

Abstract
The filoviruses, Ebola virus (EBOV), and Marburg virus (MARV), are among the most pathogenic viruses known to man and the causative agents of viral hemorrhagic fever outbreaks in Africa with case fatality rates of up to 90%. Nearly 30,000 infections were observed in the latest EBOV epidemic in West Africa; previous outbreaks were much smaller, typically only affecting less than a few hundred people. Compared to other diseases such as AIDS or Malaria with millions of cases annually, filovirus hemorrhagic fever (FHF) is one of the neglected infectious diseases. There are no licensed vaccines or therapeutics available to treat EBOV and MARV infections; therefore, these pathogens can only be handled in maximum containment laboratories and are classified as select agents. Under these limitations, a very few laboratories worldwide conducted basic research and countermeasure development for EBOV and MARV since their respective discoveries in 1967 (MARV) and 1976 (EBOV). In this review, we discuss several vaccine platforms against EBOV and MARV, which have been assessed for their protective efficacy in animal models of FHF. The focus is on the most promising approaches, which were accelerated in clinical development (phase I–III trials) during the EBOV epidemic in West Africa.
Keywords: Ebola virus, Marburg virus, Filoviruses, Vaccines, Animal models
Introduction
The discovery of two negative-strand RNA viruses, Marburg virus (MARV) and Ebola virus (EBOV), in 1967 and 1976 [1], respectively, has marked the beginning of an era of Marburg and Ebola hemorrhagic fever outbreaks (MHF and EHF, respectively) in Africa, in both humans and nonhuman primates. Both viruses are members of the Filoviridae family in the order of Mononegavirales and are classified as category A pathogens and select agents. With no licensed vaccine or treatment available for human use, these pathogens can only be handled in maximum containment laboratories, which impedes research and countermeasure development [1]. Ebolavirus is composed of five known species: Zaire ebolavirus, Sudan ebolavirus, Taï Forest ebolavirus, Bundibugyo ebolavirus, and Reston ebolavirus with some of them known for causing disease with up to 90% case fatality rates [1]. In contrast, only one species has been discovered within the genus Marburgvirus, the Marburg marburgvirus, since its initial discovery in Marburg, Germany represented by Marburg virus (MARV) and Ravn virus (RAVV). In 1967, several laboratory workers presented with flu-like symptoms that later developed into severe HF causing several of the infected individuals to die [1]. Despite more than 40 years of filovirus research, no licensed vaccine or postexposure treatment exists for either of these deadly viruses [2].
Several vaccination strategies have been developed offering complete protection from lethal challenge with either EBOV or MARV in nonhuman primates (NHPs) and rodent models [2]. While rodent models provide some insight into the efficacies of filovirus vaccination, the ‘gold standard’ model for both EHF and MHF is the rhesus and cynomolgus macaque. The macaque model displays the majority of disease hallmarks of human EHF and MHF that rodent models can only offer with limitations [3]. Considering the invaluable resource the macaque model provides as a surrogate for human EHF and MHF, the experimental vaccine strategies analyzed in this nonhuman primate model are the focus of this review (Tables 1, 2). Unless otherwise specified, NHPs were always challenged with EBOV or MARV homologous to the vaccine-expressed antigen. Other vaccine platforms with reported efficacy in rodent models (mouse, guinea pig, and hamster) are listed in Tables 3, 4, but will not be closely reviewed here.
Table 1.
Efficacy of EBOV vaccines in NHPs
| Vaccine | Challenge virus | Vaccine doses | Time to challenge (d)a | Survival (%) | Ref. |
|---|---|---|---|---|---|
| Whole virus | |||||
| inact. EBOV | EBOV | 3 | 43 | 25 | [5] |
| inact. EBOV | EBOV | 2 | 56 | 0 | [13] |
| EBOVΔVP30 | EBOV | 1, 2 | 28 | 100 | [13] |
| VLPs | |||||
| eVLPs + RIBI | EBOV | 3 | 28 | 100 | [6] |
| VLPs + QS21 | EBOV, SUDV, TAFV | 1, 2 | 28 | 67-100 | [69] |
| DNA | |||||
| EBOV GP, Filo GPs | EBOV | 3 | 56 | 83 | [31] |
| DNA + rec. Adenovirus | |||||
| DNA EBOV GP, EBOV NP + rAd5-EBOV GP | EBOV | 4 | 90 | 100 | [29] |
| DNA EBOV GP + rAd5-EBOV GP | BDBV | 5 | 49 | 100 | [70] |
| Replicon | |||||
| VEEV-EBOV GP, VEEV-EBOV NP, both | EBOV | 3 | 49 | 0 | [5] |
| VEEV-SUDV GP + VEEV-EBOV GP | SUDV, EBOV | 1 | 28, 58/63 | 100 | [27] |
| rec. Adenovirus | |||||
| rAd5-GP + rAd5-NP | EBOV | 1, 2 | 7, 28 | 100 | [71] |
| EBOV GP + NP, EBOV GPΔTM + NP | EBOV | 1 | 28 | 100 | [72] |
| CAdVax-filo GP + NP | EBOV, SUDV | 2 | 42, 114 | 100 | [40] |
| CAdVax-EBOV M7 + M8 | EBOV, SUDV | 1, 2 | 7, 28, 41, 442, 113 | 100 | [73] |
| rAd26-GP + rAd35-GP | EBOV | 2 | 28 | 100 | [36] |
| rAd5-GP | EBOV | 1 | 28 | 100 | [34, 74] |
| Ad-CAGoptZGP + Ad-IFNα | EBOV | 1 | 28 | 100 | [39] |
| ChAd63-EBOV | EBOV | 1 | 35 | 100 | [41] |
| ChAd63-EBOV + MVA-BN-filo | EBOV | 2 | 240 | 100 | [41] |
| Ad-CAGoptZGP | EBOV | 1 | 150 | 100 | [75] |
| rec. Vaccinia virus | |||||
| VACV-EBOV GP | EBOV | 3 | 45 | 0 | [5] |
| rec. Cytomegalovirus | |||||
| RhCMV/EBOV GP | EBOV | 2 | 28 | 75 | [47] |
| rec. Human parainfluenza virus | |||||
| HPIV3-EBOV GP + NP, HPIV3-EBOV GP + GM-CSF | EBOV | 1, 2 | 28 | 67–100 | [52] |
| HPIV3/EBOV GP | EBOV | 1, 2 | 27 | 100 | [54] |
| rec. Rabies virus | |||||
| BNSP(333)-GP, BNSP(333)ΔG/GP, inact. BNSP(333)-GP | EBOV | 1, 2 | 42 | 100 | [48] |
| inact. BNSP333-coEBOVGP + adjuvants | EBOV | 2 | 28 | 100 | [50] |
| FILORAB1/GLA-SE | EBOV | 2, 3 | 57, 43 | 100 | [76] |
| rec. Vesicular stomatitis virus | |||||
| VSV-EBOV | EBOV | 1 | 28 | 100 | [59, 61, 63, 77, 78] |
| VSV-EBOV, VSV-SUDV, VSV-MARV | EBOV, SUDV, TAFV | 1 | 28 | 100 | [60] |
| VSV-EBOV | EBOV | 1 | 7-28 | 100 | [65] |
| Vesiculovax-rVSV-EBOV GP | EBOV | 1 | 28 | 100 | [58] |
| Vesiculovax-rVSV-EBOV GP | EBOV | 1 | 28 | 100 | [66] |
aTime until challenge after vaccination was completed
Table 2.
Efficacy of MARV vaccines in NHPs
| Vaccine | Challenge virus | Vaccine doses | Time to challenge (d)a | Survival (%) | Ref. |
|---|---|---|---|---|---|
| Whole virus | |||||
| inact. MARV | MARV Popp | 2 | 21 | 50 | [8] |
| VLPs | |||||
| VLPs + RIBI | MARV Musoke, Ci67, RAVV | 3 | 28 | 100 | [19] |
| mVLPs + adjuvant | MARV Musoke | 3 | 28 | 100 | [20] |
| DNA | |||||
| MARV GP | MARV Musoke | 3 | 28 | 67 | [32] |
| MARV GP | MARV Angola | 4 | 42 | 100 | [33] |
| MARV GP, RAVV GP, EBOV GP, SUDV GP | MARV Musoke | 3 | 56 | 100 | [31] |
| DNA + rec. Adenovirus | |||||
| DNA MARV GP, rAD5-MARV GP | MARV Angola | 4 | 21 | 100 | [33] |
| Replicon | |||||
| VEEV-MARV GP, VEEV-MARV NP, both | MARV Musoke | 3 | 35 | 67–100 | [25] |
| rec. Adenovirus | |||||
| CAdVax-panFilo | MARV Musoke, Ci67 | 2 | 42, 112 | 100 | [40] |
| rAD5-MARV GP | MARV Angola | 1 | 28 | 100 | [33] |
| rec. Vesicular stomatitis virus | |||||
| VSV-MARV | MARV Musoke, Popp | 1 | 28, 113 | 100 | [59] |
| VSV-MARV | MARV Musoke, Angola, RAVV | 1 | 28 | 100 | [79] |
| VSV-MARV | MARV Angola | 1 | 28 | 100 | [77] |
| VSV-EBOV, VSV-SUDV, VSV-MARV | MARV Musoke | 1 | 28, 59 | 100 | [60] |
| VSV-MARV | MARV Musoke | 1 | 407 | 100 | [80] |
aTime until challenge after vaccination was completed
Table 3.
