

Abstract
Canine distemper (CD) is a highly contagious, often fatal, multisystemic, and incurable disease in dogs and other carnivores, which is caused by canine distemper virus (CDV). Although vaccines have been used as the principal means of controlling the disease, CD has been reported in vaccinated animals. The hemoagglutinin (H) protein is one of the most important antigens for inducing protective immunity against CD, and antigenic variation of recent CDV strains may explain vaccination failure. In this study, a new CDV isolate (TM-CC) was obtained from a Tibetan Mastiff that died of distemper, and its genome was characterized. Phylogenetic analysis of the H gene revealed that the CDV-TM-CC strain is unique among 20 other CDV strains and can be classified into the Asia-1 group with the Chinese strains, Hebei and HLJ1-06, and the Japanese strain, CYN07-hV. The H gene of CDV-TM-CC shows low identity (90.4 % nt and 88.9 % aa) with the H gene of the classical Onderstepoort vaccine strain, which may explain the inability of the Tibetan Mastiff to mount a protective immune response. We also performed a comprehensive phylogenetic analysis of the N, P, and F protein sequences, as well as potential N-glycosylation sites and cysteine residues. This analysis shows that an N-glycosylation site at aa 108-110 within the F protein of CDV-TM-CC is specific for the wild-type strains (5804P, A75/17, and 164071) and the Asia-1 group strains, and may be another important factor for the poor immune response. These results provide important information for the design of CD vaccines in the China region and elsewhere.
Keywords: Canine distemper virus (CDV), VerodogSLAM cells, Phylogenetic analysis, Virulence, Genotype
Introduction
Canine distemper (CD), one of the most well-known infectious diseases of wild and domestic Canidae, is a highly contagious, often fatal, multisystemic, and incurable viral disease that mainly affects the respiratory, gastrointestinal, and central nervous systems and is caused by canine distemper virus (CDV) [1]. Vaccination remains the principal means of controlling the disease. Alteration of the antigenicity of new live modified vaccines has greatly helped to control CD [2, 3]. Although the use of vaccines has reduced the incidence of CD over the past decades, it has been reported that CD (as a sporadic, endemic, or outbreak disease) can also affect vaccinated animals [4–8].
Analysis of CDV strains from various animal samples has demonstrated an important relationship with the H gene/glycoprotein, which has changed by genetic/antigenic drift. As the key protein for CDV, H is used for attachment to cell receptors as the first step of infection and mediates adequate host immune response [9]. The H protein is considered to have the highest antigenic variation and can reflect genetic changes in comparative studies of CDV strains [10–13]. This variation may affect neutralization-related sites with disruption of important epitopes. Analysis of CDV strains from different animal species and geographical settings has revealed that the geographic pattern is an important factor in the genetic/antigenic drift affecting the H gene/glycoprotein of CDV [14–20]. Therefore, the H gene may be used for identification and phylogenetic classification of CDV strains, which have been identified into seven major genetic lineages, namely America-1 and -2, Asia-1 and -2, Arctic-like, Europe, and wild-life [21, 22], as well as an indication of the antigenic response of the virus.
Three other proteins, the nucleocapsid (N) protein, the phosphoprotein (P) protein, and the fusion (F) protein, also have important roles for CDV and could provide additional sources of antigenic variability among strains. The N protein has immunosuppressive properties and is the major component of the CDV virion. The N-terminal domain of the N protein is generally well conserved, while the C-terminal end is poorly conserved and is considered hypervariable. The C-terminal tail of the N protein also contains the majority of its phosphorylation sites and antigenic sites [23, 24]. During active infection, antibodies made against the N protein in the host are predominant and account for most of the complement-fixing antibody [25, 26]. The P protein is relatively well conserved and plays a vital role in transcription and replication [27]. This protein is an essential component of the viral RNA phosphoprotein complex (vRNAP) [28] and also function as a chaperone for the N protein. The F protein is a type I integral membrane protein that mediates viral penetration by fusion between the virion envelope and the host cell plasma membrane at neutral pH. It is synthesized as an inactive precursor, F0, and must be proteolytically cleaved to produce the functionally active fusion protein, which consists of disulfide-linked F1 and F2 polypeptides [29]. Like the H protein, the F protein has high antigenic variation.
In this study, the wild-type CDV-TM-CC strain was isolated from the spleen of a 1-year-old Tibetan Mastiff that developed clinical signs of CD after having received all standard vaccines. To determine whether this occurrence may be explained by variations in specific nucleotide or amino acid residues of the CDV circulating in China, we sought to genetically characterize the CDV-TM-CC strain.
Materials and methods
Cells and viruses
VerodogSLAM cells constitutively expressing the CDV receptor dog signaling lymphocyte activation molecule (SLAM) were cultured in Dulbecco’s modified Eagle medium (DMEM; Gibco) supplemented with 10 % heat-inactivated fetal bovine serum (FBS) with an additional 8 μg of G418 per ml.
The wild-type CDV-TM-CC strain was originally isolated from spleen homogenate (10 % w/v suspension) from a Tibetan Mastiff that succumbed to naturally infection. Virus was propagated in VerodogSLAM cells and stored at −80 °C.
RNA preparation and RT-PCR
Total RNA was prepared from VerodogSLAM cells infected with CDV-TM-CC according to the manufacturer’s instructions (Total RNA Kit I, OMEGA). The reverse transcription reactions were performed using M-MLV Reverse Transcriptase (Invitrogen) with oligo d(T) and random primers. According to the complete consensus genomic sequence of CDV (GenBank), two sets of primers were designed to amplify the entire genome (Oligo6.0 design software), as shown in Table 1. Sequences were assembled and compared using DNA sequence analysis software (DNAStar), and the complete consensus genomic sequence was determined. PCR amplification was carried out using Phusion High-Fidelity DNA Polymerase (New England BioLabs). Clones (amplicons emcompassing the full-length CDV-TM-CC genome) were obtained from thirty RT-PCR reactions using CDV-specific oligonucleotides.
Table 1.
