

Abstract
Using the complete genome sequences of 35 classical swine fever viruses (CSFV) representing all three genotypes and all three kinds of virulence, we analyzed synonymous codon usage and the relative dinucleotide abundance in CSFV. The general correlation between base composition and codon usage bias suggests that mutational pressure rather than natural selection is the main factor that determines the codon usage bias in CSFV. Furthermore, we observed that the relative abundance of dinucleotides in CSFV is independent of the overall base composition but is still the result of differential mutational pressure, which also shapes codon usage. In addition, other factors, such as the subgenotypes and aromaticity, also influence the codon usage variation among the genomes of CSFV. This study represents the most comprehensive analysis to date of CSFV codon usage patterns and provides a basic understanding of the mechanisms for codon usage bias.
Electronic supplementary material
The online version of this article (doi:10.1007/s11262-008-0296-z) contains supplementary material, which is available to authorized users.
Keywords: Classical swine fever virus (CSFV), Synonymous codon usage, Mutational bias, Dinucleotide bias, Subgenotype
Introduction
Synonymous codons are not used randomly. Rather, some codons are used more frequently than others. Mutational pressure and translational selection were thought to be the main factors that account for codon usage variation among genes in different organisms [1–4]. Understanding the extent and causes of biases in codon usage is essential to the understanding of viral evolution, particularly the interplay between viruses and the immune response [5]. However, in contrast to many organisms such as bacteria, yeast, Drosophila, and mammals, where codon usage bias and nucleotide composition have been studied in great detail [6], the factors shaping synonymous codon usage bias and nucleotide composition in viruses, especially in animal viruses, have been studied only to a limited extent. For human RNA viruses, it has been observed that codon usage bias is related to mutational pressure, G + C content, the segmented nature of the genome and the route of transmission of the virus [7]. For some vertebrate DNA viruses, genome-wide mutational pressure, rather than natural selection for specific coding triplets, is the main determinant of codon usage [5]. Analysis of the bovine papillomavirus type 1 (BPV1) late genes has revealed a relationship between codon usage and tRNA availability [8]. In the mammalian papillomaviruses, it has been proposed that differences from the average codon usage frequencies in the host genome strongly influence both viral replication and gene expression [9]. Codon usage may play a key role in regulating latent versus productive infection in Epstein-Barr virus [10]. Recently, it was reported that codon usage is an important driving force in the evolution of astroviruses and small DNA viruses [11, 12]. Clearly, studies of synonymous codon usage in viruses can reveal much about the molecular evolution of viruses or individual genes. Such information would be relevant in understanding the regulation of viral gene expression.
To date, little codon usage analysis has been performed on classical swine fever virus (CSFV), which is the pathogen that causes classical swine fever (CSF), an economically important and highly contagious disease of swine. Although eradicated from many countries, CSF continues to cause serious problems in different parts of the world [13]. CSFV is an enveloped virus with a single stranded RNA genome, which contains a single open reading frame (ORF) encoding a polyprotein that, following cellular and viral protease-mediated co- and post-translational processing, gives rise to 11–12 final cleavage products [14]. Studies on the phylogenetic relationship of CSFVs have divided the viruses into 3 main genotypes and 10 subgenotypes based on sequence comparisons of 190 nt of E2 sequence [15]. Based on differences in virulence, CSFVs can also be divided into three clusters, namely, highly virulent strains, moderately virulent strains, and avirulent strains [16]. Recently, we have analyzed the positive selection pressure acting on the CSFV envelope protein genes, Erns, E1, and E2, and identified several specific codons subject to diversifying positive selection in Erns and E2 [17]. In order to better understand the characteristics of the CSFV genome and to reveal more information about the viral genome, we have analyzed the codon usage and dinucleotide composition. In this report, we sought to address the following issues concerning codon usage in CSFV: (i) the extent and causes of codon bias in CSFV; (ii) the relationship between CSFV genotype and codon usage; and (iii) how CSFV virulence might affect codon usage.
Materials and methods
Materials
Three complete genomes of CSFV were previously sequenced by our laboratory (AF407339, AF091507, and AF092448) [18, 19]. The other available complete CDS of CSFV were downloaded from GenBank in March 2008 and sequences with >99% sequence identities were excluded. A total of 35 CSFV genomes [18–33] representing 6 subgenotypes (1.1, 1.2, 2.1, 2.2, 2.3, and 3.4) and all 3 kinds of virulence (highly virulent strains, moderated virulent strains, and avirulent strains) were used in this study. The genotyping of 35 CSFV genomes was performed using the CSFV sequence database (http://viro08.tiho-hannover.de/eg/eurl_virus_db.htm) based on 190 nt of E2 sequence [34]. The serial number (SN), mononucleotide composition of each genome, GenBank accession numbers, subgenotype, virulence, and other detail information are listed in Table 1.
Table 1.
