

Abstract
Infectious bursal disease virus (IBDV) is the etiological agent of a highly contagious disease in chickens. In a recent report, proteasome inhibitor MG132 has been shown to completely inhibit IBDV-induced apoptosis. This raises the possibility that the ubiquitin–proteasome pathway may be used by the virus to promote viral replication. In this study, we examined the interplay between IBDV replication and the ubiquitin–proteasome pathway in cultured cells. Treatment of DF-1 cells with the proteasome inhibitors MG132 or lactacystin significantly decreased virus release in the supernatant and prevented virus-induced cytopathic effect. Inhibition of the ubiquitin–proteasome pathway did reduce markedly viral RNA transcription and protein translation but not affect virus internalization. We also demonstrated that IBDV activates caspase pathway via triggering the efflux of cytochrome c in mitochondria into cytosol of infected cells. This activity was dose-dependently reduced by proteasome inhibitor treatment. Taken together, our data suggest that proteasome inhibitor reduces IBDV replication through inhibition of viral RNA transcription and protein synthesis, and thus preventing IBDV-induced apoptosis.
Keywords: Infectious bursal disease virus, Proteasome inhibitor, RNA transcription, Protein translation, Apoptosis, Cytochrome c, Caspase activities, Cultured cells
Introduction
Infectious bursal disease virus (IBDV) is the etiological agent of infectious bursal disease (IBD), which is a highly contagious disease in 3- to 15-weeks-old chickens. The virus replicates in the cytoplasm of infected cells and targets the precursors of antibody-producing B cells in the bursa of Fabricius (BF). Infection by this virus leads to a severe immunosuppression characterized by the destruction of B cells present in the BF. The induced immunodepression increases susceptibility to other pathogens [1]. Two distinct serotypes (I and II) are recognized for IBDV. The serotype I virus strains differ markedly in virulence, classic and very virulent IBDV strains cause hemorrhagic inflammation of the BF, whereas variant strains cause rapid bursal atrophy without evoking an inflammation response [1, 2]. In contrast to the serotype I, the serotype II viruses are naturally avirulent for chickens [3]. IBD is one of the most important diseases affecting the poultry industry worldwide.
IBDV is a member of the Birnaviridae family, and its genome consists of two segments of double-stranded RNA, designated A (≈3.2 kb) and B (≈2.8 kb). Segment A has two partially overlapping open reading frames (ORFs). The smaller ORF encodes VP5, a nonstructural protein of 17 kDa, which is not required for viral replication [4]. The larger ORF encodes a 110-kDa polyprotein precursor in the order NH2-pVP2-VP4-VP3-COOH. The polyprotein appears to be cotranslationally processed through the proteolytic activity of the viral protease, VP4, to generate pVP2, VP4, and VP3 [5–7]. pVP2, the VP2 precursor, is further processed at its C terminus to become VP2 through the cleavage of three alanine–alanine bonds and an alanine–phenylalanine bond [8]. VP2 and VP3 are structural proteins forming the virus capsid [6]. Segment B encodes VP1 which is known as a multifunctional protein with polymerase and capping enzyme activities [9, 10].
To successfully infect a host, viruses have to both exploit the cellular resources and at the same time avoid defense reactions of the host organism. One of the common obstacles facing a virus is the overwhelming stoichiometric imbalance of the target proteins over those encoded by the virus. To overcome this, many viruses have evolved the ability to utilize the host protease machinery to direct cellular protein degradation for their survival and replication [11]. 26S proteasome is a large multi-subunit complex that selectively degrades intracellular proteins. It is involved in a wide variety of proteolytically mediated intracellular processes, such as cell cycle progression, apoptosis, metabolic regulation, and transcriptional control [12]. The majority of proteins that target the proteasome are tagged with polyubiquitin, which serves as a signal for their degradation. In the process of protein degradation, substrates are first tagged by ubiquitin by three enzymes, ubiquitin-activating enzyme (E1 enzyme), ubiquitin-conjugation enzyme (E2 enzyme), and ubiquitin ligase (E3 ligase) [13]. The polyubiquitinated proteins are then rapidly degraded by the proteasome. Recent studies have implicated that the ubiquitin–proteasome pathway might play key roles in virus infection and facilitates activities required for various aspects of the virus life cycle, from entry [14] through replication [15, 16] and enhanced cell survival [11] to viral release [17, 18]. In addition, proteasome inhibitors reduce the replication or progeny release of several viruses, including coxsackievirus B3 [19], human cytomegalovirus [20], human immunodeficiency virus type 1 [21], and Sendai virus [22].
IBDV has been previously shown to induce apoptosis both in vitro and in vivo [23–27], and recently demonstrated that IBDV-induced apoptosis was through activating caspases-3 and -9 activities which were completely inhibited by treatment with proteasome inhibitor MG132 [28], suggesting that the proteasome inhibitor blocks IBDV-induced apoptosis. But, whether inhibition of IBDV-induced apoptosis by treatment with proteasome inhibitors is associated with reduction of viral replication is still unclear. However, these results raise the possibility that the ubiquitin–proteasome pathway may be used during IBDV infection to promote viral replication and infectivity. Thus, in the present study, we attempted to explore the role of proteasome inhibition in IBDV infectivity and the mechanisms contribute to proteasome inhibitor suppression of IBDV replication in cultured cells and then the relationship between IBDV-induced apoptosis and viral replication.
