

Abstract
Fecal specimens, including swabs and litter extracts, collected from chickens, domestic ducks, turkeys, and Canadian geese were tested using degenerate primers targeting regions encoding for conserved amino acid motifs (YGDD and DY(T/S)(R/K/G)WDST) in calicivirus RNA-dependent RNA polymerases. Similar motifs are also present in other RNA viruses. Two fecal specimens and 18 litter extracts collected from chickens and turkeys yielded RT-PCR products. BLAST search and phylogenetic analysis revealed that all amplicons represented picornaviruses that clustered into two major groups. Four chicken and one turkey samples yielded 250 bp amplicons with 84–91% nucleotide identity to the recently described turkey hepatitis viruses, while 280 and 283 bp amplicons obtained from 11 chicken and 4 turkey samples represented novel picornaviruses with the closest nucleotide identity to kobuviruses (54–61%) and turdiviruses (47–54%). Analysis of 2.2–3.2 kb extended genome sequences including the partial P2 (2C) and complete P3 (3A, 3B (VPg), 3Cpro, and 3Dpol) regions of selected strains indicated that viruses yielding the 280/283 bp amplicons represent a putative new genus of Picornaviridae. The 3′-non-translated region (NTR) of the turkey hepatitis-like viruses described in this study was significantly longer (641–654 nt) than that of any of the other piconaviruses and included a putative short open reading frame (ORF). In summary, we report the molecular detection of novel picornaviruses that appear to be endemic in both chickens and turkeys.
Keywords: Picornavirus, Chicken, Turkey, Turkey hepatitis
Introduction
Picornaviruses are small, non-enveloped, single-stranded positive strand RNA viruses with a ~7–9 kb genome. All known picornavirus genomes encode a single long open reading frame (ORF) from which a long polyprotein is translated and cleaved by virus encoded proteases to yield the individual structural and non-structural viral proteins. The long ORF has been divided into three regions: P1, P2, and P3. The P1 region encodes the viral capsid proteins while the P2 and P3 regions encode proteins involved in protein processing or genome replication: 2Apro, 2B, 2C and 3A, 3B (VPg), 3Cpro, 3Dpol, respectively. Picornaviruses have been described in humans and different animal species and can be the causative agents of a wide variety of diseases. The Picornaviridae family currently consists of 12 genera: Enterovirus, Cardiovirus, Aphtovirus, Hepatovirus, Parechovirus, Erbovirus, Kobuvirus, Teschovirus, Sapelovirus, Senecavirus, Tremovirus, and Avihepatovirus [1]. Several other picornaviruses including the recently described turdiviruses and turkey hepatitis viruses still await species or genus assignment.
Turdiviruses were discovered in tracheal and cloacal swabs obtained from dead wild birds of the genus Turdus in the family Turdidae [2]. Two distinct groups representing two proposed new genera (Ortho- and Paraturdivirus) have been described. These viruses could not be propagated in cell culture or in chicken embryos and their prevalence, host range, and disease burden are unknown.
Picornaviruses that were tentatively named turkey hepatitis viruses (THV) were recently discovered in liver samples collected from diseased turkey poults with turkey hepatitis. THV was also detected in bile, intestine, serum, and cloacal swabs of diseased animals and is the candidate causative agent of turkey hepatitis [3].
Based on the morphological descriptions of small round viruses in healthy and diseased avian species [4–8] we initiated a study for the molecular detection of caliciviruses in avian fecal specimens. This study utilized a broadly reactive primer set targeting conserved amino acid motifs encoding regions present in calicivirus RNA-dependent RNA polymerases (RdRp) and are partially also present in other viral RdRps. As part of the study here we report the serendipitous detection of novel picornaviruses in chicken and turkey samples that included diagnostic cases with runting–stunting syndrome (RSS).
Materials and methods
Samples
Fecal swabs collected from 42 broiler chickens, 25 domestic ducks, 11 turkeys, 149 Canadian geese in Delaware, and 73 litter extracts collected from 4 chicken and 4 turkey farms in North Carolina were tested (Table 1). Twenty-eight of the 42 chicken swabs represented diagnostic cases with RSS. All of the other samples were collected from healthy animals. Swabs were soaked in 1 ml sterile PBS. Litter samples were saturated with sterile PBS and washes were collected. All samples were aliquoted and stored at −80°C.
Table 1.
Samples tested in this study
| Species | Sample | No. of samples | Status | Place | Year |
|---|---|---|---|---|---|
| Domestic duck | Swab | 25 | Healthy | Delaware | 2010 |
| Domestic turkey | Swab | 11 | Healthy | Delaware | 2010 |
| Chicken (broiler) | Swab | 14 | Healthy | Delaware | 2010 |
| Canadian geese | Swab | 149 | Healthy | Delaware | 2010 |
| Chicken (broiler) | Swab | 28 | Diagnostic casea | Delaware | 2007–2010 |
| Chicken (broiler)b | Litter | 9 | Not available | North Carolina | 2005 |
| Chicken (layer)c | Litter | 48 | Not available | North Carolina | 2005 |
| Turkeyd | Litter | 16 | Not available | North Carolina | 2005 |
aDiagnostic cases represent birds with runting–stunting enteritis
bLitter samples were collected from 21 to 42 days old boiler chickens housed in three separate farms (2–4 samples/farm)
cLitter samples were collected from 22 to 78 weeks old egg layers from six different houses of the same farm (6–12 samples/house)
dLitter samples were collected from 20 to 21 days old turkey poults from two different houses from each of four different farms (2 samples/house; 4 samples/farm)
RNA extraction
Equal volumes of 2–4 samples from the same sample group were pooled together and viral RNA was extracted by the QIAamp viral RNA mini kit on a QIAvac 24 plus vacuum manifold (Qiagen Inc., Valencia, CA), according to the manufacturer’s instructions. Twenty-two sample pools along with negative (deionized water) and positive (recovirus) controls were extracted at a time. The titer of tissue culture-adapted FT285 recovirus strain was adjusted to 104 pfu/ml and 150 μl aliquots were made and stored at −80°C. Extracted RNA was eluted in 30 μl buffer and stored at −80°C. RNA from individual samples of RT-PCR positive pools were extracted as described above.
