

Abstract
The complete genome of a QX-like infectious bronchitis virus (IBV) strain Sczy3 isolated recently in Sichuan was sequenced. The genome contains 27,695 nucleotides (nt), and possesses a genomic structure similar to other IBV strains. Sequence comparisons demonstrated that the Sczy3 genome had the highest nt sequence identity with QX-like IBVs and was most dissimilar to the Massachusetts type IBV. Differences in the sequences of genes present in the Sczy3 genome and other IBVs gene sequences were also identified. Phylogenic analysis showed that the entire genome and most of the Sczy3 genes were located in the same cluster as LX4. Recombination analysis showed that Sczy3 is a chimeric strain derived from LX4 (major parental sequence) and H120 (minor parental sequence) suggesting that recombination occurred in a region containing the 3′ terminal 5a sequence (83 nt), the 5′ terminal 5b sequence (222 nt), and the 5′ terminal nucleocapsid protein gene sequence (132 nt). Mutations and intergenic recombination may have played an important role in the evolution of IBVs.
Keywords: Infectious bronchitis virus, Phylogenetic analysis, Genome, Recombination
Introduction
Avian infectious bronchitis (IB) is a highly contagious, acute, and economically important disease of chickens [1]. This disease affects chickens of all ages and causes respiratory stress, interstitial nephritis in broilers, and reduces egg production. The etiologic agent of IB is the infectious bronchitis virus (IBV), a member of the Coronaviridae family, subfamily coronavirinae, genus gamma-coronavirus that replicates primarily in the respiratory tract and also in some epithelial cells of the gut, kidney, and oviduct [2]. Currently, dozens of IBV serotypes and genotypes have been described, and natural outbreaks of IBV are often the result of infections with strains that differ serologically from vaccine strains.
Infectious bronchitis virus is an enveloped, non-segmented, single-stranded, positive-sense RNA virus with a genome of approximately 27.6 kb [3–5] possessing at least ten open-reading frames (ORFs) organized 5′–3′ as follows: 5′-Cap-1a-1b-S-3a-3b-E-M-5a-5b-N-3′PolyA [6]. Six mRNAs (mRNA 1–6) are associated with viral replication. Four structural proteins including the spike glycoprotein (S), small membrane protein (E), membrane glycoprotein (M),and nucleocapsid protein (N) are encoded by mRNA 2, 3, 4, and 6, respectively [7, 8]. mRNA1 consists of ORF1a and ORF1b, which have 42 overlapping nucleotides (nt) and encode for two large polyproteins, pp1a and pp1ab, via a ribosomal frame-shift mechanism [9]. During or after synthesis, these polyproteins are cleaved into 15 non-structural proteins (nsp2–16), one of which is the polymerase. The S glycoprotein is post-translationally cleaved at protease cleavage recognition motifs into the amino-terminal S1 (92 kDa) and the carboxyl-terminal S2 (84 kDa) subunits by cellular proteases [10]. The putative protease cleavage motifs are usually associated with one or more pairs of basic amino acids (e.g., Arg–Arg–Phe–Arg–Arg M41) [11]. The multimeric coiled-coil S glycoprotein extends from the viral membrane, and the S1 subunit is anchored to the viral membrane by the S2 subunit [12]. Proteins 3a, 3b and 5a, 5b are encoded by mRNA 3 and mRNA 5 and are not essential to viral replication [3, 7, 8].
Infectious bronchitis virus genomes are continuously evolving as a result of frequent point mutations and genomic recombination events [10–12]. It is essential to characterize field isolates for the selection of appropriate vaccine strains. Our previous study revealed that isolates recently obtained from the Sichuan province were mainly of the LX4 genotype or QX-like strains [13]. Sczy3 is a QX-like IBV strain identified on the basis of S1 subunit sequence comparison [14]; however, other Sczy3 genome regions have not yet been sequenced.
To further define the Sczy3 genome and determine its potential genetic origin, the complete genomic sequence of the QX-like IBV Sczy3 strain was determined and sequence comparisons and phylogenetic and recombination analyses were carried out to compare the Sczy3 sequence to that of other IBVs. These results may also help in the development of reverse genetic approaches for the generation of vaccines against IBVs.
Methods
Virus propagation
The IBV China/Sichuan/QX-like/Sczy3/200904strain (referred to as Sczy3) was isolated from the kidneys of broilers presenting with dyspnea in April of 2009 in a farm in Yaan City, Sichuan province, China, passaged in eggs five times and shown to be a QX-like IBV based on genetic analysis. The stock virus was propagated in 9–11 day-old specific pathogen-free (SPF) chicken embryos (Beijing experimental animal center, Beijing, China), and the allantoic fluids harvested 36 h post-inoculation and stored at −70 °C.
Primer design
Twenty five primer pairs were designed based on the published IBV genome sequence using Primer Premier software (Version 5.0, Premier Biosoft International, 3786 Cornia Way, Palo Alto, CA) and used to amplify the internal fragments of the Sczy3 genome (Table 1). Four other primers including QT (5′-GCTGTCAACGATACGCTACGTAACGGCATGACAGTG(T)18-3′), GSP (5′-CTTAGTTGGTTTCCCTTGTTGATTGC-3′), QI (5′-CGCTACGTAACGGCATGACAGTG-3′), and GSP (5′-TGTATGTCTGCTCACTAAACACCACCA-3′) were specifically designed to amplify viral RNA 3′- and 5′-terminal end sequences. Primers were synthesized by Takara biotechnology (Dalian, China) Co., Ltd.
Table 1.