Materials and methods
Virus and cell culture
A DF-1 chicken embryo fibroblast (CEF) cell line [29] was maintained in Dulbecco’s modified Eagle medium (DMEM) (GIBCO) supplemented with 10% fetal bovine serum (FBS) and 100 U/ml of penicillin G, and 100 μl/ml streptomycin at 39°C in a humidified 5% CO2 incubator. The IBDV strain LM [30], a CEF cells-adapted virus, was used in the study.
Virus infection
DF-1 cells were infected at a multiplicity of infection (MOI) of 0.1–100 with IBDV strain LM for 60 min. Cells were washed with PBS and cultured in fresh DMEM medium containing 10% fetal calf serum. For inhibitor experiments, DF-1 cells after adsorption with IBDV were incubated with proteasome inhibitor MG132 or lactacysin (Calbiochem, San Diego, CA) for 6 h. Cells were then incubated with 10% DMEM medium without proteasome inhibitors.
TCID50 assay
The amount of IBDV produced was measured on monolayers of DF-1 cells. Cell supernatant was serially diluted and inoculated on monolayers of DF-1 cells. Following 1 h of incubation, fresh DMEM medium was added and incubated. Three days post-infection (p.i.), cytopathic effect (CPE) was observed under a microscopy and virus titer was determined as TCID50 per 0.1 ml.
Cell viability assay
A modified 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTS) assay, which measures mitochondrial function, was used to determine cell viability according to the manufacturer’s instruction (Promega). Twenty-one hour p.i., cells were incubated for 2 h in MTS solution, and absorbance was measured at 490 nm with an ELISA reader. The conversion of MTS into the aqueous soluble formazan product is accomplished by dehydrogenase enzymes found in metabolically active cells. The quantity of formazan product as measured is directly proportional to the number of living cells in culture. IBDV-infected DF-1 cells were also examined for morphological changes by phase-contrast microscopy.
Indirect fluorescence assay (IFA)
For infection, DF-1 cells grown on 96-well plate were incubated with the IBDV strain LM for 60 min at 39°C at a MOI of 20 TCID50 and added to DMEM for incubation. Following the incubation at 39°C, cells at 18 h p.i. were washed with PBS and fixed for 30 min at room temperature (RT) with 4% paraformaldehyde (PFA) in PBS. After fixation, the cells were blocked by PBS with 3% BSA at RT for 1 h. Primary antibody, guinea pig anti-VP2 polyclonal antibody, was diluted in PBS with 3% BSA and incubated with the cells for 1 h at 37°C. After washing with PBS, the cells were incubated with goat anti-guinea pig FITC-conjugated antibody (DAKO) diluted in PBS with 3% BSA for 1 h at 37°C. The cells were washed three times with PBS and examined using a fluorescence microscopy.
Real-time RT-PCR
Total cell RNAs were prepared from virus-infected DF-1 cells 18 h after being treated with various concentrations of MG132 by using Trizol RNA extract reagent (Invitrogen) for reverse transcription (RT)-PCR. Real-time RT-PCR assays for VP2 used the VP2-F5 (5′-CTGACTACCGGCATCGACA-3′) and VP2-R3 (5′-CCACTTGCCGACCATGA-3′) primer pair, which target a 133-nucleotide (nt) region of VP2, and were normalized with β-actin primers (sense, 5′-GAGAAATTGTGCGTGACATCA-3′; antisense, 5′-CCTGAACCTCTCATTGCCA-3′) in separate reactions. RT reactions were performed with avian myeloblastosis virus reverse transcriptase (Roche) using antisense primers. The amplification protocol followed the instructions of a LightCycler Fast Start DNA Masterplus SYBR Green I kit (Roche). The PCR parameters consisted of an initial denaturation at 94°C for 5 min, followed by 40 cycles of 94°C for 5 s, 55°C for 5 s and 72°C for 25 s. Subsequent melting curve analysis and C T value determination were performed using Roche LightCycler software version 3.5. Each sample was run in triplicate. The relative amount of target viral mRNA was normalized to that of β-actin mRNA in the same sample.
Preparation of whole cell lysates and cytosolic extracts
DF-1 cells grown in T-75 flasks at 18 h of p.i. were collected by centrifugation at 500 × g for 10 min. For whole cell lysates preparation, the cell pellets were washed twice with ice-cold PBS (pH 7.4) and lysed by vigorous vortexing in 100 μl of 6 M guanidine hydrochloride containing 250 mM Tris–HCl (pH 8.5), 10 mM EDTA, 150 mM β-mercaptoethanol, and 1 mM phenylmethylsulfonyl fluoride (PMSF). Cytosolic extracts were prepared as previously described [31]; the cell pellets were washed twice with ice-cold PBS and resuspended with 400–500 μl of buffer (250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 1 mM PMSF, pH 7.5). The cells were homogenized and centrifuged twice at 10,000 × g for 15 min at 4°C. The supernatants were further centrifuged at 10,000 × g for 1 h at 4°C, and the resulting supernatants (cytosolic extracts) were frozen as aliquots at −80°C for subsequent experiments. The whole lysates and cytosolic extracts were subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting.