RT-PCR screening and DNA sequencing
Viral RNA was amplified from 3 μl of extracted RNA template in 25 μl reactions using the AccessQuick RT-PCR system (Promega, Madison, WI) according to manufacturer’s instructions with P289/P290 as it was described in our previous studies [9–11]. Reactions were analyzed on 2% agarose gels in the presence of ethidium bromide. RT-PCR products were excised from agarose gels, recovered by the Wizard SV Gel and PCR-clean up system and cloned into pGEM-T vector (Promega, Madison, WI) according to the manufacturer’s protocols. Positive clones were identified by PCR. Plasmid DNA was isolated from 2 ml cultures by the Wizard Plus SV Miniprep DNA-purification system (Promega, Madison, WI) according to the manufacturer’s instructions and sequenced using M13 forward and reverse primers by the chain termination method on an ABI PRISM® 3730 DNA Analyzer (Applied Biosystems Inc., Foster city, CA). Each sample was sequenced in both directions from two-independent clones.
Genomic sequence extension
The genome of selected picornavirus strains representing each group was amplified to the 3′ end (~1,000 nt) with strain-specific forward primers (CTCCACTACCTCAACACTATCC for group 1, TGTGATGATTGGYGGYATG for group 2, and ATGAGATGGAAGGAGGRATG for group 3 viruses, respectively) and an oligo-dT primer. Further extension of the P3 region, encoding 3A, 3B (VPg), 3Cpro, and 3Dpol proteins was achieved by primer walking using strain-specific reverse primers and degenerate primers targeting nucleotide sequences encoding for conserved amino acid motifs DDxGQ (TTCATCGAYGACATCGGICAR) in the 2C and GxCG (CCTTCSAGGGYITSTGYGG) in the 3Cpro regions.
Sequence and phylogenetic analysis
BLAST analyses of sequences without the primers were run against NCBI databases. Multiple sequence alignments of nucleotide and amino acid sequences were created using the Omiga v2.0 software (Oxford Molecular Ltd, Oxford, UK). Dendrograms were constructed by the Unweighted Pair Group Method with Arithmetic Mean (UPGMA) and the Neighbor-Joining clustering methods of the molecular evolutionary genetics analysis (MEGA version 3.1) software with Jukes–Cantor and Poisson correction distance calculations for nucleotide and amino acid sequence alignments, respectively [12]. The confidence values of the internal nodes were obtained by performing 1,025 bootstrap analyses. Picornavirus sequences representing all established or proposed genera were included in the analyses (accession numbers are listed in Fig. 1).
Fig. 1.
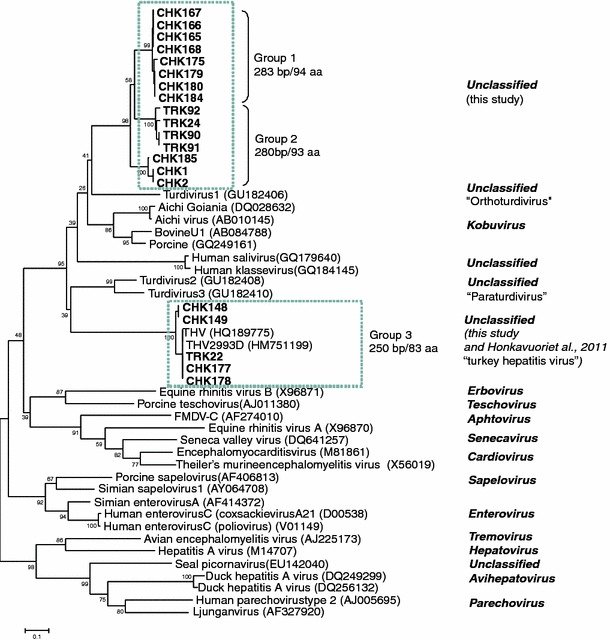
Phylogenetic analysis of partial 3Dpol protein sequences. The alignment contained 73 amino acids after columns with gaps were removed. The dendrogram was constructed by the Neighbor-Joining clustering method of MEGA version 3.1 with Poisson correction distance calculations and 1,025 bootstrap analyses
Predictions of 3′-NTR secondary structure
Secondary structure predictions for 3′-NTR regions were generated using the Webserver for Aligning non-coding RNAs (WAR, http://genome.ku.dk/resources/war/) [13]. WAR was used to generate consensus alignments and secondary structures for the terminal 240 nt of each of the 3′-NTRs (CHK148, CHK168, TRK22, TRK24, Aichi virus, and Turdivirus 1), as well as the extended 5′-portions of CHK148 and TRK22 viruses. WAR submissions are limited to 250 nt, and the greatest homology among the 3′-NTRs was in the 3′-terminal ~250 nt.