Sczy3 genome amplification primers
| Primer pairs | Locationa | Upstream primer | Downstream primer |
|---|---|---|---|
| P1 | 290–1,912 | AGCGTTGGGCTACGTTCTC | CCAAATGCCC/TTCACCAAG |
| P2 | 1,808–2,608 | GCGTACAATAGAGTGCGCAATC | GAGCAATTGTTCTACTGTGAGGTC |
| P3 | 2,506–3,617 | GGTTAGCATTGAGTGTTGTGGTG | GAGACATATGCTCATTTGCGG |
| P4 | 3,559–5,133 | GGCATTGGATGAGTTTAAAGAG | CGAACTTTAAGTGTCAG/ATGGCA |
| P5 | 4,936–6,121 | TGAAGTGGTAAAGGAAGATGTT | AAAT/ACCTACACCAGTTTGCA |
| P6 | 6,043–7,158 | GGCAGGTTTTGTTATTATTTG | AAATACTTAAGACAACTGTCAGACA |
| P7 | 7,046–8,258 | TTTTTG/AATAAATGCAGATGCT | TAGTAAATACAGTGGTTGCATAAG |
| P8 | 8,203–9,005 | AATGGTTATAAAGTTTCAAGGTGT | CAACTTCAAATCATGATTATTAGC |
| P9 | 8,843–9,777 | GCTGTTGAG/AAAGTGCATTGTT | ACACATCTACTCCAAAACCAAG |
| P10 | 9,703–10,844 | TAATCAGGTGGGTGGTGTT | TCTTAAGTATATCATCACAATACTCAC |
| P11 | 10,682–11,711 | GCAAATTCAAAAATGCATGCTTAT | CAAATTTACATGGTGGGTCTAAGTC |
| P12 | 11,560–12,822 | CAGCGTAGAGTCTAAATGTTATTA | GCCCATTTTAGCCAACAT |
| P13 | 12,760–13,580 | AATAAGGATTGGTACGACCC | CCTTCCTTAAACATACCTGCTT |
| P14 | 13,333–14,395 | GAACTTGGTGTTATTATGAATCAAGATA | CAACATTAGCAGATGTGGCTTG |
| P15 | 14,318–15,389 | GTACTAGCAGTGATGCTAC | TCAACATTTTCGCTACCTACA |
| P16 | 15,326–16,170 | TAGTATCTAATGGTACTGTTTTTGGAATTT | TGGCAAGGCATTAATAGTACTAAAAAT |
| P17 | 16,046–16,903 | AATTTCTTAAAGTTGATGATTGCACTC | TGCAAATTTTAAACAAACCTGTACCT |
| P18 | 16,792–17,872 | CGTGGTATACTTGTAGTCATGC | CTTTAGGGTTGCCTATATCATAG |
| P19 | 17,755–18,935 | CATATTGCAAATGAGGATGAAGT | ACATACAGATTCGCTCCATCT |
| P20 | 18,834–19,625 | GGTGTCTACACAGTGTTATAAGC | GGCACACACATTGTAGTTTTAG |
| P21 | 19,550–20,322 | CAAGTGGTATTATGATGAATGTGGC | AGTGACTTCCCCAACATCTCTAATC |
| P22 | 21,998–23,115 | AAGATGATAGTGCCAGAAGAAT | AACACTATGCCATTAGGTGC |
| P23 | 23,056–23,803 | TTGTGGTAGTGGAA/GACATGT | CCATTAAACAGACTTTTTAGGTC |
| P24 | 23,729–24,856 | GAATCTTCTTATTACACTACTTTTGA | ACTCTCTATAGCAAAATTACATTGT |
| P25 | 24,747–25,824 | TTAGACTCTTTAAGCGGTGTAG | GAATACTAGGGAGGTACACGC |
aPrimer positions listed according to the LX4 strain genome (AY338732)
Viral RNA extraction and RT-PCR
Genomic RNA was extracted from IBV-infected allantoic fluid with Trizol reagent (Takara biotechnology, Dalian, China) according to the manufacturer’s instructions and dissolved in 20 μl sterile diethyl diethylpyrocarbonate (DEPC)-treated water and stored at −70 °C until further use. For the reverse transcription (RT) reaction, 10 μl of template RNA and 1 μl of random 6 mers (10 μM) were added and mixed. The mixture was then heated to 65 °C for 5 min and quickly chilled on ice for 5 min. After a brief centrifugation, 4 μl of 5× RT buffer, 2 μl of dNTP mix (2.5 mM each), 1 μl of AMV reverse transcriptase (200 U/μl), and 2 μl RNase-free water were added. The reaction mixture was incubated at 37 °C for 60 min and then at 70 °C for 10 min. PCR amplifications were carried out according to the manual of Premix PrimeSTAR® HS kit (Takara) in a total volume of 50 μl using 4 μl of cDNA as template. The annealing and extension conditions of PCR reactions were based on respective primer sequences and the length of the amplification fragment.
Rapid amplification of cDNA ends (RACE) from 3′ genomic RNA
One microliter of QT primer and 10 μl of total RNA were mixed and heated to 65 °C for 5 min and quickly chilled on ice for 5 min. After a brief centrifugation, 4 μl of 5× RT buffer, 2 μl of dNTP mix (2.5 mM each), 1 μl of AMV reverse transcriptase (200 U/μl), and 2 μl of RNase-free water were added. The reaction mixture was incubated at 37 °C for 60 min and then at 70 °C for 10 min. The PCR amplification was carried out according to the manual of Premix PrimeSTAR® HS kit using 3′ GSP and QI primers. The first PCR product was diluted 100-fold and used as template for the second PCR using the same primers. The PCR conditions were as follows: 94 °C for 5 min followed by 35 cycles at 94 °C for 40 s, 62 °C for 40 s, 72 °C for 1 min, and a final extension at 72 °C for 10 min.
RACE from 5′ genomic RNA
The reverse transcription reaction was performed as described for 3′ RACE except that the primer used was 5′ GSP. cDNA was treated by ribonuclease H at 37 °C for 1 h and after purification, a poly(A) tail was added to the cDNA. PCR amplification was carried out according to the manual of Premix PrimeSTAR® HS kit using the 5′ GSP and QT primers. The first PCR product was diluted 100-fold and used as a template for the second PCR using the QI and 5′ GSP primers. The PCR conditions were as follows: 94 °C for 5 min, 35 cycles at 94 °C for 40 s, 61 °C for 40 s, 72 °C for 1 min, and a final extension at 72 °C for 10 min.
DNA cloning and sequencing
PCR products of the predicted size were isolated from agarose gels and purified using the E.Z.N.A.™ Gel Extraction Kit (Omega, USA). Purified PCR products were inserted into vector PMD18-T (TaKaRa, Japan) and transformed into DH5α competent cells. Confirmation of clones containing recombinant plasmid was carried out by PCR and restriction enzyme (RE) digestion followed by sequencing (Shanghai Sang-gong Biological Engineering Technology & Services Co., Ltd., Shanghai, China).