Western blot analysis
For the analysis of VP2 protein, the whole cell lysates were resolved on 10% SDS-PAGE and blotted onto nitrocellulose (NC) membranes (Stratagene) with a semidry transfer cell (Bio-Rad Trans-Blot SD). The membranes were blocked for 2 h at RT in blocking buffer TBST (20 mM Tris–HCl [pH 7.4], 150 mM NaCl, 0.1% Tween-20) containing 5% skim milk powder to prevent nonspecific binding, and then incubated with guinea pig anti-VP2 antibody at RT for 2 h. For cytochrome c detection, protein samples from the cytosol extracts were resolved on 12% SDS-PAGE and transferred to NC membranes as described. The rabbit anti-cytochrome c antibody (Santa Cruz Biotechnology) diluted in blocking buffer. Then, the membranes were washed three times with TBST, and incubated for 1 h at RT with horseradish peroxidase-conjugated anti-guinea pig or rabbit secondary antibody (DAKO) diluted in blocking buffer. After washing, the membrane was reacted with 3,3′-diaminobenzidine tetrahydrochloride (Pierce, Rockford, IL, USA) substrate, and then stopped with distilled water. As a load control, monoclonal antibody against β-actin (Santa Cruz Biotechnology) was also used in Western blot analysis.
Fluorimetric assay of caspase activity
Spectrofluorometric assays of proteolytic activity were carried out using synthetic fluorogenic substrates 7-amino-4-trifluoromethyl coumarin (AFC) to measure caspase-3 activity or 7-amino-methyl coumarin (AMC) to measure caspase-9 activity. BD ApoAlert Caspase Fluorescent Assay Kits (Clontech Laboratories, USA) were used to determine caspase-3 and -9 activities. In brief, 80% confluent monolayers of DF-1 cells grown on T-25 flasks were infected with IBDV at a MOI of 20 TCID50 with or without proteasome inhibitor MG132 treatment after infection. After 18 h of p.i., cells were harvested at 400 × g for 5 min. Cells (2–3 × 106) were lysed in 50 μl of lysis buffer on ice for 10 min and centrifuged at 16,000 × g for 10 min, and the supernatant was collected. A 50-μl supernatant was added to an equal volume of 2× reaction/DTT buffer supplemented with caspase-3 substrate DEVD-AFC or caspase-9 substrate LEHD-AMC, and incubated at 37°C for 2 h. The optical densities at 400 nm for caspase-3 and at 380 nm for caspase-9 were determined. The nanomoles of AFC or AMC (released) expressed per hour was calculated from the standard curve.
Results
Proteasome inhibitors reduce IBDV viral progeny release and dose dependent
Viral infections frequently result in the degradation of proteasome in the infected cells. To assess whether the proteasome–ubiquitin system plays a role in IBDV infection, two chemically distinct proteasome inhibitors, MG132 and lactacystin, were used to address the question. The peptide-aldehyde MG132 is a highly potent and reversible inhibitor of the chymotryptic-like activity of the proteasome [32]. In contrast, lactacystin is highly specific and inhibits the proteasome irreversibly by its action on the catalytic threonine of the 26S proteasome β-subunits [32]. In a preliminary experiment, DF-1 cells were infected with IBDV strain LM at a MOI of 20 TCID50 and then incubated with 50 μM of MG132 for 2, 4, or 6 h treatment as described elsewhere [28], the results indicated that viral titer from supernatant of infected cells at 21 h after infection was significantly decreased when incubated with MG132 for 6 h, as compared to that of incubation with the proteasome inhibitor for 2 or 4 h (data not shown). Therefore, in subsequent experiments, we treated infected cells with proteasome inhibitors for 6 h. To determine whether IBDV production was dose-dependently reduced by proteasome inhibitor treatment, DF-1 cells were infected with IBDV strain LM and then treated with various concentrations of proteasome-specific inhibitors. At 6 h after infection, the input proteasome inhibitors were removed and replaced by growth medium without these inhibitors. Supernatants were collected 21 h p.i., and the amount of progeny virus was determined by titration using TCID50 assays on DF-1 cells (Fig. 1A). IBDV replication in untreated cells resulted in the generation of a viral titer of 106.5 TCID50/0.1 ml after 21 h p.i., whereas proteasome inhibition reduced the titer of released virus in the supernatant in a dose-dependent manner. MG132 and lactacystin had very similar inhibitory effects on virus production. These data indicated that proteasome inhibitors significantly blocked IBDV production, suggesting that the proteasome may play an important role in IBDV production.
Fig. 1.