Primers used for amplification of the full-length genome of the CDV-TM-CC strain
| Primer | Sequence (5′ → 3′) | Position |
|---|---|---|
| A1F | ACCAGAAAAAGTTGGCTATG | 1 |
| AIR | GCFGTTFCACCCATCFGTTG | 1036 |
| A2F | GAACAAGCCTAGAAFTGCTG | 842 |
| A2R | TGAGGGCTTTGAGGCATTCC | 1858 |
| A3F | FTCAAGACCAGFGFTACAFC | 1683 |
| A3R | GCAGGTFGGCGGACATFCTC | 2627 |
| A4F | TCTGCAGTTCCCACGCAATC | 2427 |
| A4R | CCTGCAGTTCGCCTTACCAC | 3748 |
| A5F | GFGAFAGAFCCAGGACTCGG | 3540 |
| A5R | CTCACCTAATTCFGCFFTGG | 5495 |
| A6F | FAFGGTGCATFGGAATAGCC | 5296 |
| A6R | CCTACATCTAACCTCTCAAG | 6649 |
| AT | GTGTAAGTGTTATAGCACAG | 6452 |
| A7R | AAGGGTTCCCATGACGTTFG | 8271 |
| ASF | TGGATCAAGTTGAAGAGGTG | 8061 |
| ASR | CGTGCATACTCTAAAATAGC | 9127 |
| A9F | GGACTTAGGTATGATGACTG | 8928 |
| A9R | CCFGAGAGAGAAGGFGATGT | 9932 |
| A10F | FTAAFGTGTTAGTGTCFCGG | 9631 |
| A10R | CAFFFFAGCGAATAGCCTCC | 10676 |
| A11F | ATTCCTCAGGTACAACCCAC | 10478 |
| A11R | GGTCAAACCCTTTCTCAATG | 11682 |
| A12F | CCACAGAATACTTTGTAGCC | 11428 |
| A12R | AFCGAFGAGCGGAFCAFGCC | 12413 |
| A13F | CTTTAGCCGCTTTCTTGATG | 12160 |
| A13R | ACAGTCTAAATAAGTGCTCG | 13080 |
| A14F | CCGCTCATCGATCAAGACTC | 12402 |
| A14R | CTATTGATATCATCATCGCC | 13438 |
| A15F | GACTTTATAACCAGAGTCAC | 13261 |
| A15R | ACATGCAGFAGAGFTGATCC | 14321 |
| A16F | AFCACTGAFGCFGFTGGATG | 14100 |
| A16R | ACCAGACAAAGCTGGGTATG | 15690 |
| B1F | ACCAGAAAAAGTTGGCT | 1 |
| B1R | TCCGTTGTCTTGGATGCT | 1315 |
| B2F | GCTCTGGAGTTATGCTAT | 1100 |
| B2R | TTAACCTCTTCACCGCTG | 2162 |
| B3F | CCTACCATGTCAGCAAAG | 1817 |
| B3R | AAGCGATAGTGAAAGCAG | 3378 |
| B4F | TACCGCACCTTCCAAAGC | 3168 |
| B4R | GCACTCTAATCTCCATAG | 4331 |
| B5F | CGATGTACTGGTAAGATG | 4212 |
| B5R | ACCTAATTCTGCTFTGGT | 5492 |
| B6F | AATCCAATGCAACCAACT | 5251 |
| B6R | AGCAGGGCCTAAGGCAAC | 6623 |
| B7F | AGTCCTGATAAATTGCTG | 6489 |
| B7R | GACTCGAATCTTFTGCG | 7828 |
| BSF | TGGAGCTACTACTFCA | 7645 |
| B8R | ATACGGACTAAATTCTCAAC | 8874 |
| B9F | AGATGTCCTTACTGAGTC | 8584 |
| B9R | GAGAGAGAAGGTGATGTC | 9929 |
| B10F | ACTTTGCACGTCAGAATC | 9786 |
| B10R | CCTAGCTATACTTFTCAG | 11585 |
| B11F | TTGGCAGCACATGAAAGC | 11298 |
| BUR | GACCCTTCGCCAACTTAG | 12483 |
| B12F | GAGGGAGGCAATTGCTGG | 12242 |
| B12R | TGGGAGTCAAGAGAGGGT | 13705 |
| B13F | GGTCAATGTGCTGCAATC | 13491 |
| B13R | TCTCTCGTTGTCCAGTTC | 14499 |
| B14F | TACGTCTGAAGAACATGG | 14363 |
| B14R | ACCAGACAAAGCTGGGTA | 15690 |
Two sets of forward (F) and reverse (R) primers were used (sets A and B) for verification of the sequence in redundant amplicons
Sequence alignment and phylogenetic analysis
To genetically characterize the CDV-TM-CC strain, the deduced amino acid sequence was compared to F and H gene fragments of the variant field isolates shown in Table 2. A phylogenetic tree was constructed based on the deduced amino acid sequences in supplementary Table 1 using MEGA 5.0, and multiple sequence alignment was carried out using ClustalW. Statistical significance of the phylogeny was estimated by bootstrap analysis over a 1,000 pseudoreplicate data set.
Table 2.