Classical swine fever virus genomes used in this study
| SN | Strain | Genotypea | Virulenceb | GC3s | ENC | Mononucleotide frequencies (%) | Accession No. | Reference | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| G | A | U | C | ||||||||
| 1 | Alfort/187 | 1.1 | H | 0.500 | 51.84 | 0.2616 | 0.3140 | 0.2188 | 0.2056 | X87939 | [28] |
| 2 | CAP | 1.1 | H | 0.499 | 51.75 | 0.2608 | 0.3150 | 0.2190 | 0.2053 | X96550 | Unpublished |
| 3 | Alfort A19 | 1.1 | H | 0.500 | 51.82 | 0.2616 | 0.3138 | 0.2186 | 0.2060 | U90951 | Unpublished |
| 4 | Glentorf | 1.1 | H | 0.498 | 51.77 | 0.2605 | 0.3155 | 0.2189 | 0.2051 | U45478 | Unpublished |
| 5 | Riems/IVI | 1.1 | A | 0.499 | 51.93 | 0.2637 | 0.3113 | 0.2197 | 0.2054 | U45477 | Unpublished |
| 6 | Eystrup | 1.1 | H | 0.497 | 51.71 | 0.2602 | 0.3154 | 0.2184 | 0.2060 | NC002657 | [24] |
| 7 | Alfort/Tuebingen | 2.3 | M | 0.516 | 52.12 | 0.2640 | 0.3110 | 0.2144 | 0.2106 | J04358 | [25] |
| 8 | SWH | 1.1 | H | 0.494 | 51.53 | 0.2604 | 0.3157 | 0.2190 | 0.2048 | DQ127910 | [16] |
| 9 | C/HVRI | 1.1 | A | 0.503 | 51.89 | 0.2647 | 0.3107 | 0.2190 | 0.2051 | AY805221 | Unpublished |
| 10 | Shimen/HVRI | 1.1 | H | 0.496 | 51.59 | 0.2612 | 0.3149 | 0.2190 | 0.2049 | AY775178 | [33] |
| 11 | CWH | 1.1 | A | 0.503 | 51.91 | 0.2649 | 0.3101 | 0.2195 | 0.2055 | AY663656 | Unpublished |
| 12 | 94.4/IL/94/TWN | 3.4 | M | 0.514 | 52.15 | 0.2632 | 0.3121 | 0.2164 | 0.2083 | AY646427 | [23] |
| 13 | RUCSFPLUM | 1.2 | A | 0.503 | 52.03 | 0.2618 | 0.3144 | 0.2163 | 0.2075 | AY578688 | [28] |
| 14 | BRESCIAX | 1.2 | H | 0.496 | 51.43 | 0.2599 | 0.3161 | 0.2169 | 0.2071 | AY578687 | [28] |
| 15 | 0406/CH/01/TWN | 2.1 | U | 0.519 | 51.07 | 0.2637 | 0.3117 | 0.2131 | 0.2115 | AY568569 | Unpublished |
| 16 | 96TD | 2.1 | U | 0.521 | 51.34 | 0.2654 | 0.3104 | 0.2136 | 0.2106 | AY554397 | Unpublished |
| 17 | C strain | 1.1 | A | 0.505 | 51.95 | 0.2648 | 0.3102 | 0.2189 | 0.2061 | AY382481 | Unpublished |
| 18 | GXWZ02 | 2.1 | M | 0.512 | 51.19 | 0.2633 | 0.3118 | 0.2156 | 0.2093 | AY367767 | [31] |
| 19 | Riems | 1.1 | A | 0.501 | 51.88 | 0.2639 | 0.3114 | 0.2190 | 0.2056 | AY259122 | [24] |
| 20 | HCLV | 1.1 | A | 0.504 | 51.88 | 0.2648 | 0.3105 | 0.2191 | 0.2056 | AF531433 | Unpublished |
| 21 | Strain 39 | 2.2 | M | 0.506 | 51.26 | 0.2614 | 0.3137 | 0.2142 | 0.2107 | AF407339 | [18] |
| 22 | Strain cF114 | 1.1 | H | 0.497 | 51.5 | 0.2611 | 0.315 | 0.2188 | 0.2051 | AF333000 | [32] |
| 23 | Eystrup | 1.1 | H | 0.497 | 51.71 | 0.2602 | 0.3154 | 0.2184 | 0.206 | AF326963 | [24] |
| 24 | CS | 1.2 | A | 0.501 | 51.84 | 0.2613 | 0.3150 | 0.2166 | 0.2071 | AF099102 | [21] |
| 25 | Shimen | 1.1 | H | 0.498 | 51.48 | 0.2618 | 0.3144 | 0.2187 | 0.2052 | AF092448 | Unpublished |
| 26 | Brescia | 1.2 | H | 0.497 | 51.48 | 0.2605 | 0.3156 | 0.2177 | 0.2062 | AF091661 | Unpublished |
| 27 | HCLV | 1.1 | A | 0.503 | 52.00 | 0.2654 | 0.3097 | 0.2193 | 0.2057 | AF091507 | [19] |
| 28 | Thiverval | 1.1 | A | 0.498 | 51.82 | 0.2613 | 0.3142 | 0.2189 | 0.2056 | EU490425 | [20] |
| 29 | GPE | 1.1 | A | 0.498 | 51.68 | 0.2604 | 0.3150 | 0.2183 | 0.2063 | D49533 | [22] |
| 30 | ALD | 1.1 | H | 0.495 | 51.70 | 0.2613 | 0.3142 | 0.2194 | 0.2052 | D49532 | [22] |
| 31 | JL1(06) | 1.1 | H | 0.497 | 51.54 | 0.2608 | 0.3155 | 0.2187 | 0.2051 | EU497410 | Unpublished |
| 32 | B5b | 1.1 | A | 0.499 | 51.98 | 0.2637 | 0.3113 | 0.2198 | 0.2052 | Z46258 | [26] |
| 33 | Brescia | 1.2 | H | 0.495 | 51.55 | 0.2598 | 0.3157 | 0.2171 | 0.2073 | M31768 | [27] |
| 34 | LPS | 1.1 | A | 0.503 | 51.85 | 0.2636 | 0.3108 | 0.2190 | 0.2065 | AF352565 | Unpublished |
| 35 | Paderborn | 2.1 | M | 0.518 | 51.44 | 0.2646 | 0.3108 | 0.2143 | 0.2102 | AY072924 | [30] |
Note: a Genotyping of 35 CSFV genomes was performed using the CSFV sequence database (http://viro08.tiho-hannover.de/eg/eurl_virus_db.htm) based on 190 nt of E2 sequence [34]. b Virulence of CSFV strains summarized by Li [16]
H highly virulent strains; M moderately virulent strains; A avirulent strains; and U unclear
Codon usage indices