The size of 240 nt for the 3′-consensus was chosen due to this being the size of the Aichi virus 3′-NTR, the smallest of the viruses examined. For the extended 3′-NTRs of CHK148 and TRK22, the 5′-most 250 nt were used for secondary structure predication.
FASTA files of RNA alignments were uploaded to the WAR server using DNAStar MegAlign software (DNAStar, Lasergene 8.1, Madison, WI) and consensus alignments and secondary structure predictions were generated using 14 simultaneous RNA structural prediction programs (listed at http://genome.ku.dk/resources/war/).
Predictions were ranked according to free energies, highest covariance scores, average bp probability, and the fraction of canonical base-pairing. Structures predicted by more than one program are reported. Owing to differences in relatedness, consensus structures were developed using comparisons of alignments of CHK148 and TRK22, and alignments of Aichi virus, Turdivirus 1, CHK168, and TRK24 strain 3′-NTRs.
Tissue culture
RT-PCR positive samples were centrifuged at 10,000×g for 15 min and sterile filtered through 0.2 μm syringe filters (Millipore, Billerica, MA). Filtered samples were confirmed for the presence of picornaviruses by RT-PCR and sequencing and inoculated onto primary chicken embryo liver/fibroblast, LMH (chicken liver; ATCC CRL-2117), Vero (African green monkey; kidney; ATCC CRL-1586), MA104 (African green monkey kidney; ATCC CRL-2378), and LLC-MK2 (rhesus monkey kidney; ATCC CCL-7) cells at 60–70% confluent in 24-well tissue culture plates. Cultures were monitored daily for CPE, and harvested (medium and cells) at day 5 post-inoculation. After two cycles of freezing and thawing cell debris was removed by centrifugation and the supernatants were passed to fresh cultures. Sub-culturing was performed five times regardless of CPE. Passages were evaluated for the presence of picornavirus RNA by RT-PCR.
Results
RT-PCR screening of specimens
Pre-screening of pooled samples containing diagnostic cases of chicken specimens and chicken or turkey litter extracts yielded RT-PCR amplicons of the approximate expected size (~300 bp). None of the fecal samples collected from healthy broiler chickens, turkeys, domestic ducks, or Canadian geese yielded amplicons with similar size. Testing of the individual samples of the positive pools identified two chicken diagnostic cases, 13 chicken- and 5 turkey litter samples with ~300 bp amplicons (Table 2; Fig. 4).
Table 2.
Picornavirus RT-PCR
| Species | Sample | RT-PCR+ (%) | Strain | Age |
|---|---|---|---|---|
| Domestic duck | Swab | 0/25 | Not available | |
| Domestic turkey | Swab | 0/11 | Not available | |
| Chicken | Swab | 0/14 | Not available | |
| Canadian geese | Swab | 0/149 | Not available | |
| Chicken (broiler)a | Swab | 2/28 (7) | CHK1, 2 | Not available |
| Chicken (broiler) | Litter | 0/9 | 21–42 days | |
| Chicken (layer)b | ||||
| House 1 | Litter | 0/6 | 66 weeks | |
| House 2 | Litter | 2/6 (33) | CHK184, 185 | 26 weeks |
| House 3 | Litter | 5/12 (42) | CHK175, 177, 178, 179, 180 | 26 weeks |
| House 4 | Litter | 4/6 (66) | CHK165, 166, 167, 168 | 22–24 weeks |
| House 5 | Litter | 0/12 | 70–78 weeks | |
| House 6 | Litter | 2/6 (33) | CHK148, 149 | 27 weeks |
| Turkeyc | ||||
| Farm 1 | Litter | 3/4 (75) | TRK90, 91, 92 | 21 days |
| Farm 2 | Litter | 0/4 | 21 days | |
| Farm 3 | Litter | 2/4 (50) | TRK22, 24d | 20 days |
| Farm 4 | Litter | 0/4 | 21 days | |
aDiagnostic cases represent birds with runting–stunting enteritis
bLitter samples were collected from 22 to 78 weeks old egg layers from six different houses of the same farm (6–12 samples/house)
cLitter samples were collected from 20 to 21 days old turkey poults from two different houses from each of four different farms (2 samples/house; 4 samples/farm)
dTRK22 (THV) and TRK24 (new genus described in this study) were detected in two different houses of farm 3
Fig. 4.

RT-PCR products of individual chicken litter samples separated on 2% agarose gels and stained with ethidium bromide. M 1 kb Plus DNA ladder (Invitrogen); −C negative control (water); +C positive control (FT285 RNA)
Sequence and phylogenetic analysis of short RdRp amplicons
Based on their length (minus primer sequences), the amplicons could be divided into three groups: (1) 283 bp/94 aa (8 chicken samples), (2) 280 bp/93 aa (3 chicken and 4 turkey samples), and (3) 250 bp/83 aa (4 chicken and 1 turkey samples). Instead of the GLPSG amino acid motif characteristic for caliciviruses, a GMPSG motif was present in all amplicons. BLAST search and pairwise alignments indicated that all amplicons detected in this study represented picornaviruses. The 280 and 283 bp amplicons had the closest nucleotide identity (48–61%) to kobuviruses and turdivirus 1 (“Orthoturdivirus”) [2] while the 250 bp amplicons had high (84–91%) nucleotide identity to THVs [3]. None of the amplicons revealed calicivirus sequences.