Sequence comparison and phylogenetic analysis
The complete Sczy3 genome was aligned using the EDITSEQ program (Lasergene package) (DNASTAR Inc., Madison, WI, USA). The nucleotide sequences and the deduced amino acid sequences of different Sczy3 genes were compared to those of reference IBV sequences for homology analysis using the Megalign program (Lasergene package). Phylogenetic analysis of the nucleotide sequences and the deduced amino acid sequences of the target gene was performed using the neighbor-joining method using MEGA version 4.0 [15]. Bootstrap values were determined from 1,000 replicates of the original data.
The sequences of IBV reference strains were retrieved from the GenBank database and the accession numbers for the complete genomic sequences of 18 reference IBV strains used in this study are: Beaudette C (NC_001451), M41 (DQ834384), H52 (EU817497), H120 (FJ888351), ZJ971 (EU714028), Peafowl/GD/KQ6/2003 (AY641576), ArkDPI 11 (EU418976), ArkDPI101 (EU418975), Cal99 (AY514485), BJ (AY319651), CK/CH/LSD/05I (EU637854), TW2575/98 (DQ646405), SAIBK (DQ288927), A2 (EU526388), SC021202 (EU714029), partridge/GD/S14/2003 (AY646283), LX4 (AY338732), and ITA/90254/2005 (FN430414).The GenBank accession numbers for the gene sequences of other IBVs used are not indicated.
Recombination analysis
The 19 complete genome sequences were aligned using the Megalign program (DNASTAR software) and used for recombination analysis with the aid of RDP3.44 [16] and SimPlot version3.5.1 [17]. Potential recombination events were identified using the RDP [18], Maxchi [4], and GENECONV [19] methods in RDP3.44 to identify putative Sczy3 parental sequences with significance set at P values <0.05 and the sliding window size set as 30 bp. Putative potential recombination events were further identified using the similarity analysis method (SimPlot 3.5.1). Nucleotide identity was calculated using the Kimura 2-parameter method with a transition–transversion ratio of 2 in each window of 200 bp, and the window was successively extended by 20 bp increments. The recombination breakpoints identified using Simplot 3.5.1 were expressed as a vertical line, and the region between vertical lines represented the recombinant fragment. Phylogenetic trees of the putative recombinant fragments were also drawn using the neighbor-joining method and the UPGMA method implemented in RDP3.44.
Results
The Sczy3 genomic sequence
The complete Sczy3 genome sequence obtained in this study was submitted to GenBank under accession number of JF732903. The complete genome of Sczy3 consists of 27,695 nt including poly (A) tail and encodes six different genes flanked by 5′(525 nt) and 3′(532 nt) un-translated regions (UTRs). The nucleotide sequence length of the different ORFs1a, 1b, S1, S2, 3a, 3b, E, M, 5a, 5b, and N were 11856, 7959, 1620, 1878, 174, 189, 327, 678, 198, 249, and 1230 nt long, respectively (Fig. 1). The nucleotide sequence length of putative 15 non-structural proteins (nsp2–16) formed by the cleavage of polyproteins pp1a and pp1ab were 2,022 nt (526–2,547 nt), 4,770 nt (2,548–7,317 nt), 1,542 nt (7,318–8,859 nt), 921 nt (8,860–9,780 nt), 882 nt (9,781–10,662 nt), 328 nt (10,663–10,911 nt), 630 nt (10,912–11,541), 333 nt (11,542–11,874 nt), 435 nt (11,875–12,309 nt), 72 nt (12,310–12,381 nt), 2,820 nt (12,310–12,351 nt, 12,351–15,128 nt), 1,800 nt (15,129–16,928 nt), 1,563 nt (16,929–18,491 nt), 1,014 nt (18,492–19,505 nt), and 909 nt (19,506–20,414 nt), respectively. The predicted PLpro and Mpro cleavage sites that define the nsp2–15 boarders are shown in Table 2. In addition, genes with overlapping sequences, that is, gene 1 (526–20,414 nt) and gene 2 (20,305–23,862 nt), gene 2 and gene 3 (23,831–24,536 nt), gene 3 and gene 4 (24,423–245,185 nt), gene 5 (25,178–25,991 nt) and gene 6 (25,833–27,163 nt) have overlaps of 110, 32, 114, and 159 nt, respectively.
Fig. 1.
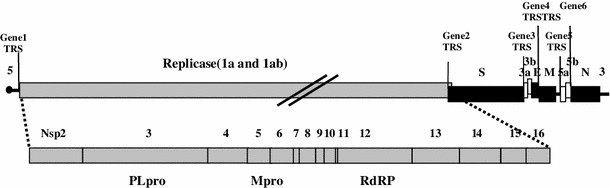
Genomic organization of strain Sczy3. The complete genome consists of 27,695 bp single-strand (+) RNA (including poly-A tails). The replicase gene is shown in gray with nonstructural protein domains (nsp) 2–16 indicated. The transcription-regulating sequence (TRS) is located in front of each gene. The PLpro is located in polyprotein G675–G2264 and the C-end cleavage site is AG/GV.Mpro is located in polyprotein A2279–Q3085 and the C-end cleavage site is LQ/SS. RDRP is located on polyprotein S3929–Q4868, and the C-end cleavage site is LQ/SC
Table 2.