Relative synonymous codon usage (RSCU) values of each codon in each ORF were used to measure the synonymous codon usage [35]. RSCU values are largely independent of amino acid composition and are particularly useful in comparing codon usage between genes, or sets of genes that differ in their size and amino acid composition. The effective number of codons (ENC) was used to quantify the codon usage bias of an ORF [36], which is the best overall estimator of absolute synonymous codon usage bias [37]. The ENC values range from 20 to 61. The larger the extent of codon preference in a gene, the smaller the ENC value is. In an extremely biased gene where only one codon is used for each amino acid, this value would be 20; in an unbiased gene, it would be 61. The index GC3s was used to calculate the fraction of the nucleotides G + C at the synonymous third codon position (excluding Met, Trp, and the termination codons). Similarly, GC12s is the fraction of the nucleotide G + C at the synonymous first and second positions. The general average hydrophobicity (GRAVY) score and the frequency of aromatic amino acids (Aromo) in the hypothetical translated gene product were also computed. All the indices mentioned above were calculated using the program CodonW, version 1.4.
Correspondence analysis (COA)
The relationships between variables and samples can be explored using multivariate statistical analysis. Correspondence analysis (COA) was used to study the major trend in codon usage variation among ORFs. In order to minimize the effects of amino acid composition on codon usage, each ORF is represented as a 59-dimensional vector; each dimension corresponds to the RSCU value of one sense codon (excluding AUG, UGG, and stop codons). Major trends within this dataset can be determined using measures of relative inertia and genes ordered according to their positions along the axis of major inertia.
Relative dinucleotide abundance in CSFV ORFs
The relative abundance of dinucleotides in the CSFV ORFs was assessed using the method described by Karlin and Burge [38]. The odds ratio ρ xy = f xy/f x f y, where f x denotes the frequency of the nucleotide X and f xy the frequency of the dinucleotide XY, etc., for each dinucleotide were calculated. As a conservative criterion, for P xy > 1.23 (or <0.78), the XY pair is considered to be of high (or low) relative abundance compared with a random association of mononucleotides [38].
Statistical analysis
Correlation analysis was carried out using Spearman’s rank correlation analysis method. All statistical analyses, as well as cluster analysis, were carried out using the statistical analysis software SPSS Version 15.0.
Results
Synonymous codon usage variation in CSFV
In order to investigate the extent of codon bias in CSFV, the RSCU values of different codon in each ORF was calculated. The details of each ORF and the overall RSCU values of 59 codons in 35 CSFV genomes are shown in Table 1 and supplemental material, respectively. The preferentially used codons were A-ended, C-ended, and G-ended codons (see supplement material). It is interesting to note that no U-ended codons were used as preferential codons. In order to investigate if these 35 coding sequences of CSFV display similar compositional features, ENC and GC3s values were calculated (Table 1). The ENC values of different CSFV genes vary from 51.07 to 52.15, with a mean of 51.703 and S.D. of 0.2635. We found that all the ENC values for CSFV ORFs are high. Based on this finding, together with published data on codon usage bias among some RNA viruses [39–43], we conclude that the codon usage bias in CSFV genome is slight. Similarly, the GC3s values of each CSFV strain also confirm the homogeneity of synonymous codon usage among different CSFV viruses, which range from 49.4% to 52.1%, with a mean of 50.23% and S.D. of 0.735%.
Correspondence analysis of codon usage
To investigate synonymous codon usage variation among CSFV viruses, COA was implemented for all 35 CSFV ORFs selected for this study. Figure 1 depicts the position of each ORF on the plane defined by the first and second principal axes generated by COA on RSCU values of ORFs. The first principal axis accounts for 36.87% of the total variation. The next three axes account for 19.54%, 8.79%, and 7.54% of the variation, respectively. This observation indicates that although the first major axis explains a substantial amount of variation in trends in codon usage, the second major axis also has an appreciable impact on total variation in synonymous codon usage. It is worth noting that several CSFV Chinese C strains that can replicate efficiently in rabbits but not in swine have similar coordinates (Fig. 1) to two CSFV Riems strains, which can replicate efficiently in swine. This suggests that the host may not influence the codon usage bias between the CSFV C strain and other CSFV strains. In fact, our study demonstrated that a 12-nt insertion (CUUUUUUCUUUU) at position 61 of 3′ UTR may be responsible for the characteristics of the CSFV Chinese C strain [44].
Fig. 1.