Efficacy of EBOV vaccines in rodents
| Vaccine | Species | Challenge virus | Vaccine doses | Time to challenge (d)a | Survival (%) | Ref. |
|---|---|---|---|---|---|---|
| Whole virus | ||||||
| inact. EBOV (formalin, heat) | Guinea pig | EBOV | 1, 2 | 21 | 100 | [4] |
| inact. EBOV | Mouse | MA-EBOV | 3 | 42 | 25 | [81] |
| INA-inact. MA-EBOV | Mouse | MA-EBOV | 1 | 3 | 100 | [7] |
| EBOVΔVP30 | Mouse | MA-EBOV | 2 | 56 | 100 | [12] |
| EBOVΔVP30 | Guinea pig | GPA-EBOV | 2 | 42 | 100 | [12] |
| VLPs | ||||||
| eVLP | Mouse | MA-EBOV | 3 | 42 | 100 | [81] |
| eVLP | Guinea pig | GPA-EBOV | 1 | 28 | 100 | [22] |
| eVLP + QS21 | Mouse | MA-EBOV | 2 | 42 | 100 | [82] |
| eVLP, eVLPs + QS21 | Mouse | MA-EBOV | 2 | 28 | 100 | [7, 101] |
| eVLP + poly-ICLC | Guinea pig | GPA-EBOV | 2 | 28 | 100 | [23] |
| Subunit | ||||||
| EBOV GP-Fc + Freund’s adjuvant | Mouse | MA-EBOV | 4 | 14 | 88 | [9] |
| EBOV GP-Fc + poly-ICLC | Guinea pig | GPA-EBOV | 4 | 14 | 100 | [10] |
| EBOV GP + adjuvant | Mouse | MA-EBOV | 3 | 30 | 100 | [11] |
| EBOV GP + VP24 + VP40 + adjuvant | Mouse | MA-EBOV | 3 | 30 | 100 | [11] |
| DNA | ||||||
| EBOV GP | Mouse | MA-EBOV | 1, 3, 4 | 28 | 100 | [21, 84] |
| EBOV GP, EBOV NP, both | Guinea pig | GPA-EBOV | 3 | 90 | 100 | [29] |
| EBOV GP | Guinea pig | GPA-EBOV | 3 | 91 | 67 | [32] |
| EBOV GP, SUDV GP, MARV GP | Mouse | MA-EBOV | 2, 3 | 28 | 100 | [83] |
| EBOV GP | Guinea pig | GPA-EBOV | 2 | 28 | 100 | [84] |
| EBOV GP | Mouse | MA-EBOV | 1, 2 | 28 | 60–63 | [85] |
| DNA + rec. Adenovirus | ||||||
| DNA-EBOV GP, rAD5-EBOV GP | Mouse | MA-EBOV | 4 | 84 | n/a | [71] |
| DNA-EBOV GP, rAD5-EBOV GP | Mouse | MA-EBOV | 2 | 28 | 88–100 | [85] |
| DNA-EBOV GP, AAV-po6-EBOV GP | Mouse | MA-EBOV | 2 | 28 | 25–75 | [85] |
| Replicons | ||||||
| VEEV-EBOV GP, VEEV-EBOV NP, both | Mouse | MA-EBOV | 2, 4 | 28 | 90–100 | [24] |
| VEEV-EBOV GP, VEEV-EBOV NP, both | Guinea pig | GPA-EBOV | 2, 3 | 28 | 40–100 | [24] |
| VEEV-EBOV GP | Guinea pig | GPA-EBOV | 3 | 28 | 100 | [24] |
| VEEV-EBOV VP24,30,35,40 | Mouse | MA-EBOV | 2, 3 | 28 | 0–95 | [26] |
| VEEV-EBOV NP | Mouse | MA-EBOV | 2, 3 | 28 | 75–80 | [86] |
| Kunjin virus—EBOV GP | Guinea pig | GPA-EBOV | 2 | 20 | 86 | [87] |
| rec. Adenovirus | ||||||
| Ad5-EBOV GP | Mouse | MA-EBOV | 1, 2 | 28, 7 | n/a | [71] |
| Ad5-EBOV GP | Guinea pig | GPA-EBOV | 1 | 28, 7 | 100 | [34] |
| simian AdC5/C1-EBOV GP | Mouse | MA-EBOV | 1 | 21 | 100 | [35] |
| CAdVax-EBOV GP | Mouse | MA-EBOV | 2 | 30 | 100 | [35] |
| Ad5-EBOV GP | Mouse | MA-EBOV | 1 | 28 | 100 | [88] |
| AdCMV-EBOV GP, CAGopt-EBOV GP | Mouse | MA-EBOV | 1 | 28 | 100 | [64, 89] |
| AdCMV-EBOV GP, CAGopt-EBOV GP | Guinea pig | GPA-EBOV | 1 | 28 | 100 | [64, 90] |
| Ad5-EBOV GP | Mouse | MA-EBOV | 1, 2 | 28 | 38–90 | [85] |
| AAV-po6-EBOV GP | Mouse | MA-EBOV | 1, 2 | 28 | 38–100 | [85] |
| AAV-po6-EBOV GP, Ad5-EBOV GP | Mouse | MA-EBOV | 2 | 28 | 0–75 | [85] |
| rec. Vaccinia virus | ||||||
| VACV-EBOV GP | Guinea pig | GPA-EBOV | 1 | 30 | 60 | [91] |
| rec. Cytomegalovirus | ||||||
| CMV-NPctl | Mouse | MA-EBOV | 2 | 42 | 100 | [45] |
| MCMV/ZEBOV-NPctl | Mouse | MA-EBOV | 1 | 119 | 100 | [46] |
| rec. Human parainfluenza virus | ||||||
| HPIV3-GP, HPIV3GP-NP | Guinea pig | GPA-EBOV | 1 | 28 | 100 | [92] |
| HPIV3/ΔF-HN/GP, HPIV3-GP | Guinea pig | GPA-EBOV | 1 | 25 | 100 | [93] |
| rec. Rabies virus | ||||||
| BNSP(333)ΔG/GP, inact. BNSP(333)-GP | Mouse | MA-EBOV | 1 | 77 | 100 | [48] |
| rec. Vesicular stomatitis virus | ||||||
| VSV-EBOV | Mouse | MA-EBOV | 2 | 14 | 100 | [57] |
| VSV-EBOV | Mouse | MA-EBOV | 1 | 1 | 100 | [94] |
| VSV-EBOV | Guinea pig | GPA-EBOV | 1 | 1 | 67 | [7] |
| VSV-EBOV | Mouse | MA-EBOV | 1 | 28 | 100 | [95] |
| VSV-EBOV, VSV-SUDV | Guinea pig | GPA-EBOV | 1 | 21 | 100 | [96] |
| VSV-EBOV/ANDV | Hamster | MA-EBOV | 1 | 14, 7, 3 | 100 | [97] |
| VSV-EBOV | Mouse | MA-EBOV | 1 | 6.5, 12, 18 months | 80–100 | [98] |
| VSV-EBOV | Guinea pig | GPA-EBOV | 1 | 7, 12, 18 months | 83–100 | [98] |
| Vesiculovax-rVSV-EBOV GP | Guinea pig | GPA-EBOV | 2 | 21 days | 100 | [58] |
aTime until challenge after vaccination was completed
Table 4.
Efficacy of MARV vaccines in rodents
| Vaccine | Species | Challenge virus | Vaccine doses | Time to challenge (d)a | Survival (%) | Ref. |
|---|---|---|---|---|---|---|
| Whole virus | ||||||
| inact. MARV, RAVV | Guinea Pig | MARV Musoke, RAVV | 2 | 14 | 100 | [7] |
| inact. MARV | Guinea Pig | GPA-MARV | 3 | 30 | 100 | [99] |
| inact. MARV + RIBI | Guinea Pig | GPA-MARV, RAVV | 3 | 30 | 100 | [19] |
| VLPs | ||||||
| mVLPs | Guinea pig | GPA-MARV | 3 | 30 | 100 | [99] |
| eVLP + mVLP or m/eVLP | Guinea pig | GPA-MARV | 1 | 28 | 100 | [22] |
| VLPs + RIBI | Guinea pig | GPA-MARV, RAVV | 3 | 30 | 100 | [19] |
| mVLP + poly-ICLC | Guinea pig | GPA-MARV | 2 | 28 | 100 | 23 |
| Subunit | ||||||
| MARV GPΔTM | Guinea Pig | GPA-MARV, RAVV | 2 | 14 | 80–100 | [7] |
| DNA | ||||||
| MARV GP, RAVV GP | Guinea Pig | GPA-MARV, RAVV | 3, 4 | 28 | 100 | [32] |
| MARV GP, RAVV GP | mouse | MA-RAVV | 2 | 28 | 100 | [83] |
| MARV GP | Guinea Pig | GPA-MARV | 2 | 28 | 100 | [84] |
| Replicon | ||||||
| VEEV-MARV GP, NP, GPΔTM, VP35 | Guinea Pig | GPA-MARV | 1, 2, 3 | 28 | 83–100 | [25] |
| rec. Adenovirus | ||||||
| cAdVax-MARV GP | Guinea Pig | MARV Musoke, Ci67, RAVV | 2 | 28 | 67–100 | [100] |
aTime until challenge after vaccination was completed
Replication-incompetent vaccines
Inactivated virus and subunit vaccines
The first vaccine platform explored for EBOV was published in 1980 using both heat- and formalin-inactivated, whole-virus particles of the EBOV E-178 isolate [4]. Both inactivation methods resulted in protection of guinea pigs from lethal challenge; however, another study using gamma-irradiated, inactivated viral particles (with and without liposome encapsulation) was unsuccessful in providing protection in NHPs [5]. In addition to conventional methods, other inactivation treatments such as the photoinduced alkylation probe 1,5-iodonaphthylazide (INA) have shown similar success in the EBOV rodent models, but have not been evaluated in NHPs [6].
Similarly, successful protection of guinea pigs with irradiated, whole MARV, both strains Musoke and RAVV, has been demonstrated [7], but limited data have been shown to be efficacious in NHPs for the inactivated virus vaccine platform [8]. While promising data for inactivated, whole-virus vaccines have been reported in filovirus rodent models, the limited immunogenicity in NHPs vaccinated by this strategy has not justified further NHP studies [3].
Limited data describing the use of subunit vaccines produced from recombinant baculovirus systems have been published. Experimental data determining the efficacy for a MARV Musoke glycoprotein (GP) variant lacking its transmembrane domain (GPΔTM) produced by recombinant baculovirus were used to determine efficacy in guinea pigs [7]. When guinea pigs were vaccinated with 0.5 µg/dose of GPΔTM and lethally challenged with either MARV Musoke or RAVV, only 4/5 and 0/5 guinea pigs survived, respectively [4]. With such limited efficacy for protection from MHF, subunit baculovirus vaccines have not been further developed.
