, CH/HNQX-3/14 shared the highest nucleotide identity (99.7 %) with CH/ZMDZY/11 strain, a strain from the same province with CH/HNQX-3/14, and lowest identity (96.9 %) with attenuated DR13 strain. It exhibits 98.9 % nucleotide identity with that of two German strains (GER/L00719/2014 and L00721/GER/2014) and 98.6 % similar to the US PEDV variant, OH851, and 97.7, 97.4, 97.1, and 97.0 % nucleotide sequence similarity to those of attenuated CH/S, CHGD-01, AJ1102, and CH/FJND-3/2001, respectively. In addition, CH/HNQX-3/14 shared 97.4 % nucleotide identity with a AH-M, a new variant recently causing disease in China, with a higher similarity (98.7 %) to the LZC, a historic strain from China, but it was less similar (96.9–97.1 %) to the isolates from America (USA/Indiana12.83/2013, USA/Minnesota52/2013, USA/Ohio69/2013, USA/Ohio60/2013, USA/Illinois63/2013, USA/Tennesse56/2013, USA/Colorado/2013, USA/Iowa96/2013, OH1414, MN and IA2).
Phylogenetic and recombination analysis
Phylogenetic analysis was performed to investigate genetic relationship of the CH/HNQX-3/14 with the historic strains. In the NJ tree based on the PEDV whole genomes showed that all of the PEDV strains could be divided into seven clusters (C1–C7). The strain CH/HNQX-3/14 was clustered in group C3, together with two Chinese strain (CH/GDZQ/2014 and CH/ZMDZY/11), three American strains (OH851, USA/Minnesota52/2013, and USA/Indiana12.83/2013), and two Germany strains (L00721/GER/2014 and GER/L00719/2014) (Fig. 1).
Fig. 1.
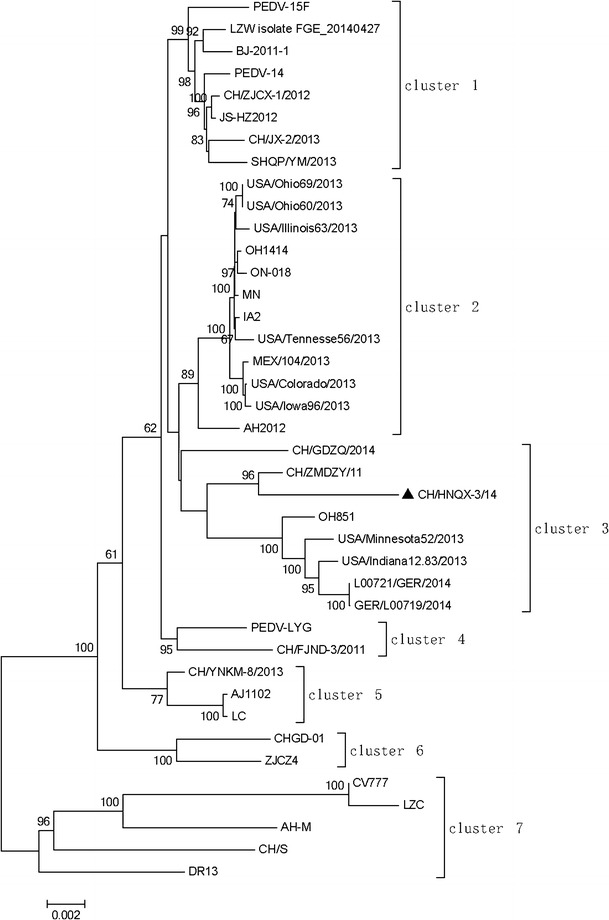
Phylogenetic analysis based on the full-length genome sequences of CH/HNQX-3/14 isolate in this study and PEDV reference strains available in GenBank. The tree was constructed using the neighbor-joining method in MEGA 5.2.1 software. Numbers above branches are bootstrap values computed from 1000 replications. The scale bar represents nucleotide substitutions per site. Filled triangle indicates the CH/HNQX-3/14 strain from this study
To determine the involvement of recombinant events in evolution of the CH/HNQX-3/14 strain, we performed recombinant detection using RDP4 software based on comparing CH/HNQX-3/14 with the representative Chinese historical PEDV strains from different clusters in the phylogenetic tree, including strains from cluster 1 (BJ-2011-1 and CH/JX-2/2013), cluster 2 (AH2012), cluster 3 (CH/ZMDZY/11), cluster 5 (LC), and cluster 7 (CV777 and DR13). The results showed that four putatively major recombinant breakpoints in the CH/HNQX-3/14 genome with a high degree of reliability were detected, which were located, respectively, at the ORF1a (nt 2, 769), S (nt 20, 691 and nt 22, 176), and N (nt 27, 252) genes (Fig. 2a; Table 2). Notably, the zones before breakpoint nt 2, 769 and after the breakpoint nt 2, 7252 exhibited greater similarity with CV777 (an attenuated PEDV vaccine strain used in China), and the zone between breakpoint nt 2, 0691 and nt 2, 2176 had high levels of similarity with DR13 (an attenuated PEDV vaccine strain used in Korea). The other zones spanned ORF1b, partial S, ORF3, E, M, and partial N between the strain CV777 (as the major parent) and the CH/ZMDZY/11 (as the minor parent) (Fig. 2a; Table 2). Additionally, phylogenetic trees based on the complete genome sequences displayed evolutionary relationships and potential recombination events between CH/ZMDZY/11 and the putative parental strains (Fig. 2b). Overall, these findings revealed that CH/HNQX-3/14 may be a mosaic strain derived from 3 putative parental-like strains (CV777, DR13, and CH/ZMDZY/11) through recombination.