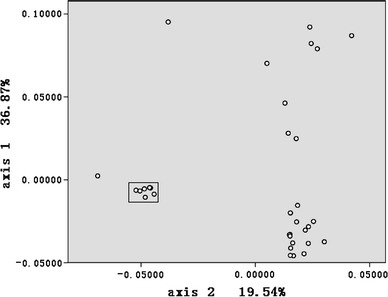
A plot of value of the first and second axis of each ORF in COA. The first axis accounts for 36.89% of all variation among ORFs and the second axis accounts for 19.54% of total vibrations. Box indicates that CSFV Chinese C strains and CSFV Riems strains were clustered together
Mutational pressure is the main factor accounting for codon usage variation in CSFV
Mutational pressure and translational selection are thought to be the main factors that account for codon usage variation in different organisms [1–4]. Hence, in order to establish which factor in CSFV can explain their codon usage, first, the G + C content at the first and second codon positions (GC12s) was compared with that at the synonymous third position (GC3s). It was found that GC12s and GC3s are significantly correlated (r = 0.483, P < 0.01). This suggests that they are most likely the result of mutational pressure, as natural selection would be expected to act differently on different codon positions. Additionally, Wright [36] suggested that the ENC-plot (ENC plotted against GC3s) be used as part of a general strategy to investigate patterns of synonymous codon usage. Genes, whose codon choice is constrained only by a G + C mutation bias, will lie on or just below the curve of the predicted values. As shown in Fig. 2, all of the spots lie below the expected curve, indicating that the codon usage bias in these 35 genomes is greatly influenced by the G + C compositional constraints. Furthermore, the correlation between the first or second axis values in COA and GC12s or GC3s values of each strain was analyzed. As shown in Table 4, the first axis value in COA of each selected genome, which contains most of the variation in synonymous codon usage bias between these genomes, is closely correlated with the GC composition at the first, second, and third codon position. The second axis in the COA of each gene is also closely correlated with the GC12s. This analysis indicated that most of the codon usage bias among different ORFs is directly related to the nucleotide composition. Therefore, the compositional constraint is the main determinant of the variation in synonymous codon usage among different CSFV ORFs.
Fig. 2.
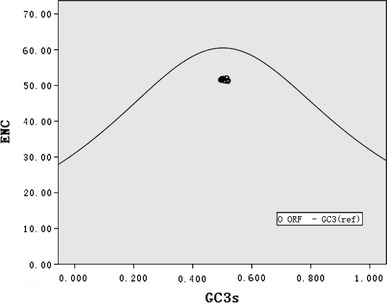
Effective number of codons used in each ORF plotted against the GC3s. The continuous curve plots the relationship between GC3s and NEC in the absence of selection. All of spots lie below the expected curve
Table 4.
Summary of correlation analysis between the first two axes in COA and GC12s, GC3s, GRAVY, or aromaticity in the selected 35 CSFV ORFs
| GRAVY | Aromaticity | GC3s | GC12s | ||
|---|---|---|---|---|---|
| Axis 1 | r | −0.51 | −0.526** | 0.867** | 0.614** |
| P | 0.386 | 0.001 | <0.001 | <0.001 | |
| Axis 2 | r | −0.51 | 0.473** | −0.244 | −0.368* |
| P | 0.386 | 0.002 | 0.079 | 0.015 | |
* P-value ≤ 0.05
** P-value ≤ 0.01
The relative abundance of dinucleotide and CpG suppression also shape the codon usage in CSFV
It has been reported that dinucleotide biases can affect codon bias. To study the possible effect of the composition of dinucleotides on codon usage in CSFV, the relative abundances of the 16 dinucleotides in the 35 CSFV genomes were calculated. As shown in Table 2, the frequencies of occurrence for dinucleotides were not randomly distributed and no dinucleotides were present at the expected frequencies. The relative abundance of CpG showed the most marked deviation from the “normal range” (mean ± S.D. = 0.426 ± 0.018). The relative abundance of UpG and CpC also showed slight deviation from the “normal range” (mean ± S.D. = 1.250 ± 0.018 and 1.262 ± 0.019, respectively). Among the 16 dinucleotides, 6 are correlated with the first axis value in COA; 8 are correlated with the second axis value in COA (Table 3). These observations indicated that the composition of dinucleotides, which are independent of the overall base composition but still the result of differential mutational pressure, also determines the variation in synonymous codon usage among different CSFV ORFs. To study the possible effects of CpG under-representation on codon usage bias, the RSCU value of the eight codons that contain CpG (CCG, GCG, UCG, ACG, CGC, CGG, CGU, and CGA) were analyzed. Of these eight codons, seven [GCG (mean 0.375), UCG (mean 0.125), ACG (mean 0.406), CGC (mean 0.141) CGG (mean 0.200), CGU (mean 0.0794), and CGA (mean 0.139)] were markedly suppressed, while CCG (mean 0.676) is slightly suppressed. To study the possible effects of UpG and CpC over-representation on codon usage bias, codons that contain UpG (UUG, CUG, GUG, and UGC) or CpC (UCC, CCU, CCC, CCA, CCG, ACC, GCC) were analyzed. Of these five UpG containing codons, three [CUG (mean 1.677), GUG (mean 1.408), and UUG (mean 1.366)] were markedly over-used. Since both two cysteine codons [UGC (mean 1.082), UGU (mean 0.918)] begin with UpG, these two UpG containing codons are almost equally used. Of seven CpC containing codons, two [ACC (mean 1.342) and GCC (mean 1.347)] were over-used. UCC (mean 0.745) is slightly suppressed. In the rest four CpC containing codons for proline, CCA (mean 1.520) is markedly over-used; CCG (mean 0.676), which also is a CpG containing codon, is slightly suppressed; CCU (mean 0.933) and CCC (mean 0. 871) are almost equally used.