Nucleotide sequence accession numbers of CDV strains used in this study
| Strain | Country | Organ | GenBank | Reference |
|---|---|---|---|---|
| 00-2601 | USA | Raccoon brain | AY443350.1 | [40] |
| 01-2689 | USA | Raccoon brain | AY649446.1 | Unpublished |
| 007Lm | Japan | Canine pulmonary | AB474397.1 | Unpublished |
| 011C | Japan | Canine cerebellum | AB476401.1 | [41] |
| 50Con | Japan | Canine conjunctiva | AB476402.1 | [41] |
| 55L | Japan | Canine lung | AB475099.1 | [41] |
| 98-2646 | USA | Raccoon brain | AY542312.2 | Unpublished |
| 98-2654 | USA | Raccoon brain | AY466011.2 | [40] |
| 5804P | USA | – | AY386316.1 | [42] |
| 164071 | USA | Blood lymphocytes | EU716337.1 | Unpublished |
| A75/17 | Switzerland | – | AF164967.1 | Unpublished |
| CDV3 | China | Mink | EU726268.1 | Unpublished |
| CYN07-hv | Japan | Macaca fascicularis | AB687721.2 | [7, 43] |
| Hebei | China | Mink lung | KC427278.1 | Unpublished |
| HLJ1-06 | China | Fox | JX681125.1 | [44] |
| M25CR | Japan | Canine cerebrum | AB475097.1 | [41] |
| Onderstepoort | USA | – | AF014953.1 | [34, 45] |
| Phoca/Caspoian/2007 | Kazakhstan | Vero cells | HM046486.1 | Unpublished |
| Shuskiy | Kazakhstan | – | HM063009.1 | Unpublished |
| Snyder Hill | Canada | Cerebrospinal fluid | AF259552.1 | [16] |
A GenBank number is provided for each of the strains of CDV that were compared with CDV-TM-CC in this study. The geographical location of strain isolation and the species/organ of isolation are also indicated, as well as the clade into which the strains are categorized
Results
Phylogenetic analysis of the nucleotide and deduced amino acid sequences of the H gene of CDV-TM-CC
The wild-type CDV-TM-CC strain was isolated from the spleen of a 1-year-old Tibetan Mastiff in Jilin province that had succumbed to CD after having received all standard vaccines (6 weeks first immunization, 8 weeks second immunization, 10 weeks third immunization with Distemper, adenovirus type 2, parvovirus, parainfluenza quadruple vaccine; Canine coronavirus disease killed virus vaccine portion, USA). The virus was propagated in VerodogSLAM cells and the virulence of the strain was confirmed (data not shown). To identify sequence features that may explain the failure of the vaccine strain to protect the dog against CD, we sequenced the entire genome, using two sets of overlapping primers (Table 1).
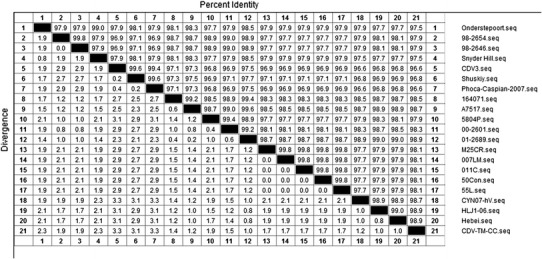
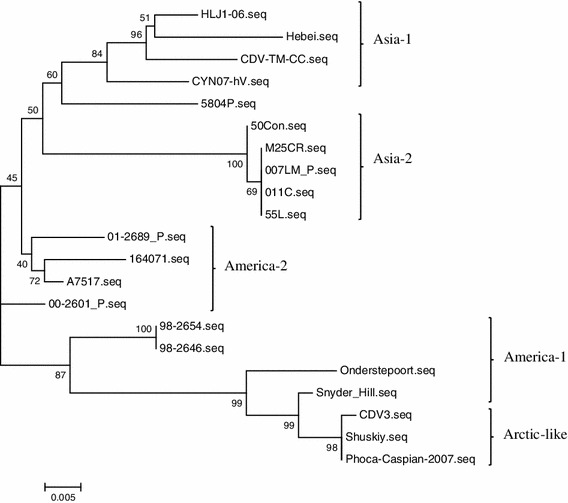
Within the CDV genome, the H gene is a major causative disease determinant and also has one of the highest rates of mutation. Consequently, the phylogenetic relationship of CDV strains is often based on the deduced amino acid sequence of the H protein. The H gene of the CVT-TM-CC strain has 1,824 nucleotides and the inferred protein sequence has 607 amino acids, similar to the other CDV strains. Amino acid analysis of the H protein from CDV-TM-CC and 20 other CDV strains in GenBank (Table 2) identified seven clades of CDV strains (America-1, America-2, Asia-1, Asia-2, Europe, Arctic-like, and Europe wild-life). CDV-TM-CC was classified into the Asia-1 group with the strains CYN07-hV (Japan), Hebei (China), and HLJ-06 (China) (Fig. 1). CDV-TM-CC has high conservation of both the H gene nucleotide sequence (97.4 % nt identity with CYN07-hV, 98.1 % with HLJ1-06, and 98.2 % with Hebei) and the amino acid sequence (97.4 % aa identity with CYN07-hV, 97.4 % with HLJ1-06, and 98.0 % with Hebei). On the other hand, the America-1 group (strains Onderstepoort, 98-2654, 98-2646, and Snyder Hill) was less similar to CDV-TM-CC in both the nucleotide sequence (90.4 % nt identity with Onderstepoort, 91.1 % nt with 98-2654, 91.3 % with 98-2646, and 90.7 % with Snyder Hill) and the amino acid sequence (88.9 % identity with Onderstepoort, 90.6 % with 98-2654, 90.8 % with 98-2646, and 90.6 % with Snyder Hill) (Fig. 2a, b).
Fig. 1.
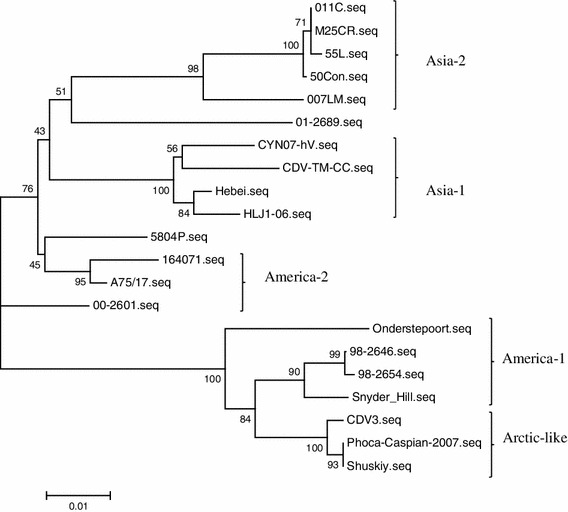
Phylogenetic relationship between different CDV strains on the basis of the amino acid alignment of the H protein of the CDV-TM-CC strain in comparison with other strains from GenBank. Results show that CDV-TM-CC is an Asia-1 strain
Fig. 2.