Fig. 2.
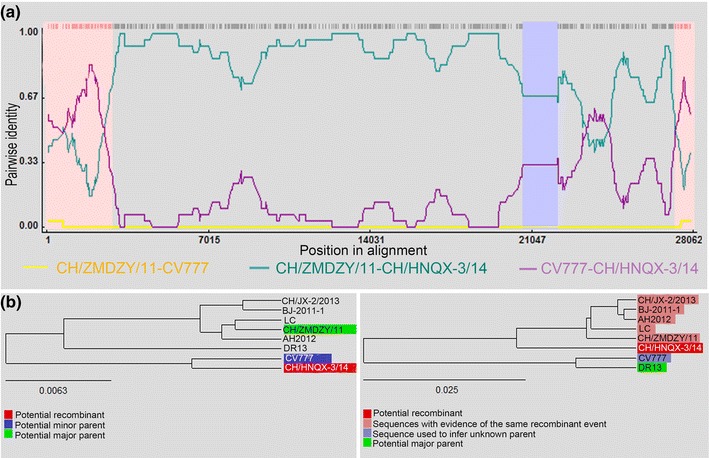
Detection of potential recombination events in CH/HNQX-3/14 by RDP4. a Four major recombination breakpoints were located, respectively, at the ORF1a (nt 2769), S (nt 20,691 and nt 22,176), and N (nt 27,252) genes. The analysis was performed with an F84 distance model, a window size of 1000 base pairs and a step size of 200 base pairs. b Evolutionary relationships between CH/HNQX-3/14 and the putative parental strains (CH/ZMDZY/11, DR13. and CV777) based on the full-length genomes. Phylogenetic trees were generated by the distance-based neighbor-joining method in RDP4 program
Table 2.
Summary of recombination events identified by the Recombination Detection Program v.4.16 (RDP4)
| Breakpoint position in recombinant sequence | Parental sequence(s) | Score for the six detection methods in RDP4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Recombinant segment | Begin | End | Minor | Major | RDP | GENECONV | BOOTSCAN | MaxChi | Chimaera | SiSCAN |
| ORF1a | 1 | 2769 | CH/ZMDZY/11 | CV777 | 3.783E−3 | 3.172E−9 | 2.601E−2 | 1.337E−10 | 1.648E−12 | NS |
| S gene | 20,691 | 22,176 | CV777 | DR13 | 3.864E−3 | NS | 1.319E−2 | 2.799E−3 | 2.583E−10 | 1.992E−24 |
| N gene | 27,252 | 28,020 | CH/ZMDZY/11 | CV777 | 1.855E−3 | NS | 2.425E−3 | 1.297E−6 | 2.491E−6 | 1.489E−12 |
The abbreviation “NS” indicates that the recombination event is not significant
Discussion
PEDV infection in pig populations has been reported in Asia, Europe, and the United States. Licensed PED vaccines are currently available in South Korea, Japan, and China. However, the CV777-based vaccine is not effective against the new PEDV variants circulated in China since 2011 [21]. Genetic recombination plays a critical role in virus evolution and presents one of the greatest challenges to prevent PEDV infection and combating PED because it introduces genetic variation that complicates vaccine development [22]. Divergence of PEDV is driven by genetic recombination, as in other coronaviruses. By recombination analysis, the PEDV strains emerging in the United States have been demonstrated originating from China [23]. A previous study demonstrated that the PEDV circulating strains in China may be originated from either South Korean and/or Chinese ancestors that underwent some genetic variation [24]. Another previous report about a possible recombinant PEDV strain (JS2008) from eastern China provided no evidence on the origin of the recombinant, the recombinant breakpoints and locations in genome [25]. PEDV is still currently circulating in many swine farms in central China, but little is known on the role of recombination in the PEDV evolution. In the present study, we identified a novel recombinant PEDV strain from Henan, a province of central China, and found new combination patterns in PEDV evolution.