Enzymatic pp1a/pp1b sites and characteristics of IBV strain Sczy3 nonstructural proteins
| Cleavage Products | Position on polyprotein | C-end cleavage | Size (amino acid) | Cleavage by |
|---|---|---|---|---|
| Nsp2 | M1–G674 | AG/GK | 674 | Plpro |
| Nsp3 | G675–G2264 | AG/GV | 590 | Plpro |
| Nsp4 | G2265–Q2778 | LQ/AG | 514 | Plpro |
| Nsp5 | A2779–Q3085 | LQ/SS | 307 | Mpro |
| Nsp6 | S3086–Q3379 | VQ/SK | 294 | Mpro |
| Nsp7 | S3380–Q3462 | LQ/SV | 83 | Mpro |
| Nsp8 | S3463–Q3672 | LQ/NN | 210 | Mpro |
| Nsp9 | N3673–Q3783 | LQ/SK | 111 | Mpro |
| Nsp10 | S3784–Q3928 | VQ/SD | 145 | Mpro |
| Nsp11 | S3929–G3951 | – | 23 | Mpro |
| Nsp12 | S3929–Q4868 | LQ/SC | 940 | Mpro |
| Nsp13 | S4869–Q5468 | LQ/GT | 600 | Mpro |
| Nsp14 | G5469–Q5989 | LQ/SI | 521 | Mpro |
| Nsp15 | S5960–Q6327 | LQ/SA | 338 | Mpro |
| Nsp16 | S6327–M6629 | – | 302 | Mpro |
A alanine, C cysteine, D aspartic acid, G glycine, I isoleucine, K Lysine, L leucine, M methionine, N asparagine, Q glutamine, S serine, T threonine, V valine
The complete genome sequence of strain Sczy3 was compared to 18 available complete IBV genomic sequences available from GenBank. Sequence comparison analysis demonstrated that the genomic nucleotide identity between Sczy3 and other IBV strains ranged from 86.1 % (H52 and Cal99) to 91.4 % (LX4). In addition, different sequence identities corresponding to different genes or regions between Sczy3 and other IBVs were observed. Specifically, the nucleotide identity between the 5′ UTR, gene 1, S1, S2, 3a, 3b, E, M, 5a, 5b, N, and the 3′ UTR between Sczy3 and other IBVs were 90.8–96.8, 86.4–91.3, 60.1–96.5, 75.3–95.5, 78.2–96.0, 74.1–93.0, 82.9–94.2, 89.7–96.0, 80.8–89.4, 90.4–98.4, 86.0–94.1, and 77.9–97.9 %, respectively. Gene 1 and 3a of Sczy3 had the highest nucleotide identity with LX4 strain at 91.3 and 96.0 %, respectively. The S1 and S2 Sczy3 subunits had the highest nucleotide identity with QXIBV strain at 96.5 and 95.5 %, respectively, and the 5′ UTR, 3b, E, M, 5a, 5b, N, and the 3′ UTR had the highest nucleotide identity with other IBVs (Table 3).
Table 3.
Nucleotide and amino acid identity of different regions between strain Sczy3 and other IBV strain genomes
| Strains | Complete genome | 5′-UTR | Gene1 | S1 | S2 | 3a | 3b | E | M | 5a | 5b | N | 3′-UTR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Beaudette | 86.4 | 94.1 | 86.9 | 77.8(77.5) | 85.7(88.0) | 82.2 | 78.8 | 87.8(87.2) | 92.9(93.4) | 87.4 | 94.4 | 86.8(90.0) | 86.7 |
| M41 | 86.6 | 94.6 | 86.9 | 77.9(76.9) | 86.0(88.2) | 82.2 | 79.4 | 87.8(88.1) | 94.2(94.7) | 80.8 | 93.6 | 87.1(91.2) | 96.5 |
| H52 | 86.1 | 94.8 | 86.6 | 78.2(77.1) | 85.7(88.3) | 81.6 | 79.4 | 87.5(88.1) | 93.8(94.2) | 86.9 | 94.4 | 86.5(91.0) | 81.7 |
| H120 | 86.2 | 94.8 | 86.6 | 77.8(77.5) | 85.8(88.6) | 82.2 | 79.4 | 86.5(83.5) | 93.8(94.2) | 86.4 | 98.4 | 87.4(91.7) | 82.5 |
| ZJ971 | 86.2 | 95.2 | 86.7 | 77.7(77.3) | 85.8(89.0) | 82.2 | 79.4 | 86.2(83.5) | 93.8(94.2) | 86.4 | 98.4 | 87.6(92.0) | 82.1 |
| Peafowl/GD/KQ6/2003 | 86.6 | 94.5 | 86.8 | 78.0(76.7) | 85.7(87.7) | 82.2 | 79.3 | 87.8(86.2) | 89.7(94.2) | 86.4 | 98.4 | 88.4(92.2) | 95.8 |
| ArkDPI 11 | 86.6 | 95.4 | 86.8 | 77.6(76.3) | 87.0(90.2) | 84.5 | 74.6 | 83.5(83.2) | 93.3(93.8) | 85.9 | 95.2 | 88.0(92.9) | 87.3 |
| ArkDPI101 | 86.5 | 95.4 | 86.7 | 77.8(76.3) | 86.9(89.9) | 84.5 | 74.6 | 83.5(83.2) | 93.3(93.8) | 85.9 | 94.8 | 87.9(92.7) | 87.1 |
| Cal99 | 86.1 | 95.2 | 86.4 | 77.1(77.5) | 85.9(88.2) | 81.0 | 74.1 | 88.1(84.4) | 93.4(93.8) | 85.9 | 94.4 | 87.9(92.0) | 86.1 |
| BJ | 90.2 | 96.8 | 90.2 | 85.1(83.4) | 86.9(89.6) | 85.1 | 91.9 | 88.7(88.1) | 92.5(92.9) | 89.4 | 92.7 | 93.6(94.4) | 97.3 |
| CK/CH/LSD/05I | 86.9 | 94.5 | 86.7 | 80.9(81.1) | 90.4(93.0) | 83.0 | 74.6 | 89.3(86.2) | 96.0(96.5) | 85.4 | 95.6 | 88.0(92.9) | 91.0 |
| TW2575/98 | 86.2 | 94.5 | 86.5 | 78.0(79.8) | 87.7(91.4) | 82.2 | 78.3 | 89.3(91.7) | 95.1(95.6) | 82.8 | 93.6 | 86.7(89.5) | 91.6 |
| Partridge/GD/S14/2003 | 90.5 | NA | 90.4 | 85.5(84.7) | 94.2(95.8) | 85.6 | 93.0 | 94.2(95.4) | 94.2(94.7) | 85.4 | 92.0 | 94.1(96.1) | 97.3 |
| LX4 | 91.4 | NA | 91.3 | 96.3(94.6) | 92.6(95.7) | 96.0 | 91.5 | 92.7(94.5) | 95.6(96.0) | 88.9 | 90.4 | 93.0(95.9) | 77.9 |
| SAIBK | 88.9 | 90.8 | 89.3 | 81.5(80.3) | 93.5(95.8) | 90.2 | 79.8 | 87.2(88.1) | 94.7(95.1) | 84.8 | 92.8 | 87.4(91.7) | 97.9 |
| A2 | 90.2 | 96.6 | 90.2 | 80.8(79.9) | 92.5(93.8) | 83.9 | 89.4 | 93.9(93.6) | 94.2(94.7) | 85.4 | 93.2 | 93.4(94.4) | 87.6 |