Recently, the extracellular domain of EBOV GP fused to the fc fragment of human IgG1 was tested in mice and guinea pigs for protection against lethal challenge [9, 10]. While this vaccine showed only partial protection in mice [9], when guinea pigs were immunized with the vaccine plus adjuvant, up to 100% protection could be observed [10]. Another group reported protective efficacy of EBOV GP with adjuvant or GP, VP24, and VP40 in combination and with adjuvant in mice [11]. These are promising results for these safe and easy-to-produce vaccination strategies and hopefully efficacy will soon be evaluated in the EBOV NHP model.
EBOVΔVP30
A promising vaccine strategy recently developed by Halfmann, Marzi, and colleagues uses a replication-incompetent, whole EBOV vaccine completely protective in mice, guinea pigs, and cynomolgus macaques from EBOV challenge [12, 13]. The vaccine, EBOVΔVP30, is a replication-incompetent, nearly whole-virus vaccine derived from the Mayinga strain of EBOV, where the coding region for the viral transcription activator VP30 has been deleted [14]. The recombinant virus is only able to replicate in cells expressing VP30, rendering the virus unable to propagate in the absence of VP30 [15].
Despite convincing data showing no recombination events or mutations surrounding the VP30 region after seven serial passages on VeroVP30 cells [14], and its inability to replicate in regular cells (not expressing VP30) and rodents [12], there are still concerns about the vaccine’s safety. To address these apprehensions, EBOVΔVP30 particles were subjected to hydrogen peroxide (H2O2) inactivation. H2O2 has been shown to successfully inactivate infectivity while retaining antigenicity for several viruses such as vaccinia virus [16], influenza virus [17], West Nile virus [16], and lymphocytic choriomeningitis [16, 18]. EBOVΔVP30 was shown to be completely inactivated by this method, which was confirmed by plaque assay in VP30-expressing cells [13].
The EBOVΔVP30 vaccine was able to fully protect cynomolgus macaques from lethal challenge when administered with a prime/boost vaccination using 107 focus-forming units (FFU) 4 weeks apart in both the EBOVΔVP30 and H2O2-inactivated groups. When only one administration of 107 FFU of EBOVΔVP30 was given to NHPs, protection from lethal challenge was achieved, yet viremia and clinical signs of illness (fever) were observed [13]. This nearly whole-virus vaccine offers an interesting approach to vaccination for EBOV. The viral RNA and the majority of viral proteins (with the exception of VP30) are presented to contribute to a more complete EBOV-specific immune response [15]. The retention of antigenicity through H2O2 inactivation, in addition to the replication deficiency allows for the EBOVΔVP30 to be manufactured in BSL-3 conditions, which is an additional benefit to its platform yet not as attractive as BSL-2 production [13].
Virus-like particles
Virus-like particles (VLPs) have been assessed as an immunization approach for both EBOV and MARV. VLPs offered homologous protection in nonhuman primates, with selected studies producing broad protection to several strains of MARV. For both EBOV and MARV, VLPs were produced by expression of just two viral genes, the matrix protein VP40, as well as the respective GP in either mammalian or insect cells [6, 19]. Marburg Musoke VLPs (mVLPs) [19] were able to provide protection against three strains of MARV, MARV Musoke, MARV Ci67 and RAVV, in rodents and NHPs. A more advanced version of these VLPs was produced by the addition of the nucleoprotein (NP). These three-component VLPs were tested in cynomolgus macaques, where after three intramuscular injections of 1 mg mVLPs with 0.1 ml of QS-21 adjuvant in 42-day intervals, all animals were protected from lethal challenge with 1000 plaque-forming units (PFU) of MARV Musoke, MARV Ci67, or RAVV [19]. More recently, Dye and colleagues showed that mVLPs with QS-21 or poly I:C adjuvant are also protective against aerosol challenge with MARV in NHPs [20]. Generation of broadly protective immunity against several diverse MARV strains through a VLP system offers a promising candidate for a vaccine in humans.
Similar results have been achieved for the EBOV vaccine, when using VLPs consisting of VP40, NP, and GP. While the only essential components to produce Ebola VLPs (eVLPs) are VP40 and GP, NP is often added to eVLP productions as several studies have shown that NP-specific antibodies can protect rodents from lethal EBOV challenge [21]. Warfield and colleagues achieved complete protection from lethal EBOV challenge of 1000 PFU in NHPs when three intramuscular injections spaced 42 days apart were administered with only 250 µg of eVLPs including 0.5 mL of RIBI adjuvant [6] After challenge, all animals survived, and no clinical signs were observed; in addition, no viremia could be detected at any time point by plaque assay [6]. In this study, the adjuvant greatly reduced the necessity to provide higher doses of eVLPs [22].
While Warfield and colleagues have shown encouraging results in EBOV and MARV vaccination in NHPs using both mammalian and insect cell-derived VLPs, attempts to produce a pan-filovirus vaccine were unable to provide consistent protection to heterologous challenge [22]. Attempts to produce hybrid VLPs containing the GP from both MARV and EBOV were not successful in providing protection from lethal challenge of either virus. For the hybrid VLPs, GP but not VP40 was important to provide protection from homologous virus challenge [19, 23]. However, a boosting strategy increased the heterologous antibody response, and the administration of additional doses of the VLP mixture may be able to provide an efficacious pan-filovirus vaccine.
Considering the fairly easy production of VLPs in both mammalian and insect cell lines, the impressive safety profile, along with similar morphology and antigenicity to wild-type viruses, the absence of pre-existing antivector immunity, and the extensive research shown on their ability to produce innate, humoral, and cellular immunity, all while being shown safely administered in humans [6], VLPs are a promising vaccine platform to prevent FHF outbreaks.
Virus-like replicon particles (VRPs)
Similar to the VLP system, virus-like replicon particle (VRP) systems are typically generated using either a flavivirus or an alphavirus vector to generate replication-incompetent particles through the deletion of the key structural proteins to limit the infection of VRPs to one cycle [24]. A majority of research concerning VRP vaccination against EBOV or MARV infections has been focused on using the Venezuelan equine encephalitis virus (VEEV), an alphavirus vector. Replicon systems such as VEEV are used to produce a protein of interest (antigen) in place of the structural proteins for the replicon vector, thus generating a self-replicating RNA molecule, which additionally encodes proteins necessary for RNA replication [25]. The resulting VRPs can infect cells for one cycle. While the absence of VEEV structural proteins inhibits production of progeny virus, the infected cell produces ample amounts of the desired protein [25]. For both MARV and EBOV, the antigen inserted into the VRP is typically the GP, which is the target of the majority of neutralizing antibodies during filoviral infection. However, other EBOV proteins have been examined in the past as antigens using this system and it was found that, indeed, only the GP could provide complete protection in the context of the VRP system [26].
A VRP system based on the VEEV described by Hevey and colleagues [25] was one of the earlier vaccine platforms to show complete protection against MARV in NHPs. Subsequent testing of multiple replicons expressing various MARV proteins was first assessed in guinea pigs. Vaccinations with VRPs encoding the antigens MARV virion protein (VP) 40, VP35, VP30, VP24, as well as GPΔTM resulted in partial protection, but not to the degree of efficacy as GP and NP [25]. In a similar study, VRPs expressing EBOV VP24, VP30, VP35, and VP40 were tested in comparison to EBOV GP and NP in mice, but again only EBOV GP and NP antigens could confer protection.
The promising results with the VEEV replicons expressing GP or NP warranted further efficacy studies using the NHP model. Cynomolgus macaques were exposed to 10-fold higher doses of replicons expressing GP, NP, or a combination of both. After the same three-dose vaccination schedule, animals were challenged with 8000 PFU of MARV Musoke, wherein all animals vaccinated with VRP-GP alone or in combination with VRP-NP, survived with minimal clinical signs of disease. VRP-NP by itself was not nearly as efficacious, as one of three NHPs succumbed to illness, with the two survivors showing clinical symptoms ranging from mild to severe disease [25].
In 2013, Herbert and colleagues published promising results using a single dose of VEEV replicons in NHPs [27]. This updated vaccine was bivalent for both EBOV and Sudan virus (SUDV), another species in the genus Ebolavirus. In this study, six cynomolgus macaques were intramuscularly injected with a combination of 1010 FFU of VRP-SUDV Boniface GP and VRP-EBOV Kikwit GP. Two groups of three monkeys were then challenged with either 1000 PFU of either EBOV or SUDV. All vaccinated macaques showed no clinical signs of disease and were protected from initial challenge with EBOV or SUDV, respectively. All six surviving animals were then back-challenged with heterologous virus at 1000 PFU 28 or 30 days after initial lethal viral challenge. All NHPs back-challenged with EBOV survived without developing any clinical signs of disease. However, the animals back-challenged with SUDV developed disease and only 67% of the animals survived. The data presented by Herbert and colleagues demonstrate the ability for VRP-EBOV GP to confer cross-protection to SUDV, while VRP-SUDV GP is insufficient in providing complete protection to EBOV challenge [27]. The ability for this VRP platform to cross-protect against SUDV and EBOV is promising, as these two viruses have caused the largest number of human Ebolavirus infections [28].
DNA vaccines
One of the first successful vaccination strategies to provide full protection in NHPs was a DNA vaccine in conjunction with a recombinant adenovirus vector [29], even progressing into a phase I clinical trial [30]. DNA vaccines have several benefits; they are safe to use, easy to produce, the host–cell protein synthesis allows for endogenous presentation of the desired antigen, and finally, the DNA itself provided in the vaccine can induce immune-stimulatory responses [29].
Previous studies using DNA vaccines expressing the EBOV GP in rodents resulted in both humoral and cell-mediated immune responses with a correlation of survival from lethal challenge and antibody titer [29]. Underlining the importance of the humoral immune response in protection from EBOV challenge, the immunogenicity of DNA vaccines was improved using a DNA prime, followed by a viral vector boost [29]. In a preliminary mouse study, priming with DNA and boosting with a replication-incompetent adenoviral (Ad) vector expressing EBOV GP (Ad-GP(Z)) increased antibody titers about 10–100 fold, compared to DNA vaccination alone [29]. These results were followed up by a NHP study for which the animals were immunized with three multicomponent DNA injections four weeks apart, followed by an additional Ad-GP(Z) boost several months later. Infection with a lethal dose of EBOV occurred three months after the final vaccination. All vaccinated macaques survived challenge showed no clinical signs of disease, and had no detectable viremia even when examined more than six months after challenge [29]. This encouraging result led to a phase I clinical trial, in which all adults [21] who completed the vaccination schedule showed T-cell responses as well as antibodies directed to EBOV GP and NP antigens; the vaccination was well tolerated and deemed safe. A more detailed report of the trial can be found elsewhere [30]. Recently, a novel approach using codon-optimized DNA plasmids and electroporation for vaccination has shown 67% survival by itself in NHPs [31].