Alignment of the nucleotide and amino acid sequences of H protein of different CDV strains. The percent identity to and divergence from the nucleotide sequence (a) and protein sequence (b) of CDV-TM-CC is shown
N-linked glycosylation sites of the H protein of the CDV-TM-CC strain
Glycosylation is an important factor in determining the antigenicity of many proteins [30]. Prediction of the glycosylation sites of the H gene (http://www.cbs.dtu.dk/services/NetNGlyc/, NetNGlyc 1.0 Servera) identified a total of eight potential glycosylation sites at positions 19NSS, 149NFT, 309NGS, 391NQT, 422NIS, 456NGT, 584NIT, and 587NST (Fig. 3). Among them, six glycosylation sites (positions 19–21, 149–151, 391–393, 422–424, 456–458, and 587–590) were conserved for all of the CDV strains. Notably, the 309–311 N-glycosylation site is specific for virulent strains [14, 18] with the exception of A75/17. The 584 N-glycosylation site is specific for the Asian-1 strains, suggesting that it was acquired later [18, 20]. CDV-TM-CC has both of these predicted glycosylation sites, which could explain its virulence properties.
Fig. 3.
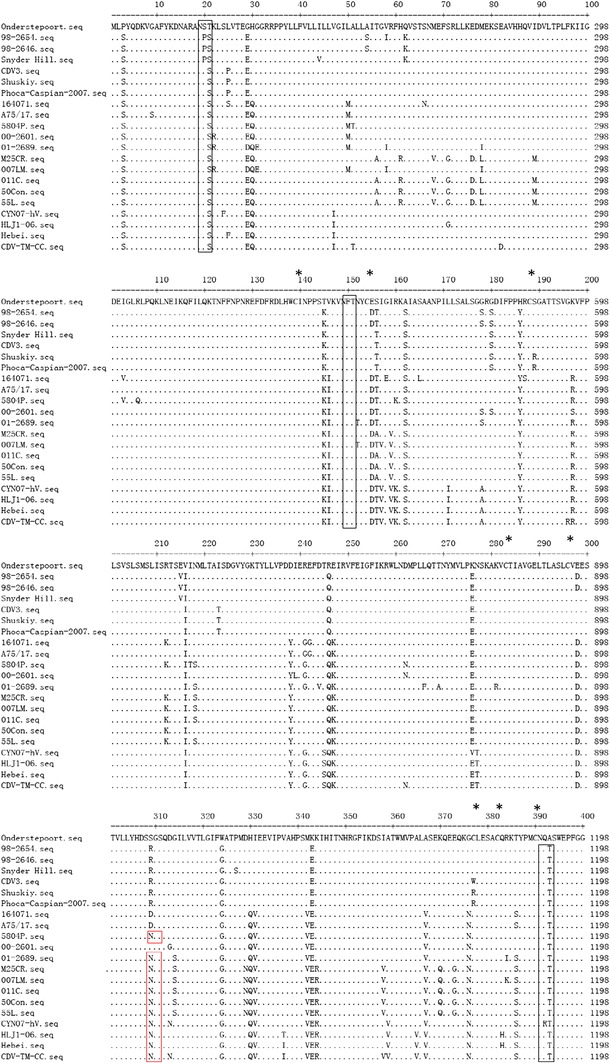
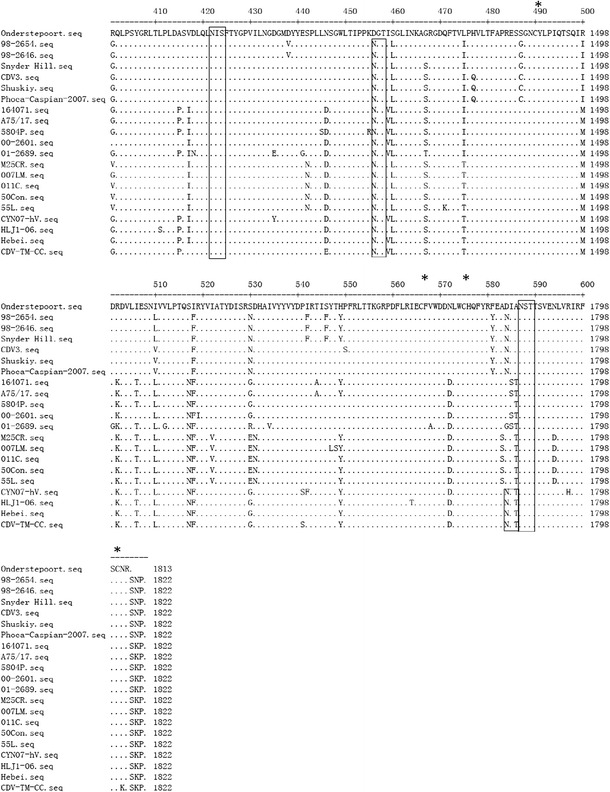
Conservation of the amino acid sequence of the H protein. The protein sequences of each of the strains in this study are compared to the sequence of the vaccine strain, Onderstepoort. Potential N-link glycosylation sites (N-X-S/T) are boxed. Among them, the 309–311 N-glycosylation site, which is specific for the wild-type strain, is boxed in red, and the 584–586 N-glycosylation site, acquired in the Asian-1 strains, is boxed in blue. The position of cysteine residues is indicated by asterisks
Phylogenetic analyses of the amino acid sequence of the N and P proteins
To determine whether the conservation of CDV-TM-CC also extends to other proteins within the virus, we assessed the similarity of the N and P proteins. Consistent with the results for the H protein, the homology of the deduced CDV-TM-CC amino acid sequence of the N protein to the Asia-1 strains (CYN07-hV, HLJ1-06, and Hebei) was high with 98.7-98.9 % identity, as shown in Fig. 4. The N protein sequence of CDV-TM-CC also showed 98.1 % identity with the Asia-2 group (strains M25CR, 007Lm, 011C, 50Con, and 55L), and 97.5 % identify with the Onderstepoort strain. Moreover, CDV-TM-CC had high similarity (98.5, 98.7, and 97.9 % identity) with wild-type strains 164071, A75/17, and 5804P. The lowest homology of the CDV-TM-CC N protein sequence (96.6–96.8 % aa identity) was found with Arctic-like strains CDV3, Shuskiy, and Phoca-Caspian-2007. This relatively high similarity between the N protein of CDV-TM-CC and other CDV strains is consistent with the generally high conservation among N proteins.