| SC021202 | 89.4 | 92.3 | 89.9 | 81.0(79.3) | 93.5(95.8) | 90.2 | 76.5 | 87.2(89.0) | 95.6(96.0) | 85.9 | 92.0 | 86.0(91.2) | 97.9 |
| ITA/90254/2005 | 89.1 | 96.5 | 88.1 | 95.3(95.0) | 94.1(96.2) | 82.8 | 92.1 | 90.5(89.9) | 92.9(93.4) | 86.9 | 93.6 | 88.0(91.7) | 88.0 |
| Conn | NA | NA | NA | 77.6(76.4) | NA | 81.0 | 74.1 | 83.8(84.1) | 92.0(92.5) | NA | NA | 87.8(92.7) | NA |
| Jilin | NA | NA | NA | 77.4(76.2) | 86.9(89.9) | 84.5 | 74.6 | 83.5(83.2) | 93.3(93.8) | 85.9 | 94.8 | 88.0(92.9) | NA |
| HK | NA | NA | NA | 77.8(77.3) | 86.0(89.3) | 83.9 | 74.6 | 83.5(83.2) | 92.9(93.3) | 85.9 | 94.8 | 87.7(92.4) | NA |
| W93 | NA | NA | NA | 77.6(77.2) | 85.8(88.8) | 82.2 | 78.8 | 86.2(84.3) | 94.0(94.0) | 86.4 | 98.4 | 87.5(91.7) | NA |
| Ma5 | NA | NA | NA | 77.6(77.6) | NA | NA | NA | NA | 94.2(94.7) | NA | NA | 92.2(94.6) | NA |
| DE072 | NA | NA | NA | 60.1(51.1) | 75.3(74.7) | 81.0 | 75.7 | 86.2(84.4) | 93.8(94.2) | 84.8 | 94.8 | 87.5(91.2) | NA |
| Ark99 | NA | NA | NA | 77.4(77.0) | 85.9(89.6) | 81.0 | 75.7 | 82.9(81.3) | 94.2(94.6) | NA | NA | 87.8(92.0) | NA |
| Vic | NA | NA | NA | 77.8(75.8) | 84.9(87.7) | 80.5 | 74.1 | 84.4(83.2) | 90.6(91.1) | 84.3 | 91.6 | 86.3(91.2) | NA |
| QXIBV | NA | NA | NA | 96.5(95.3) | 95.5(96.8) | NA | NA | NA | 91.6(92.0) | NA | 91.6 | 92.4(93.7) | NA |
| Gray | NA | NA | NA | 75.3(73.8) | 86.9(90.4) | NA | NA | 84.4(85.0) | 94.2(94.6) | 84.3 | 94.8 | 87.2(92.4) | NA |
| CU-T2 | NA | NA | NA | 77.5(76.2) | 84.7(87.7) | 78.2 | 74.6 | 83.5(84.4) | 84.1(85.0) | 84.3 | 94.8 | 87.2(90.5) | NA |
| Md27 | NA | NA | NA | 76.1(76.4) | 86.5(89.9) | 79.3 | 75.1 | 84.7(84.1) | 92.9(93.3) | 86.4 | 94.8 | 87.9(92.0) | NA |
| LDT3 | NA | NA | NA | 85.1(83.8) | 94.2(96.0) | 85.6 | 93.0 | 94.5(95.4) | 94.2(94.7) | 85.4 | 91.6 | 94.1(96.1) | NA |
| LKQ3 | NA | NA | NA | 77.8(77.5) | 85.6(88.2) | 81.0 | 79.4 | 87.8(89.0) | 93.8(94.2) | 81.3 | 93.6 | 87.0(91.0) | NA |
| S | NA | NA | NA | 78.0(75.8) | 84.9(87.5) | 80.5 | 74.1 | 84.4(83.2) | 91.1(91.5) | 83.8 | 91.2 | 86.4(91.2) | NA |
| TW1171/92 | NA | NA | NA | 78.6(79.2) | 88.9(92.8) | 83.3 | 80.4 | 87.8(85.3) | 94.7(95.1) | 82.3 | 95.6 | 86.3(88.5) | NA |
Sequences with the highest or lowest identity with Sczy3 at the nt and aa level indicated in bold
() indicates amino acid sequence identity, NA Not analyzed
Analysis of the amino acid sequence identity between different nsps encoded by the Sczy3 gene 1 and other IBVs demonstrated that the Sczy3 nsp4, nsp9, nsp12, nsp13, and nsp14 had the highest identity with corresponding proteins encoded by strain LX4 at 94.2–99.2 %, while the remaining nsps had the highest identity with the same proteins expressed by other IBVs (data not shown).
The putative cleavage site sequence of the Sczy3 spike protein was His–Arg–Arg–Arg–Arg that was similar to the sequence observed for LX4 type IBVs from China and one IBV strain ITA/90254/2005 from South Africa. Gene variation was observed throughout the S1 subunit of analyzed IBVs although the highest variation was observed in the HVRs of the S1 subunit (data not shown).
For the transcription-regulating sequence (TRS) located at the beginning of each gene, that is, the core sequence (CS)CT(T/G)AACAA was conserved for most of the TRS present in the Sczy3 genome. However, the CS of gene 5 of strains SAIBK, SC021202, and S14 had an additional three nucleotide insertion, that is, AAA, and the CS of gene 5 of strains Sczy3, BJ, A2, and LX4 had an additional six nucleotide insertion, that is, (A/G)AGAAA compared to homologous sequences in other IBV strains. The distances between CS of Sczy3 and the initiation codons of gene 1–6 were 461 nt (65–525), 52 nt (20,313–20,364), 23 nt (23,839–23,861), 77 nt (24,431–24,507), 7 nt (25,542–25,548), 93 nt (25,840–25,933), respectively (data not shown).
Phylogenetic analysis of Sczy3 and other IBVs
Phylogenetic trees were generated based on the complete Sczy3 genome compared to the genomes of reference IBV strains. This analysis identified six genotypes defined as LX4, Ark99, TW, A2, and Massachusetts type (Mass-type) based on S1 subunit analysis. The entire Sczy3 genome, E, S1, N, PLpro, Mpro, and RDRP are located in the same cluster as LX4, while the Sczy3 S2 and M clustered differently from LX4 and Mass-type IBVs (Fig. 2). The Sczy3 strain had a well-defined evolutionary distance to the Mass-type IBVs. Interestingly, the Sczy3 and Mass-type vaccine strain (i.e., H120 and W93) 5b gene are located in the same cluster.
Fig. 2.