Table 2.
Relative abundance of the 16 dinucleotides in 35 Classical swine fever virus with complete genomes available
| Relative abundance of the 16 dinucleotides | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| TT | TC | TA | TG | CT | CC | CA | CG | AT | |
| Rangea | 0.986–1.126 | 0.752–0.867 | 0.837–0.918 | 1.176–1.277 | 1.152–1.265 | 1.218–1.301 | 1.115–1.194 | 0.399–0.486 | 0.855–0.934 |
| Mean ± S.Db | 1.052 ± 0.036 | 0.828 ± 0.027 | 0.865 ± 0.019 | 1.250 ± 0.018 | 1.207 ± 0.029 | 1.262 ± 0.019 | 1.176 ± 0.019 | 0.426 ± 0.018 | 0.901 ± 0.016 |
| Relative abundance of the 16 dinucleotides | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| AC | AA | AG | GT | GC | GA | GG | |||
| Rangea | 1.046–1.086 | 0.953–0.992 | 1.047–1.083 | 0.877–0.947 | 0.841–0.900 | 0.986–1.033 | 1.134–1.185 | ||
| Mean ± S.Db | 1.068 ± 0.009 | 0.972 ± 0.009 | 1.063 ± 0.009 | 0.908 ± 0.016 | 0.868 ± 0.014 | 1.009 ± 0.010 | 1.158 ± 0.009 | ||
Note: a The range of 35 CSFVs’ relative dinucleotide ratios
bMean values of 35 CSFVs relative dinucleotide ratios ± S.D
Table 3.
Summary of correlation analysis between the first two axes in COA and sixteen dinucleotides in the selected viruses
| Sixteen dinucleotides | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TT | TC | TA | TG | CT | CC | CA | CG | AT | AC | AA | AG | GT | GC | GA | GG | ||
| Axis 1 | r | −0.069 | −0.399** | 0.327* | 0.309* | 0.235 | −0.079 | −0.824** | 0.531** | −0.159 | 0.104 | 0.282 | −0.111 | 0.223 | 0.153 | 0.02 | −0.313* |
| P | 0.346 | 0.009 | 0.028 | 0.035 | 0.087 | 0.326 | <0.001 | 0.001 | 0.181 | 0.276 | 0.05 | 0.262 | 0.99 | 0.19 | 0.455 | 0.033 | |
| Axis 2 | r | −0.716** | 0.559** | 0.543** | 0.067 | 0.705** | −0.456 | 0.195 | −0.727** | 0.237 | −0.028 | 0.054 | 0.079 | 0.281 | −0.79** | −0.528** | 0.314* |
| P | <0.001 | <0.001 | <0.001 | 0.351 | <0.001 | 0.003 | 0.131 | <0.001 | 0.085 | 0.437 | 0.38 | 0.327 | 0.051 | <0.001 | 0.001 | 0.033 | |
* P-value ≤ 0.05
** P-value ≤ 0.01
The effect of selection pressure on codon usage
As shown in Fig. 2, the majority of the actual ENC values are slightly lower than the expected ENC values. This implies that although codon bias is mainly explained by mutational pressure, there are other factors, with less of an effect, that also influence the codon bias. To test that whether any selection pressure contributes to the codon usage variation between these CSFVs, we performed a correlation analysis between axis values in COA and aromaticity or GRAVY score of each polyprotein. It was found that both axis 1 and axis 2 are significantly correlated with the aromaticity score (r = − 0.526, P < 0.01, r = 0.473, P < 0.01, respectively), indicating that the frequency of aromatic amino acids (Phe, Tyr, Trp) in the hypothetical translated gene product of each ORF is also related to the observed variation in codon bias. No significant relationship was found between axis values in COA and GRAVY using Spearman’s correlation (Table 4).
The effect of CSFV genotype and virulence on codon usage
Beyond the factors mentioned above, we were also concerned with how CSFV genotype and virulence might affect codon usage. Based on the variation in RSCU values among the 35 CSFV genomes, a cluster tree was generated by the hierarchical clustering method. As shown in Fig. 3, these 35 CSFV genomes were divided into 7 sublineages. Sublineages I-1 and I-2 contain all subgenotype 1.1 strains, and sublineage I-2 contains almost all avirulent strains in genotype 1.1. Sublineages I-3, II-1, II-2, II-3, and II-4 contain the subgenotypes 1.2, 2.1, 2.3, 3.4, and 2.2, respectively. It should be noted that the distance between sublineages II-2 and II-3 is closer than the distance between sublineages II-2 and II-4 (Fig. 3). Since sublineages II-2 and II-4 contain the subgenotypes 2.3 and 2.2, respectively, which, in turn, belong to genotype 2, the distance between two sublineages is closer than the distance between sublineage II-2 and sublineage II-3 (contains the subgenotype 3.4). This may be because of the special characteristics of strain 39 in subgenotype 2.2 (see Discussion).
Fig. 3.