Two separate experiments described the efficacy of DNA vaccination against MARV infection using expression plasmids for MARV GP. NHPs were vaccinated three times at one-month intervals with DNA expressing either MARV Musoke GP. Only partial protection was achieved after lethal challenge with MARV as 33% of the animals succumbed to MHF. All animals showed clinical signs of disease at seven and ten days after challenge. While this study was the first to report efficacy data for a vaccine against MARV challenge in NHPs [32], the lack of complete protection from disease leaves room for improvement for this vaccine.
A study published several years later showed greater efficacy to protect against MARV challenge in NHPs using DNA vaccinations in combination with recombinant Ad5 (rAd5) vectors [33]. Delivery and expression of DNA plasmids containing the MARV GP from the most recent MARV outbreak in Angola were improved by codon optimization and the addition of enhanced promotor regulatory elements described previously to increase the protein expression and, therefore, the immune response [33]. Vaccination with either DNA, DNA/rAd5 boost, or rAd5 alone all protected the animal from fatal disease after challenge with a lethal dose of MARV Angola. While none of the animals developed MARV Angola viremia, the DNA/rAd5 combination group as well DNA only group developed mild clinical signs such as rash, lymphocyte depletion, and anorexia [33]. The results described by Geisbert and colleagues provide insight into the importance of both CD4 and CD8 T-cell responses; a qualitative difference in T-cell responses seems to correlate with protection of the NHPs from both mortality and morbidity using the DNA and/or rAd5 vaccine, whereas antigen-specific antibodies alone are not sufficient for protection from MHF [33].
Recombinant adenovirus vectors
A considerable amount of research has been conducted using recombinant Ad vectors as a vaccine strategy to prevent filovirus infections, as briefly mentioned above. The use of Ad-based vectors has had success in preventing disease and mortality from EBOV and MARV infection in NHPs, and several Ad vector strategies have been proposed in order to increase immunization potential. However, recently a group updated the previous rAd5-EBOV vaccine to express GP from the most recent outbreak strain EBOV-Makona and demonstrated its protective efficacy in guinea pigs and NHPs [34]. The concerns of pre-existing immunity related to the rAd5 vector were not addressed in this study.
In an effort to circumvent the pre-existing vector immunity to human Ad vectors, particularly human Ad5 vectors, which have been widely and successfully used in preclinical studies for EBOV vaccines [15], Roy and colleagues designed a chimeric Ad vector expressing two separate chimpanzee Ads, SAdV-21 and SAdV-22 [35]. While this vaccine was completely protective in mice against lethal challenge, only NHP immunogenicity data are presented in this publication. This chimeric chimpanzee vector did not yield the same magnitudes of CD8 T-cell activation in rhesus macaques compared to human Ad5 [35], and is therefore still in the development stage.
Another group attempted to bypass pre-existing immunity to human Ad5 vectors by selecting human Ad26 and Ad35 [36]. Ad35 has been shown to be one of the most scarcely neutralized adenoviruses by human serum samples collected from several distinct geographic regions; in contrast, Ad26 was selected for its ability to evade Ad5 pre-existing immunity. As proof-of-concept, Geisbert and colleagues demonstrated that the ADV-GP(Z) vector can no longer protect NHPs from lethal EBOV infection when these animals were exposed to Ad5 expressing no transgene prior to vaccination and challenge. Both recombinant Ad26-expressing EBOV GP (rAd26-GP) and rAd35-GP were tested individually to assess the potency of their immune responses. While both, rAd26-GP and rAd35-GP, induced neutralizing antibody responses after vaccination, neither vector alone could protect the NHPs from lethal challenge with EBOV. When the vaccine doses were increased to a range from 1010 to 1012 particles of either vector, partial survival was achieved. In a last experiment, NHPs were vaccinated with 1011 particles of each rAd26-GP(Z) and –GP(S) (SUDV Gulu), then boosted one month later with 1011 particles of rAd35-GP(Z & S). Antibody titers specific to GP as well as CD8 T-cell responses were significantly increased post-boost with rAd35 vectors, resulting in complete protection from mortality and morbidity after lethal EBOV challenge [36]. The rAd26-GP vector together with a Modified Vaccinia Ankara expressing filovirus antigens (MVA-BN-filo) was used in the UK in a phase I clinical trials and demonstrated good safety and immunogenicity with humoral immune responses lasting for over 12 months [37, 38].
Another group looked at intranasal/airway delivery of the human Ad5 vector to circumvent pre-existing immunity [39]. For this, a codon-optimized EBOV GP was expressed in a second generation Ad5 vector (Ad-CAGoptZGP) and delivered both intramuscularly (i.m.) and intranasally (i.n.)/intratracheally (i.t.) into either Ad5-naïve and Ad5-immune NHPs. 1010 IFU Ad-CAGoptZGP were used in combination with 109 PFU Ad-IFNα, an Ad vector expressing Interferon α (IFNα) acting as an antiviral and adjuvant [39]. When challenged with a lethal dose of EBOV, all Ad5-naïve i.m. vaccinated NHPs survived; in contrast, all Ad5-immune i.m. vaccinated NHPs succumbed to EBOV infection. The i.n./i.t. vaccination route did not fully protect Ad5-naïve NHPs (67% survival); however, 3 of the 4 Ad5-immune i.n./i.t. vaccinated NHPs were protected from lethal disease [39]. This is very encouraging data and demonstrates the importance of the route of vaccine administration for efficacy.
Swenson and colleagues developed a complex Ad (CAdVax) system expressing multiple antigens from both EBOV and MARV in an attempt to generate a pan-filovirus vaccine. The CAdVax system expresses the following filovirus antigens: EBOV GP, EBOV NP, SUDV GP, MARV NP, two MARV GPs (Ci67 and Musoke), and RAVV GP [40]. Filovirus NPs were incorporated into the system as they have been described to increase the cell-mediated immune responses [40]. Several constructs expressing different combinations of the antigens mentioned above were generated: EBO2, expressing two copies of EBOV NP; M8, expressing MARV GP Ci67 and RAVV GP; M11, containing MARV GP Musoke and MARV NP; EBO7, expressing SUDV GP and EBOV GP. NHPs were vaccinated twice (days 0 and 63) with 4 × 1010 PFU of the CAdVax-Panfilo vaccine (containing 1 × 1010 PFU of each EBO2, EBO7, M8, and M11) and separated in two groups. Group 1 was challenged with a lethal dose of MARV, whereas group 2 was infected with a lethal dose of EBOV. All NHPs in both groups survived without developing any signs of disease, including no detectable viremia. In order to assess the potency of this pan-filovirus vaccine, the two groups were back-challenged with a heterologous species of filovirus. Ten weeks after the initial MARV challenge, group 1 was challenged with a lethal dose of SUDV; in a similar manner, the EBOV survivors in group 2 were infected with a lethal dose of MARV. The back-challenge resulted again in 100% protection from disease and fatal outcome for all the animals. Several studies have showed that there are no cross-reactive antibodies between EBOV and MARV, indicating that indeed the pan-filovirus vaccine conferred protection against the different filovirus challenges [40].
The newest approach involving an Ad-based vector for protection from EBOV challenge was developed by Stanley and colleagues; this vaccine is based on chimpanzee Ad3 (ChAd3), expresses EBOV GP, and has shown complete protection from lethal EBOV infection in NHPs [41]. Without the limitations of pre-existing immunity by being a ChAd vector, this vaccine was accelerated for human phase I clinical trials during the 2013–2016 Ebola virus epidemic in West Africa. The vaccine was well tolerated in all participants and the antibody titers in human reached a similar level as observed in NHPs protected from EHF [42, 43]. The only concerning factor was the durability of the protective immune responses, as antibody titers in the vaccines started to drop by 6 months after vaccination [43] In order to circumvent this, a boost vaccination with a the MVA-BN-filo was implemented as this strategy had improved the immunogenicity and was previously shown to protect NHPs [41]. This strategy was first tested in a human phase I clinical trial in Mali with promising results [44], but no long-term durability data are available yet.
Replication-competent vaccine vectors
Recombinant vaccinia virus
A recombinant vaccinia virus (VACV) expressing EBOV GP was one of the first recombinant vaccine vectors tested for efficacy against EHF in NHPs [5]. Three NHPs were vaccinated with three doses of the VACV-GP and developed EBOV neutralizing antibodies. Challenge with 1000 PFU of EBOV at 45 days after the last vaccination resulted in all animals succumbing to disease at 7 days post challenge. Without protective efficacy in NHPs, this vaccine approach was no longer regarded as a viable vaccine option against EHF [5].
Recombinant cytomegalovirus
The cytomegalovirus (CMV)-based vaccine vector has been developed with the potential of being used as a wildlife immunization strategy. Like all herpesviruses, CMV is strictly species-specific and as a disseminating vaccine platform can spread from vaccinated animals to the entire population while establishing a persistent infection in the host. The strict species-specificity of CMV is of great safety benefit to this platform, as animal to human, or species to species transmission is very unlikely [15].
While EBOV NP has been shown to act as a protective antigen in the context of the mouse CMV vaccine in mice [45, 46], a proof-of-concept study in NHPs was needed to demonstrate the potential of this platform for wildlife vaccination, e.g., apes in Africa. Therefore, in a recent study, recombinant rhesus macaque CMV (RhCMV) expressing a codon-optimized EBOV GP (optZGP) was used in a prime/boost vaccination scheme in 4 rhesus macaques. Vaccination with RhCMV/EBOV GP was sufficient to protect 75% of the macaques from lethal EHF [47]. The surviving animals showed mild clinical signs of disease, and total antigen-specific IgG correlated with survival. Despite the lack of complete protection, this vaccine platform has a great potential as a disseminating wildlife vaccine among animal species that are thought to introduce EHF into the human population, e.g., bats and great apes [47].