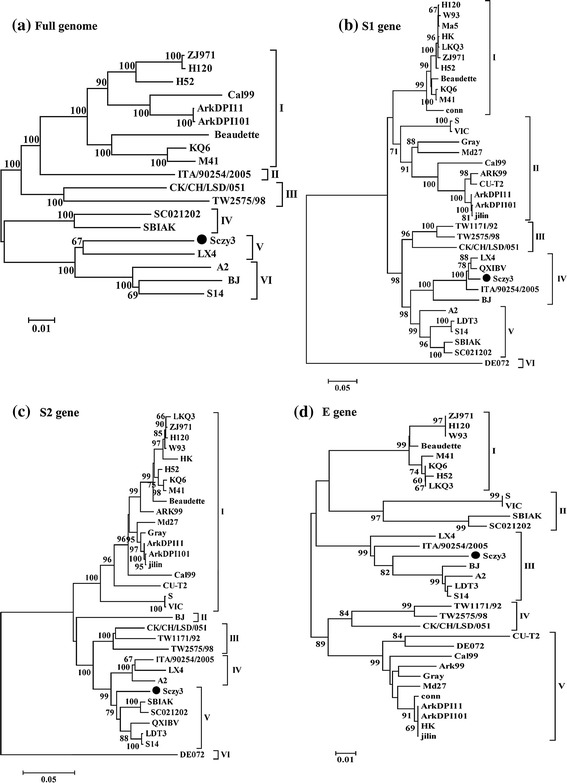
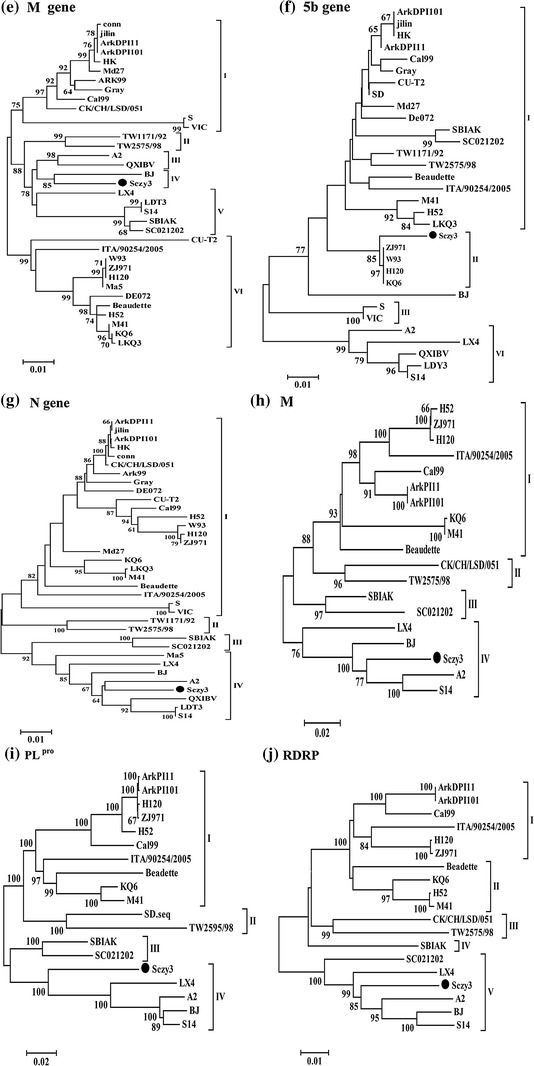
Phylogenetic relationship by neighbor-joining method (bootstrapping for 1,000 replicates with values >60 %) based on nucleotide sequences of complete genomes (a), 1–27,695 nt; S1 subunit (b), 20,865–21,894 nt; S2 subunit (c), 21,985–23,862 nt; E gene (d), 24,210–24,536 nt; M gene (e), 24,508–25,185 nt; 5b gene (f), 25,743–25,991 nt; N gene (g), 25,934–27,163 nt; M pro gene (h), 8,860–9,780 nt; PLpro region (i), 2,548–7,317 nt; and RDRP region (j), 12,310–15,128 nt. The Sczy3 sequence is indicated by a filled circle
Recombination analysis
Analysis of recombination events determined that Sczy3 was a chimeric strain with the major parental sequence derived from LX4 and the minor parental sequence derived from H120 (P < 2.558 × 10−7) with the recombination region located between 25,892 and 26,192 nt containing an 83 nt long sequence of the 3′ terminal ORF 5a, a 222 nt long sequence of the 5′ terminal ORF 5b, and a 132 nt long sequence of the 5′ terminal nucleocapsid protein gene. The aligned full genome sequence of 27,963 nt was used as the reference to identify the recombinant position breakpoint, and this recombination region corresponded to the 25,664–25,964 nt of the Sczy3 genome (Fig. 3).
Fig. 3.
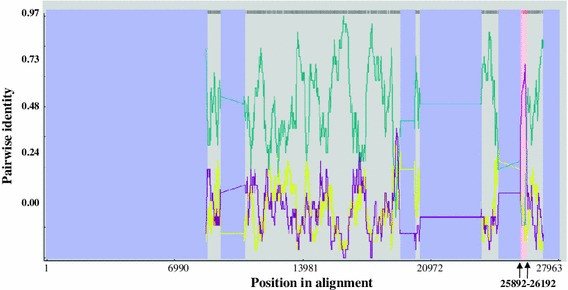
RDP screenshot displaying the possible recombination events associated with Sczy3. Each line displays the pairwise sequence identity between two of three IBVs relevant to recombination. Pairwise identity refers to the average pairwise sequence identity within a 30 nt sliding window moved 1 nt at a time along the alignment of the three sequences. The pink regions demarcate potential recombination regions. Crossover sites are indicated by arrows with nt positions above. The purple line indicates the percentage identity between the minor parent (H120) and recombinant (Sczy3); the yellow line indicates the percentage identity between the minor parent (H120) and major parent (LX4); the green line indicates the percentage identity between the major parent (LX4) and the recombinant (Sczy3). Potential recombination regions are identified as the region where the percentage identity between the minor parent (H120) and the recombinant (Sczy3) is higher than that between the major parent (LX4) and the recombinant (Sczy3) (Color figure online)
To further confirm the recombination event, comparative analysis of the genomic LX4, H120, and Sczy3 sequences were carried out, and the Sczy3 sequence was used as a putative chimeric strain (similarity scanning analysis was performed using Simplot 3.5.1) resulting in the identification of the recombination signal and the breakpoint positions at 25,892 and 26,192 nt. The region between these two breakpoints was the putative recombinant region which was also consistent with the RDP results (Fig. 4). The phylogenetic tree of complete genomic sequences and those of the detected recombinant region were also conducted and analyzed demonstrating that the recombinant region had a topological alternation which further confirmed the recombination event (Fig. 5).