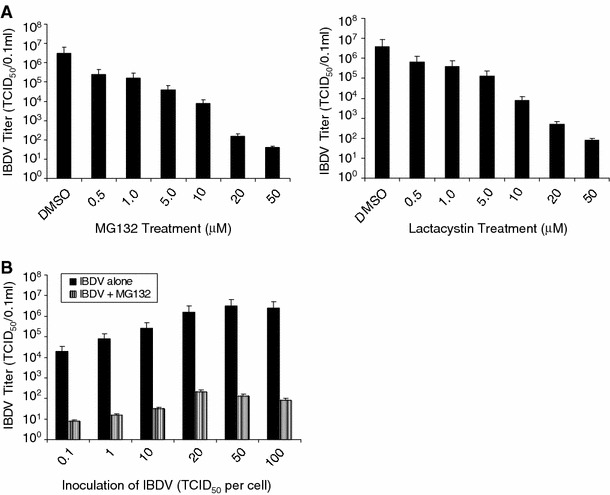
Proteasome inhibition reduces IBDV release. (A) DF-1 cells were infected with IBDV (MOI = 20) at 39°C for 1 h, washed with PBS, and incubated with different concentrations of proteasome inhibitors, MG132 and lactacystin, for 6 h, and washed with PBS, and then incubated with 10% DMEM medium without proteasome inhibitors. Viral titers in the supernatants of IBDV-infected DF-1 cells were determined after 21 h p.i. by TCID50 assay. (B) DF-1 cells were infected with different amounts of IBDV and then treated with 20 μM of MG132 for 6 h, and continued to incubate at 39°C. Virus titers of supernatants collected at 21 h p.i. as determined by TCID50 assay are shown. Values are means ± standard errors (SE) of three independent experiments
In addition, we also used different amounts of IBDV to infected DF-1 cells to determine the effect of proteasome inhibitor MG132 on viral replication. Viral titers in the supernatants harvested at 21 h p.i. were significantly reduced by MG132 treatment (Fig. 1B), implying that the effect is due to a reduction in virus growth rather than to a delay in virus release. Different amounts of input virus change the absolute amount of progeny virus but do not affect significantly the percentage of reduction of viral titers, indicating that the antiviral properties of proteasome inhibitors are independent of the multiplicity of infection.
Proteasome inhibitors reduce IBDV-induced CPE
Many viruses are capable of inducing cell death, leading to lysis of infected cells. In late stages of IBDV infections, morphological changes, commonly known as CPE, can be microscopically observed. IBDV-induced CPE is characterized by cell rounding, shrinkage, deformation of nuclei, chromatin condensation, detachment of infected cells from the culture flask, and cell lysis and death.
We used MTS-based assay to test the cytotoxicity of the proteasome inhibitor MG132. It was noted that throughout all doses of the proteasome inhibitor used in this study, cell viability assays and morphological assessment showed no detectable cell death in DF-1 cells (data not shown). In contrast, IBDV-induced CPE and cell death were dramatically blocked after the addition of MG132 (Fig. 2A).
Fig. 2.
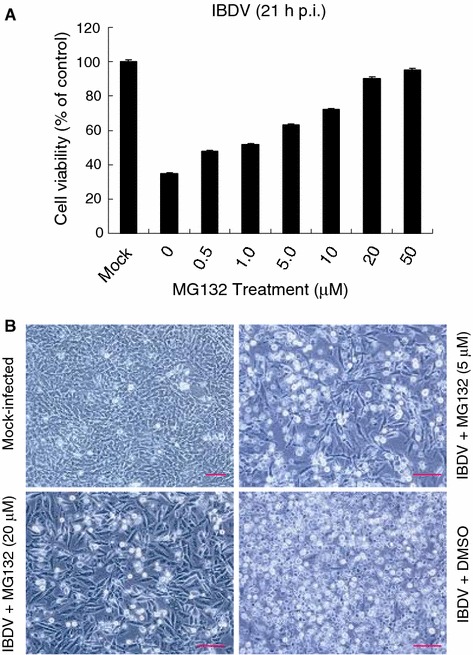
Proteasome inhibitors inhibit IBDV-induced CPE. (A) DF-1 cells were infected with IBDV (MOI = 50) and then treated with various concentrations of MG132 as described in Materials and methods. Cell viability was assessed by the MTS assay at 21 h p.i. The values of MTS in mock-infected cells in the absence of MG132 were defined as 100% survival. Values are means ± SE of three independent experiments. (B) Representative morphological changes of DF-1 cells treated with MG132 or DMSO at 21 h p.i. by phase-contrast microscopy. Bars, 10 μm
During infections of DF-1 cells with IBDV strain LM using 50 TCID50/cell, a clear CPE was visible within 21 h p.i. Figure 2B shows the morphology of DF1 cells infected with IBDV. Mock-infected cells or cells treated with various amounts of MG132 showed typical spread-out shapes and normal morphology. Addition of MG132 upon infection inhibited the formation of a visible CPE. Thus, the CPE of the virus infection is prevented by the presence of MG132.
The ubiquitin–proteasome system is involved in early step of virus replication but does not block virus internalization
To elucidate the mechanism of inhibition, we took advantage of the reversible nature of MG132 to dissect the potential targets of the proteasome inhibitor. DF-1 cells were infected with IBDV and MG132 (20 μM) was added and kept in the media for three different intervals, as shown in Fig. 3A. The cells were incubated until 18 h p.i., and the supernatants were collected for virus titration. Treatment for 0–6 h resulted in significant reduction of virus yield (3-log-unit reduction compared to the titer of the no-treatment control). The virus titer of the group treated for 6–12 h was 1 log unit lower than the titer of the control sample. For virus accumulate from 12 to 18 h p.i., only a slight reduction of the virus titer was observed. The virus titers from the 0 to 6 h, 6 to 12 h, and 12 to 18 h treatment groups were compared to that of the no-treatment control, indicating that MG132 does not have significant effects on the late steps of the viral life cycle. The result suggests that the ubiquitin–proteasome system is most likely involved at an early step in the virus life cycle, since the inhibitory effect of MG132 was seen primarily in the 0–6 treatment group.
Fig. 3.