A dendrogram representing the extent of divergence in synonymous codon usage in 35 CSFV strains constructed with the hierarchical clustering method. SG subgenotype; SL sublineage
Discussion
Studies of synonymous codon usage in viruses can reveal much about viral genomes. In the present study, we analyzed synonymous codon usage and dinucleotide composition in CSFV. We found that, as for other viruses such as H5N1 influenza virus (mean ENC = 50.91) [39, 43], SARS-covs (mean ENC = 48.99) [40], human Bocavirus (mean ENC = 44.45) [41], and foot-and-mouth virus (mean ENC = 51.53) [42], the ENC values for CSFV are high (mean ENC = 51.7), indicating that the overall extent of codon usage bias in CSFV genomes is low. In fact, Jenkins et al. [7] have previously reported that the overall extent of codon usage bias in RNA viruses is low with an average ENC value close to 45. Nevertheless, we still wished to determine the factors that constrain codon usage in CSFV. According to the selection–mutation–drift model [35, 45], mutational pressure and translational selection are generally thought to be the main factors that account for codon usage variation between genes in different organisms [1–4]. In our study, the general correlation between codon usage bias and base composition we observed suggests that mutational pressure is the main factor that determines codon usage bias in CSFV; this conclusion is also supported by the highly significant correlation between GC12s and GC3s (r = 0.483, P < 0.01), and the result of ENC-plot (Fig. 2). Since mutation rates in RNA viruses are much higher than those in DNA viruses [46], it is understandable that mutational pressure is the major cause of codon usage bias in the 35 CSFV strains included in this study.
The majority of the actual ENC values are slightly lower than the expected ENC values (Fig. 2), indicating that there are other factors, albeit with smaller effects, that also influence codon bias. We then asked how CSFV genotype and virulence might affect codon usage. Our cluster analysis revealed that the CSFV genotype also constrains codon usage, since different CSFV strains with the same genotype were clustered together with only one exception, CSFV strain 39 (Fig. 3). CSFV strain 39 (AF407339) was, however, postulated to be a recombinant virus by He et al. [47]. To date phylogenetic analyses have been performed largely on one or three genomic regions but not the complete genome, which might limit it to genotype recombinant viruses. On the other hand, our RSCU-based cluster was based on the complete CDS of each virus. Therefore, it is expected that differences will arise between phylogenetic analyses of recombinant viruses using the two different clustering methods. Our results suggest that CSFV strain 39 might indeed be a recombinant virus and also raised interesting questions about CSFV evolution and the relative contribution of intertypic recombination to the generation of CSFV genetic diversity. Furthermore, our results indicate that virulence is not significantly influenced by codon bias, since not all avirulent strains were clustered together. Although 9 of the 11 avirulent strains of subgenotype 1.1 were clustered together (Fig. 3 subgenotype 1.1B), the other avirulent strains were clustered with highly virulent strains, and 5 moderately virulent strains were also not clustered together (Fig. 3). At present, however, only small numbers of complete CDS of CSFV are available, and these only six cover subgenotypes. Clearly, more complete sequences are needed to allow us to make more precise judgments.
Due to a previous report about CpG under-representation in RNA and small DNA viruses [10], we wanted to determine if the relative abundances of dinucleotides in CSFV affects codon usage. The frequencies of occurrence for dinucleotides were not randomly distributed and no dinucleotides were present at the expected frequencies (Table 2). The general correlation between the axis values in COA and the relative dinucleotide abundances (Table 3) suggests that codon usage in CSFV can also be strongly influenced by underlying biases in dinucleotide frequencies. As a case in point, all CpG containing codons are markedly suppressed. The marked CpG deficiency is a common phenomenon in small eukaryotic viruses [48, 49]. The CpG deficiency was proposed to be related to the immunostimulatory properties of unmethylated CpGs, which were recognized by the host’s innate immune system as a pathogen signature [5, 49]. Indeed, unmethylated CpG motifs in DNA sequences can be recognized by TLR9 [50], and unmethylated CpG motifs in ssRNA may stimulate monocytes through a novel mechanism [51]. This notion was further supported by the fact that CpG is not suppressed in the genomes of most large viruses [48, 49] because they might encode a range of proteins that interfere with cellular pathogen recognition. As a case in point, vaccinia poxvirus encodes agonists of TLRs [52]. In CSFV, Ruggli et al. and our group have shown that Npro and Erns protein can prevent both poly(IC)-and NDV-mediated IFN-α/β induction [53–56]. Inhibition by Npro protein is thought to involve an inactivation of interferon regulatory transcription factor 3 (IRF-3) [57]. However, no evidence has been found to support the notion that Npro and Erns proteins interfere with ssRNA through the recognition of unmethylated CpG motifs. It is most likely that the codon usage bias in CSFV may be also related to its host’s innate immune selective forces.
Taken together, our study reveals that codon usage bias in CSFV is slight and mutational pressure is the main factor that affects codon usage variation in CSFV. Other factors, such as dinucleotide composition, genotype, aromaticity, and even innate immune selective forces also significantly influence codon usage bias. However, due to a lack of sequence data and detailed information about these isolations, it is currently impossible to performance an exhaustive analysis about CSFV codon usage. Clearly, a more comprehensive analysis is needed, based on more available data, to reveal more about the viral genome. To our knowledge, this work is the first report of codon usage analysis in CSFV, and it provides a basic understanding of the mechanisms that give rise to codon usage bias. The results we have reported are also useful in understanding the processes involved in CSFV evolution.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by Key Projects in the National Science & Technology Pillar Program of China (2006BAD06A03).