Fig. 4.
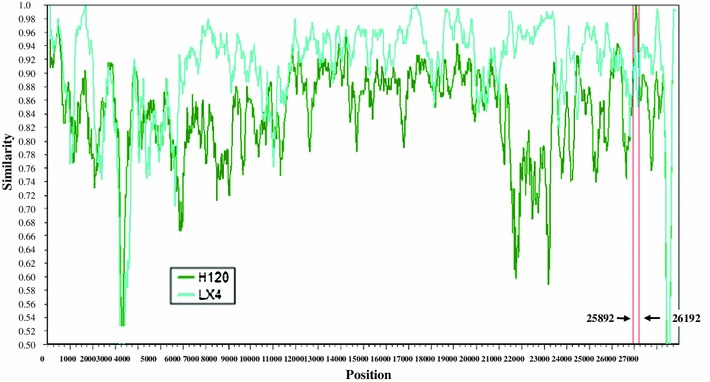
Simplot analyses of strain Sczy3. Similarity refers to sequence identity (green line) between the minor putative parent H120 and the putative recombinant Sczy3 and that (blue line) between the major putative parent (LX4) and putative recombinant Sczy3. The nucleotide identity was calculated using the Kimura 2-parameter method with a transition–transversion ratio of 2 in each window of 200 bp. The window was successively extended with a 20 bp increment. LX4 and H120 were used as putative parental strains, while Sczy3 was regarded as the putative recombinant strain. The breakpoints where the minor and major parental strains have equal sequence identity with Sczy3 were the predicted recombination sites. Putative recombinant region was the region between the recombination sites (red line) (Color figure online)
Fig. 5.

Phylogenic analysis of topological alternations in the Sczy3 recombinant sequence. (a) Phylogenetic tree of the complete genome sequence. (b) Phylogenetic tree of recombinant fragments (25,892–26,192 nt). Topological changes of recombinant fragments were observed. Phylogenetic trees were drawn by Maximum likelihood method in RDP3.44. Red highlight represents the recombinant sequence, blue highlight represents the minor parental sequence, and green highlight represents the major parental sequence (Color figure online)
Discussion
Infectious bronchitis is currently one of the most important contagious diseases significantly impacting poultry production. Because the genomes of respective IBV strains are continuously evolving as a result of frequently occurring point mutations and genomic recombination events, it is necessary that genomic sequence analysis of prevalent IBV strains be available to establish evolutionary patterns that can be used in the prevention and control of IB.
Genomic comparison of the recently identified Sczy3 isolate to genomes of other IBV strains demonstrated that Sczy3 was most similar to LX4-type IBVs and least similar to Mass-type IBVs, including the widely used Mass-type vaccine strains (e.g., H52, H120, Ma5, and W93). Nucleotide variability was observed across the entire genome; however, different regions varied more or less between strain Sczy3 and other IBVs studied. For example, the Sczy3 gene 1 and 3a had the highest nt identity with LX4 strain at 91.3 and 96.0 %, respectively, and the Sczy3 S1 and S2 subunits had the highest nt identity with QXIBV strain at 96.5 and 95.5 %, respectively, while the Sczy3 5b gene had the highest nt identity with that of Mass-type IBVs such as H120, ZJ971, KQ6, and W93 at 98.4 % (Table 3). Different amino acid sequence identities between different nsps between Sczy3 and other IBVs were also observed. Phylogenic analysis showed that the entire Sczy3 genome (and most Sczy3 genes) located in the same cluster as LX4, but several genes or regions such as the S2 subunit, M gene, and the PLpro region located in a cluster different from that of LX4 and Mass-type IBVs demonstrating that evolution of different IBV genes was not synchronous.
The putative spike protein cleavage site sequence His–Arg–Arg–Arg–Arg had been regarded as unique to IBVs from China [20] even though this sequence was also found in IBV strain ITA/90254/2005 isolated in Africa, suggesting that this sequence was not unique to IBV isolates from China. However, phylogenetic analysis showed that the IBV isolates that clustered with Sczy3 were mainly Chinese isolates. We previously demonstrated that Sczy3 was a QX-like isolate that is prevalent in China and has been identified in other countries in recent years. These results further indicated that IBV isolates that are prevalent in China were significantly evolutionarily distant from Mass-type strains. However, IBV vaccine strains commercially used in China today are primarily derived from Mass-type strains such as H120, H52, Ma5, and W93, which may not be able to provide efficient protection against field strains. Thus, it will be necessary to rapidly develop new vaccines against the QX-like field strains.
Several factors can affect IBV evolution, such as high error rates associated with RNA replication, recombination between strains (resulting from widespread use of live vaccines), immunologic pressure, and frequent mixed infections [21–23]. This study demonstrated that recombination events occurred between non-vaccine and the vaccine strains. In China, the H120 strain is a widely used live vaccine; however, since the early 1980s, IBV has been diagnosed in China and in 1999, the LX4 was identified as a cause of IB [13]. Based on the widespread use of live vaccines and isolation of field IBVs, we hypothesize that Sczy3 represents a chimeric strain derived from vaccine strain H120 and field strain LX4 that evolved independently by point mutations and recombination events.
Recombination results in the development of new IBV strains [24, 25], and the unique discontinuous transcription mechanism can often cause template switching in IBVs [16, 26]. Many recombination regions have been detected for IBV isolates, including E, RdRp, PLpro [5], the 3′ S1 terminus, the 3a gene [27], TRS [28], the 5′ M terminus [29], the 3′ S2 terminus, N regions, and the 3′ UTR region [11, 30]. Presently, ‘hot spots’ are generally believed to be located adjacent to putative crossover recombination sites, including C(T/G)TAACAA [28, 30], CTTTTG [31], CTTTT(C/T) [29], and other A-T-rich regions.