According to phylogenetic analysis of the deduced short 3Dpol aa sequences, the 20 sequences obtained in our study fell into two distinct clusters, with distances suggesting the existence of two new genera (genus 1 and genus 2 in this study) within Picornaviridae. Genus 1 included viruses yielding 280 and 283 bp amplicons and genus 2 was comprised of the THV-like viruses, yielding 250 bp amplicons (Fig. 1).
Analyses of the P3 regions and 3′-NTR
Genome amplification of selected strains from each group was extended from the P3 region to the poly-A tail. The P3 region of picornaviruses encode proteins 3A, 3B (VPg), 3Cpro (protease), and 3Dpol (RNA-dependent RNA polymerase). For strains CHK165, CHK175, TRK90, and TRK91 a segment (~1 kb) stretching from the P290 primer binding site of 3Dpol to the poly-A tail was amplified. For strains CHK148, CHK168, TRK22, and TRK24 a segment (~1.8–2.2 kb) stretching from the GxCG motif of 3Cpro to the poly-A tail was amplified. Finally, for strain CHK1 a segment (~3.2 kb) stretching from the DDxGQ motif of the 2C protein to the poly-A tail was amplified. Phylogenetic and distance analyses of the complete 3Dpol amino acid sequences placed genus 1 viruses closer to Turdivirus 1 (“Orthoturdivirus”) than our original analyses based on partial RdRp sequences, indicating that genus 1 viruses may represent a highly divergent species of “Orthoturdiviruses” rather than a new genus (Fig. 2a). However, analyses of the partial 2C and the 3A–3C region supported their classification as a new genus (Fig. 2b; Table 3).
Fig. 2.
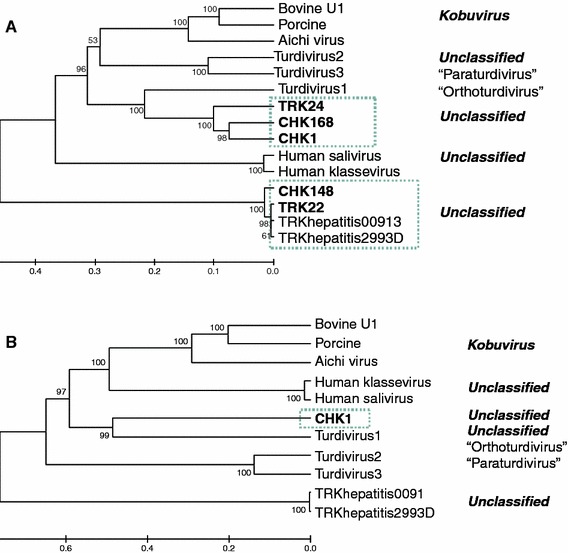
Phylogenetic analysis of complete 3Dpol (a), and 3A–3Cpro (b) protein sequences. The length of the individual proteins are listed in Table 3. The dendrogram was constructed by the UPGMA clustering method of MEGA version 3.1 with Poisson correction distance calculations and 1,025 bootstrap analyses
Table 3.
Comparison of amino acid homologies
| Number of nt or aa residues (% homology) | ||||
|---|---|---|---|---|
| 3′-NTR | 3Dpol | 3A–3Cpro | Partial 2Cb | |
| CHK1 | 309 | 477 | 302 | 177 |
| CHK168 | 310 | 478 (84) | NA | NA |
| TRK24 | 311 | 477 (80) | NA | NA |
| Aichi virus | 240 | 468 (54) | 311 (21) | 140 (35) |
| Bovine kobuvirus | 177 | 469 (55) | 316 23) | 140 (35) |
| Procine kobuvirus | 170 | 468 (55) | 316 (25) | 173 (24) |
| Turdivirus 1 | 320 | 475 (64) | 326 (28) | 154 (41) |
| Turdivirus 2 | 224 | 470 (49) | 314 (19) | 134 (36) |
| Turdivirus 3 | 242 | 470 (47) | 307 (20) | 134 (38) |
| Human salivirus | 151 | 474 (47) | 301 (24) | 148 (29) |
| Human klassevirus | 161 | 473 (46) | 301 (23) | 148 (29) |
| THV 0091a | 137 | 472 (35) | 379 (15) | 174 (24) |
| THV 2993D | 172 | 472 (35) | 379 (15) | 173 (24) |
| CHK148 | 641 | 472 (36) | NA | NA |
| TRK22 | 654 | 472 (35) | NA | NA |
The 3′-NTRs of CHK165, CHK175, TRK90, and TRK91 strains are 310, 308, 314, and 312 nt long, respectively. The 3′-NTR of CHK178 is 641 nt long
aComplete 3′-NTRs of turkey hepatitis viruses were not available
bLength of partial 2Cs represent compatible regions to the 177 nt partial 2C of CHK1 starting from the deduced 2C/3A cleavage sites
The 3′-NTR sequences of genus 2 (THV-like) viruses obtained in this study were significantly longer than any of the other picornaviruses (Table 3). These long 3′-NTRs contained a short open reading frame (ORF) encoding a putative protein (98–114 aa) (Fig. 3). BLAST searches of these ORFs did not reveal any similarity to known proteins in public databases.