Recombinant rabies virus
The development of a recombinant rabies virus (RABV) vaccine has provided yet another potential platform for the use in both human and animal species for protection from EHF. Blaney and colleagues have developed both replication-competent and -deficient RABV vectors expressing the EBOV GP as the antigen for protection from EHF. This vector was designed in hopes of being a bivalent vaccine protecting from both rabies and EHF. As RABV kills an estimated 24,000 people yearly in Africa, a vaccine that could protect from two life-threatening diseases in Africa is highly desirable. The RABV vectors are regarded as safe and efficacious vaccines providing immunity to HIV, SARS-CoV, and hepatitis C virus [48]. The parental RABV strain used to generate the recombinant vaccine, SAD B19, is attenuated and effective and has been used for wildlife vaccination [48]. Therefore, this platform seems to be a good approach to prevent RABV and EHF in both humans and at-risk animal populations [48].
Several RABV-based EBOV vaccine vectors have been shown to be protective in mice [49]. In a next step, three different RABV vectors were tested for efficacy against EHF in NHPs; single dose of BNSP333-GP (RABV with EBOV GP), single dose of BNSPΔG-GP (RABV with EBOV GP in place of its own GP), or two doses of inactivated BNSP333-GP particles. One dose of the replication-competent BNSP333-GP vector was able to fully protect all NHPs from lethal EHF when challenged 70 days after immunization. The two other RABV vectors only protected 50% of the NHPs from lethal EBOV infection [48]. The authors found that antibodies are critical for protection and they provided evidence that antibodies in the IgG1 class, in particular, seem to matter most.
In order to improve the protective efficacy of this platform, codon-optimized EBOV GP was inserted into BNSP333 to achieve increased antigen expression. When two doses of an inactivated version of this vaccine were administered with adjuvant to NHPs, all NHPs were protected from low dose challenge with 100 PFU EBOV [50]. Despite a challenge dose that is 10-fold lower than the majority of NHP challenge studies, the protective efficacy of this RABV vaccine platform has been encouraging. Recently, this vaccine was orally given to captive chimpanzees and resulted in good immunogenicity data underlining its potential use as a wildlife vaccine [51]. In addition, clinical trial grade vaccine production is currently ongoing and human clinical trials will hopefully start soon.
Recombinant paramyxoviruses
The human parainfluenza type 3 (HPIV3) recombinant vaccine vector, belonging to the Paramyxoviridae family, provides the ability for intranasal and topical respiratory tract vaccinations, which are effective for inducing both systemic and mucosal immune responses [52]. HPIV3 replicates within the superficial layer of the respiratory epithelium and does not typically disseminate past the respiratory tract. Applying an intranasal vaccination for a disease such as EHF is an intriguing concept, as an induced mucosal immunity would provide additional protection in the event of a bioterrorism attack [52].
A single-dose vaccination administered intranasal/intratracheal with recombinant HPIV3 vectors expressing EBOV GP, EBOV GP & NP, or EBOV GP with the human cytokine adjuvant GM-CSF partially protected NHPs from lethal EBOV challenge on 28 days post immunization [52]. One of the two animals vaccinated with HPIV3-EBOV GP & NP succumbed to disease 8 days post challenge; in contrast, all animals vaccinated with HPIV3-EBOV GP or HPIV3-EBOV GP + GM-CSF were protected from lethal disease, but mild symptoms such as fever were observed in each group [52]. A second study compared the protective efficacy of a single vs. two doses of the HPIV3-EBOV GP vaccine in NHPs. Two vaccination doses protected all NHPs from morbidity and mortality, whereas signs of disease and only 88% survival were recorded when only a single dose of the vaccine was administered [52]. Recently, another group developed a similar HIPV1-based product expressing EBOV GP for intranasal application and showed immunogenicity in African green monkeys; however, no challenge data are provided [53].
Furthermore, an aerosolized HPIV3-EBOV GP vaccine was tested and was shown to provide full protection from lethal EHF with a single-dose vaccination when challenged 28 days post immunization [54]. The induction of a strong, systemic IgG, and IgA response specific to EBOV GP marks the HPIV3 vaccine platform an effective recombinant vector for protection against EHF. Furthermore, CD8 T-cell responses were induced primarily in the lung and seemingly contributed to protection from lethal EHF. Protection against the EBOV infection through aerosolized vaccination represents a noninvasive way of efficient vaccine delivery and would add further to the reduction of outbreak potential as well as a preventative immunization from natural or bioterrorism threats.
Recombinant vesicular stomatitis virus
One of the most promising vaccination strategies against both EHF and MHF is based on recombinant vesicular stomatitis virus (rVSV). Similar to RABV, VSV belongs to the Rhabdoviridae family, and contains a negative-strand RNA genome encoding five proteins. Rose and colleagues developed the reverse genetics system for VSV allowing easy manipulation of the virus leading to optimal expression of antigens from this vector [55]. The ability for VSV to grow to very high titers in vivo, as well as in vitro in most mammalian cell lines makes it an easy-to-produce vaccine. Additionally, VSV has been shown to be a strong inducer of innate and adaptive immune responses, both advantageous properties of a vaccine vector [56]. VSV naturally infects livestock and various animals, making infection in humans very rare and disease often asymptomatic resulting in very limited pre-existing immunity in the human population. If pre-existing immunity occurs, it is mainly directed against the VSV surface protein G, which has been described as a pathogenicity factor for this virus [55, 57]. In order to circumvent complications by the presence of VSV G, two approaches have been explored; first, truncating G’s cytoplasmic domain which is mainly associated with pathogenicity [58], or second, replacing the G protein with a heterologous viral surface protein, e.g., the EBOV GP [55]. Garbutt and colleagues described the generation of rVSV vectors replacing VSV G with either EBOV Mayinga GP, or MARV Musoke GP, or Lassa virus (LASV) Josiah glycoprotein [57]. The VSV-EBOV vector has been termed VSVΔG-EBOVgp, VSV/ZEBOV-GP or similar. For the rest of this manuscript, we will refer to them as VSV-EBOV, VSV-MARV, and VSV-LASV.
Jones and colleagues performed the first efficacy studies using a single dose of each the VSV-EBOV or the VSV-MARV vaccine in NHPs, which fully protected all animals from lethal EBOV or MARV challenge 28 days after vaccination [59]. When the MARV survivors were rechallenged with the related MARV Popp strain 113 days after the first MARV Musoke challenge, 100% survival was observed. In contrast, only 25% of the EBOV-surviving NHPs survived the rechallenged with SUDV 234 days after the EBOV challenge [59].
In order to overcome this lack of cross-protection particularly between Ebolavirus species, VSV-EBOV, VSV-SUDV, and VSV-MARV were blended into a single-dose vaccine and NHPs survived lethal challenge 28 days later with either Tai Forest virus (TAFV), EBOV, SUDV, or MARV, respectively [60]. This study demonstrated for the first time that there is the potential for cross-protection between the Ebolavirus species using this vaccine platform, as TAFV GP was not part of the vaccine, yet the animals survived the lethal TAFV infection [60]. Furthermore, Marzi and colleagues showed recently that consecutive vaccination with the VSV-LASV followed 3 months later with VSV-EBOV does not compromise immunogenicity as the animals were 100% protected from lethal LASV and EBOV challenge [61]. A number of additional studies have been performed which are summarized in [62].
A mechanistic study by Marzi and colleagues identified EBOV GP-specific antibodies elicited by the VSV-EBOV as critical for protection [63], and Wong and associates have shown in both rodents and NHPs that EBOV GP-specific serum IgG levels correlate with survival [64]. Most recently, a study demonstrated the fast-acting potential of the VSV-EBOV vaccine against an EBOV-Makona isolate from the 2013-2016 West African EBOV epidemic outbreak strain. NHPs vaccinated 7 days prior to EBOV challenge survived the lethal challenge without developing disease; however, when the vaccine was administered only 3 days prior to challenge, only 67% of the NHPs survived and all the animals developed mild, moderate, or severe disease, respectively [65].
In addition to VSV-EBOV, there is a second VSV-based platform developed which includes the VSV G gene with a truncated cytoplasmic tail, rVSVNCT1. Mire and colleagues demonstrated that two versions of this novel rVSVNCT1 vector expressing EBOV GP protect 100% of the NHPs when challenged with a lethal dose of EBOV-Makona 28 days after a single-dose vaccination. Neither clinical signs of disease nor viremia were observed in any of the animals [66].
The preclinical studies performed using the rVSV vaccine platform against EHF and MHF in the NHP model have supported the acceleration of VSV-EBOV clinical trials during the 2013–2016 West African EBOV epidemic. Phase I and II clinical trials took place worldwide and yielded promising immunogenicity results, despite some concerning side-effects with high-dose vaccinations in particularly one cohort (reviewed in [3]). However, the most encouraging news resulted from a Phase III clinical trial in Guinea where the VSV-EBOV was used in a ring vaccination strategy to stop EBOV transmission and control the epidemic. This study showed that the VSV-EBOV is indeed a fast-acting vaccine and showed 100% efficacy after 10 days of vaccination with no new cases occurring after that time in the cohort [67, 68]. This is very encouraging data underlining the great potential of this vaccine for emergency use. In addition, the study confirms the NHP data presented earlier [65] validating the NHP model as the ‘gold standard’ for EBOV and MARV countermeasure development.