Fig. 4.
Alignment of the amino acid sequences of the N protein of different CDV strains. The percent identity to and divergence from the protein sequence of CDV-TM-CC is shown
The phylogenetic relationship of CDV-TM-CC based on the deduced amino acid sequence of the P protein was also analyzed (Fig. 5). Similar to the results for the H protein, CDV-TM-CC classified into the Asia-1 group, but was in a separate branch from the classical Onderstepoort vaccine strains. These results verify the classification of CDV-TM-CC as an Asia-1 group virus.
Fig. 5.
Phylogenetic analysis based on the amino acid sequences of the P protein. Results verify the categorization of CDV-TM-CC as an Asia-1 strain
Phylogenetic analysis of the signal peptide region of the F protein
The signal peptide is a short amino acid sequence at the N-terminus of the majority of newly synthesized proteins that are destined towards the secretory pathway and is a highly divergent region [31]. Analysis of the 1–135 aa signal peptide region of the F protein of CDV-TM-CC demonstrated the same set of amino acid variations in comparison with the Onderstepoort strain as for the other Asia-1 strains (CYN07-hV, HLJ1-06, and Hebei): 8S/8K, 11T/11P, 19R/19P, 81G/81R, 101Q/101W, 102I/102F, and 112S/112A (Fig. 6). Among the Asia-2 strains (M25CR, 007Lm, 011C, 50Con, and 55L), variations in comparison with the Onderstepoort strain were found in 30T/30S, 53S/53A, 55R/55W, 59S/59Y, 62N/62K, 99R/99K, 110I/110V, and 111N/111K. Additionally, both the Asia-1 and Asia-2 strains had clade-specific amino acid variation in 21P/21Q. Moreover, the CDV-TM-CC strain had characteristic additional variations in 107P/107Y and 116C/116Y. Therefore, the signal peptide region of CDV-TM-CC has both Asia group-specific and individual variations.
Fig. 6.
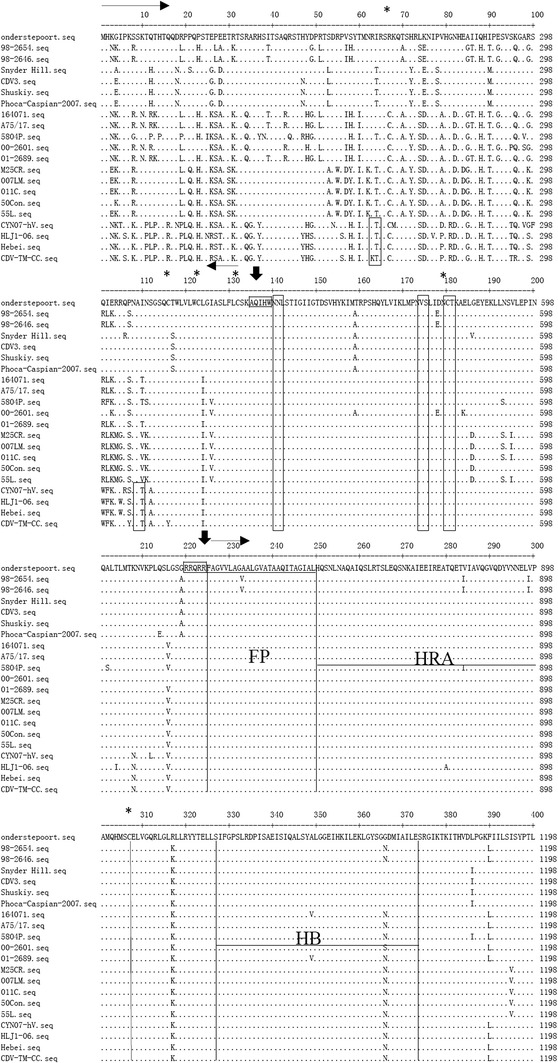

Alignment of the amino acid sequence of the F protein of different CDV strains. The sequences of the F proteins of each of the strains in this study are compared to the sequence of the vaccine strain, Onderstepoort. The potential N-linked glycosylation sites (N-X-S/T) are boxed. The positions of the fusion peptide (FP), heptad repeats (HRA and HRB), helical bundles (HB), trans-membrane (TM) and cytoplasmic tail (CT) regions are indicated. The positions of cysteine residues are marked by asterisks, and cleavage sites are marked by arrows
Phylogenetic analysis of the F2 and F1 regions
Among the CDV strains, amino acid variation was also found in 208K/208N in the F2 region (aa 136–224) for the Asia-1 group. Generally, there was high conservation within the hydrophobic fusion peptide (FP) domain at the N-terminus of the membrane anchored F1 subunit, with the exception of 233A/233V in the 98-2654 and 98-2646 strains (Fig. 6). Amino acid variations between the Asia-1 and Asia-2 groups were also found in a region between the helical bundles (HB) and heptad repeats B (HRB) at 394V/394S, 429R/429K, and 466L/466I; within the trans-membrane (TM) domain at 627C/627Y, 634Q/634R, and 637H/637F; and within the cytoplasmic tail (CT) domain, at 656R/656K. Among the Asia group strains, the HRA (aa 250-307) and HB (aa 328-374) domains were highly conserved, with the exception of a 280Q/280A variation in the HRA domain. Likewise, the amino acids were highly conserved in the HRB (aa 557-601) domain in all CDV strains except for Hebei (583D/583N) and 5804P (587V/587I). Common amino acid changes in other regions of CDV strains in comparison to the Onderstepoort strain were found at 317K/317R and 556S/556G.