References
- 1.Karlin S, Mrazek J. J. Mol. Biol. 1996;262:459–472. doi: 10.1006/jmbi.1996.0528. [DOI] [PubMed] [Google Scholar]
- 2.Lesnik T, Solomovici J, Deana A, Ehrlich R, Reiss C. J. Theor. Biol. 2000;202:175–185. doi: 10.1006/jtbi.1999.1047. [DOI] [PubMed] [Google Scholar]
- 3.Sharp PM, Li WH. Nucleic Acids Res. 1986;14:7737–7749. doi: 10.1093/nar/14.19.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharp PM, Tuohy TM, Mosurski KR. Nucleic Acids Res. 1986;14:5125–5143. doi: 10.1093/nar/14.13.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shackelton LA, Parrish CR, Holmes EC. J. Mol. Evol. 2006;62:551–563. doi: 10.1007/s00239-005-0221-1. [DOI] [PubMed] [Google Scholar]
- 6.Mooers AO, Holmes EC. Trends Ecol. Evol. 2000;15:365–369. doi: 10.1016/S0169-5347(00)01934-0. [DOI] [PubMed] [Google Scholar]
- 7.Jenkins GM, Holmes EC. Virus Res. 2003;92:1–7. doi: 10.1016/S0168-1702(02)00309-X. [DOI] [PubMed] [Google Scholar]
- 8.Zhou J, Liu WJ, Peng SW, Sun XY, Frazer I. J. Virol. 1999;73:4972–4982. doi: 10.1128/jvi.73.6.4972-4982.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao KN, Liu WJ, Frazer IH. Virus Res. 2003;98:95–104. doi: 10.1016/j.virusres.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 10.Karlin S, Blaisdell BE, Schachtel GA. J. Virol. 1990;64:4264–4273. doi: 10.1128/jvi.64.9.4264-4273.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sewatanon J, Srichatrapimuk S, Auewarakul P. Intervirology. 2007;50:123–132. doi: 10.1159/000098238. [DOI] [PubMed] [Google Scholar]
- 12.van Hemert FJ, Berkhout B, Lukashov VV. Virology. 2007;361:447–454. doi: 10.1016/j.virol.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edwards S, Fukusho A, Lefevre PC, Lipowski A, Pejsak Z, Roehe P, Westergaard J. Vet. Microbiol. 2000;73:103–119. doi: 10.1016/S0378-1135(00)00138-3. [DOI] [PubMed] [Google Scholar]
- 14.C.M. Rice, Flaviviridae: The Viruses and their Replication (Lippincott Raven, Philadelphia, 1996)
- 15.Paton DJ, McGoldrick A, Greiser-Wilke I, Parchariyanon S, Song JY, Liou PP, Stadejek T, Lowings JP, Bjorklund H, Belak S. Vet. Microbiol. 2000;73:137–157. doi: 10.1016/S0378-1135(00)00141-3. [DOI] [PubMed] [Google Scholar]
- 16.Li X, Xu Z, He Y, Yao Q, Zhang K, Jin M, Chen H, Qian P. Virus Genes. 2006;33:133–142. doi: 10.1007/s11262-005-0048-2. [DOI] [PubMed] [Google Scholar]
- 17.Tang F, Pan Z, Zhang C. Virus Res. 2008;131:132–135. doi: 10.1016/j.virusres.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 18.Wu HX, Zhang CY, Zheng CY, Guo JQ. Wuhan Univ. J. Nat. Sci. 2001;6:864–866. doi: 10.1007/BF02850922. [DOI] [Google Scholar]
- 19.Wu HX, Wang JF, Zhang CY, Fu LZ, Pan ZS, Wang N, Zhang PW, Zhao WG. Virus Genes. 2001;23:69–76. doi: 10.1023/A:1011187413930. [DOI] [PubMed] [Google Scholar]
- 20.Fan Y, Zhao Q, Zhao Y, Wang Q, Ning Y, Zhang Z. Virus Genes. 2008;36:531–538. doi: 10.1007/s11262-008-0229-x. [DOI] [PubMed] [Google Scholar]
- 21.Grebennikova T.V., Zaberezhnyi A.D., Sergeev V.A., Biketov S.F., Aliper T.I., Nepoklonov E.A. Mol. Gen. Mikrobiol. Virusol. 1999;2:34–40. [PubMed] [Google Scholar]
- 22.Ishikawa K, Nagai H, Katayama K, Tsutsui M, Tanabayashi K, Takeuchi K, Hishiyama M, Saitoh A, Takagi M, Gotoh K, et al. Arch. Virol. 1995;140:1385–1391. doi: 10.1007/BF01322665. [DOI] [PubMed] [Google Scholar]
- 23.Lin YJ, Chien MS, Deng MC, Huang CC. Virus Genes. 2007;35:737–744. doi: 10.1007/s11262-007-0154-4. [DOI] [PubMed] [Google Scholar]
- 24.Mayer D, Thayer TM, Hofmann MA, Tratschin JD. Virus Res. 2003;98:105–116. doi: 10.1016/j.virusres.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 25.Meyers G, Rumenapf T, Thiel HJ. Virology. 1989;171:555–567. doi: 10.1016/0042-6822(89)90625-9. [DOI] [PubMed] [Google Scholar]
- 26.Moormann RJ, van Gennip HG, Miedema GK, Hulst MM, van Rijn PA. J. Virol. 1996;70:763–770. doi: 10.1128/jvi.70.2.763-770.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moormann RJ, Warmerdam PA, van der Meer B, Schaaper WM, Wensvoort G, Hulst MM. Virology. 1990;177:184–198. doi: 10.1016/0042-6822(90)90472-4. [DOI] [PubMed] [Google Scholar]
- 28.Risatti GR, Borca MV, Kutish GF, Lu Z, Holinka LG, French RA, Tulman ER, Rock DL. J. Virol. 2005;79:3787–3796. doi: 10.1128/JVI.79.6.3787-3796.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruggli N, Moser C, Mitchell D, Hofmann M, Tratschin JD. Virus Genes. 1995;10:115–126. doi: 10.1007/BF01702592. [DOI] [PubMed] [Google Scholar]