Conclusion
With the discovery of the filoviruses now over four decades old, one wonders why there is still no approved therapeutic or vaccine available for human use. While several experimental vaccine approaches have been tested for EBOV and, to a lesser extent, for MARV (Fig. 1), almost no progress has been made towards clinical trials. Taking into consideration that filoviruses can only be handled in high containment facilities available in a few countries worldwide and that, until 2014, they had not caused more than a few thousand human fatalities, there was never enough commercial interest or funding available for the development of licensed countermeasures. The situation changed following the EBOV epidemic that devastated West Africa from 2013 to 2016—clinical trials for the most promising countermeasure approaches were accelerated and funding was made available for the licensure process. Almost one year after West Africa was declared free of human EBOV cases, the progress of countermeasure licensure continues to be slow, although promising data from a few phase I–III clinical trials have raised the hope that protocols can be established to at least allow the use of pre-IND vaccines and treatments in emergency outbreaks. Ideally, we should be better prepared to act quickly after an infectious disease outbreak starts by knowing the status of experimental or pre-IND countermeasure approaches that could be used in emergencies. The West African EBOV epidemic is one of the most devastating examples of a modern-day emerging infectious disease outbreak that, thanks to continual—albeit slow—progress in the development of vaccines and countermeasures, will hopefully never be repeated.
Fig. 1.
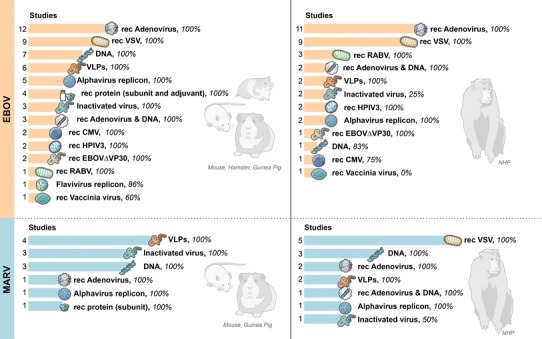
Vaccine efficacy for Ebola virus (EBOV) and Marburg virus (MARV) in rodents and NHPs. The vaccine platform, number of studies performed, and protective efficacy are summarized for EBOV and MARV in rodents and NHPs, respectively
Acknowledgements
The authors thank Ryan Kissinger (NIAID) for assistance with figure production, and Heinz Feldmann (NIAID) for his critical review of the manuscript.
References
- 1.Feldmann H, Sanchez A, Geisbert TW. In: Fields Virology. Knipe DM, Howley PM, editors. Philadelphia: Lippincott Williams & Wilkins; 2013. pp. 923–956. [Google Scholar]
- 2.Geisbert TW, Bausch DG, Feldmann H. Rev. Med. Virol. 2010;20:344–357. doi: 10.1002/rmv.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.C.E. Mire, T.W. Geisbert, H. Feldmann, A. Marzi, Expert Rev. Vaccines (2016) [DOI] [PMC free article] [PubMed]
- 4.Lupton HW, Lambert RD, Bumgardner DL, Moe JB, Eddy GA. Lancet. 1980;2:1294–1295. doi: 10.1016/S0140-6736(80)92352-1. [DOI] [PubMed] [Google Scholar]
- 5.Geisbert TW, Pushko P, Anderson K, Smith J, Davis KJ, Jahrling PB. Emerg. Infect. Dis. 2002;8:503–507. doi: 10.3201/eid0805.010284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warfield KL, Posten NA, Swenson DL, Olinger GG, Esposito D, Gillette WK, Hopkins RF, Costantino J, Panchal RG, Hartley JL, Aman MJ, Bavari S. J. Infect. Dis. 2007;196(Suppl 2):S421–S429. doi: 10.1086/520612. [DOI] [PubMed] [Google Scholar]
- 7.Hevey M, Negley D, Geisbert J, Jahrling P, Schmaljohn A. Virology. 1997;239:206–216. doi: 10.1006/viro.1997.8883. [DOI] [PubMed] [Google Scholar]
- 8.Ignatyev GM, Agafonov AP, Streltsova MA, Kashentseva EA. J. Biotechnol. 1996;44:111–118. doi: 10.1016/0168-1656(95)00104-2. [DOI] [PubMed] [Google Scholar]
- 9.Konduru K, Bradfute SB, Jacques J, Manangeeswaran M, Nakamura S, Morshed S, Wood SC, Bavari S, Kaplan GG. Vaccine. 2011;29:2968–2977. doi: 10.1016/j.vaccine.2011.01.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Konduru K, Shurtleff AC, Bradfute SB, Nakamura S, Bavari S, Kaplan G. PLoS ONE. 2016;11:e0162446. doi: 10.1371/journal.pone.0162446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.A.T. Lehrer, T.S. Wong, M.M. Lieberman, T. Humphreys, D.E. Clements, R.R. Bakken, M.K. Hart, W.D. Pratt, J.M. Dye, Vaccine (2017) [DOI] [PMC free article] [PubMed]
- 12.Halfmann P, Ebihara H, Marzi A, Hatta Y, Watanabe S, Suresh M, Neumann G, Feldmann H, Kawaoka Y. J. Virol. 2009;83:3810–3815. doi: 10.1128/JVI.00074-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marzi A, Halfmann P, Hill-Batorski L, Feldmann F, Shupert WL, Neumann G, Feldmann H, Kawaoka Y. Science. 2015;348:439–442. doi: 10.1126/science.aaa4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halfmann P, Kim JH, Ebihara H, Noda T, Neumann G, Feldmann H, Kawaoka Y. Proc. Natl. Acad. Sci. USA. 2008;105:1129–1133. doi: 10.1073/pnas.0708057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marzi A, Feldmann H. Expert Rev. Vaccines. 2014;13:521–531. doi: 10.1586/14760584.2014.885841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amanna IJ, Raue HP, Slifka MK. Nat. Med. 2012;18:974–979. doi: 10.1038/nm.2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dembinski JL, Hungnes O, Hauge AG, Kristoffersen AC, Haneberg B, Mjaaland S. J. Virol. Methods. 2014;207:232–237. doi: 10.1016/j.jviromet.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 18.Walker JM, Raue HP, Slifka MK. J. Virol. 2012;86:13735–13744. doi: 10.1128/JVI.02178-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swenson DL, Warfield KL, Larsen T, Alves DA, Coberley SS, Bavari S. Expert Rev. Vaccines. 2008;7:417–429. doi: 10.1586/14760584.7.4.417. [DOI] [PubMed] [Google Scholar]
- 20.Dye JM, Warfield KL, Wells JB, Unfer RC, Shulenin S, Vu H, Nichols DK, Aman MJ, Bavari S. Viruses. 2016;8:94. doi: 10.3390/v8040094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vanderzanden L, Bray M, Fuller D, Roberts T, Custer D, Spik K, Jahrling P, Huggins J, Schmaljohn A, Schmaljohn C. Virology. 1998;246:134–144. doi: 10.1006/viro.1998.9176. [DOI] [PubMed] [Google Scholar]
- 22.Swenson DL, Warfield KL, Negley DL, Schmaljohn A, Aman MJ, Bavari S. Vaccine. 2005;23:3033–3042. doi: 10.1016/j.vaccine.2004.11.070. [DOI] [PubMed] [Google Scholar]
- 23.Martins K, Carra JH, Cooper CL, Kwilas SA, Robinson CG, Shurtleff AC, Schokman RD, Kuehl KA, Wells JB, Steffens JT, van Tongeren SA, Hooper JW, Bavari S. Viral Immunol. 2015;28:62–70. doi: 10.1089/vim.2014.0071. [DOI] [PubMed] [Google Scholar]
- 24.Pushko P, Bray M, Ludwig GV, Parker M, Schmaljohn A, Sanchez A, Jahrling PB, Smith JF. Vaccine. 2000;19:142–153. doi: 10.1016/S0264-410X(00)00113-4. [DOI] [PubMed] [Google Scholar]
- 25.Hevey M, Negley D, Pushko P, Smith J, Schmaljohn A. Virology. 1998;251:28–37. doi: 10.1006/viro.1998.9367. [DOI] [PubMed] [Google Scholar]
- 26.Wilson JA, Bray M, Bakken R, Hart MK. Virology. 2001;286:384–390. doi: 10.1006/viro.2001.1012. [DOI] [PubMed] [Google Scholar]
- 27.Herbert AS, Kuehne AI, Barth JF, Ortiz RA, Nichols DK, Zak SE, Stonier SW, Muhammad MA, Bakken RR, Prugar LI, Olinger GG, Groebner JL, Lee JS, Pratt WD, Custer M, Kamrud KI, Smith JF, Hart MK, Dye JM. J. Virol. 2013;87:4952–4964. doi: 10.1128/JVI.03361-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feldmann H, Geisbert TW. Lancet. 2011;377:849–862. doi: 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sullivan NJ, Sanchez A, Rollin PE, Yang ZY, Nabel GJ. Nature. 2000;408:605–609. doi: 10.1038/35046108. [DOI] [PubMed] [Google Scholar]
- 30.Martin JE, Sullivan NJ, Enama ME, Gordon IJ, Roederer M, Koup RA, Bailer RT, Chakrabarti BK, Bailey MA, Gomez PL, Andrews CA, Moodie Z, Gu L, Stein JA, Nabel GJ, Graham BS. Clin. Vaccine Immunol. 2006;13:1267–1277. doi: 10.1128/CVI.00162-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grant-Klein RJ, Altamura LA, Badger CV, Bounds CE, Van Deusen NM, Kwilas SA, Vu HA, Warfield KL, Hooper JW, Hannaman D, Dupuy LC, Schmaljohn CS. Hum. Vaccines Immunother. 2015;11:1991–2004. doi: 10.1080/21645515.2015.1039757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riemenschneider J, Garrison A, Geisbert J, Jahrling P, Hevey M, Negley D, Schmaljohn A, Lee J, Hart MK, Vanderzanden L, Custer D, Bray M, Ruff A, Ivins B, Bassett A, Rossi C, Schmaljohn C. Vaccine. 2003;21:4071–4080. doi: 10.1016/S0264-410X(03)00362-1. [DOI] [PubMed] [Google Scholar]