However, our study showed that recombination occurred in a region containing the 3′ 5a terminus (83 nt), the 5′ 5b terminus (222 nt), and the 5′ N terminus (132 nt) sequences. Recombination event breakpoint analysis showed that sequences C(T/G)TAACAA, CTTTTG, and CTTTTG(C/T) were not found near recombination regions. Therefore, it was difficult to determine whether these motifs were associated with recombination. Further analyses will be needed to determine the roles of these motifs in recombination events; however, previous studies have suggested that recombination in IBV occurred randomly rather than at specific sites [32].
In summary, the complete genome and individual Sczy3 strain gene sequences had different levels of sequence identity with sequences from other IBVs. Sczy3 was most similar to LX-type (QX-type) IBVs. Most Sczy3 genes clustered with LX4-type (QX-type) IBVs, and following phylogenic and recombination analysis, it was shown that Sczy3 was a chimeric strain. Work presented in this report demonstrated that gene mutations and intergenic recombination may play an important role in the evolution of IBVs.
Acknowledgments
This work was financially supported by the Program for Changjiang Scholars and Innovative Research Team in Universities “PCSIRT” (Grant No: IRTO848). The authors thank Medjaden Bioscience Limited for assisting in the preparation of this manuscript.
References
- 1.D. Cavanagh, J. Gelb Jr., in Disease of Poultry, ed. by A.M. Faldy, Y.M. Saif, J.R. Glisson, L.R. McDougald, L.K. Nolan, D.E. Swayne (Blackwell, Ames, 2008), pp. 117–135
- 2.Cavanagh D. Arch. Virol. 1997;142(3):629–633. [PubMed] [Google Scholar]
- 3.Boursnell ME, Brown TD, Foulds IJ, Green PF, Tomley FM, Binns MM. J. Gen. Virol. 1987;68(Pt 1):57–77. doi: 10.1099/0022-1317-68-1-57. [DOI] [PubMed] [Google Scholar]
- 4.Liu XL, Su JL, Zhao JX, Zhang GZ. Virus Genes. 2009;38(1):56–65. doi: 10.1007/s11262-008-0282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Wang HN, Wang T, Fan WQ, Zhang AY, Wei K, Tian GB, Yang X. Virus Genes. 2010;41(3):377–388. doi: 10.1007/s11262-010-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sawicki SG, Sawicki DL, Siddell SG. J. Virol. 2007;81(1):20–29. doi: 10.1128/JVI.01358-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casais R, Davies M, Cavanagh D, Britton P. J. Virol. 2005;79(13):8065–8078. doi: 10.1128/JVI.79.13.8065-8078.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hodgson T, Britton P, Cavanagh D. J. Virol. 2006;80(1):296–305. doi: 10.1128/JVI.80.1.296-305.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fang S, Chen B, Tay FP, Ng BS, Liu DX. Virology. 2007;358(1):136–147. doi: 10.1016/j.virol.2006.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cavanagh D, Davis PJ, Cook JK. Avian Pathol. 1992;21(3):401–408. doi: 10.1080/03079459208418858. [DOI] [PubMed] [Google Scholar]
- 11.Kottier SA, Cavanagh D, Britton P. Virology. 1995;213(2):569–580. doi: 10.1006/viro.1995.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gelb J, Jr, Weisman Y, Ladman BS, Meir R. Avian Pathol. 2005;34(3):194–203. doi: 10.1080/03079450500096539. [DOI] [PubMed] [Google Scholar]
- 13.Liu S, Kong X. Avian Pathol. 2004;33(3):321–327. doi: 10.1080/0307945042000220697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou NL, Zhao FF, Wang YP, Liu P, Cao SJ, Wen XT, Huang Y. Virus Genes. 2010;41(2):202–209. doi: 10.1007/s11262-010-0500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tamura K, Dudley J, Nei M, Kumar S. Mol. Biol. Evol. 2007;24(8):1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 16.Lai MM. Curr. Top. Microbiol. Immunol. 1992;176:21–32. doi: 10.1007/978-3-642-77011-1_2. [DOI] [PubMed] [Google Scholar]
- 17.Posada D. Mol. Biol. Evol. 2002;19(5):708–717. doi: 10.1093/oxfordjournals.molbev.a004129. [DOI] [PubMed] [Google Scholar]
- 18.Martin D, Rybicki E. Bioinformatics. 2000;16(6):562–563. doi: 10.1093/bioinformatics/16.6.562. [DOI] [PubMed] [Google Scholar]
- 19.Padidam M, Sawyer S, Fauquet CM. Virology. 1999;265(2):218–225. doi: 10.1006/viro.1999.0056. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Xue C, Chen F, Qin J, Xie Q, Bi Y, Cao Y. Vet. Microbiol. 2010;143(2–4):145–154. doi: 10.1016/j.vetmic.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cavanagh D, Picault JP, Gough R, Hess M, Mawditt K, Britton P. Avian Pathol. 2005;34:20–25. doi: 10.1080/03079450400025414. [DOI] [PubMed] [Google Scholar]
- 22.Lai MM, Cavanagh D. Adv. Virus Res. 1997;48:1–100. doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Yu X, Collisson EW. Virus Res. 1997;49:139–145. doi: 10.1016/S0168-1702(97)01466-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen HW, Huang YP, Wang CH. Virus Res. 2009;140(1–2):121–129. doi: 10.1016/j.virusres.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Denison MR, Graham RL, Donaldson EF, Eckerle LD, Baric RS. RNA Biol. 2011;8(2):270–279. doi: 10.4161/rna.8.2.15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kusters JG, Jager EJ, Niesters HG, van der Zeijst BA. Vaccine. 1990;8(6):605–608. doi: 10.1016/0264-410X(90)90018-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ammayappan A, Upadhyay C, Gelb J, Jr, Vakharia VN. Virol. J. 2008;5:157. doi: 10.1186/1743-422X-5-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee CW, Jackwood MW. Arch. Virol. 2000;145(10):2135–2148. doi: 10.1007/s007050070044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mondal SP, Cardona CJ. Virus Genes. 2007;34(3):327–341. doi: 10.1007/s11262-006-0014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia W, Karaca K, Parrish CR, Naqi SA. Arch. Virol. 1995;140(2):259–271. doi: 10.1007/BF01309861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, Junker D, Collisson EW. Virology. 1993;192(2):710–716. doi: 10.1006/viro.1993.1093. [DOI] [PubMed] [Google Scholar]
- 32.Brooks JE, Rainer AC, Parr RL, Woolcock P, Hoerr F, Collisson EW. Virus Res. 2004;100(2):191–198. doi: 10.1016/j.virusres.2003.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]