To characterize the virus isolate, we determined the complete genome sequence of CH/HNQX-3/14 and analyzed the phylogenetic relationships with other PEDV strains at the genomic level. CH/HNQX-3/14 had three unique amino acid mutations (S548A, G749S, and K766L) in the neutralizing epitope regions compared to CV777, DR13, and CH/ZMDZY/11, which located in the epitope region A (aa 499–638), B (aa 748–755), and C (aa 764–771), respectively (Fig. 3). We infer that these mutations might alter the antigenicity of vaccine based on the CV777 and consequently resulted in inefficient vaccination in many swine farms of China. In addition, previous studies confirmed that the neutralizing epitopes of PEDV are mostly present at position aa 499–638, 748–755, 764–771, and 1368–1374 of S protein [2]. In the present study, four remarkable deletions were found in the nucleotide sequence of CH/HNQX-3/14 genome. However, only the deletion nt 22,252–22,257 (encoded 539AS540) occurred within the epitope region A (aa 499–638), but other three deletions were all out of the epitope regions. In addition, the deletions at position 59QGVN62 were similar to attenuated strains CV777 and DR13 (Fig. 3). Further, the sequence analysis showed that CH/HNQX-3/14 had a continuous 27-bp deletion (nt 25,027–25,053) in the ORF3 gene, which is similar to CV777. These results indicated that CH/HNQX-3/14 is a novel field strain with the characterization of attenuated and vaccine strains.
Fig. 3.

Amino acid sequences alignment, recombination sites, and location of epitope regions in the partial S protein of CH/HNQX-3/14 recombinant and the putative parental strains (CH/ZMDZY/11, DR13, and CV777). The shade regions of the S amino acid sequences marked by yellow, pink, and gray indicate the neutralizing epitope region A (aa 499–638), B (aa 748–755), and C (aa 764–771) reported previously [3], respectively. Recombination breakpoints are marked by black arrowhead (Color figure online)
To investigate thse evolutionary process by which CH/HNQX-3/14 hypothetically descended from precursors in Chinese historical strains, we conducted phylogenetic and recombination analysis based on the PEDV complete genome. Interestingly, the strain CH/HNQX-3/14 was grouped into the same subcluster with CH/ZMDZY/11) in phylogenetic tree, and it was nearest to the subcluster formed by three US strains (OH851, USA/Minnesota52/2013, and USA/Indiana12.83/2013) [26], as well as two Germany strains (L00721/GER/2014 and GER/L00719/2014). Further, the recombination analysis suggested that CH/HNQX-3/14 may be a recombinant strain derived from the CV777, DR13, and CH/ZMDZY/11. It was noted that the CV777 is a parental strain of PEDV vaccine being used in China. DR13, isolated from South Korean, is an attenuated PEDV strain derived from its virulent strain counterpart by serial propagation in Vero cells, which has been used as vaccine strain in Korean [27]. CH/ZMDZY/11 is a historical virulent PEDV strain isolated from Henan province, where CH/HNQX-3/14 is circulating [28]. Additionally, a retrospective survey found that, since 2011 outbreak, several large-scale transportations of pigs had happened between Henan and neighboring provinces including Anhui. Furthermore, it was surveyed that the CV777 vaccine, as well as DR13 vaccine from Korean, had been used in the swine farms CH/HNQX-3/14 circulating. We speculate that these factors may accelerate the combination between different PEDV strains and be critical to understand the role of recombination in a complex and dynamic PEDV evolution.
In summary, this study provides further information on the origin of the PEDV variants in China by identifying the full-length genome of a PEDV strain (CH/HNQX-3/14) from Henan province of China. Comparative genomic, phylogenetic, and recombination analysis using CH/HNQX-3/14 and known genome sequences demonstrated that the CH/HNQX-3/14 is a natural recombination between the vaccine and highly virulent PEDV strains in the field. Our findings add the knowledge on genetic diversity and newly emerging PEDV variants in China. Further studies will be required to determine the immunogenicity and pathogenicity of this recombinant PEDV strain.
Acknowledgments
We gratefully acknowledge the invaluable comments and suggestion on the manuscript of Dr Gregson, UCL-ION, London University. This work was supported by grants from China Agriculture Research System (No. CARS-36), and the Pig Industry Technology System Innovation Team Project of Henan Province (S2012-06).
Contributor Information
Songlin Qiao, Email: cdj565@gmail.com.
Gaiping Zhang, Email: zhanggaiping2003@163.com.