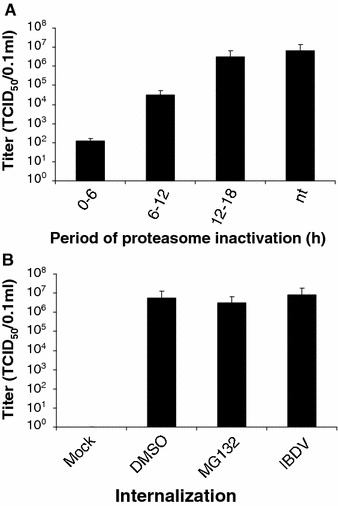
(A) Effect of proteasome inactivation at increasing periods of time after infection. DF-1 cells were infected with IBDV at a MOI of 20 TCID50/cell. Proteasomes were blocked at the indicated times by the reversible MG132 (20 μM). The cells were washed to remove the drug and further incubated until 18 h. The supernatants from the indicated time points were collected and determined for virus titration by TCID50 assay. Values represent the mean of three independent experiments. nt, not treated. (B) Effect of MG132 on virus internalization. DF-1 cells preincubtaed for 3 h with or without MG132 were incubated with IBDV at 4°C for 1 h (MOI of 20). After cells were washed with medium, they were shifted to 39°C for one more hour for virus internalization. Afterwards, the infected cells were further incubated until 18 h. The supernatants were collected and determined for virus titration by TCID50 assay. Values represent the mean of three independent experiments
As described above, the proteasome inhibitor has an inhibitory effect on IBDV infection at the early step. To further reveal the possible mechanism of inhibition by MG132, the virus internalization assay was performed. After 3-h pretreatment with MG132, the cells were incubated with IBDV at 4°C for 1 h to allow virus binding and then incubated at 39°C for an additional hour to allow virus internalization. Afterwards, the fresh medium without MG132 was added and the cells were further incubated at 39°C until 18 h p.i. The supernatants were collected for virus titration. There was no difference in virus titer in the presence or absence of the proteasome inhibitor (Fig. 3B). This result suggests that the ubiquitin–proteasome system is not involved in the virus internalization.
Reduction of IBDV viral transcription and protein expression by protease inhibitor
To confirm that the effect of proteasome inhibitor on IBDV viral production was through the reduction of IBDV RNA levels, we performed real-time RT-PCR analysis with RNA extracted from DF-1 cells 18 h after infection with IBDV in the presence or absence of proteasome inhibitor MG132. The amount of each viral mRNA was normalized to that of β-actin mRNA in the same sample. The abundance of VP2 mRNA was dose-dependently decreased in IBDV-infected cells treated with the proteasome inhibitor MG132 (Fig. 4A). To further study the inhibitory effect of proteasome inhibitor on viral protein expression, capsid protein VP2 of IBDV was analyzed in IBDV-infected DF-1 cells by IFA detection as well as Western blotting. For IFA detection, a large amount of cells positive for IBDV VP2 protein expression were detected at 18 h p.i., whereas the MG132-treatment groups showed significant reductions in amounts of cells positive for IBDV VP2 protein expression in a dose-dependent manner (Fig. 4B and data not shown). For Western blotting analysis, significant amount of the IBDV protein VP2 in protein extracts of infected DF-1 cells was detectable at 18 h p.i. At the time point, treatment with proteasome inhibitor MG132 reduces dose-dependently the expression of the capsid protein (Fig. 4C). These results suggest that the proteasome inhibitor decreased viral replication via suppression of viral RNA transcription and protein translation.
Fig. 4.
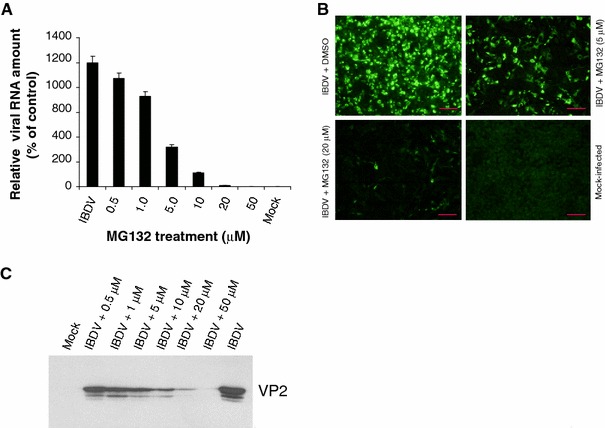
Proteasome inhibition decreases viral RNA transcription and protein translation in IBDV-infected DF-1 cells. (A) The effects of proteasome inhibitor on IBDV RNA level. Total RNAs isolated from IBDV-infected cells 18 h after treatment with MG132 were subject to a real-time RT-PCR analysis of VP2 RNA. The data were normalized to the amount of β-actin mRNA and are expressed as percentages of the normalized value for IBDV-infected cells in mock-infected cells (control); they are means ± SD of values from at least three independent experiments. (B) Representative IFA staining of IBDV-infected cells used to detect VP2 protein expression. Infected DF-1 cells 18 h after treatment with various concentrations of MG132 were fixed and analyzed by immunofluorescence staining with guinea pig anti-VP2 serum. Bars, 10 μm. (C) Whole-cell extracts prepared from infected cells 18 h after treatment with the indicated proteasome inhibitors were assayed by Western blotting for the presence of VP2 expression in IBDV-infected cells
Reduction of caspase activities induced by IBDV after proteasome inhibitor treatment
IBDV has been shown to induce apoptosis in cultured cells through activation of caspase-9 followed by activation of caspase-3 pathway [28]. As demonstrated above, IBDV replication was strongly prevented after treatment with proteasome inhibitors. To determine whether IBDV-induced apoptosis was associated with viral replication, caspase-3 and -9 activities in DF-1 cells with IBDV infection after treatment with various amounts of proteasome inhibitor MG132. As expected, the IBDV strain LM induced activation of caspase-9 and caspase-3 in infected cells, whereas their activities were significantly reduced when the infected cells were treated with Ac-DEVD-CHO, a peptide inhibitor of caspase-3 activity, or LEHD-CHO, an inhibitor of caspase-9 activity (Fig. 5). Furthermore, activation of caspase-3 and caspase-9 was dose-dependently decreased in infected cells when treated with increasing amounts of MG132 (Fig. 5). The results indicated that IBDV-induced apoptosis was closely related to viral replication.