Fig. 3.
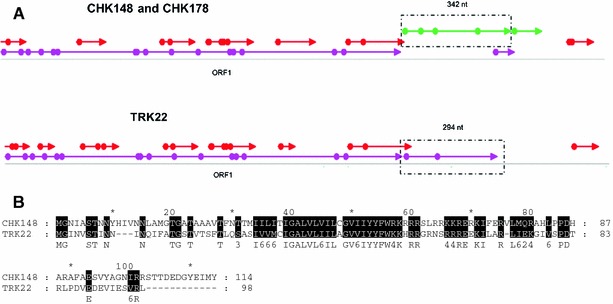
ORF maps of CHK148, CHK178, and TRK22 were generated in Omiga v2.0 software (Oxford Molecular Ltd, Oxford, UK). Dots represent start codons (ATG), arrow heads represent stop codons (TAA, TAG, or TGA). Frames are shown in both orientations (+1, +2, +3, −1, −2, −3). The 342 and 294 nt 3′-NTR putative ORFs are boxed (a). Alignment of deduced amino acid sequences of CHK148 and TRK22 3′-NTR putative ORFs were generated in GeneDoc. Identical or similar amino acids are shaded (b)
Secondary structure predication analysis was performed on these 3′-NTR sequences using a structural and alignment-based collection of RNA structure prediction programs (Webserver for Aligning non-coding RNAs, WAR) (Figs. 5, 6). The 3′-most 240 nt of CHK148 and TRK22 were predicted to form a nearly identical series of stem-loops predicted by RNAForester (Fig. 5b; ∆g = −52.2), MURLET (∆g = −50.8), and MAFFT-RNAalifold (∆g = −51.0). The 3′-NTR of Aichi virus, and 240 nt of the 3′NTRs of CHK148, TRK22, and Turdivirus 1 were predicted to form a common set of stem-loops predicted by MAFFT-RNAalifold (∆g = −31.8) (Fig. 5c).
Fig. 5.
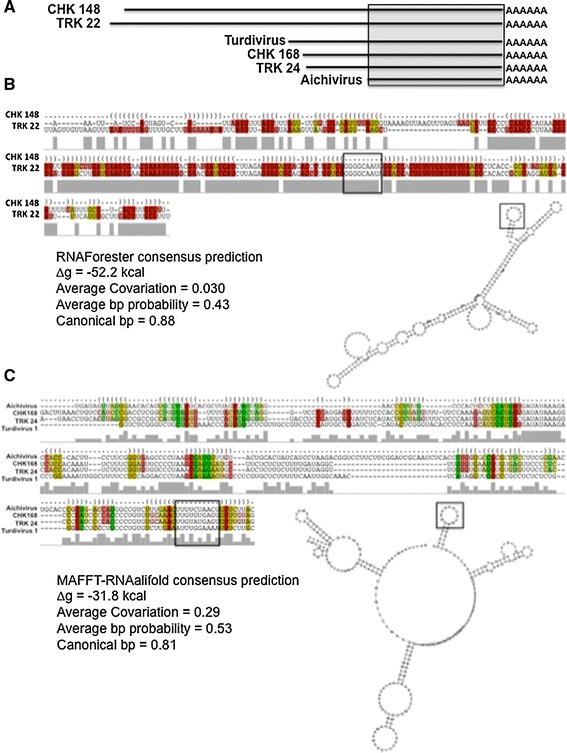
Secondary structural analysis of avian picornavirus 3′-NTRs. a Map of the 3′-NTRs and their relative sizes. The gray box indicates the region aligned by secondary structure predictions in b and c. b The secondary structure alignment of CHK 148 and TRK 22 3′-NTRs (terminal 240 nt), the predicted stem-loop structure (below right), and derived thermodynamic and statistical values for the proposed structure (∆g, avg covariation, avg bp probability and canonical bp). The structure shown was predicted by RNAForester using the Webserver for Aligning non-structural RNAs (WAR, http://genome.ku.dk/resources/war/). c The alignment based on structural prediction for the 3′-NTRs of CHK168, TRK 24, Aichi virus, and Turdivirus, the predicted stem-loop structure (bottom right) and values generated in the derivation of this structure (bottom left). Structural alignments were generated using the IUPAC nucleotide ambiguity system. Boxed sequences in alignments b and c correlate with the boxed loops in the secondary structure predications and are provided for reference and orientation
Fig. 6.