N-linked glycosylation sites and cysteine residues of the F protein
The potential N-glycosylation sites (N-X-S/T) of the F protein were highly conserved at 141NLS, 173NVS, 179NCT, and 517NQS in the F1 region among all CDV strains as reported previously [32–34] (Fig. 6). Moreover, the Asia-1 group (strains CYN07-hV, HLJ1-06 Hebei, and CDV-TM-CC) had specific potential N-glycosylation sites at 62NRT and 108NAT in the signal peptide region, with the exception of the CDV-TM-CC strain, which had the sequence 62NKT. Five of these six potential glycosylation sites of the CDV-TM-CC strain were at the same positions within the known virulent CDV strains (A75/17, 5804P and 164071) at aa 108–110, 141–143, 173–175, 179–181, and 517–519, whereas 62NKT was unique for CDV-TM-CC, and 62NRT and 38NIT were unique for 5804P.
Cysteine is an α-amino acid that plays an important role in intramolecular disulfide bond formation and the steric structure of proteins. In the F protein of CDV-TM-CC, a total of 16 cysteine residues were detected. Among them, 14 residues (aa 123, 132, 180, 307, 446, 455, 470, 478, 502, 507, 509, 531, 628, and 629) were located at identical positions in all CDV strains; however, several amino acid(s) were characteristic to individual strain(s), such as 67R/67C in the America group (strains Onderstepoort, 98-2654, 98-2646, Snyder Hill, CDV3, Shuskiy and Phoca-Caspian-2007) and 116Y/116C in CDV-TM-CC. The presence of amino acid variations, as well as specific N-glycosylation sites and cysteine residues within the F protein, could affect the immune response to CDV-TM-CC.
Discussion
Improved vaccination has reduced the frequency and magnitude of CD [35]. Distemper vaccination failures are uncommon, but outbreaks of CD continue to occur among vaccinated individuals and populations [4, 5, 36, 37]. The most common factor in CD occurrence is a lack of appropriate vaccination, while the residual virulence of the vaccine strains is another possibility. Furthermore, genetic/antigenic drift of wild-type CDV strains driven mainly by geographic variation is an important factor in vaccine failure. In this study, we isolated a new virulent CDV strain, CDV-TM-CC, from a Tibetan Mastiff that died due to natural infection in Jilin province. According to the complete consensus genomic sequence of CDV, at least two sets of primers were designed within different CDV genes, leading to the identification of the complete CDV-TM-CC sequence.
The H protein, a major structural protein of CDV, mediates host selection and pathogenicity, and the rate of genetic variation for its gene is greater than for other genes. With geographically distinct lineages, many studies have demonstrated that phylogenetic analysis can be carried out in accordance with the deduced amino acid sequences of the H protein [14, 18, 21, 38]. In this study, phylogenetic analysis based on the H protein identified seven clades of CDV strains (America-1, America-2, Asia-1, Asia-2, Europe, Arctic-like, and Europe wild-life), and CDV-TM-CC was classified into the Asia-1 group, with the highest identity to the Chinese strains, HLJ1-6 and Hebei, and the Japanese strain, CYN07-hV. Potential N-glycosylation sites may differ for the H protein of the wild-type and vaccine strains of CDV. Usually, only 4–7 potential sites are found within vaccine strains (such as Onderstepoort), in comparison with 8–9 sites in wild-type CDV strains (for example, 5804P). In particular, the 309–311 N-glycosylation site, which is specific for the wild-type strain [14, 18], is suggestive of the pathogenicity of the CDV-TM-CC strain. Furthermore, the 584–586 N-glycosylation site has been acquired in the Asian-1 strains [18, 20]. Further study may determine whether these differences in glycosylation may contribute to the failure of the vaccine to protect against virulent strains of CDV.
The N protein is a highly conserved immunogenic protein that can elicit cellular and humoral immunity [39]. Based on sequence differences between the gene of the wild strains and vaccine strain, the N protein may affect the seroprotection rate of the host and lead to immune failure. Like the H protein, the N protein of CDV-TM-CC showed the highest homology with the Asia-1 group. High homology was also observed with the Asia-2 group (strains M25CR, 007Lm, 011C, 50Con, and 55L) and wild-type strains (164071, A75/17, and 5804P). Moreover, the lowest homology was found between CDV-TM-CC and the Onderstepoort strain. Variation in the immunodominant epitope of the virus may change the structure, and therefore, we can speculate that the T cell-mediated immune response may be altered by variations in this protein. The P gene is extremely well conserved and, therefore, is particularly important in the phylogenetic classification. Based on the phylogenetic relationship of the deduced amino acid sequence of the P protein, CDV-TM-CC was also classified into the Asia-1 group. These results highlight the importance of considering the geographical setting to control the occurrence of the disease in a more efficient manner.
The F protein is a surface glycoprotein that mediates viral entry into the host cell by fusion of the virion envelope and the host cell plasma membrane at a neutral pH. Within the F protein, the signal peptide region (aa 1–135) has the lowest amino acid homology, especially at positions 13–37 and 72–112. However, our analysis shows that the signal peptide region is relatively well conserved among the Asia-1 group, except for specific individual amino acids, indicating that the signal peptide of the F protein is geographically distinct. In addition, three amino acids specific to the CDV-TM-CC strain (62K, 107Y, and 116Y) are located in the signal peptide region. The previous study reported that the amino acids 208K and 216L are specific for the CDV vaccine strains; however, we also found 208K in the wild-type strains in the America group (A75/17, 164071, and 5804P) and Asia-2 group (011C, M25CR, 55L,50Con, and 007Lm). The F protein of the CDV-TM-CC strain has six potential glycosylation sites. Among them, differences were found to reside mainly in the signal peptide region, but no clear rule could obviously explain the differences in the wide-type and vaccine strains or the geographical variation, including the occurrence of a strain-specific site (62–64 NKT) for CDV-TM-CC. Four additional potential glycosylation sites were recognized at positions 141–143, 173–175, 179–181 in the F2 region and 517–519 in the F1 region, as reported previously [32–34]. The 108–110 N-glycosylation site is specific for the wild-type strains (5804P, A75/17, and 164071) and the Asia-1 group (Hebei, HLJ1-06, and CYN07-hV), and may be another important factor in vaccination failure. The fusion peptide (FP) domain also was found to be highly conserved among all CDV strains, except for 233A/233V in 98-2654 and 98-2646. In short, the genetic/antigenic drift observed in the currently circulating CDV strains should be considered as a possible factor leading to the resurgence of CD cases. Analysis of CDV strains detected globally and from a variety of host species will provide a more in-depth understanding of the global ecology of CDV and will provide the basis for the improvement of current CDV vaccines.