Fig. 5.
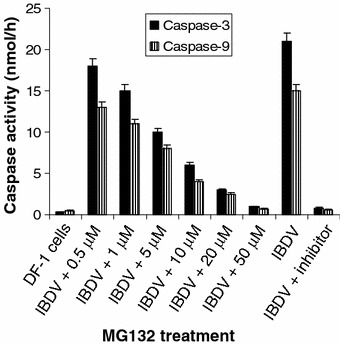
Dose-dependent inhibition of IBDV-induced caspase activities by treatment with proteasome inhibitor. DF-1 cell lysates harvested from infected cells 18 h p.i. after treatment with various concentrations of MG132 were assayed for DEVDase or LEHDase activity using specific substrate DEVD-AFC (caspase-3) or LEHD-AMC (caspase-9), respectively. Simultaneously, the cells at 18 h p.i. were treated with inhibitor DEVD-CHO (for caspase-3) or LEHD-CHO (for caspase-9), respectively. In the above caspase activity assays, mock-infected cells were used as a negative control for each of the infections. Values shown are means from duplicated experiments
To further assess the mechanism of the apoptosis induced by IBDV and determine the effect of proteasome inhibitor on cytochrome c release of infected cells, the cytosol fractions from IBDV-infected cells with or without MG132 treatment were used to demonstrate the localization of the cytochrome c by Western blotting analysis. As shown in Fig. 6, IBDV induced the release of cytochrome c into the cytoplasm of infected cells. Level of cytochrome c thus rose in cytosol fractions of the infected cells (Fig. 6), whereas they could only be sequestered in the mitochondria of the mock-infected cells (data not shown). However, the release of cytochrome c from the mitochondria into the cytosol was dose-dependently decreased in the IBDV-infected cells when treated with proteasome inhibitor MG132 (Fig. 6 and data not shown). Therefore, the results indicate that IBDV could trigger the efflux of cytochrome c in the mitochondria into the cytoplasm after infection, which activates series of caspases in turn induces apoptosis. Furthermore, this apoptotic activity induced by IBDV could be strongly reduced by proteasome inhibitor which is due to their inhibition to viral replication.
Fig. 6.
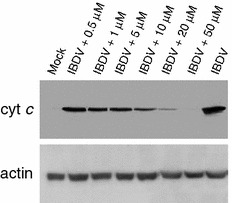
Dose-dependent reduction of release of cytochrome c into the cytoplasm of the IBDV-infected cells after treatment with proteasome inhibitor. Cells were infected with IBDV (MOI = 20) and then treated with various concentrations of MG132 for 6 h and continued to incubate. Eighteen hours p.i., cells were harvested. Cytosol (cyto) fractions were then prepared and used as lysates for Western blot analysis. Lysates were probed with antibody directed against cytochrome c. Levels of β-actin were used to control for protein loading
Discussion
Recently, several viruses were found to require active proteasomes. Retroviruses require the proteasome for budding from cells [33–35], the human cytomegalovirus for inducing cell cycle progression [36, 37], the simian virus 5 and human parainfluenza virus type 2 for blocking IFN signaling [38, 39], Epstein–Barr virus and herpes simplex virus 1 for regulating viral latency [40, 41], minute virus of mice for nuclear translocation [42], and murine coronavirus for its transfer from endosome to cytoplasm during viral entry [43]. Adenovirus requires active proteasomes to promote late gene expression [44] and coxsackievirus for viral RNA transcription and protein synthesis [19]. In the present study, we showed that the proteasome inhibitor decreased IBDV replication via suppression of viral RNA transcription (Fig. 4A) and protein translation (Fig. 4B, C), and further showed that the ubiquitin–proteasome system is involved in an early step of IBDV replication but does not block virus internalization (Fig. 3A, B). However, whether the ubiquitin–proteasome pathway-mediated IBDV replication is related to ubiquitination process and/or the proteasome degradation is not clear. We examined the proteasome activity following IBDV infection and to determine the effect of proteasome inhibitor on its activity. 26S proteasome activities were unchanged in IBDV-infected DF-1 cells, whereas proteasome inhibitor MG132 dramatically inhibited proteasome activity in both mock- and IBDV-infected cells (data not shown). In addition, we also observed a significant accumulation of polyubiquitinated proteins after MG132 treatment by Western blot analysis (data not shown). Therefore, the ubiquitination process may play a role in the ubiquitin–proteasome pathway-mediated IBDV replication or protein degradation.