- 33.Geisbert TW, Bailey M, Geisbert JB, Asiedu C, Roederer M, Grazia-Pau M, Custers J, Jahrling P, Goudsmit J, Koup R, Sullivan NJ. J. Virol. 2010;84:10386–10394. doi: 10.1128/JVI.00594-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu S, Kroeker A, Wong G, He S, Hou L, Audet J, Wei H, Zhang Z, Fernando L, Soule G, Tran K, Bi S, Zhu T, Yu X, Chen W, Qiu X. J. Infect. Dis. 2016;214:S326–S332. doi: 10.1093/infdis/jiw250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roy S, Zhi Y, Kobinger GP, Figueredo J, Calcedo R, Miller JR, Feldmann H, Wilson JM. J. Gen. Virol. 2006;87:2477–2485. doi: 10.1099/vir.0.81989-0. [DOI] [PubMed] [Google Scholar]
- 36.Geisbert TW, Bailey M, Hensley L, Asiedu C, Geisbert J, Stanley D, Honko A, Johnson J, Mulangu S, Pau MG, Custers J, Vellinga J, Hendriks J, Jahrling P, Roederer M, Goudsmit J, Koup R, Sullivan NJ. J. Virol. 2011;85:4222–4233. doi: 10.1128/JVI.02407-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Milligan ID, Gibani MM, Sewell R, Clutterbuck EA, Campbell D, Plested E, Nuthall E, Voysey M, Silva-Reyes L, McElrath MJ, De Rosa SC, Frahm N, Cohen KW, Shukarev G, Orzabal N, van Duijnhoven W, Truyers C, Bachmayer N, Splinter D, Samy N, Pau MG, Schuitemaker H, Luhn K, Callendret B, Van Hoof J, Douoguih M, Ewer K, Angus B, Pollard AJ, Snape MD. JAMA. 2016;315:1610–1623. doi: 10.1001/jama.2016.4218. [DOI] [PubMed] [Google Scholar]
- 38.Winslow RL, Milligan ID, Voysey M, Luhn K, Shukarev G, Douoguih M, Snape MD. JAMA. 2017;317:1075–1077. doi: 10.1001/jama.2016.20644. [DOI] [PubMed] [Google Scholar]
- 39.Richardson JS, Pillet S, Bello AJ, Kobinger GP. J. Virol. 2013;8(7):3677–3688. doi: 10.1128/JVI.02864-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swenson DL, Wang D, Luo M, Warfield KL, Woraratanadharm J, Holman DH, Dong JY, Pratt WD. Clin. Vaccine Immunol. 2008;15:460–467. doi: 10.1128/CVI.00431-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stanley DA, Honko AN, Asiedu C, Trefry JC, Lau-Kilby AW, Johnson JC, Hensley L, Ammendola V, Abbate A, Grazioli F, Foulds KE, Cheng C, Wang L, Donaldson MM, Colloca S, Folgori A, Roederer M, Nabel GJ, Mascola J, Nicosia A, Cortese R, Koup RA, Sullivan NJ. Nat. Med. 2014;20:1126–1129. doi: 10.1038/nm.3702. [DOI] [PubMed] [Google Scholar]
- 42.J.E. Ledgerwood, A.D. DeZure, D.A. Stanley, L. Novik, M.E. Enama, N.M. Berkowitz, Z. Hu, G. Joshi, A. Ploquin, S. Sitar, I.J. Gordon, S.A. Plummer, L.A. Holman, C.S. Hendel, G. Yamshchikov, F. Roman, A. Nicosia, S. Colloca, R. Cortese, R.T. Bailer, R.M. Schwartz, M. Roederer, J.R. Mascola, R.A. Koup, N.J. Sullivan, B.S. Graham, V.R.C.S.T, New Engl. J. Med. (2014)
- 43.De Santis O, Audran R, Pothin E, Warpelin-Decrausaz L, Vallotton L, Wuerzner G, Cochet C, Estoppey D, Steiner-Monard V, Lonchampt S, Thierry AC, Mayor C, Bailer RT, Mbaya OT, Zhou Y, Ploquin A, Sullivan NJ, Graham BS, Roman F, De Ryck I, Ballou WR, Kieny MP, Moorthy V, Spertini F, Genton B. Lancet Infect. Dis. 2015;20:43. doi: 10.1016/S1473-3099(15)00486-7. [DOI] [PubMed] [Google Scholar]
- 44.Tapia MD, Sow SO, Lyke KE, Haidara FC, Diallo F, Doumbia M, Traore A, Coulibaly F, Kodio M, Onwuchekwa U, Sztein MB, Wahid R, Campbell JD, Kieny MP, Moorthy V, Imoukhuede EB, Rampling T, Roman F, De Ryck I, Bellamy AR, Dally L, Mbaya OT, Ploquin A, Zhou Y, Stanley DA, Bailer R, Koup RA, Roederer M, Ledgerwood J, Hill AV, Ballou WR, Sullivan N, Graham B, Levine MM. Lancet Infect. Dis. 2016;16:31–42. doi: 10.1016/S1473-3099(15)00362-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsuda Y, Caposio P, Parkins CJ, Botto S, Messaoudi I, Cicin-Sain L, Feldmann H, Jarvis MA. PLoS Negl. Trop. Dis. 2011;5:e1275. doi: 10.1371/journal.pntd.0001275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsuda Y, Parkins CJ, Caposio P, Feldmann F, Botto S, Ball S, Messaoudi I, Cicin-Sain L, Feldmann H, Jarvis MA. Vaccine. 2015;33:2261–2266. doi: 10.1016/j.vaccine.2015.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marzi A, Murphy AA, Feldmann F, Parkins CJ, Haddock E, Hanley PW, Emery MJ, Engelmann F, Messaoudi I, Feldmann H, Jarvis MA. Sci. Rep. 2016;6:21674. doi: 10.1038/srep21674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blaney JE, Marzi A, Willet M, Papaneri AB, Wirblich C, Feldmann F, Holbrook M, Jahrling P, Feldmann H, Schnell MJ. PLoS Pathog. 2013;9:e1003389. doi: 10.1371/journal.ppat.1003389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blaney JE, Wirblich C, Papaneri AB, Johnson RF, Myers CJ, Juelich TL, Holbrook MR, Freiberg AN, Bernbaum JG, Jahrling PB, Paragas J, Schnell MJ. J. Virol. 2011;85:10605–10616. doi: 10.1128/JVI.00558-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Willet M, Kurup D, Papaneri A, Wirblich C, Hooper JW, Kwilas SA, Keshwara R, Hudacek A, Beilfuss S, Rudolph G, Pommerening E, Vos A, Neubert A, Jahrling P, Blaney JE, Johnson RF, Schnell MJ. J. Infect. Dis. 2015;212(Suppl 2):S414–S424. doi: 10.1093/infdis/jiv251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walsh PD, Kurup D, Hasselschwert DL, Wirblich C, Goetzmann JE, Schnell MJ. Sci Rep. 2017;7:43339. doi: 10.1038/srep43339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bukreyev A, Rollin PE, Tate MK, Yang L, Zaki SR, Shieh WJ, Murphy BR, Collins PL, Sanchez A. J. Virol. 2007;81:6379–6388. doi: 10.1128/JVI.00105-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.M. Lingemann, X. Liu, S. Surman, B. Liang, R. Herbert, A.D. Hackenberg, U.J. Buchholz, P.L. Collins, S. Munir, J. Virol. (2017) [DOI] [PMC free article] [PubMed]
- 54.Meyer M, Garron T, Lubaki NM, Mire CE, Fenton KA, Klages C, Olinger GG, Geisbert TW, Collins PL, Bukreyev A. J Clin Invest. 2015;125:3241–3255. doi: 10.1172/JCI81532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rose NF, Roberts A, Buonocore L, Rose JK. J. Virol. 2000;74:10903–10910. doi: 10.1128/JVI.74.23.10903-10910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rose NF, Marx PA, Luckay A, Nixon DF, Moretto WJ, Donahoe SM, Montefiori D, Roberts A, Buonocore L, Rose JK. Cell. 2001;106:539–549. doi: 10.1016/S0092-8674(01)00482-2. [DOI] [PubMed] [Google Scholar]
- 57.Garbutt M, Liebscher R, Wahl-Jensen V, Jones S, Moller P, Wagner R, Volchkov V, Klenk HD, Feldmann H, Stroher U. J. Virol. 2004;78:5458–5465. doi: 10.1128/JVI.78.10.5458-5465.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matassov D, Marzi A, Latham T, Xu R, Ota-Setlik A, Feldmann F, Geisbert JB, Mire CE, Hamm S, Nowak B, Egan MA, Geisbert TW, Eldridge JH, Feldmann H, Clarke DK. J. Infect. Dis. 2015;212(Suppl 2):S443–S451. doi: 10.1093/infdis/jiv316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jones SM, Feldmann H, Stroher U, Geisbert JB, Fernando L, Grolla A, Klenk HD, Sullivan NJ, Volchkov VE, Fritz EA, Daddario KM, Hensley LE, Jahrling PB, Geisbert TW. Nat. Med. 2005;11:786–790. doi: 10.1038/nm1258. [DOI] [PubMed] [Google Scholar]
- 60.Geisbert TW, Geisbert JB, Leung A, Daddario-DiCaprio KM, Hensley LE, Grolla A, Feldmann H. J. Virol. 2009;83:7296–7304. doi: 10.1128/JVI.00561-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marzi A, Feldmann F, Geisbert TW, Feldmann H, Safronetz D. Emerg. Infect. Dis. 2015;21:305–307. doi: 10.3201/eid2102.141649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.A. Marzi, H. Feldmann, T.W. Geisbert, D. Falzarano, J. Bioterror. Biodef. (2011) [DOI] [PMC free article] [PubMed]
- 63.Marzi A, Engelmann F, Feldmann F, Haberthur K, Shupert WL, Brining D, Scott DP, Geisbert TW, Kawaoka Y, Katze MG, Feldmann H, Messaoudi I. Proc. Natl. Acad. Sci. USA. 2013;110:1893–1898. doi: 10.1073/pnas.1209591110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wong G, Richardson JS, Pillet S, Patel A, Qiu X, Alimonti J, Hogan J, Zhang Y, Takada A, Feldmann H, Kobinger GP. Science translational medicine. 2012;4:158ra146. doi: 10.1126/scitranslmed.3004582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marzi A, Robertson SJ, Haddock E, Feldmann F, Hanley PW, Scott DP, Strong JE, Kobinger G, Best SM, Feldmann H. Science. 2015;349:739–742. doi: 10.1126/science.aab3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mire CE, Matassov D, Geisbert JB, Latham TE, Agans KN, Xu R, Ota-Setlik A, Egan MA, Fenton KA, Clarke DK, Eldridge JH, Geisbert TW. Nature. 2015;520:688–691. doi: 10.1038/nature14428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Henao-Restrepo AM, Longini IM, Egger M, Dean NE, Edmunds WJ, Camacho A, Carroll MW, Doumbia M, Draguez B, Duraffour S, Enwere G, Grais R, Gunther S, Hossmann S, Konde MK, Kone S, Kuisma E, Levine MM, Mandal S, Norheim G, Riveros X, Soumah A, Trelle S, Vicari AS, Watson CH, Keita S, Kieny MP, Rottingen JA. Lancet. 2015;386:857–866. doi: 10.1016/S0140-6736(15)61117-5. [DOI] [PubMed] [Google Scholar]