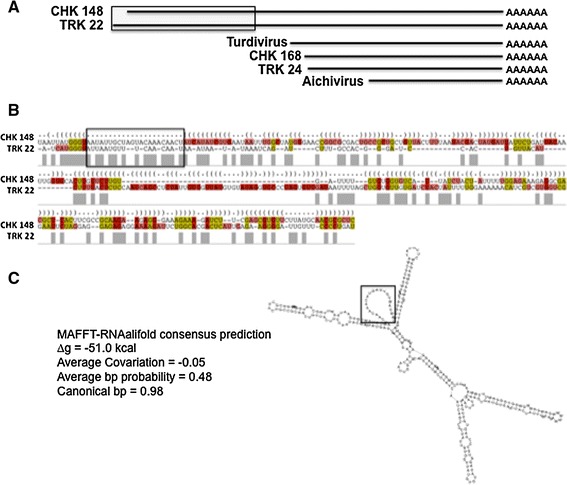
Secondary structural analysis of the CHK 148 and TRK 22 3′-NTRs. a Map of the region of the CHK 148 and TRK 22 3′-NTRs used for analysis. The gray box indicates the region aligned by secondary structure predictions in b and c. b The secondary structure alignment of the 5′-regions of the CHK 148 and TRK 22 3′-NTRs (250 nt). Structural alignments were generated using the IUPAC nucleotide ambiguity system. As in Fig. 5, the structure shown was predicted by MAFFT-RNAalifold program using the Webserver for Aligning non-structural RNAs (WAR, http://genome.ku.dk/resources/war/). c The predicted stem-loop structure (below right), and derived thermodynamic and statistical values for the proposed structure (∆g, avg covariation, avg bp probability, and canonical bp). Boxed sequences in the alignment in b correlate with the boxed loop in the secondary structure predication in c and is provided for reference and orientation
The additional ~300 nt upstream of the 3′-NTR stem-loop structures in CHK148, 178, and TRK22, were similarly examined for structurally homologous stem-loops (Fig. 6). This region was found to form a series of stem-loops predicted by RNAForester (∆g = −43.2), MAFFT-RNAalifold (∆g = −24.4), and LaRA (∆g = −23.86) programs. The function of these stem-loops upstream of those common to the picornavirus 3′-NTR stem-loops associated with replication, is currently unknown.
Tissue culture
After inoculation with RT-PCR positive swabs or litter extracts, no cytopathic effect (CPE) was observed in the non-human primate cell line cultures tested (LLC-MK2, MA104, and Vero) up to five blind passages. In some of the primary chicken embryo liver/fibroblast and LMH cultures, however, CPE characterized by rounding of the cells developed and was transferable to subsequent passages even after chloroform extraction, indicating the presence of non-enveloped viruses. However, all cultures with or without CPE were negative for the presence of picornavirus RNA after the second and subsequent passages.
Discussion
Based on the previous description of calicivirus-like particles in avian species [5–7], including chickens with RSS [4, 8] the initial goal of our study was the molecular detection of caliciviruses in avian fecal specimens. Fecal swabs collected from broiler chickens, domestic ducks, turkeys, and Canadian geese in Delaware, and litter extracts collected from chicken and turkey farms in North Carolina were tested using P289/P290 (Table 1). With the exception of the 28 swabs collected from RSS positive chickens in Delaware, all of the fecal specimens represented healthy animals. Data on the health status of North Carolina flocks (litter extracts) were not available. The primers (P289/P290) used in this study are targeting nucleotide sequences encoding for conserved amino acid motifs (YGDD and DY(T/S)(R/K/G)WDST) in the calicivirus RdRps. However, RdRps of RNA viruses because of their common evolutionary origin share several conserved motifs. Indeed, using P289/P290 for the detection of caliciviruses in several previous studies resulted in the unintentional detection of RNA viruses including rotavirus, porcine kobuvirus and astrovirus [14–16]. Similarly, in this study novel picornaviruses were serendipitously amplified with amplicons that were indistinguishable from the calicivirus positive control by size (Fig. 4). Analysis of the complete 3Dpol sequences of CHK1, CHK148, CHK168, TRK22, TR24, and the THVs revealed ~16–18 nt match in the 23 nt P290 binding site, with DYSCFDST and DYSCFDSS amino acid motifs for genus 1 and genus 2 viruses, respectively.
Recently, two reports describing the molecular detection of caliciviruses in avian species were published. Day et al. [17] reported a partial calicivirus sequence (936 nt) identified in a metagenomic analysis of turkey gut RNA virus community and Wolf et al. [18] reported the full genome sequence (7908 nt) of a chicken calicivirus detected in two clinically normal and one RSS chicken. Both of these caliciviruses are genetically related to but distinct from Sapovirus and represent two putative new genera of Caliciviridae. Unfortunately, there is no published data on the prevalence of these avian caliciviruses and their role in disease still needs to be established. Surprisingly, despite the relatively large number and diverse samples tested, caliciviruses were not detected in any of the samples including the 28 chicken samples collected from chickens with RSS. The primers used in our study could not be evaluated directly for their ability to detect the avian caliciviruses but sequence analysis of the chicken calicivirus [18] indicated a good match for primer binding at the sites encoding for the DYSGWDST and YGDD amino acid motifs.
Picornaviruses were detected both in chicken and turkey samples including two fecal swabs collected from chickens with RSS, 13 litter samples collected from egg layers and 5 litter samples collected from turkey farms (Table 2). Phylogenetic analyses of partial 3Dpol sequences divided the 20 picornaviruses into two distinct clusters (Fig. 1). Both clusters contained viruses detected in both chicken and turkey samples suggesting that these picornaviruses can infect both avian species.
Fifteen samples including the two positive swabs from chickens with RSS, 9 litter samples collected from egg layers, and 4 litter samples collected from turkeys contained novel picornaviruses (genus 1) with no closely related sequences in public databases.
Recently, in a metagenomic analysis of the turkey gut RNA virus community Day et al. [17] reported the identification of RNA sequences with homology to seven of the nine recognized picornavirus genera with the largest number of sequences bearing homology to Kobuvirus. Unfortunately, these sequences are not available from public databases for comparison with sequences obtained in our study.