In a recent report, Liu and Vakharia [28] demonstrated that the proteasome inhibitor MG132 at 50 μM was completely able to inhibit NF-κB activity as well as IBDV-induced DNA laddering and caspase-3 activation, and thus suggested that MG132 prevents IBDV-induced apoptosis via strong inhibition of NF-κB activity required for IBDV-induced apoptosis. However, the IBDV RNA transcription and protein synthesis in the infected cells in the presence of MG132 was not available in the study [28]. In this study, IBDV RNA transcription and protein synthesis were significantly inhibited in the IBDV-infected cells after treatment with MG132 at 50 μM (Fig. 4A–C), and activations of caspase-9 and caspase-3 were dose-dependently reduced after treatment with various concentrations of MG132 (Fig. 5). Therefore, we suggested that proteasome inhibitors decrease IBDV infectivity and the anti-apoptotic effect of the proteasome inhibitor may be due to reduced viral replication.
Research data showed that mitochondrial apoptotic pathways are activated following infection with a wide variety of viruses [45]. Release of cytochrome c from mitochondria is a well-known event in apoptosis that is often required formation of downstream caspases. In this study, we for the first time demonstrated that the cytochrome c was also located in the cytoplasm of IBDV-infected cells as assayed by Western-blotting (Fig. 6). Furthermore, levels of cytochrome c in cytosol fractions were dose-dependently reduced after treatment with various concentrations of MG132 (Fig. 6). Released cytochrome c activates caspase-9 in concert with the cytoplasmic factors dATP and Apaf-1, as a result, leading consequently to activation of caspase-3 [46]. Therefore, IBDV-induced apoptosis in cultured cells suggests that IBDV is capable of influencing mitochondrial functions, leading to the release of cytochrome c into the cytoplasm, which is related to the level of viral replication, triggering the latent activity of caspases and activation of apoptosis.
In conclusion, the present study demonstrates that inhibition of the ubiquitin–proteasome pathway effectively reduces IBDV replication in cultured cells as determined by decreases in both viral RNA transcription and protein synthesis, and suggests that the anti-apoptotic effect of the proteasome inhibitor may be due to reduced IBDV viral replication.
Acknowledgments
This work was supported by a grant from the Institute of Animal Husbandry and Veterinary Medicine, Beijing Municipal Academy of Agriculture and Forestry Sciences, Beijing, the People’s Republic of China.
References
- 1.P.D. Lukert, Y.M. Saif, in Diseases of Poultry, 10th edn. (Iowa State University Press, Ames, IA, 1997), pp. 721–738
- 2.Snyder D.B., Vakharia V.N., Savage P.K. Arch. Virol. 1992;127:89. doi: 10.1007/BF01309577. [DOI] [PubMed] [Google Scholar]
- 3.Ismail N.M., Saif Y.M., Moorhead P.D. Avian Dis. 1988;32:757. doi: 10.2307/1590995. [DOI] [PubMed] [Google Scholar]
- 4.Mundt E., Beyer J., Müller H. J. Gen. Virol. 1995;76:437. doi: 10.1099/0022-1317-76-2-437. [DOI] [PubMed] [Google Scholar]
- 5.Azad A.A., Barrett S.A., Fahey K.J. Virology. 1985;143:35. doi: 10.1016/0042-6822(85)90094-7. [DOI] [PubMed] [Google Scholar]
- 6.Hudson P.J., McKern N.M., Power B.E., Azad A.A. Nucleic Acids Res. 1986;14:5001. doi: 10.1093/nar/14.12.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jagadish M.N., Staton V.J., Hudson P.J., Azad A.A. J. Virol. 1988;62:1084. doi: 10.1128/jvi.62.3.1084-1087.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Da Costa B., Chevalier C., Henry C., Huet J.-C., Petit S., Lepault J., Boot H., Delmas B. J. Virol. 2002;76:2393. doi: 10.1128/jvi.76.5.2393-2402.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spies U., Müller H., Becht H. Virus Res. 1987;8:127. doi: 10.1016/0168-1702(87)90024-4. [DOI] [PubMed] [Google Scholar]
- 10.Spies U., Müller H. J. Gen. Virol. 1990;71:977. doi: 10.1099/0022-1317-71-4-977. [DOI] [PubMed] [Google Scholar]
- 11.Thomas M., Pim D., Banks L. Oncogene. 1999;18:7690. doi: 10.1038/sj.onc.1202953. [DOI] [PubMed] [Google Scholar]
- 12.W. Hilt, D.H. Wolf, in Proteasomes: The World of Regulatory Proteolysis, eds. by W. Hilt, D.H. Wolf (Eurekah.com/Austin and Landes Biosciences, Georgetown, TX, 2000)
- 13.Pickart C.M. Annu. Rev. Biochem. 2001;70:503. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 14.Galinier R., Gout E., Lortart-Jacob H., Wood J., Chroboczek J. Biochemistry. 2002;41:14299. doi: 10.1021/bi020125b. [DOI] [PubMed] [Google Scholar]