Conclusion
The wild-type CDV-TM-CC strain, originally isolated from spleen homogenate from a fully vaccinated Tibetan Mastiff in China, was classified into the Asia-1 group cluster of CDV strains based on the sequence of its H protein and verified by the sequence of its P protein. Variations in specific amino acid residues, N-glycosylation sites, and cysteine residues throughout the CDV-TM-CC genome may explain the failure of the dog to mount vaccine-mediated protection against CD. These results provide the foundations for the global improvement in current CDV vaccines.
Acknowledgments
This work was supported by Ecology of Zoonoses and Research of Infection and Immunity mechanisms (2012CB722501).
Footnotes
Weike Li and Tiansong Li contributed equally to this paper.
Contributor Information
Weike Li, Email: weikeduo0535@163.com.
Zhigao Bu, Phone: +86-451-51997173, FAX: +86-451-51997166, Email: zgbu@yahoo.com.
Xianzhu Xia, Phone: +86-431-86985808, FAX: +86-431-86985828, Email: xia_xzh@yahoo.com.cn.
References
- 1.Vandevelde M, Zurbriggen A. Vet. Microbiol. 1995;44:271–280. doi: 10.1016/0378-1135(95)00021-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Appel MJ, Summers BA. Vet. Microbiol. 1995;44:187–191. doi: 10.1016/0378-1135(95)00011-X. [DOI] [PubMed] [Google Scholar]
- 3.Appel MJ. Virus Infections of Carnivores. Amsterdam: Elsevier Science Publishers B V; 1987. pp. 133–159. [Google Scholar]
- 4.Glardon O, Stockli R. Schweiz. Arch. Tierheilkd. 1985;127:707–716. [PubMed] [Google Scholar]
- 5.Blixenkrone-Moller M, Svansson V, Have P, Orvell C, Appel M, Pedersen IR, Dietz HH, Henriksen P. Vet. Microbiol. 1993;37:163–173. doi: 10.1016/0378-1135(93)90190-I. [DOI] [PubMed] [Google Scholar]
- 6.Maes RK, Wise AG, Fitzgerald SD, Ramudo A, Kline J, Vilnis A, Benson C. J. Vet. Diagn. Investig. 2003;15:213–220. doi: 10.1177/104063870301500302. [DOI] [PubMed] [Google Scholar]
- 7.Sakai K, Yoshikawa T, Seki F, Fukushi S, Tahara M, Nagata N, Ami Y, Mizutani T, Kurane I, Yamaguchi R, Hasegawa H, Saijo M, Komase K, Morikawa S, Takeda M. J. Virol. 2013;87:7170–7175. doi: 10.1128/JVI.03479-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garde E, Pérez G, Acosta-Jamett G, Bronsvoort B. Animals. 2013;3:843–854. doi: 10.3390/ani3030843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan P, Thompson TB, Wurzburg BA, Paterson RG, Lamb RA, Jardetzky TS. Structure. 2005;13:803–815. doi: 10.1016/j.str.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 10.Blixenkrone-Moller M, Svansson V, Appel M, Krogsrud J, Have P, Orvell C. Arch. Virol. 1992;123:279–294. doi: 10.1007/BF01317264. [DOI] [PubMed] [Google Scholar]
- 11.Harder TC, Klusmeyer K, Frey HR, Orvell C, Liess B. J. Virol. Methods. 1993;41:77–92. doi: 10.1016/0166-0934(93)90164-M. [DOI] [PubMed] [Google Scholar]
- 12.Iwatsuki K, Tokiyoshi S, Hirayama N, Nakamura K, Ohashi K, Wakasa C, Mikami T, Kai C. Vet. Microbiol. 2000;71:281–286. doi: 10.1016/S0378-1135(99)00172-8. [DOI] [PubMed] [Google Scholar]
- 13.Orvell C, Blixenkrone-Moller M, Svansson V, Have P. J. Gen. Virol. 1990;71(Pt 9):2085–2092. doi: 10.1099/0022-1317-71-9-2085. [DOI] [PubMed] [Google Scholar]
- 14.Bolt G, Jensen TD, Gottschalck E, Arctander P, Appel MJ, Buckland R, Blixenkrone-Moller M. J. Gen. Virol. 1997;78(Pt 2):367–372. doi: 10.1099/0022-1317-78-2-367. [DOI] [PubMed] [Google Scholar]
- 15.Carpenter MA, Appel MJ, Roelke-Parker ME, Munson L, Hofer H, East M, O’Brien SJ. Vet. Immunol. Immunopathol. 1998;65:259–266. doi: 10.1016/S0165-2427(98)00159-7. [DOI] [PubMed] [Google Scholar]
- 16.Haas L, Martens W, Greiser-Wilke I, Mamaev L, Butina T, Maack D, Barrett T. Virus Res. 1997;48:165–171. doi: 10.1016/S0168-1702(97)01449-4. [DOI] [PubMed] [Google Scholar]
- 17.Harder TC, Kenter M, Vos H, Siebelink K, Huisman W, van Amerongen G, Orvell C, Barrett T, Appel MJ, Osterhaus AD. J. Gen. Virol. 1996;77(Pt 3):397–405. doi: 10.1099/0022-1317-77-3-397. [DOI] [PubMed] [Google Scholar]
- 18.Iwatsuki K, Miyashita N, Yoshida E, Gemma T, Shin YS, Mori T, Hirayama N, Kai C, Mikami T. J. Gen. Virol. 1997;78(Pt 2):373–380. doi: 10.1099/0022-1317-78-2-373. [DOI] [PubMed] [Google Scholar]
- 19.Martella V, Pratelli A, Cirone F, Zizzo N, Decaro N, Tinelli A, Foti M, Buonavoglia C. Mol. Cell. Probes. 2002;16:77–83. doi: 10.1006/mcpr.2001.0387. [DOI] [PubMed] [Google Scholar]