- 30.Uttenthal A, Le Potier MF, Romero L, De Mia GM, Floegel-Niesmann G. Vet. Microbiol. 2001;83:85–106. doi: 10.1016/S0378-1135(01)00409-6. [DOI] [PubMed] [Google Scholar]
- 31.Wu XS, Luo TR, Liao SH, Liu QZ, Huang WJ. Chin. J. Vet. Sci. 2003;25:125–128. [Google Scholar]
- 32.Nie Y, Ke Y, Chen J, Ding M. Wei Sheng Wu Xue Bao. 2001;41:452–456. [PubMed] [Google Scholar]
- 33.Zhao JJ, Cheng D, Li N, Sun Y, Shi Z, Zhu QH, Tu C, Tong GZ, Qiu HJ. Vet. Microbiol. 2008;126:1–10. doi: 10.1016/j.vetmic.2007.04.046. [DOI] [PubMed] [Google Scholar]
- 34.Dreier S, Zimmermann B, Moennig V, Greiser-Wilke I. J. Virol. Methods. 2007;140:95–99. doi: 10.1016/j.jviromet.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 35.Sharp PM, Li WH. J. Mol. Evol. 1986;24:28–38. doi: 10.1007/BF02099948. [DOI] [PubMed] [Google Scholar]
- 36.Wright F. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- 37.Comeron JM, Aguade M. J. Mol. Evol. 1998;47:268–274. doi: 10.1007/PL00006384. [DOI] [PubMed] [Google Scholar]
- 38.Karlin S, Burge C. Trends Genet. 1995;11:283–290. doi: 10.1016/S0168-9525(00)89076-9. [DOI] [PubMed] [Google Scholar]
- 39.Ahn I, Jeong BJ, Bae SE, Jung J, Son HS. Eur. J. Epidemiol. 2006;21:511–519. doi: 10.1007/s10654-006-9031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu W, Zhou T, Ma J, Sun X, Lu Z. Virus Res. 2004;101:155–161. doi: 10.1016/j.virusres.2004.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao S, Zhang Q, Liu X, Wang X, Zhang H, Wu Y, Jiang F. Biosystems. 2008;92:207–214. doi: 10.1016/j.biosystems.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong J, Li Y, Zhao S, Liu S, Zhang Z. Virus Genes. 2007;35:767–776. doi: 10.1007/s11262-007-0159-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou T, Gu W, Ma J, Sun X, Lu Z. Biosystems. 2005;81:77–86. doi: 10.1016/j.biosystems.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Wang Q, Lu X, Zhang C, Fan X, Pan Z, Xu L, Wen G, Ning Y, Tang F, Xia Y. Virology. 2008;374:390–398. doi: 10.1016/j.virol.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 45.Bulmer M. Genetics. 1991;129:897–907. doi: 10.1093/genetics/129.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drake JW, Holland JJ. Proc. Natl. Acad. Sci. USA. 1999;96:13910–13913. doi: 10.1073/pnas.96.24.13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He CQ, Ding NZ, Chen JG, Li YL. Virus Res. 2007;126:179–185. doi: 10.1016/j.virusres.2007.02.019. [DOI] [PubMed] [Google Scholar]
- 48.Karlin S, Doerfler W, Cardon LR. J. Virol. 1994;68:2889–2897. doi: 10.1128/jvi.68.5.2889-2897.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woo PC, Wong BH, Huang Y, Lau SK, Yuen KY. Virology. 2007;369:431–442. doi: 10.1016/j.virol.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wagner H. Trends Immunol. 2004;25:381–386. doi: 10.1016/j.it.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 51.Sugiyama T, Gursel M, Takeshita F, Coban C, Conover J, Kaisho T, Akira S, Klinman DM, Ishii KJ. J. Immunol. 2005;174:2273–2279. doi: 10.4049/jimmunol.174.4.2273. [DOI] [PubMed] [Google Scholar]
- 52.Harte MT, Haga IR, Maloney G, Gray P, Reading PC, Bartlett NW, Smith GL, Bowie A, O’Neill LA. J. Exp. Med. 2003;197:343–351. doi: 10.1084/jem.20021652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen L, Xia YH, Pan ZS, Zhang CY. Protein Expr. Purif. 2007;55:379–387. doi: 10.1016/j.pep.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 54.Ruggli N, Bird BH, Liu L, Bauhofer O, Tratschin JD, Hofmann MA. Virology. 2005;340:265–276. doi: 10.1016/j.virol.2005.06.033. [DOI] [PubMed] [Google Scholar]
- 55.Ruggli N, Tratschin JD, Schweizer M, McCullough KC, Hofmann MA, Summerfield A. J. Virol. 2003;77:7645–7654. doi: 10.1128/JVI.77.13.7645-7654.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xia YH, Chen L, Pan ZS, Zhang CY. J. Biochem. Mol. Biol. 2007;40:611–616. doi: 10.5483/bmbrep.2007.40.5.611. [DOI] [PubMed] [Google Scholar]
- 57.La Rocca SA, Herbert RJ, Crooke H, Drew TW, Wileman TE, Powell PP. J. Virol. 2005;79:7239–7247. doi: 10.1128/JVI.79.11.7239-7247.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.