References
- 1.Pensaert MB, de Bouck P. Arch. Virol. 1978;58:243–247. doi: 10.1007/BF01317606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ge JW, Li BX, Tang LJ, Li YJ. Virus Genes. 2006;33:215–219. doi: 10.1007/s11262-005-0059-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song D, Park B. Virus Genes. 2012;44:167–175. doi: 10.1007/s11262-012-0713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park SJ, Moon HJ, Yang JS, Lee CS, Song DS, Kang BK, et al. Virus Genes. 2007;35:321–332. doi: 10.1007/s11262-007-0096-x. [DOI] [PubMed] [Google Scholar]
- 5.Oh J, Lee KW, Choi HW, Lee C. Arch. Virol. 2014;159:2977–2987. doi: 10.1007/s00705-014-2163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puranaveja S, Poolperm P, Lertwatcharasarakul P, Kesdaengsakonwut S, Boonsoongnern A, Urairong K, et al. Emerg. Infect. Dis. 2009;15:1112–1115. doi: 10.3201/eid1507.081256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hou XL, Yu LY, Liu J. Vet. Microbiol. 2007;123:86–92. doi: 10.1016/j.vetmic.2007.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang DQ, Ge FF, Ju HB, Wang J, Liu J, Ning K, et al. Arch. Virol. 2014;159:2777–2785. doi: 10.1007/s00705-014-2102-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bridgen A, Duarte M, Tobler K, Laude H, Ackermann M. J. Gen. Virol. 1993;74:1795–1804. doi: 10.1099/0022-1317-74-9-1795. [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Wang C, Shi H, Qiu H, Liu S, Chen X, et al. Arch. Virol. 2010;155:1471–1476. doi: 10.1007/s00705-010-0720-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang YW, Dickerman AW, Pineyro P, Li L, Fang L, Kiehne R, et al. mBio. 2013;4:e00737-13. doi: 10.1128/mBio.00737-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Theuns S, Conceição-Neto N, Christiaens I, Zeller M, Desmarets LMB, Roukaerts IDM, et al. Genome Announc. 2015;3:e00506–e00515. doi: 10.1128/genomeA.00506-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grasland B, Bigault L, Bernard C, Quenault H, Toulouse O, Fablet C, et al. Genome Announc. 2015;3:e00535-15. doi: 10.1128/genomeA.00535-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xuan H, Xing D, Wang D, Zhu W, Zhao F, Gong H. Chin. J. Vet. Sci. 1984;4:202–208. [Google Scholar]
- 15.Gao Y, Kou Q, Ge X, Zhou L, Guo X, Yang H. Arch. Virol. 2013;158:711–715. doi: 10.1007/s00705-012-1541-2. [DOI] [PubMed] [Google Scholar]
- 16.Li R, Qiao S, Yang Y, Su Y, Zhao P, Zhou E, et al. Arch. Virol. 2014;159:1057–1065. doi: 10.1007/s00705-013-1929-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan Y, Tian X, Li W, Zhou Q, Wang D, Bi Y, et al. Virol. J. 2012;9:195. doi: 10.1186/1743-422X-9-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Bioinformatics (Oxford, England) 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 19.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin D, Rybicki E. Bioinformatics (Oxford, England) 2000;16:562–563. doi: 10.1093/bioinformatics/16.6.562. [DOI] [PubMed] [Google Scholar]
- 21.Li W, Li H, Liu Y, Pan Y, Deng F, Song Y, et al. Emerg. Infect. Dis. 2012;18:1350–1353. doi: 10.3201/eid1803.120002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith SE, Showers-Corneli P, Dardenne CN, Harpending HH, Martin DP, Beiko RG. PLoS One. 2012;7:e50070. doi: 10.1371/journal.pone.0050070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tian PF, Jin YL, Xing G, Qv LL, Huang YW, Zhou JY. Emerg. Infect. Dis. 2014;20:1735–1738. doi: 10.3201/eid2010.140338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang XM, Niu BB, Yan H, Gao DS, Yang X, Chen L, et al. Arch. Virol. 2013;158:2487–2494. doi: 10.1007/s00705-013-1767-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li B, Liu H, He K, Guo R, Ni Y, Du L, et al. Genome Announc. 2013;1:e0010513. doi: 10.1128/genomeA.00105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Byrum B, Zhang Y. Emerg. Infect. Dis. 2014;20:917–919. doi: 10.3201/eid2005.140195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park SJ, Kim HK, Song DS, An DJ, Park BK. J. Virol. 2012;86:5964. doi: 10.1128/JVI.00557-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang XM, Niu BB, Yan H, Gao DS, Huo JY, Chen L, et al. Genome Announc. 2013;1:e00243-12. doi: 10.1128/genomeA.00243-12. [DOI] [PMC free article] [PubMed] [Google Scholar]