- 15.Everett R.D., Earnshaw W.C., Findlay J., Lomonte P. EMBO J. 1999;18:1526. doi: 10.1093/emboj/18.6.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parkinson J., Everett R.D. J. Virol. 2001;75:5357. doi: 10.1128/JVI.75.11.5357-5362.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harty R.N., Brown M.E., McGettigan J.P., Wang G., Jayakar H.R., Huibregtse J.M., Whitt M.A., Schnell M.J. J. Virol. 2001;75:10623. doi: 10.1128/JVI.75.22.10623-10629.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yasuda J., Hunter E., Nakao M., Shida H. EMBO Rep. 2002;3:636. doi: 10.1093/embo-reports/kvf132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo H., Zhang J., Cheung C., Suarez A., McManus B.M., Yang D. Am. J. Pathol. 2003;163:381. doi: 10.1016/S0002-9440(10)63667-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prosch S., Priemer C., Hoflich C., Liebenthaf C., Babel N., Kruger D.H., Volk H.D. Antivir. Ther. 2003;8:555. [PubMed] [Google Scholar]
- 21.Bultmann A., Eberle J., Hass J. J. Virol. 2000;74:5373. doi: 10.1128/JVI.74.11.5373-5376.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watanable H., Tanaka Y., Shimasu Y., Sugahara F., Kuwayama M., Hiramatsu A., Kiyotani K., Yoshida T., Sakaguchi T. Microbiol. Immunol. 2005;49:835. doi: 10.1111/j.1348-0421.2005.tb03672.x. [DOI] [PubMed] [Google Scholar]
- 23.Lam K. J. Comp. Pathol. 1997;116:367. doi: 10.1016/S0021-9975(97)80053-9. [DOI] [PubMed] [Google Scholar]
- 24.Ojeda F., Skardova I., Guarda M.I., Ulloa J., Folch H. Avian Dis. 1997;41:312. doi: 10.2307/1592183. [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez-Lecompte J.C., Nino-Fong R., Lopez A., Frederick Markham R.J., Kibenge F.S. Cop. Immunol. Microbiol. Infect. Dis. 2005;28:321. doi: 10.1016/j.cimid.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 26.Tanimura N., Sharma J.M. J. Comp. Pathol. 1998;118:15. doi: 10.1016/S0021-9975(98)80024-8. [DOI] [PubMed] [Google Scholar]
- 27.Vasconcelos A.C., Lam K.M. J. Gen. Virol. 1994;75:1803. doi: 10.1099/0022-1317-75-7-1803. [DOI] [PubMed] [Google Scholar]
- 28.Liu M.H., Vakharia V.N. J. Virol. 2006;80:3369. doi: 10.1128/JVI.80.7.3369-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Himly M., Foster D.N., Bottoli I., Iacovoni J.S., Vogt P.K. Virology. 1998;248:295. doi: 10.1006/viro.1998.9290. [DOI] [PubMed] [Google Scholar]
- 30.Liu J., Zhou J., Kwang J. Virus Genes. 2002;24:135. doi: 10.1023/A:1014568532292. [DOI] [PubMed] [Google Scholar]
- 31.Liu J., Wei T., Kwang J. Virology. 2004;318:169. doi: 10.1016/j.virol.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 32.Lee D.H., Goldberg A.L. Trends Cell Biol. 1998;8:397. doi: 10.1016/S0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 33.Patnaik A., Chau V., Wills J.W. Proc. Natl. Acad. Sci. USA. 2000;97:13069. doi: 10.1073/pnas.97.24.13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schubert U., Ott D.E., Chertova E.N., Welker R., Tessmer U., Princiotta M.F., Bennink J.R., Krausslich H.G., Yewdell J.W. Proc. Natl. Acad. Sci. USA. 2000;97:13057. doi: 10.1073/pnas.97.24.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strack B., Calistri A., Accola M.A., Palu G., Gottlinger H.G. Proc. Natl. Acad. Sci. USA. 2000;97:13063. doi: 10.1073/pnas.97.24.13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalejta R.F., Shenk T. Proc. Natl. Acad. Sci. USA. 2003;100:3263. doi: 10.1073/pnas.0538058100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalejta R.F., Bechtel J.T., Shenk T. Mol. Cell Biol. 2003;23:1885. doi: 10.1128/MCB.23.6.1885-1895.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andrejeva J., Young D.F., Goodbourn S., Randall R.E. J. Virol. 2002;76:2159. doi: 10.1128/jvi.76.5.2159-2167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Didcock L., Young D.F., Goodbourn S., Randall R.E. J. Virol. 1999;73:9928. doi: 10.1128/jvi.73.12.9928-9933.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dantuma N.P., Masucci M.G. Semin. Cancer Biol. 2003;13:69. doi: 10.1016/S1044-579X(02)00101-3. [DOI] [PubMed] [Google Scholar]
- 41.Eom C.Y., Lehman I.R. Proc. Natl. Acad. Sci. USA. 2003;100:9803. doi: 10.1073/pnas.1733876100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ros C., Kempf C. Virology. 2004;324:350. doi: 10.1016/j.virol.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 43.Yu G.-Y., Lai M.M.C. J. Virol. 2005;79:644. doi: 10.1128/JVI.79.1.644-648.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Corbin-Lickfett K.A., Bridge E. Virology. 2003;315:234. doi: 10.1016/S0042-6822(03)00527-0. [DOI] [PubMed] [Google Scholar]
- 45.Boya P., Pauleau A.L., Poncet D., Gonzalez-Polo R.A., Zamzami N., Kroemer G. Biochim. Biophys. Acta. 2004;1659:178. doi: 10.1016/j.bbabio.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 46.Li P., Nijhawan D., Budihardjo I., Srinivasula S.M., Ahmad M., Alnemri E.S., Wang X.D. Cell. 1997;91:479. doi: 10.1016/S0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]