- 68.Henao-Restrepo AM, Camacho A, Longini IM, Watson CH, Edmunds WJ, Egger M, Carroll MW, Dean NE, Diatta I, Doumbia M, Draguez B, Duraffour S, Enwere G, Grais R, Gunther S, Gsell PS, Hossmann S, Watle SV, Konde MK, Keita S, Kone S, Kuisma E, Levine MM, Mandal S, Mauget T, Norheim G, Riveros X, Soumah A, Trelle S, Vicari AS, Rottingen JA, Kieny MP. Lancet. 2016;389:505–518. doi: 10.1016/S0140-6736(16)32621-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Warfield KL, Dye JM, Wells JB, Unfer RC, Holtsberg FW, Shulenin S, Vu H, Swenson DL, Bavari S, Aman MJ. PLoS ONE. 2015;10:e0118881. doi: 10.1371/journal.pone.0118881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hensley LE, Mulangu S, Asiedu C, Johnson J, Honko AN, Stanley D, Fabozzi G, Nichol ST, Ksiazek TG, Rollin PE, Wahl-Jensen V, Bailey M, Jahrling PB, Roederer M, Koup RA, Sullivan NJ. PLoS Pathog. 2010;6:e1000904. doi: 10.1371/journal.ppat.1000904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sullivan NJ, Geisbert TW, Geisbert JB, Xu L, Yang ZY, Roederer M, Koup RA, Jahrling PB, Nabel GJ. Nature. 2003;424:681–684. doi: 10.1038/nature01876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sullivan NJ, Geisbert TW, Geisbert JB, Shedlock DJ, Xu L, Lamoreaux L, Custers JH, Popernack PM, Yang ZY, Pau MG, Roederer M, Koup RA, Goudsmit J, Jahrling PB, Nabel GJ. PLoS Med. 2006;3:e177. doi: 10.1371/journal.pmed.0030177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pratt WD, Wang D, Nichols DK, Luo M, Woraratanadharm J, Dye JM, Holman DH, Dong JY. Clin. Vaccine Immunol. 2010;17:572–581. doi: 10.1128/CVI.00467-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sullivan NJ, Hensley L, Asiedu C, Geisbert TW, Stanley D, Johnson J, Honko A, Olinger G, Bailey M, Geisbert JB, Reimann KA, Bao S, Rao S, Roederer M, Jahrling PB, Koup RA, Nabel GJ. Nat. Med. 2011;17:1128–1131. doi: 10.1038/nm.2447. [DOI] [PubMed] [Google Scholar]
- 75.Choi JH, Jonsson-Schmunk K, Qiu X, Shedlock DJ, Strong J, Xu JX, Michie KL, Audet J, Fernando L, Myers MJ, Weiner D, Bajrovic I, Tran LQ, Wong G, Bello A, Kobinger GP, Schafer SC, Croyle MA. Mol. Pharm. 2015;12:2712–2731. doi: 10.1021/mp500646d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Johnson RF, Kurup D, Hagen KR, Fisher C, Keshwara R, Papaneri A, Perry DL, Cooper K, Jahrling PB, Wang JT, Ter Meulen J, Wirblich C, Schnell MJ. J. Infect. Dis. 2016;214:S342–S354. doi: 10.1093/infdis/jiw231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Geisbert TW, Daddario-Dicaprio KM, Geisbert JB, Reed DS, Feldmann F, Grolla A, Stroher U, Fritz EA, Hensley LE, Jones SM, Feldmann H. Vaccine. 2008;26:6894–6900. doi: 10.1016/j.vaccine.2008.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qiu X, Fernando L, Alimonti JB, Melito PL, Feldmann F, Dick D, Stroher U, Feldmann H, Jones SM. PLoS ONE. 2009;4:e5547. doi: 10.1371/journal.pone.0005547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Daddario-DiCaprio KM, Geisbert TW, Geisbert JB, Stroher U, Hensley LE, Grolla A, Fritz EA, Feldmann F, Feldmann H, Jones SM. J. Virol. 2006;80:9659–9666. doi: 10.1128/JVI.00959-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mire CE, Geisbert JB, Agans KN, Satterfield BA, Versteeg KM, Fritz EA, Feldmann H, Hensley LE, Geisbert TW. PLoS ONE. 2014;9:e94355. doi: 10.1371/journal.pone.0094355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Warfield KL, Bosio CM, Welcher BC, Deal EM, Mohamadzadeh M, Schmaljohn A, Aman MJ, Bavari S. Proc. Natl. Acad. Sci. USA. 2003;100:15889–15894. doi: 10.1073/pnas.2237038100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Warfield KL, Olinger G, Deal EM, Swenson DL, Bailey M, Negley DL, Hart MK, Bavari S. J Immunol. 2005;175:1184–1191. doi: 10.4049/jimmunol.175.2.1184. [DOI] [PubMed] [Google Scholar]
- 83.Grant-Klein RJ, Van Deusen NM, Badger CV, Hannaman D, Dupuy LC, Schmaljohn CS. Hum. Vaccines Immunother. 2012;8:1703–1706. doi: 10.4161/hv.21873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shedlock DJ, Aviles J, Talbott KT, Wong G, Wu SJ, Villarreal DO, Myles DJ, Croyle MA, Yan J, Kobinger GP, Weiner DB. Mol. Ther. 2013;21:1432–1444. doi: 10.1038/mt.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aviles J, Bello A, Wong G, Fausther-Bovendo H, Qiu X, Kobinger G. J. Infect. Dis. 2015;212(Suppl 2):S389–S397. doi: 10.1093/infdis/jiv175. [DOI] [PubMed] [Google Scholar]
- 86.Wilson JA, Hart MK. J. Virol. 2001;75:2660–2664. doi: 10.1128/JVI.75.6.2660-2664.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Reynard O, Mokhonov V, Mokhonova E, Leung J, Page A, Mateo M, Pyankova O, Georges-Courbot MC, Raoul H, Khromykh AA, Volchkov VE. J. Infect. Dis. 2011;204(Suppl 3):S1060–S1065. doi: 10.1093/infdis/jir347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Croyle MA, Patel A, Tran KN, Gray M, Zhang Y, Strong JE, Feldmann H, Kobinger GP. PLoS ONE. 2008;3:e3548. doi: 10.1371/journal.pone.0003548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Richardson JS, Yao MK, Tran KN, Croyle MA, Strong JE, Feldmann H, Kobinger GP. PLoS ONE. 2009;4:e5308. doi: 10.1371/journal.pone.0005308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Richardson JS, Abou MC, Tran KN, Kumar A, Sahai BM, Kobinger GP. J. Infect. Dis. 2011;204(Suppl 3):S1032–S1042. doi: 10.1093/infdis/jir332. [DOI] [PubMed] [Google Scholar]
- 91.Gilligan JKGJ, Jahrling PB, Anderson K. In: Vaccines. Brown FBD, Doherty P, Mekalanos J, Norrby E, editors. Cold Spring Habor: Cold Spring Harbor Lanoratory Press; 1997. pp. 87–92. [Google Scholar]
- 92.Bukreyev A, Yang L, Zaki SR, Shieh WJ, Rollin PE, Murphy BR, Collins PL, Sanchez A. J. Virol. 2006;80:2267–2279. doi: 10.1128/JVI.80.5.2267-2279.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bukreyev A, Marzi A, Feldmann F, Zhang L, Yang L, Ward JM, Dorward DW, Pickles RJ, Murphy BR, Feldmann H, Collins PL. Virology. 2009;383:348–361. doi: 10.1016/j.virol.2008.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Feldmann H, Jones SM, Daddario-DiCaprio KM, Geisbert JB, Stroher U, Grolla A, Bray M, Fritz EA, Fernando L, Feldmann F, Hensley LE, Geisbert TW. PLoS Pathog. 2007;3:e2. doi: 10.1371/journal.ppat.0030002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jones SM, Stroher U, Fernando L, Qiu X, Alimonti J, Melito P, Bray M, Klenk HD, Feldmann H. J. Infect. Dis. 2007;196(Suppl 2):S404–S412. doi: 10.1086/520591. [DOI] [PubMed] [Google Scholar]
- 96.Marzi A, Ebihara H, Callison J, Groseth A, Williams KJ, Geisbert TW, Feldmann H. J. Infect. Dis. 2011;204(Suppl 3):S1066–S1074. doi: 10.1093/infdis/jir348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tsuda Y, Safronetz D, Brown K, LaCasse R, Marzi A, Ebihara H, Feldmann H. J. Infect. Dis. 2011;204:S1090–S1097. doi: 10.1093/infdis/jir379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wong G, Audet J, Fernando L, Fausther-Bovendo H, Alimonti JB, Kobinger GP, Qiu X. Vaccine. 2014;32:5722–5729. doi: 10.1016/j.vaccine.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Warfield KL, Swenson DL, Negley DL, Schmaljohn AL, Aman MJ, Bavari S. Vaccine. 2004;22:3495–3502. doi: 10.1016/j.vaccine.2004.01.063. [DOI] [PubMed] [Google Scholar]
- 100.Wang D, Hevey M, Juompan LY, Trubey CM, Raja NU, Deitz SB, Woraratanadharm J, Luo M, Yu H, Swain BM, Moore KM, Dong JY. Virology. 2006;353:324–332. doi: 10.1016/j.virol.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 101.Sun Y, Carrion R, Jr, Ye L, Wen Z, Ro Y-T, Brasky K, Ticer AE, Schwegler EE, Patterson JL, Compans RW, Yang C. Virology. 2008;383:12–21. doi: 10.1016/j.virol.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]