Phylogenetic analysis of the entire P3 region including 3A, 3B (VPg), 3Cpro, and 3Dpol of CHK1 placed genus 1 viruses closer to “Orthoturdivirus” than our original analysis of the partial 3Dpol sequences (Fig. 1). Phylogeny of the complete 3Dpol sequences separately indicated that genus 1 viruses might represent a highly divergent species of “Orthoturdivirus” (Fig. 2a), however, this was not supported by analyses of the partial 2C and the 3A–3C regions which placed genus 1 viruses further apart from “Orthoturdivirus” supporting their classification as a new genus (Fig. 2b; Table 3). According to the genus definition recommended by Picornavirus Study Group of the International Committee on Taxonomy of Viruses (ICTV), members of a picornavirus genus should share >40, >40, and >50% amino acid identity in the P1, P2, and P3 regions, respectively. CHK1 shares 47% amino acid identity with Turdivirus 1 in the P3 region, which indicates that the genus 1 viruses described in this study represent a new genus. In accordance with the results of the phylogenetic distance analysis, alignments of the separate P3 proteins revealed that CHK1 3Dpol region alone shared a higher (62%), while the 3A–3Cpro region of P3 and the available partial 2C region of P2 shared a lower (28 and 41%, respectively) amino acid identity with Turdivirus 1 (Table 3). Ortho- and paraturdiviruses were discovered recently in tracheal and cloacal swabs obtained from dead wild birds of the genus Turdus in the family Turdidae [2]. Turdiviruses could not be propagated in cell culture or in chicken embryos and their prevalence, host range, and disease burden are unknown. Based on our analysis, genus 1 viruses described in this study represent a putative new picornavirus genus with the closest evolutionary roots to Orthoturdivirus. Since genus 1 viruses were described in chicken and turkey samples we propose the tentative name “Gallivirus” for the genus. For the final classification and nomenclature of genus 1 viruses, analysis of complete genome sequences, their host range, pathogenicity, and antigenic relationships needs to be determined.
The remaining five picornavirus sequences (genus 2) clustered separately from genus 1 viruses and together with the recently described THVs [3] (Fig. 1). Pairwise amino acid alignments of the complete 3Dpol revealed a high (97–99%) homology between TRK22, CHK148, and the THVs. The published sequences of the turkey hepatitis viruses did not include the complete 5′ and 3′-NTRs. In this study complete 3′-NTR sequences of three genus 2 viruses (CHK148, CHK178, and TRK22) were obtained revealing a significantly longer 3′-NTR region (641–654 nt) than that of any other picornaviruses (Table 3). Aligments of the 3′-NTRs with the partial (137 and 172 nt) 3′-NTR regions that were published for THVs clearly separated the chicken and turkey viruses into two groups. CHK148 and CHK178 had a 52–55% nucleotide identity with THV0091 and THV2993D, while TRK22 exhibited a 82–89% identity to THV0091 and THV2993D, respectively. Moreover, an eight nucleotide deletion was clearly conserved among THV0091 and THV2993D and TRK22.
The full length 3′-NTRs of genus 2 viruses obtained in our study contained a putative short ORF: 342 nt (114 aa) for CHK148, and CHK178 and 294 nt (98 aa) for TRK22, respectively (Fig. 3). The putative short ORF sequences of CHK148 and CHK178 had 93% nucleotide and 99% amino acid identity to each other but only 53% nucleotide and 34% amino acid identity and 50% amino acid similarity to the TRK22 short ORF. None of these proteins showed homology to any viral proteins available in public databases. Whether these ORFs encode for a functional protein or the relevance of the unusually long 3′-NTR of these viruses remains to be established in future studies.
Structures at the extreme 3′-ends of picornavirus genomes define the OriR (3′-NTR origin of replication) typically include stem-loops (X, Y, and Z) important for circularization of the genome during minus strand (antigenome) replication. Secondary and tertiary structures in the 3′-NTR vary in overall complexity, but have been described as having tRNA-like folds with the “kissing” of stem-loops in a higher order folded pseudoknot [19, 20].
In our analysis of predicted consensus RNA secondary structures of the viruses reported here, complex secondary structures identified for both the extreme 3′-NTRs (Fig. 5) and the additional sequences found in CHK148 and TRK22 3′-NTRs (Fig. 6). As sequences at the extreme 3′-ends of picornaviruses, arteriviruses, and coronaviruses are important for antigenome and subsequent genome synthesis [21], and given the limited sequence identity among the strains examined, we used a structural alignment-based set of programs to predict common structural elements of these 3′-NTRs. As several common structures were predicted using different algorithms, it seems likely that these structures provide the basis for future structure/function studies with respect to their role in genome replication.
While the THVs were described in samples collected from turkey poults with symptoms of turkey hepatitis [3], in our study similar viruses were detected in 4 litter samples collected from egg layer chickens and in 1 litter sample collected from turkey poults. Since THVs are the proposed causative agents of turkey hepatitis, evaluation of the pathogenicity of these viruses in chickens is important. Preliminary studies indicate that both CHK148 and TRK22 can be propagated in embryonated chicken eggs (unpublished data). Efforts for the tissue culture adaptation of the picornaviruses described in this study were unsuccessful. Virus isolation from fecal material can be difficult due to low virus load, toxicity of the material, and the abundance of diverse viral agents that often overgrow the target virus. Many enteric viruses require polarized epithelial cells for replication and may have species-specific requirements. More cell lines and primary cell cultures should be evaluated in future studies.