- 20.Mochizuki M, Hashimoto M, Hagiwara S, Yoshida Y, Ishiguro S. J. Clin. Microbiol. 1999;37:2936–2942. doi: 10.1128/jcm.37.9.2936-2942.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martella V, Cirone F, Elia G, Lorusso E, Decaro N, Campolo M, Desario C, Lucente MS, Bellacicco AL, Blixenkrone-Moller M, Carmichael LE, Buonavoglia C. Vet. Microbiol. 2006;116:301–309. doi: 10.1016/j.vetmic.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 22.McCarthy AJ, Shaw MA, Goodman SJ. Proc. Biol. Sci. 2007;274:3165–3174. doi: 10.1098/rspb.2007.0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu CH, Kingsbury DW. Virology. 1982;120:225–234. doi: 10.1016/0042-6822(82)90020-4. [DOI] [PubMed] [Google Scholar]
- 24.Ryan KW, Portner A, Murti KG. Virology. 1993;193:376–384. doi: 10.1006/viro.1993.1134. [DOI] [PubMed] [Google Scholar]
- 25.Graves M, Griffin DE, Johnson RT, Hirsch RL, de Soriano IL, Roedenbeck S, Vaisberg A. J. Virol. 1984;49:409–412. doi: 10.1128/jvi.49.2.409-412.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Norrby E, Gollmar Y. Infect. Immun. 1972;6:240–247. doi: 10.1128/iai.6.3.240-247.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curran J, Marq JB, Kolakofsky D. Virology. 1992;189:647–656. doi: 10.1016/0042-6822(92)90588-G. [DOI] [PubMed] [Google Scholar]
- 28.Horikami SM, Curran J, Kolakofsky D, Moyer SA. J. Virol. 1992;66:4901–4908. doi: 10.1128/jvi.66.8.4901-4908.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scheid A, Choppin PW. Virology. 1974;57:475–490. doi: 10.1016/0042-6822(74)90187-1. [DOI] [PubMed] [Google Scholar]
- 30.Takeuchi H., and Haltiwanger R.S. Role of Glycosylation of Notch in Development. Elsevier, New York, pp. 638–645, %@ 1084–9521 (2010) [DOI] [PMC free article] [PubMed]
- 31.Plemper RK, Hammond AL, Cattaneo R. J. Biol. Chem. 2001;276:44239–44246. doi: 10.1074/jbc.M105967200. [DOI] [PubMed] [Google Scholar]
- 32.Iwatsuki K, Miyashita N, Yoshida E, Shin YS, Ohashi K, Kai C, Mikami T. J. Vet. Med. Sci. 1998;60:381–385. doi: 10.1292/jvms.60.381. [DOI] [PubMed] [Google Scholar]
- 33.Visser IK, van der Heijden RW, van de Bildt MW, Kenter MJ, Orvell C, Osterhaus AD. J. Gen. Virol. 1993;74(Pt 9):1989–1994. doi: 10.1099/0022-1317-74-9-1989. [DOI] [PubMed] [Google Scholar]
- 34.Barrett T, Clarke DK, Evans SA, Rima BK. Virus Res. 1987;8:373–386. doi: 10.1016/0168-1702(87)90009-8. [DOI] [PubMed] [Google Scholar]
- 35.Chappuis G. Vet. Microbiol. 1995;44:351–358. doi: 10.1016/0378-1135(95)00028-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mori T, Shin YS, Okita M, Hirayama N, Miyashita N, Gemma T, Kai C, Mikami T. J. Gen. Virol. 1994;75(Pt 9):2403–2408. doi: 10.1099/0022-1317-75-9-2403. [DOI] [PubMed] [Google Scholar]
- 37.Lan NT, Yamaguchi R, Inomata A, Furuya Y, Uchida K, Sugano S, Tateyama S. Vet. Microbiol. 2006;115:32–42. doi: 10.1016/j.vetmic.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 38.Zhao JJ, Yan XJ, Chai XL, Martella V, Luo GL, Zhang HL, Gao H, Liu YX, Bai X, Zhang L, Chen T, Xu L, Zhao CF, Wang FX, Shao XQ, Wu W, Cheng SP. Vet. Microbiol. 2010;140:34–42. doi: 10.1016/j.vetmic.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 39.Cardoso A.I., Blixenkrone-Moller M., Fayolle J., Liu M., Buckland R., and Wild T.F., Virology 225, 293–299, %@ 0042–6822 (1996) [DOI] [PubMed]
- 40.Lednicky JA, Meehan TP, Kinsel MJ, Dubach J, Hungerford LL, Sarich NA, Witecki KE, Braid MD, Pedrak C, Houde CM. J. Virol. Methods. 2004;118:147–157. doi: 10.1016/j.jviromet.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 41.Sultan S, Charoenvisal N, Lan NT, Yamaguchi R, Maeda K, Kai K. Virol. J. 2009;6:157. doi: 10.1186/1743-422X-6-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.von Messling V, Springfeld C, Devaux P, Cattaneo R. J. Virol. 2003;77:12579–12591. doi: 10.1128/JVI.77.23.12579-12591.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakai K, Nagata N, Ami Y, Seki F, Suzaki Y, Iwata-Yoshikawa N, Suzuki T, Fukushi S, Mizutani T, Yoshikawa T, Otsuki N, Kurane I, Komase K, Yamaguchi R, Hasegawa H, Saijo M, Takeda M, Morikawa S. J. Virol. 2013;87:1105–1114. doi: 10.1128/JVI.02419-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang Q., Hu X., Ge Y., Lin H., Jiang Y., Liu J., Guo D., Si C., and Qu L., Genome announcements 1 (2013) [DOI] [PMC free article] [PubMed]
- 45.Sidhu MS, Husar W, Cook SD, Dowling PC, Udem SA. Virology. 1993;193:66–72. doi: 10.1006/viro.1993.1103. [DOI] [PubMed] [Google Scholar]