In summary, we described the molecular detection of novel picornaviruses in chicken and turkey samples, including viruses that were recently suggested to be the causative agents of turkey hepatitis. These viruses represent two possible new genera of Picornaviridae that appear to be endemic in both chickens and turkeys. Further characterization of these viruses including their host range and prevalence and studies to link infection to clinical disease such as hepatitis or RSS are necessary.
Acknowledgments
We thank Dr. Carolyne Price for providing the LMH cell line, Bryan Donnelly for providing the primary chicken embryo fibroblast cells and Nicole Farkas for helping with sample transport. We also thank Dr. Margaret K. Hostetter for her support. The Infectious Disease Scholar Fund of CCHMC to T. F. was used to fund this study.
Footnotes
The GenBank/EMBL/DDBJ accession numbers for the nucleotide sequences determined in this study are: JF424824–JF424832.
References
- 1.N.J. Knowles, T. Hovi, T. Hyypiä, A.M.Q. King, M. Lindberg, M.A. Pallansch, A.C. Palmenberg, P. Simmonds, T. Skern, G. Stanway, T. Yamashita, R. Zell, in Virus taxonomy: classification and nomenclature of viruses: Ninth Report of the International Committee on Taxonomy of Viruses, ed. by A.M.Q. King, M.J. Adams, E.B. Carstens, E.J. Lefkowitz (Elsevier, San Diego, 2011), pp. 855–880
- 2.Woo PC, Lau SK, Huang Y, Lam CS, Poon RW, Tsoi HW, Lee P, Tse H, Chan AS, Luk G, Chan KH, Yuen KY. J. Gen. Virol. 2010;91:2433. doi: 10.1099/vir.0.021717-0. [DOI] [PubMed] [Google Scholar]
- 3.Honkavuori KS, Shivaprasad HL, Briese T, Street C, Hirschberg DL, Hutchison SK, Lipkin WI. Emerg. Infect. Dis. 2011;17:480. doi: 10.3201/eid1703.101410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cubitt WD, Barrett AD. J. Gen. Virol. 1985;66(Pt 7):1431. doi: 10.1099/0022-1317-66-7-1431. [DOI] [PubMed] [Google Scholar]
- 5.Gough RE, Drury SE, Bygrave AC, Mechie SC. Vet. Rec. 1992;131:290. doi: 10.1136/vr.131.13.290. [DOI] [PubMed] [Google Scholar]
- 6.Poet SE, Skilling DE, Megyesl JL, Gilmartin WG, Smith AW. J. Wildl. Dis. 1996;32:461. doi: 10.7589/0090-3558-32.3.461. [DOI] [PubMed] [Google Scholar]
- 7.Sironi G. Vet. Rec. 1994;134:196. doi: 10.1136/vr.134.8.196. [DOI] [PubMed] [Google Scholar]
- 8.Wyeth JP, Chettle NJ, Labram J. Vet. Rec. 1981;109:477. doi: 10.1136/vr.109.21.477-b. [DOI] [PubMed] [Google Scholar]
- 9.Farkas T, Cross RW, Hargitt E, III, Lerche NW, Morrow AL, Sestak K. J. Virol. 2010;84:8617. doi: 10.1128/JVI.00630-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farkas T., Zhong W.M., Jing Y., Huang P.W., Espinosa S.M., Martinez N., Morrow A.L., Ruiz-Palacios G.M., Pickering L.K., Jiang X. Arch. Virol. 2004;149(7):1309. doi: 10.1007/s00705-004-0296-9. [DOI] [PubMed] [Google Scholar]
- 11.Jiang X, Huang PW, Zhong WM, Farkas T, Cubitt DW, Matson DO. J. Virol. Methods. 1999;83:145. doi: 10.1016/S0166-0934(99)00114-7. [DOI] [PubMed] [Google Scholar]
- 12.Kumar S, Tamura K, Nei M. Brief Bioinform. 2004;5:150. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- 13.Torarinsson E, Lindgreen S. Nucleic Acids Res. 2008;36:W79. doi: 10.1093/nar/gkn275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ludert JE, Alcala AC, Liprandi F. J. Clin. Microbiol. 2004;42:835. doi: 10.1128/JCM.42.2.835-836.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reuter G, Boldizsar A, Kiss I, Pankovics P. Emerg. Infect. Dis. 2008;14:1968. doi: 10.3201/eid1412.080797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reuter G, Pankovics P, Boros A. Arch. Virol. 2011;156:125. doi: 10.1007/s00705-010-0827-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Day JM, Ballard LL, Duke MV, Scheffler BE, Zsak L. Virol. J. 2010;7:313. doi: 10.1186/1743-422X-7-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolf S, Reetz J, Otto P. Arch. Virol. 2011;156:1143. doi: 10.1007/s00705-011-0964-5. [DOI] [PubMed] [Google Scholar]
- 19.Melchers WJ, Hoenderop JG, Bruins Slot HJ, Pleij CW, Pilipenko EV, Agol VI, Galama JM. J. Virol. 1997;71:686. doi: 10.1128/jvi.71.1.686-696.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pilipenko EV, Maslova SV, Sinyakov AN, Agol VI. Nucleic Acids Res. 1992;20:1739. doi: 10.1093/nar/20.7.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Wimmer E, Paul AV. Biochim. Biophys. Acta. 2009;1789:495. doi: 10.1016/j.bbagrm.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]